
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe structural diversity of heterocycle-fused potassium cyclopentadienides†
Ilya E. Nifant'ev *ab,
Alexander A. Vinogradova,
Mikhail E. Minyaeva,
Pavel D. Komarova,
Konstantin A. Lyssenkobc,
Kirill P. Birind,
Viktor P. Dyadchenkob and
Pavel V. Ivchenkoab
*ab,
Alexander A. Vinogradova,
Mikhail E. Minyaeva,
Pavel D. Komarova,
Konstantin A. Lyssenkobc,
Kirill P. Birind,
Viktor P. Dyadchenkob and
Pavel V. Ivchenkoab
aA. V. Topchiev Institute of Petrochemical Synthesis, Russian Academy of Sciences, Leninsky Pr. 29, Moscow 119991, Russian Federation. E-mail: ilnif@yahoo.com
bChemistry Department, M. V. Lomonosov Moscow State University, Leninskie Gory, 1-3, Moscow 119991, Russian Federation
cPlekhanov Russian University of Economics, Stremyanny Per. 36, Moscow 117997, Russian Federation
dA. N. Frumkin Institute of Physical Chemistry and Electrochemistry, Russian Academy of Sciences, Leninsky Pr. 31, Moscow 119071, Russian Federation
First published on 17th September 2019
Abstract
Cyclopentadienides of d- and f-elements are highly important complexes with undoubted potential for practical applications. Annelation of a heterocyclic fragment with an η5-ring results in substantial improvement of the catalytic properties of these compounds, called “heterocenes”; the investigation of metal coordination with these specific ligands is a highly important problem. We prepared potassium derivatives 5–8 of heterocycle-annelated cyclopentadienes with different structures – derivatives of cyclopenta[1,2-b:4,3-b′]dithiophene (1), indeno[2,1-b]indole (2), indeno[1,2-b]indole (3), and indeno[1,2-b]indolizine (4) and studied the crystal and molecular structures of these salts by X-ray diffraction. We found that heterocycle-fused cyclopentadienides demonstrate remarkable diversity in metal–ligand coordination modes and crystal packing, with formation of two-dimensional polymeric (5), linear polymeric (6), tetrameric (7) and monomeric (8) structures. The NMR spectral data and results of DFT modeling indicate an increase in electron density in the cyclopentadienyl fragment, and this effect was found to be larger in the derivative of the new indolizine ligand precursor 4. The results of our study will be used in the design of next-generation catalysts of α-olefin polymerization.
Introduction
Sandwich and half-sandwich complexes of transition metals and f-elements are actual catalysts of industrially important processes, such as olefin1–3 and diene4 polymerization, olefin epoxidation,5,6 hydrosilylation,7 etc. The remarkable variety of cyclopentadiene-type ligands obtained to date is due to the catalytic application of the corresponding complexes: substituted cyclopentadiene, indene and fluorene are η5-bound ligands that successfully provide the “basic” steric environment of the catalytic center and affect the catalytic activity, regioselectivity and stereoselectivity.8 Annelation of a five-membered carbon ring with a heterocyclic fragment is a promising direction in the design of the heteroanalogs of indene and fluorene (Scheme 1) which represent prospective η5-ligands. Such ligands have been successfully applied in the design of an effective epoxidation catalyst;9 sandwich and half-sandwich complexes of group 4 metals (so-called “heterocenes”) represent next-generation metallocene catalysts for α-olefin polymerization.10–15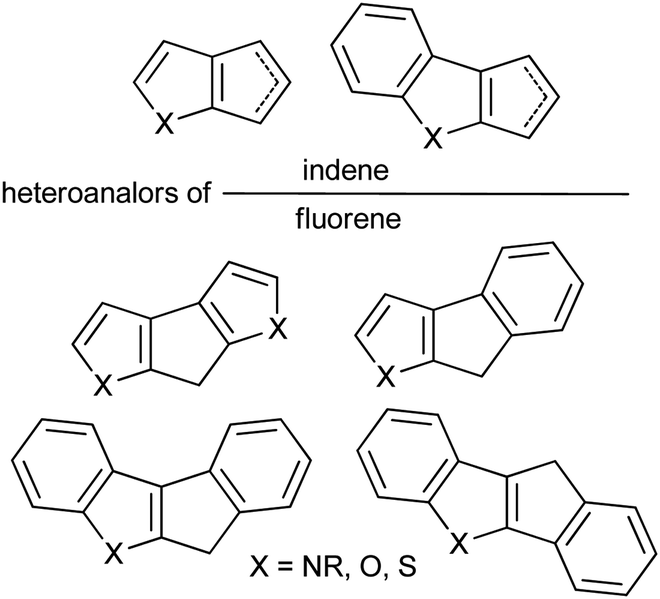 | ||
| Scheme 1 Donor heterocycle – fused cyclopentadienes. | ||
Recently, we demonstrated the high efficiency of cyclopenta[b]thiophene Zr complexes in oligomerization of C6–C10 α-olefins,16 and the promising catalytic properties of Zr ansa-complexes based on cyclopenta[1,2-b:4,3-b′]dithiophene, indeno[1,2-b]indole and indeno[2,1-b]indole (Scheme 2, top) in polymerization of 1-octene.17 In our experiments, heterocenes outperformed traditional cyclopentadienyl, indenyl and fluorenyl sandwich complexes. This behavior indicates that heterocene ligands much more effectively stabilize the Zr cationic reaction center relative to traditional cyclopentadienides due to their electron-donor nature,18 which entails increasing in catalytic activity and feasibility of using heterocenes at elevated temperatures. The structural diversity of heterocycle-fused cyclopentadienes permits a more variety in stereocontrol of α-olefin insertion: rac-forms of ansa-zirconocenes based on heteroanalogs of indene allows to obtain isotactic polypropylene19 or polypropylene elastomers;20,21 Zr complexes based on heteroanalogs of fluorene were successfully used in the synthesis of polypropylene elastomers,11,14,15 isotactic poly(1-butene),12 high MW atactic polyolefins17 and other prospective materials.
