DOI:
10.1039/C9RA04513A
(Paper)
RSC Adv., 2019,
9, 25318-25325
Novel vinyl-modified RGD conjugated silica nanoparticles based on photo click chemistry for in vivo prostate cancer targeted fluorescence imaging†
Received
16th June 2019
, Accepted 2nd August 2019
First published on 13th August 2019
Abstract
Molecular imaging is a powerful tool for non-invasive visualization of tumors that plays an important role in their diagnosis and treatment. The specificity of molecular imaging probes for cancer cells is important for accurate tumor visualization, with antibody and polypeptide nanoprobe conjugates having often been used as targeting agents for tumor detection. However, many traditional chemical conjugation methods employ complex conjugation reactions that result in poor efficiency and poor bioactivity. Herein, we describe the use of photo click methodology for the rapid synthesis of nanoprobes comprised of silica nanoparticles functionalized with RGD targeting units (SiO2@T1-RGDk NPs) (∼80 nm) for in vivo prostate cancer fluorescent imaging applications. These SiO2@T1-RGDk NPs exhibit a maximum absorption wavelength of 380 nm in their UV absorption spectra with a maximum fluorescence emission wavelength of 550 nm. Confocal immunofluorescent imaging reveal that SiO2@T1-RGDk NPs exhibit excellent targeting ability for visualizing cancer cells, with in vivo fluorescence imaging intensity in a subcutaneous tumor model of prostate cancer reaching a maxima after 4 h. Biosafety assessments showed that SiO2@T1-RGDk NPs demonstrate no obvious toxicity in mice, thus demonstrated that these novel NPs may prove to be promising fluorescent imaging agents for the accurate detection and treatment of tumors.
Introduction
Cancer is a leading cause of death worldwide, with the total number of cancer patients diagnosed globally continuing to grow annually.1 Molecular imaging technology is a powerful tool for the non-invasive visualization of tumors,2 which can radically affect their diagnosis and treatment. Various imaging techniques are used in clinical research, such as computed tomography (CT), positron emission tomography (PET), magnetic resonance imaging (MRI), single-photon emission computed tomography (SPECT), photoacoustic imaging (PAI) and fluorescence imaging. Amongst these approaches, fluorescence imaging is considered to be one of the most promising methods for early tumor diagnosis and treatment, because it is cost effective, highly sensitive, radiation free, enables rapid imaging and is easy to quantify.3–6 This approach requires the availability of high-quality molecular probes for the selective visualization and early diagnosis of tumors in vivo. Selective tumor targeting has often been achieved through conjugation of a molecular imaging nanoprobe to tumor directing antibodies or polypeptides. A range of traditional chemical conjugation methods have been used to prepare these type of imaging probes for in vivo biomedical application, including reductive amination reactions,7,8 chemical functionalization of specific groups on the surface of nanoparticles (NPs),9–11 and Cu(I)-catalyzed azide–alkyne cycloaddition reactions.12–14 However, these conjugation reactions often require strict temperature control, long-time reactions and employ complex functionalization protocols that can adversely affect the bioactivity of the targeting unit to compromise the probes' cancer cell targeting ability. Consequently, there is a growing demand for the development of efficient and rapid conjugation methods that do not adversely affect the bioactivity of antibody or polypeptide targeting units in fluorescent probes used for tumor diagnosis and treatment.
Photo click chemistry has been reported to provide a fast and efficient conjugation method for the conjugation of antibodies and polypeptides,15–18 with the click reaction being initiated via irradiation with light (e.g. no Cu(I) required). These click reactions involve irradiation of a tetrazole compound with ultraviolet light, which results in elimination of nitrogen gas to afford a nitrile imine dipole that can cyclize onto an alkene bond to afford a pyrazoline ring system.19–21 This cycloaddition methodology has the following characteristics: (1) the tetrazole compound only reacts with an olefin in the presence of light which enables it to be used for specific biomolecule labelling applications; (2) the photo click reaction generally exhibits good solvent compatibility, functional group tolerance, stereoselectivity and proceeds in high yield; (3) photochemical initiated decomposition of the tetrazole is rapid, with the rate of formation of the nitrile diamine exhibiting a rate constant of around 0.14 S−1, and the secondary cycloaddition reaction rate of the nitrile diamine with an alkene of around 11.0 M−1 S−1. (4) The pyrazoline cycloaddition product that is formed is fluorescent, so the progress of the cycloaddition reaction employed for its formation can be monitored using fluorescence analysis. Consequently, several research groups have used this type of photo click reaction to functionalize molecular probes for imaging applications. For example, Zhou et al. used this type of photo-click reaction to construct a probe for two-photon imaging of cell apoptosis pathways.22 Alternatively, Gai et al. employed photo-click chemistry to prepare a bifunctional chelating-based scaffold for PET/CT multi-modality imaging of U87MG tumor-bearing mice.23 However, conjugation of small molecules to polypeptides often results in probes that exhibit short lifecycle in vivo, making it impossible to use them for long-term tumor monitoring.