 | ||
| Scheme 2 Heterocenes – promising precatalysts for α-olefin polymerization (top); racemo-selective transmetallation of dilithium salt (bottom).25 | ||
Heterocenes have been extensively studied in α-olefin polymerization since the early 2000s. The molecular structure of these compounds is poorly studied; to date, only a few dichlorozirconium ansa-complexes have been characterized by X-ray diffraction analysis.10,17,19–24 Obviously, these data do not reflect the entire spectrum of possible metal–ligand interactions in catalytic species due to the predominantly covalent nature of bonding in neutral LZrCl2 complexes.
We believe that a detailed study of the molecular structure of alkali metal derivatives of heterocycle-fused cyclopentadienes is an effective tool to find such interactions. These studies also have an important practical aspect, since the structure of alkali metal cyclopentadienides is crucially important for the transmetallation that is used in the synthesis of metallocenes; recently, we detected high racemo-selectivity in the synthesis of zirconium complex (Scheme 2, bottom), and this selectivity could be attributed to effects of crystal packing in the corresponding dilithium salt.25
On the other hand, heterocycle-fused cyclopentadienides may be considered as a “bridge” between traditional derivatives of alkyl- or aryl-substituted cyclopentadiene, indene, and fluorene26 – and side-chain functionalized cyclopentadienides27 – thus complementing and completing our knowledge of the nature of alkali metal π-ligand coordination.
In this paper, we report the synthesis and crystallographic study of four heterocene-fused potassium cyclopentadienides.
Results and discussion
The synthesis of heterocycle-fused cyclopentadienes
2,5-Dimethyl-7H-cyclopenta[1,2-b:4,3-b′]dithiophene 1,14,28 5-methyl-5,6-dihydroindeno[2,1-b]indole 2 (ref. 29) and 5-methyl-5,10-dihydroindeno[1,2-b]indole 3 (ref. 30) were synthesized according to previously reported procedures (see also17 and Section S1 in the ESI†). 11-Phenyl-6H-indeno[1,2-b]indolizine 4 is a new compound that was prepared by Scheme 3 (see Section S1 in the ESI† for NMR spectra). The synthesis of 4 was based on the quaternization of 2-benzylpyridine with 2-bromo-2,3-dihydro-1H-inden-1-one31 followed by cyclization in basic aqueous media.32 Note that 4 is the first example of a cyclopentadienyl-type ligand precursor with indolizine fragment. | ||
| Scheme 3 Synthesis of 11-phenyl-6H-indeno[1,2-b]indolizine (4). | ||
Preparation and NMR study of potassium salts
Potassium salts were obtained from corresponding heterocycle-fused cyclopentadienes 1–4 by metallation with benzyl potassium in THF medium. This efficient method of metallation of substituted cyclopentadienes, which we recently proposed,33–35 significantly increased the availability of potassium derivatives that are synthetic precursors of cyclopentadienyl complexes. Crystalline salts 5–8 were obtained by slow (∼1 week) diffusion of n-hexane into solutions of metallation products in THF (5) or in THF/diglyme mixture (for 6–8, see Scheme 4). The crystals obtained were suitable for X-ray diffraction analysis.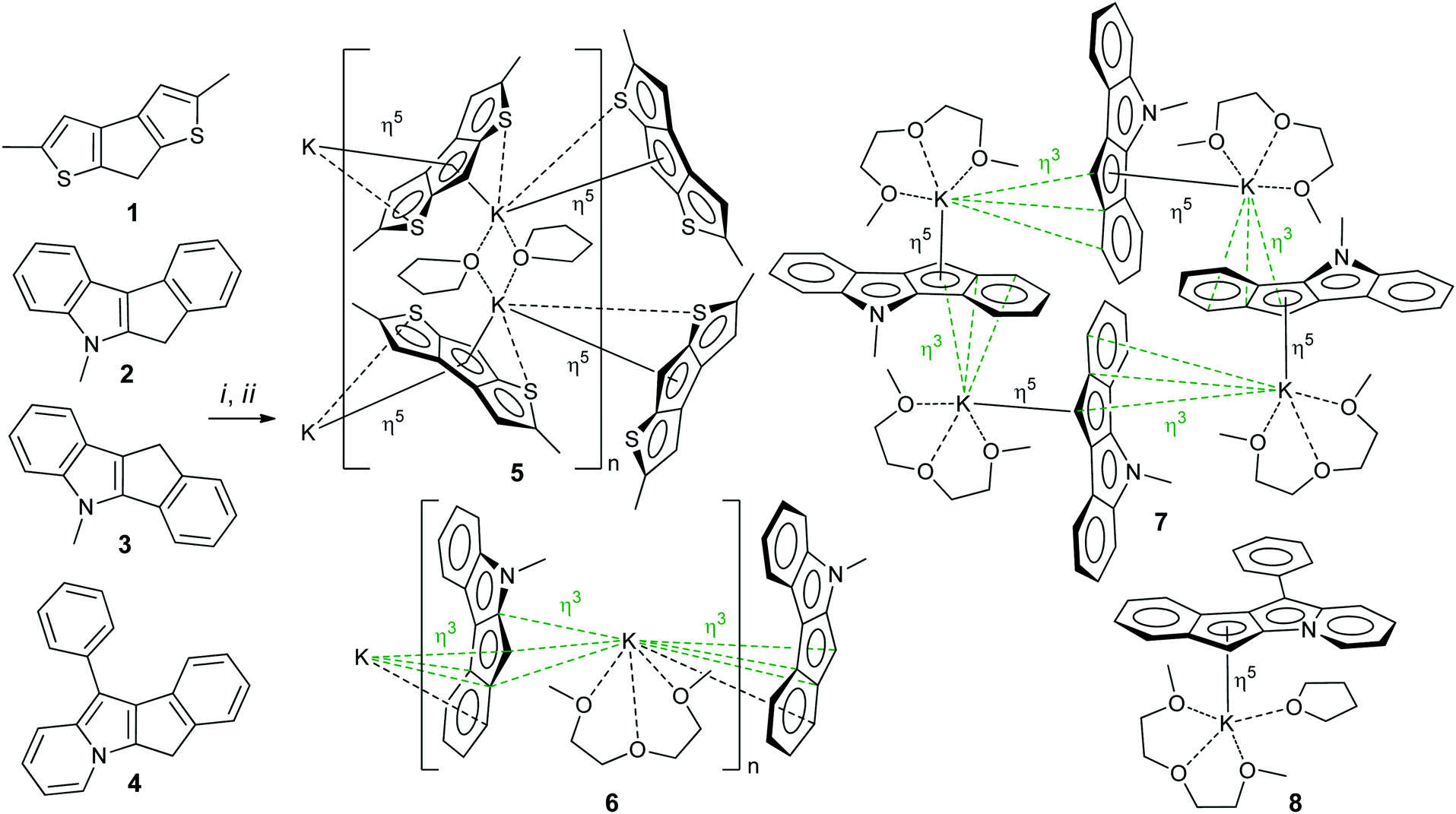 | ||
| Scheme 4 Preparation of 5–8. (i) BnK/THF; (ii) hexane (5) or hexane/diglyme (6–8). | ||
Using correlation NMR techniques, we accomplished complete signal assignment in 1H and 13C NMR spectra of 5–8 (see Section S1 in the ESI†). We conclude that the values of 13C NMR chemical shifts related to C(H) carbon atom correlate to electron density in the C5 fragment of the cyclopentadienide anion. In comparison with such values in cyclopentadienides and fluorenides, the chemical shifts for heterocycle-fused cyclopentadienides 5–8 are substantially lower (Table 1). The degree of shielding is minimal for 5 and maximal for indolizine derivative 8, which correlates with results of DFT calculations.