This study describes the use of photo click chemistry for the preparation of RGD conjugated dense silica nanoparticles (SiO2@T1-RGDk NPs) for in vivo molecular imaging of prostate cancer. In our previous study, RGD-modified biodegradable mesoporous silica fluorescent probes were synthesized through photo-click chemistry for in vivo breast cancer imaging.24 However, in this study, SiO2 was selected as a nanocarrier instead of mesoporous silica for its good dispersibility and stability, and smaller particle size (within 80 nm) compared with mesoporous silica. Moreover, as for the choice of intermediate tetrazole compound, [methyl 4-(2-(4-methoxyphenyl)-2H-tetrazol-5-yl) benzoate (T1)] was chose to provide a tetrazole group for photo-click reaction due to its simpler synthesis method, longer UV absorption wavelength, and higher absorbance. Based on previous reports, we have synthesized SiO2 NPs with large surface areas and uniform particle sizes,25–27 that are modified with RGD peptides that act as specific targeting ligands for tumor receptors.28–30 This has been achieved by first attaching a tetrazole compound T1 to the surface of amino-modified SiO2 NPs (SiO2–NH2 NPs). Subsequent photolysis of this T1 modified NP in the presence of a vinyl modified RGD (RGD-Ack) then results in a photo-click reaction to afford the desired fluorescent SiO2@T1-RGDk NPs. In vitro cytotoxicity tests have revealed that these monodisperse SiO2@T1-RGDk NPs are non-toxic and exhibit excellent targeted cellular uptake levels in tumor cells, where they were found to accumulate in the cytoplasm. These SiO2@T1-RGDk NPs have been shown to be useful for imaging and detecting tumors in vivo in an established subcutaneous model for prostate cancer in mice.
Experimental
Reagents
Tetraethyl orthosilicate (TEOS, 99%), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, crystalline), N-hydroxysuccinimide (NHS, 98%), 3-aminopropyltriethoxysilane (APS) and p-anisidine (≥99%) were purchased from Sigma Aldrich. Ammonia solution (≥25%), benzenesulfonyl hydrazide (98%) and sodium nitrite (NaNO2, 99%) were purchased from Aladdin. Methyl 4-formylbenzoate (99%) was purchased from J&K Scientific, whilst pyridine was purchased from Macklin. RGD-Ack was obtained from GL Biochem (Shanghai) Ltd. All reagents and solvents were used as received, without further purification.
Synthesis of SiO2 NPs
35.7 mL of absolute ethanol, 5 mL of water and 0.785 mL of ammonia solution were stirred for 10 minutes, 1 mL of TEOS was then added and the reaction stirred at room temperature for a further 1 h. The desired NPs were then collected by centrifugation and washed with deionized (DI) water and ethanol to remove unreacted TEOS, before then being dispersed in absolute ethanol.
Synthesis of T1
One equivalent of benzenesulfonyl hydrazide (1.72 g, 10.0 mmol) was added to a stirred solution of methyl 4-formylbenzoate (1.64 g, 10.0 mmol) in 100 mL of dry ethanol and the reaction stirred at room temperature overnight. The resultant white solid precipitate was filtered off to afford a Schiff base that was used directly in the next reaction without further purification. p-Methoxyaniline (615 mg, 5.0 mmol) was dissolved in a mixture of absolute ethanol and water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 10 mL), concentrated hydrochloric acid (1.0 mL) added, and the reaction mixture stirred at 0 °C for 10 min. An aqueous solution of NaNO2 (363 mg, 5.25 mmol) in 5 mL of water was then slowly added over a period of 1 hour at 0 °C. This solution was then added to a solution of the previously prepared Schiff base (1.59 g, 5.0 mmol) in 20 mL of pyridine. After the reaction was complete an equivalent volume of water was added which resulted in precipitation of a large amount of solid that was filtered off and washed with diethyl ether/ethyl acetate (1:1) to give a pale pink solid.
:1, 10 mL), concentrated hydrochloric acid (1.0 mL) added, and the reaction mixture stirred at 0 °C for 10 min. An aqueous solution of NaNO2 (363 mg, 5.25 mmol) in 5 mL of water was then slowly added over a period of 1 hour at 0 °C. This solution was then added to a solution of the previously prepared Schiff base (1.59 g, 5.0 mmol) in 20 mL of pyridine. After the reaction was complete an equivalent volume of water was added which resulted in precipitation of a large amount of solid that was filtered off and washed with diethyl ether/ethyl acetate (1:1) to give a pale pink solid.
Synthesis of SiO2@T1-RGDk NPs
10 mL of SiO2 NPs solution (5 mg mL−1), 10 mL of absolute ethanol and 1 mL of APS were mixed together and the resultant reaction mixture was stirred at 88 °C for 48 h. The resultant SiO2–NH2 NPs were obtained by centrifugation and washed with absolute ethanol, before being dissolved in DI water. 10.8 × 10−6 mol of T1 were dispersed in 2 mL of dimethyl sulphoxide (DMSO), followed by addition of 10.8 × 10−6 mol of EDC and 5.4 × 10−5 mol of NHS. This reaction mixture was stirred for 0.5 h, then 100 μL SiO2–NH2 NPs solution (20 mg mL−1) was added and the reaction mixture was stirred for further 3 h, with any unreacted reagents then being separated off by centrifugation, and the precipitate was washed 3 times with DMSO, then the T1 conjugated SiO2–NH2 NPs (SiO2@T1 NPs) were prepared. SiO2@T1 NPs in 1 mL of DI water and 1 μmol RGD-Ack in 1 mL of DI water were mixed and placed in a glass vessel that was irradiated at 254 nm for 2 h at 0 °C. The resultant SiO2@T1-RGDk NPs were then obtained by centrifugation and washed with DI water, before then being dissolved in PBS.
Characterization of NPs
The morphology and size of the SiO2 NPs were characterized using a JEOL JEM-2100 transmission electron microscope (TEM). The hydrodynamic size and surface zeta potential were measured using a Malvern Zetasizer Nano ZS. Their UV absorption spectra were measured using a UV-visible spectrophotometer. Fluorescent spectra were obtained by a fluorescence spectrometer (F-7000), with fluorescent intensities and regions of interest (ROIs) measured using an IVIS imaging system. The tetrazole compound T1 was characterized using 1H NMR spectroscopy and mass spectrometry (MS). Fourier transform infrared spectroscopy (FTIR) was obtained by infrared spectrometer (Thermo Fisher, Nicolet IS50FT-IR).
Cell lines and animal model
PC-3 cell lines were provided by Xi'an Medical University and maintained in complete DMEM solution supplemented with 10% (w/w) FBS and penicillin–streptomycin (76 and 36 U mL−1, respectively). Cells were incubated under 5% CO2 at 37 °C in a cell incubator (MCO-18AIC, Sanyo, Japan), prior to being used for in vitro tests or establishment of mouse model. Nude male mice (4 weeks old, about 18 g) were purchased from the Department of Experimental Animals, Health Science Center, Xi'an Jiaotong University. The mice were fed in a SPF animal room whose temperature was maintained at 20 °C, with all mice kept under these conditions for 1 week prior to use. PC-3 luciferase tumor-bearing nude mice model were obtained by inoculating the left and right sides of their crotches with 5 × 105 of the required cells in 20 μL of PBS. These mice were then used for in vivo probe distribution experiments after their tumors had grown to an appropriate volume. All animal tests were conducted using protocols approved by the University of Xi'an Jiaotong Animal Care and Use Committee (number XJTULAC 2016-412).
Stability of SiO2@T1-RGDk NPs
The stability of the SiO2@T1-RGDk NPs was determined by measuring the size distribution of NPs in PBS or 10% FBS at different temperatures over a period of 5 days, with all data presented as mean values with ± SD (n = 3).