| Cyclopentadienide | 1H NMR, HCp | 13C NMR, C(H)Cp | Reference |
|---|---|---|---|
| a This work.b C9H7 = indenide.c C13H9 = fluorenide. | |||
| 5 | 5.64 | 82.4 | a |
| 6 | 5.51 | 69.3 | a |
| 7 | 5.92 | 75.9 | a |
| 8 | 5.88 | 67.6 | a |
| (C5H5)Na | 5.60 | 103.3 | 36 |
| (C5H5)K | 5.57 | 102.1 | 37 |
| (tBuC5H4)K | 5.36, 5.42 | 104.1, 103.8 | 37 |
| (tBu3C5H2)K | 5.56 | 101.5 | 37 |
| (C9H7)Lib | 6.51, 5.89 | 116.6, 92.7 | 38 |
| (C13H9)Kc | 6.93 | 84.1 | 39 |
| (C13H9)K(diglyme)c | 6.04 | 83.0 | 40 |
Crystallographic study of potassium salts
Potassium derivatives of heterocycle-fused cyclopentadienes demonstrated a remarkable versatility in metal coordination and crystal packing (Scheme 4). | ||
| Fig. 1 The structure of [K2(THF)2(Me2C9H3S2)2]∞ (5). Displacement ellipsoids are drawn at the 50% probability level. All H atoms are omitted, and THF disorder is not shown for clarity. | ||
 | ||
| Scheme 5 The C–C, C–S and C–N bond distances in fluorenide anion ((C13H9)K(diglyme)40) and in anions of 5–8. | ||
| a Symmetry codes: (i) −x + 1, y − 1/2, −z + 1; (ii) x + 1, y, z. | |||
|---|---|---|---|
| K1–O1 | 2.7543 (17) | K2–O1 | 2.7998 (17) |
| K1–O2 | 2.8042 (18) | K2–O2 | 2.9013 (18) |
| K1–S2 | 3.7814 (8) | K2–S4 | 3.6343 (8) |
| K1–C1 | 3.173 (2) | K2–C12 | 3.434 (2) |
| K1–C2 | 2.937 (2) | K2–C13 | 3.332 (2) |
| K1–C3 | 2.989 (2) | K2–C14 | 2.976 (2) |
| K1–C4 | 3.300 (2) | K2–C15 | 2.907 (2) |
| K1–C5 | 3.425 (2) | K2–C16 | 3.214 (2) |
| K1–C12i | 3.097 (2) | K2–C1ii | 3.018 (2) |
| K1–C13i | 3.222 (2) | K2–C2ii | 3.001 (2) |
| K1–C14i | 3.162 (2) | K2–C3ii | 3.142 (2) |
| K1–C15i | 3.084 (2) | K2–C4ii | 3.290 (2) |
| K1–C16i | 3.043 (2) | K2–C5ii | 3.197 (2) |
| K2–C19 | 3.353 (2) | ||
| K1-centroid C1⋯C5 | 2.8526 (13) | K2-centroid C1ii⋯C5ii | 2.8882 (11) |
| K1-centroid C12i⋯C16i | 2.8790 (12) | K2-centroid C12⋯C16 | 2.9405 (11) |
| K2-centroid C12⋯C16, C19, S4 | 2.8308 (9) | ||
Interactions of K+ cations with the ligand via its middle ring are symmetrical in cases of K1–C12i⋯C16i and K2–C1ii⋯C5ii interactions [symmetry codes: (i) −x + 1, y − 1/2, −z + 1; (ii) x + 1, y, z], which is confirmed by very similar values of K+-centroid(C5) distances and normals from K+ to the ligand planes (Table 2). In the case of less symmetrical K1–C1⋯C5 interaction, the difference between the mentioned distances (∼0.078 Å) is explained by the fact that the intersection point of the normal and the C5-plane is shifted from the C5-centroid towards atom C2, which is likely due to the presence of a relatively long K1–S2 contact (Fig. 1 and S27 in the ESI†). Moreover, the interaction K2–C12⋯C16 is far from symmetrical, since cation K2 is also coordinated by C19 and S4 atoms. The normal from K2 to the C12⋯C16 plane intersects the plane close to the middle of the C14–C15 bond and the position of the centroid defined by atoms C13⋯C16, C19 and S4.
Therefore, the anionic ligand displays two similar coordination modes: μ-η5:η6 (C1⋯C11, S1, S2) and μ-η5:η7 (C12⋯C22, S3, S4). The [K2(μ-THF)2]2+ moiety exhibits bonds with four [2,5-Me2C9H3S2]− anions, thus forming a 2D coordination polymer (Fig. 2, S29 and S30 in the ESI†).
 | ||
| Fig. 2 A 2D coordination polymer sheet of 5, formed parallel to the ab plane. Displacement ellipsoids are set to the 50% probability level, H atoms are omitted. | ||
All K+ cations lie nearly in one plane in the 2D sheet with the K–K–K angles ranging from 99.74 (1)° to 139.35 (1)° (Table S2 in the ESI†). A simplified scheme of Coulomb interactions between dications [K2(THF)2]2+ and anions [Me2C9H3S2]− within the 2D layer is presented in Fig. S31 in the ESI.†
| a Symmetry code: (i) x, −y + 1/2, z + 1/2. | |||
|---|---|---|---|
| K1–C2 | 3.1916 (12) | K1–C8i | 3.2182 (12) |
| K1–C6 | 3.0119 (13) | K1–C12i | 3.5107 (14) |
| K1–C7 | 3.3135 (13) | K1–O19 | 2.7784 (10) |
| K1–C6i | 3.2869 (13) | K1–O22 | 2.7611 (9) |
| K1–C7i | 3.0399 (12) | K1–O25 | 2.8821 (10) |
| K1-centroid C2, C6, C7 | 3.0115 (8) | K1-normal N1, C1⋯C17 | 2.9962 (6) |
| K1-centroid C6i, C7i, C8i | 3.0071 (7) | K1-normal N1i, C1i⋯C17i | 2.9841 (5) |
 | ||
| Fig. 3 The structure of [K(diglyme)(C16H12N)]∞ (6) (top) and 1D coordination polymer chains of 6, formed along the c direction (bottom). Displacement ellipsoids are set to the 50% probability level, H atoms are omitted. | ||
The mentioned interionic interactions between the [K(diglyme)]+ and [μ-η3:η3-C16H12N]− moieties provide formation of a 1D coordination polymer structure [K(diglyme)(C16H12N)]∞, a part of which is shown in Fig. 3, with the K1i–K1–K1ii angle of 127.65 (1)° [symmetry code: (ii) x, −y + 1/2, z − 1/2]. The formed polymer chains are aligned along the c axis (Fig. 3).