Cellular uptake and affinity assay
The cellular uptake of the SiO2@T1-RGDk NPs was measured using a confocal microscope (TCS SP5 II, Leica, Germany). Cells were cultured for 48 h in confocal dishes containing SiO2@T1-RGDk NPs coated onto the glass surfaces of each well. A confocal microscope with laser excitation wavelengths of 410 nm was used for detection of SiO2@T1-RGDk NPs. The endocytosis of the SiO2@T1-RGDk NPs was detected by monitoring cellular uptake behavior using a confocal microscope. For confocal microscopy studies, PC-3 cells were incubated with SiO2@T1-RGDk NPs in 6-well plates for a predetermined time, after which time the cells were rinsed with PBS (pH 7.4) and fixed with 4% paraformaldehyde solution. The effect of RGD-Ack blocking on PC-3 cells was determined using cellular affinity analysis. PC-3 cells were cultured for 48 h in 24-well flat-bottomed plates, with the cells in control group were incubated with SiO2@T1-RGDk NPs for 24 h. The cells in blocking group were incubated with RGD-Ack for 24 h prior to addition of SiO2@T1-RGDk NPs, followed by a further incubation period of 24 h. The fluorescent intensities of PC-3 cells were measured by staining their nuclei using 4′,6-diamidino-2-phenylindole (DAPI), with laser excitation wavelengths of 340 nm was used for detection.
Cytotoxicity assay
An MTT method was used to detect the viability of PC-3 cells in the presence of SiO2@T1-RGDk NPs, with cells that had not been exposed to NPs being used as a control. Cells were seeded onto 96-well plates (with a density of 1 × 105 cells per well) and incubated under 5% CO2 atmosphere at 37 °C. Cells were washed with PBS after 24 h and the complete medium containing different concentrations of SiO2@T1-RGDk NPs was added to each well. After 72 h, their media was replaced with fresh culture media, before adding 20 μL of MTT solution (5 mg mL−1) to each well. These cells were then incubated for further 4 h under the same conditions, with the media replaced with 100 μL of DMSO. The absorbances were measured at 590 nm using an ELISA plate reader (Infinite® 200 Pro, Tecan, Switzerland). The cytotoxicity of SiO2@T1-RGDk NPs was determined using a maximum concentration of SiO2@T1-RGDk NPs of 81 μg mL−1. The in vitro cytotoxicity of SiO2@T1-RGDk NPs, T1, RGD-Ack and SiO2 NPs were measured using the MTT assay described above.
In vivo biotolerance of SiO2@T1-RGDk NPs
In vivo biotolerance levels of the SiO2@T1-RGDk NPs were investigated using an acute toxicity method employing twenty male and twenty female BALB/c mice that were divided randomly into two groups with the same sex ratio. These mice were purchased from the Department of Experimental Animals, Health Science Center, Xi'an Jiaotong University and fed under SPF conditions. After 5 days of adaptation, the treatment group were intravenously injected with a dosage of 25 mg kg−1 SiO2@T1-RGDk NPs (10 times more than in vivo fluorescence imaging dosage), with the control group injected with saline. Food and water were provided 2 h after injection and the mortality rate of the mice determined 14 days after injection. Heart, liver and kidney tissues were taken for histopathology analysis. All animal experiments were approved by the Laboratory Animal Administration Committee of Xi'an Medical University and performed according to the Guidelines for Animal Experimentation of Xi'an Medical University and the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85-23, revised 1996).
In vivo fluorescence imaging and image analysis
A subcutaneous transplanted prostate tumor model in nude mice was used for in vivo probe distribution experiments, with mice being divided into three groups: targeted, non-targeted and blocking groups. The tail veins of mice in the targeted and non-targeted groups were injected with 200 μL of SiO2@T1-RGDk NPs and SiO2@T1-AM NPs (AM: acrylamide), respectively. The blocking group were injected with RGD-Ack 0.5 h before subsequently being injected with 200 μL of SiO2@T1-RGDk NPs. In vivo fluorescence images were obtained at 0, 0.5, 1, 2, 4, 8 and 12 h after injection with NPs, using a 420 nm excitation light source and a 570 nm band-pass emission filter (exposure time of 1 s). Injection and image acquisition were carried out on mice that had been anesthetized using isoflurane, with their body temperatures being maintained using a thermostat-controlled thermal heater. Fluorescence images of mice were acquired, with their heart, liver, spleen, lungs, kidneys and tumors then collected and analyzed using an IVIS imaging system. Tissue distribution of SiO2@T1-RGDk NPs in mice was obtained and analyzed using image processing software, with ROIs extracted and visualized using Living Image 4.5 software. Tumor ROIs were determined using white light images, with mean values of fluorescence intensities for each ROI calculated after background values had been subtracted from each fluorescence image. This analysis enabled fluorescence intensity versus time curves of tumor ROIs from these fluorescent images to be determined accurately.