The isomeric red crystalline complex [K(diglyme)(C16H12N)] (7) based on 5-methyl-5,10-dihydroindeno[1,2-b]indole has a similar asymmetric unit that includes one cation [K(diglyme-κO,O′,O′′)]+ and one anion [μ-η3:η5-C16H12N]− (Fig. S36 in the ESI†). The C–C and C–N bond distances in the anion are given in Scheme 5 and in Table S4 (ESI†). These values are expected, but the C7–C8 bond (1.4711(19) Å) that is common to C5 and C6 rings is elongated as in complex 6. Atom K1 is η5-coordinated by one ligand (atoms C4–C8) and η3-coordinated by the other ligand (atoms C6i, C7i and C12i) as shown in Fig. 4 [symmetry code: (i) y, −x + 3/2, −z + 1/2]. The normal from K1 to the plane defined by atoms N1, C2–C17 is 2.9430(5) Å, which is close to the K1-centroid(C4⋯C8) distance of 2.9243(7) Å (Table 4).
 | ||
| Fig. 4 The [K(diglyme)(C16H12N)]4 unit of 7. Displacement ellipsoids are drawn at the 50% probability level, H atoms are omitted for clarity. | ||
The [K(diglyme)(C16H12N)] moiety is located near a 4-fold rotoinversion axis, which results in formation of the coordination tetramer [K(diglyme)(C16H12N)]4 (Fig. 5 and S37 in the ESI†) displaying the rare S4 Schoenflies point group in a crystal structure. All four K+ cations are located nearly but not quite in the same plane, with the K1i–K1–K1ii angle of 89.65° [symmetry code: (ii) −y + 3/2, x, −z + 1/2]. The tetramer forms layers parallel to the ab plane (Fig. S38 in the ESI†). Packing of two such layers parallel to the ac plane is shown in Fig. S39 in the ESI.† A direct analogy can be traced between 7 and the previously described potassium fluorenide, which forms a trimeric solvate with diglyme (3-fold rotoinversion axis).40
 | ||
| Fig. 5 The structure of [K(diglyme)(THF)(C21H14N)] (8). Displacement ellipsoids are drawn at the 50% probability level. All H atoms are omitted for clarity, minor disorder components are not shown. | ||
Hence, a small change in the anion geometry (and consequently in its electronic structure), namely, altering the position of the CHCp (or NCH3) fragment, results in dramatic structural changes and formation of either the 1D coordination polymer 6 or the tetramer 7.
| K1–C4 | 3.1598 (19) | K1–O24 | 2.7209 (17) |
| K1–C5 | 3.2236 (19) | K1–O27 | 2.7374 (17) |
| K1–C6 | 3.156 (2) | K1–O30 | 2.7342 (18) |
| K1–C7 | 2.9824 (19) | K1–O32 | 2.686 (4) |
| K1–C8 | 2.9827 (19) |
The [K(diglyme)(THF)(C21H14N)] unit is sterically crowded enough not to form coordination polymers, yet the K+ coordination sphere is insufficiently saturated, and K+ has a low formal coordination number (7). Therefore, the anion [C21H14N]− (atoms N1, C16, C15, C5, C6) and the cation [K(diglyme)(THF)]+ of two different molecules, which are related by an inversion center, demonstrate weak electrostatic intermolecular interactions that lead to association into a dimeric unit (Fig. S41 in the ESI†). Packing plots of 8, demonstrating the molecular lattice, are given in Fig. S42 and S43 in the ESI.†
DFT calculations
Taking into account the results of NMR studies of compounds 5–8, which indicate significant shielding of the C(H) carbon atom in the C5 ring, we calculated the electronic structure of the corresponding anions at the PBE0/def-2-TZVP level of theory.41 Additionally, we estimated the influence of the potassium cation on the view and energy of orbitals by DFT optimization of model complex [K(Me2O)(diglyme)]+[C16H12N]− and found that such influence is negligible (see Fig. S44 in the ESI†).The plots for highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbitals (LUMO) are presented in Fig. 6. The atoms of C5-rings provide substantive input to the formation of HOMO for all heterocycle-fused cyclopentadienides, which affects the stabilization of metal cation in catalytic center. High symmetry in HOMO and LUMO in 5 explains the extremely high stability and catalytic productivity of cationic Zr complexes based on derivatives of cyclopenta[1,2-b:4,3-b′]dithiophene. In addition, the impact of HOMO on π-electron density at the C(H) carbon atom in 5 is minimal, which correlates with the maximal chemical shift for this atom in the 13C NMR spectrum of 5. Substantial contribution of electrons at the N atom in 7 to HOMO allows us to assume the possibility of coordination of electrophiles at this atom in reactions catalyzed by indeno[1,2-b]indolyl metal complexes; this significant aspect of catalytic behavior of these compounds is being explored in our laboratory.
 | ||
| Fig. 6 HOMO (left) and LUMO (right) plots for anions 5–8 (PBE0/def-2-TZVP). | ||
Experimental
General experimental remarks
Preparation of heterocycle-fused cyclopentadienes
Optimized synthetic protocols and NMR spectra of compounds 1–3 are given in Section S1 in the ESI.† Indolizine derivative 4 was prepared as described below.A solution of 2-benzylpyridine (16.9 g, 0.1 mol) in acetone (50 mL) was added to the solution of 2-bromo-2,3-dihydro-1H-inden-1-one (21.1 g, 0.1 mol) in acetone (100 mL) at 0 °C. The mixture was allowed to warm to room temperature and was stirred for 12 h. Acyl pyridinium bromide was filtered off, washed by hexane, dried, and transferred into a 500 mL flask containing K2CO3 (30 g, >0.2 mol) in water (200 mL). The mixture was stirred for 8 h at 80 °C and cooled. The product was filtered off, recrystallized from EtOH and dried in vacuo. The yield of 4 was 17.4 g (62%), beige crystalline powder. Calculated for C21H15N: C, 89.65; H, 5.37; N, 4.98. Found: C, 89.61; H, 5.44; N, 4.95. 1H NMR (293 K, 400 MHz, CDCl3) δ: 3.78 (bs, 2H, –CH2–), 6.56 (t, 1H, 3JHH = 6.9 Hz), 6.68 (m, 1H), 7.21 (t, 1H, 3JHH = 6.9 Hz), 7.29 (t, 1H, 3JHH = 7.5 Hz), 7.37 (t, 1H, CHAr, 3JHH = 7.4 Hz), 7.49 (d, 1H, CHAr, 3JHH = 7.4 Hz), 7.55 (t, 1H, CHAr, 3JHH = 7.9 Hz), 7.67 (d, 1H, CHAr, 3JHH = 8.1 Hz), 7.76 (m, 4H). 13C{1H} NMR (293 K, 101 MHz, CDCl3) δ: 29.38, 106.03, 108.31, 111.11, 116.50, 118.83, 120.61, 122.55, 124.69, 125.34, 125.81, 126.91, 128.44, 128.78, 129.12, 130.54, 131.18, 133.84, 135.73, 139.19, 144.26 (see Fig. S1 and S2 in the ESI†).
Synthesis of potassium salts
Crystallography
The experimental intensities of single crystal reflections were measured on a Bruker SMART APEX II platform at 120 K, using graphite monochromatized Mo-Kα radiation (λ = 0.71073 Å) in ω-scan mode. The collected data were integrated with the SAINT program.43 Absorption corrections based on measurements of equivalent reflections were carried out by SADABS.44 The structures were solved by direct methods with the SHELXS program45 and refined by full matrix least-squares on F2 with SHELXL.46 Crystal data, data collection and structure refinement details are summarized in Table S1 in the ESI.† The positions of all non-hydrogen atoms were found from electron difference density maps. Non-hydrogen atoms were refined with individual anisotropic displacement parameters. Positions of all hydrogen atoms (with the exception of disordered THF fragments in 5 and 8) were also found from the difference map, but hydrogen atoms were positioned geometrically and refined as riding atoms with relative isotropic displacement parameters. The SHELXTL program suite47 and the Mercury program48 were used for molecular graphics. Selected bond distances and angles, as well as more detailed data refinement and crystal structure description, are presented in Section S2 in the ESI.† CCDC numbers are 1920202–1920205 for compounds 5–8, respectively.†Quantum chemical calculations
All quantum chemistry computations were performed with the Gaussian 09 program (Revision D.01)49 using density functional theory (PBE0)41 and the def-2-TZVP basis set. Topological analyses of the ρ(r) function integration over interatomic zero-flux surfaces were performed using the AIMAll program.50 All expected critical points were found, and the whole set of critical points in each system satisfies the Poincaré–Hopf rule.Conclusions
In the present paper, we report the results of comparative study of crystal, molecular and electronic structures of four potassium cyclopentadienides that contain C5 rings annelated with thiophene (5), indole (6, 7) and indolizine (8) fragments. Depending on the type of ligand, the metal atoms exhibit a wide range of coordination, from traditional η5-Cp to allyl-like η3 with involvement of carbon atoms of the benzene rings. The variety of coordination modes resulted in an amazing diversity in crystal packing. Potassium derivatives of four different ligands formed four different types of crystal packing, from de facto molecular crystal for indolizine derivative 8 via tetrameric association (indeno[1,2-b]indolide salt 7) to one-dimensional (indeno[2,1-b]indolide complex 6) and two-dimensional (cyclopenta[1,2-b:4,3-b′]dithiophenide 5) coordination polymers. These results are important for understanding the transmetallation mechanisms in the synthesis of sandwich metal complexes.NMR spectra and results of DFT modeling allowed us to conclude that the presence of annelated electron-donor heterocyclic fragments increases the π-electron density in the Cp ring. This effect is enhanced for N-containing ligands and maximal for indolizine derivative 8. Thus, the novel compound 11-phenyl-6H-indeno[1,2-b]indolizine 4, the synthesis of which was reported in this manuscript, represents a prospective ligand for the design of metallocene catalysts with potentially high thermal stability and excellent productivity in α-olefin polymerization. The preparation and study of indeno[1,2-b]indolizine complexes is now considered a promising area for our future research.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Russian Science Foundation (Grant No. 18-13-00351). This work was carried out within the State Program of TIPS RAS in part of NMR study of 4–8.Notes and references
- M. Delferro and T. J. Marks, Chem. Rev., 2011, 111, 2450–2485 CrossRef CAS PubMed.