Histology
Tumors from the three groups of mice were frozen and cryo-sectioned for histological analysis, with frozen tissue slices of 10 μm thickness stained with DAPI and images acquired using a confocal laser scanning microscope (CLSM).
Statistical analysis
Data are given as mean values ± SD of independent repeat experiments, with a P-value < 0.05 considered to represent a statistically significant difference between comparative data.
Results and discussion
Synthesis and characterization of SiO2@T1-RGDk NPs
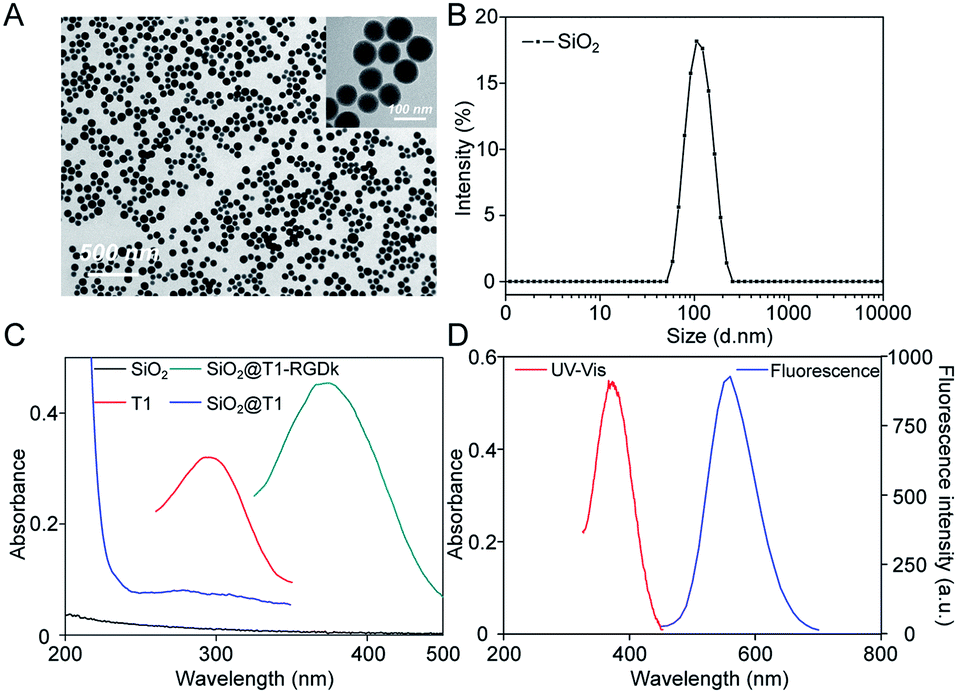
NPs have been widely used for molecular imaging applications,31–33 with silica nanoparticles having proven to be one of the most popular nanocarriers used for cancer therapy.27,34–36 In this study, SiO2@T1-RGDk NPs were prepared using the protocol described in Scheme 1A, and the reaction between amino-modified SiO2, tetrazole compound and RGD-Ack is shown in the Scheme S1,† with SiO2 NPs prepared using a previously reported method.37 As shown in Fig. 1A, TEM results reveal that the SiO2 NPs exhibited uniform particle sizes and good monodispersities, with an average diameter of ∼80 nm. DLS measurements verified that the SiO2 NPs had an average hydrodynamic diameter of 105.7 ± 9.6 nm (Fig. 1B), with particle sizes measured using TEM analysis found to be smaller because of loss of solvent. The SiO2 NPs were functionalized with APS, with the zeta potential of the resultant SiO2–NH2 NPs found to be +30.7 ± 0.57 mV (Fig. S1A†) indicating that their amino-groups had been successfully modified. The tetrazole compound T1 was synthesized in high purity according to a previous literature report (Fig. S2†),38 whose mass spectra and 1H NMR spectra are shown in Fig. S3 and S4,† respectively [1H NMR (600 MHz, d-DMSO) δ = 3.87 (s, 3H), 3.91 (s, 3H), 7.23 (d, J = 8.1 Hz, 2H), 8.09 (d, J = 8.1 Hz, 2H), 8.17 (d, J = 7.6 Hz, 2H), 8.30 (d, J = 7.6 Hz, 2H)]. Ultraviolet irradiation of the tetrazole fragment of T1 results in a reactive nitrile imine dipole that undergoes cycloaddition with the alkene bond of RGD-Ack to generated the fluorescent pyrazoline ring system of T1-RGDk (Scheme 1B). This functionalization method has the advantage of being simple to operate, mild reaction conditions and good efficiency when compared to traditional conjugation methods. T1 was conjugated onto the surface of SiO2–NH2 NPs using a condensation reaction, with any unreacted T1 then being separated by ultrafiltration and its concentration was determined. This