- R. Zhao, J. Ma, H. Zhang and J. Huang, Molecules, 2017, 22, 856 CrossRef PubMed.
- M. Stürzel, S. Mihan and R. Mülhaupt, Chem. Rev., 2016, 116, 1398–1433 CrossRef PubMed.
- L. Friebe, O. Nuyken and W. Obrecht, Adv. Polym. Sci., 2006, 204, 1–154 CrossRef CAS.
- S. A. Hauser, M. Cokoja and F. E. Kühn, Catal. Sci. Technol., 2013, 3, 552–561 RSC.
- I. I. E. Markovits, M. H. Anthofer, H. Kolding, M. Cokoja, A. Pöthig, A. Raba, W. A. Herrmann, R. Fehrmann and F. E. Kühn, Catal. Sci. Technol., 2014, 4, 3845–3849 RSC.
- M. Zaranek and P. Pawluc, ACS Catal., 2018, 8, 9865–9876 CrossRef CAS.
- Y. Nakayama and T. Shiono, Molecules, 2005, 10, 620–633 CrossRef CAS PubMed.
- M. E. Minyaev, A. A. Vinogradov, A. V. Churakov, B. Kimmich, I. E. Nifant'ev, S. M. Nagy and P. V. Ivchenko, Mendeleev Commun., 2018, 28, 508–510 CrossRef CAS.
- J. A. Ewen, M. J. Elder, R. L. Jones, A. L. Rheingold, L. M. Liable-Sands and R. D. Sommer, J. Am. Chem. Soc., 2001, 123, 4763–4773 CrossRef CAS PubMed.
- C. De Rosa, F. Auriemma, A. Di Capua, L. Resconi, S. Guidotti, I. Camurati, I. E. Nifant'ev and I. P. Laishevtsev, J. Am. Chem. Soc., 2004, 126, 17040–17049 CrossRef CAS PubMed.
- C. De Rosa, F. Auriemma and L. Resconi, Angew. Chem., Int. Ed., 2009, 48, 9871–9874 CrossRef CAS PubMed.
- S. H. Kim, J. H. Park, B. G. Song, S.-W. Yoon, M. J. Go, J. Lee and B. Y. Lee, Catalysts, 2013, 3, 104–124 CrossRef CAS.
- I. E. Nifant'ev, I. P. Laishevtsev, P. V. Ivchenko, I. A. Kashulin, S. Guidotti, F. Piemontesi, I. Camurati, L. Resconi, P. A. A. Klusener, J. J. H. Rijsemus, K. P. de Kloe and F. M. Korndorffer, Macromol. Chem. Phys., 2004, 205, 2275–2291 CrossRef.
- L. Resconi, S. Guidotti, I. Camurati, R. Frabetti, F. Focante, I. E. Nifant'ev and I. P. Laishevtsev, Macromol. Chem. Phys., 2005, 206, 1405–1438 CrossRef CAS.
- I. E. Nifant'ev, A. A. Vinogradov, A. A. Vinogradov, I. V. Sedov, V. G. Dorokhov, A. S. Lyadov and P. V. Ivchenko, Appl. Catal., A, 2018, 549, 40–50 CrossRef.
- I. E. Nifant'ev, A. A. Vinogradov, A. A. Vinogradov, A. V. Churakov, V. V. Bagrov, I. A. Kashulin, V. A. Roznyatovsky, Y. K. Grishin and P. V. Ivchenko, Appl. Catal., A, 2019, 571, 12–24 CrossRef.
- I. E. Nifant'ev, L. Y. Ustynyuk and D. N. Laikov, Organometallics, 2001, 20, 5375–5393 CrossRef.
- J. A. Ewen, R. L. Jones, M. J. Elder, A. L. Rheingold and L. M. Liable-Sands, J. Am. Chem. Soc., 1998, 120, 10786–10787 CrossRef CAS.
- F. van Baar, A. D. Horton, K. P. de Kloe, E. Kragtwijk, S. G. Mkoyan, I. E. Nifant'ev, P. A. Schut and I. V. Taidakov, Organometallics, 2003, 22, 2711–2722 CrossRef.
- S. Deisenhofer, T. Feifel, J. Kukral, M. Klinga, M. Leskelä and B. Rieger, Organometallics, 2003, 22, 3495–3501 CrossRef CAS.
- A. N. Ryabov, D. V. Gribkov, V. V. Izmer and A. Z. Voskoboynikov, Organometallics, 2002, 21, 2842–2855 CrossRef CAS.
- V. V. Izmer, A. Y. Lebedev, M. V. Nikulin, A. N. Ryabov, A. F. Asachenko, A. V. Lygin, D. A. Sorokin and A. Z. Voskoboynikov, Organometallics, 2006, 25, 1217–1229 CrossRef CAS.
- Z. M. Dzhabieva, S. V. Topilin, G. V. Shilov and T. S. Dzhabiev, Russ. J. Inorg. Chem., 2012, 57, 46–51 CrossRef CAS.
- I. E. Nifant'ev, A. A. Vinogradov, M. E. Minyaev, K. A. Lyssenko and P. V. Ivchenko, Mendeleev Commun., 2019, 29, 266–268 CrossRef.
- R. Michel, R. Herbst-Irmer and D. Stalke, Organometallics, 2011, 30, 4379–4386 CrossRef CAS.
- G. Erker, G. Kehr and R. Fröhlich, Organometallics, 2008, 27, 3–14 CrossRef CAS.
- I. E. Nifant'ev, I. A. Kashulin, P. V. Ivchenko, P. A. Klusener, F. M. Korndorffer, K. P. De Kloe and J. J. H. Rijsemus, U.S. Pat. 7635781 (B2), 2009.
- D. Brown, P. Graupner, M. Sainsbury and H. Shertzer, Tetrahedron, 1991, 47, 4383–4408 CrossRef CAS.
- I. E. Nifant'ev and V. V. Bagrov, U.S. Pat. 6451724 (B1), 2002.
- R.-B. Hu, S. Sun and Y. Su, Angew. Chem., Int. Ed., 2017, 56, 10877–10880 CrossRef CAS PubMed.
- V. S. Venturella, J. Pharm. Sci., 1963, 52, 868–871 CrossRef CAS PubMed.
- M. E. Minyaev, A. A. Vinogradov, D. M. Roitershtein, R. S. Borisov, I. V. Ananyev, A. V. Churakov and I. E. Nifant'ev, J. Organomet. Chem., 2016, 818, 128–136 CrossRef CAS.
- D. M. Roitershtein, L. N. Puntus, A. A. Vinogradov, K. A. Lyssenko, M. E. Minyaev, M. D. Dobrokhodov, I. V. Taidakov, E. A. Varaksina, A. V. Churakov and I. E. Nifant'ev, Inorg. Chem., 2018, 57, 10199–10213 CrossRef CAS PubMed.
- J. E. Ellis, M. E. Minyaev, I. E. Nifant'ev and A. V. Churakov, Acta Crystallogr., Sect. C: Struct. Chem., 2018, 74, 769–781 CrossRef CAS PubMed.
- T. K. Panda, M. T. Gamer and P. W. Roesky, Organometallics, 2003, 22, 877–878 CrossRef CAS.
- T. Kinoshita, N. Murata, R. Fujita, Y. Yanagi and K. Takeuchi, J. Organomet. Chem., 1994, 471, 19–21 CrossRef CAS.
- R. E. Dinnebier, S. Neander, U. Behrens and F. Olbrich, Organometallics, 1999, 18, 2915–2918 CrossRef CAS.
- A. Zaeni, U. Behrens, P. Liebing, F. Olbrich and F. T. Edelmann, J. Organomet. Chem., 2017, 830, 141–145 CrossRef CAS.
- S. Neander, J. Kornich and F. Olbrich, J. Organomet. Chem., 2002, 656, 89–96 CrossRef CAS.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- L. Lochmann and J. Trekoval, J. Organomet. Chem., 1987, 326, 1–7 CrossRef CAS.
- Bruker, APEX2, Bruker AXS Inc., Madison, Wisconsin, USA, 2008 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- D. J. F. M. Frisch, J. G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian and J. V. Ort, Gaussian 09, Revision D.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- T. Keith, AIMAll ver. 16.08.17 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of 1–3, NMR spectra of 4–8, details of X-ray diffraction study of 5–8, results of DFT calculations (table of charges and common .xyz file). CCDC 1920202–1920205. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra04587b |
| This journal is © The Royal Society of Chemistry 2019 |