analysis revealed that around 70% of the reactive groups of total T1 input had been successfully conjugated to the surface of the SiO2–NH2 NPs, with UV and fluorescent analysis confirming that T1 had been successfully attached to the surface of the NPs. In addition, the zeta potential of SiO2@T1 changed to 0.006 mV (Fig. S1B†), which also confirmed that T1 was conjugated on the SiO2 surface successfully. Furthermore, the optimum reaction time of cycloaddition reaction between T1 and RGD-Ack was confirmed by UV absorption and fluorescence spectra (Fig. S5†). Fig. 1C shows the UV absorption spectra of SiO2 NPs, T1, SiO2@T1 NPs and SiO2@T1-RGDk NPs, with maximum UV absorption wavelengths for SiO2@T1 NPs and SiO2@T1-RGDk NPs observed at 290 and 380 nm, respectively. The UV absorption and fluorescence spectra of T1-RGDk (Fig. 1D) revealed maximum UV absorption and fluorescence emission wavelengths at 380 and 550 nm respectively, consistent with the presence of their pyrazoline fragments. Furthermore, the successful conjugation of T1 and RGD-Ack was also proved by infrared spectroscopy. As can be seen from Fig. S6,† the carboxyl group (1720 cm−1) on T1 reacted with the amino group on the surface of SiO2–NH2 (3400 cm−1), and after the photo-click reaction with RGD-Ack, the vinyl group (1650 cm−1) of RGD-Ack disappeared. NP stability is an important factor when carrying out long-term imaging studies in vivo, and so the stability of these SiO2@T1-RGDk NPs was determined by monitoring their size distributions in different solvents at different temperatures over a period of 5 days (Fig. S7†). These results indicated that the particle sizes of SiO2@T1-RGDk NPs did not change significantly over 5 days, with solutions remaining clear with no evidence of any precipitation having occurred.
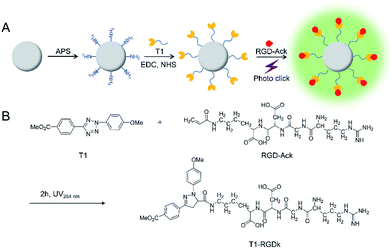 |
| | Scheme 1 Synthesis of SiO2@T1-RGDk NPs. (A) Synthesis schematic of SiO2@T1-RGDk NPs; (B) the photo click reaction between tetrazole compound T1 and RGD. | |
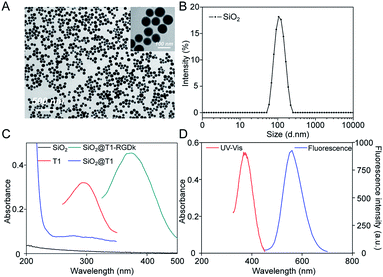 |
| | Fig. 1 Characterization of SiO2@T1-RGDk NPs. (A) TEM images of SiO2 NPs; (B) size distribution of SiO2 NPs determined through DLS; (C) UV absorption spectra of T1, SiO2 NPs, SiO2@T1 NPs and SiO2@T1-RGDk NPs; (D) UV absorption and fluorescence spectra of T1-RGDk. | |
Cellular uptake and affinity assay
The in vitro cellular uptake ability of the model cell line PC-3 towards SiO2@T1-RGDk NPs was first investigated (Fig. 2A). Confocal microscopy results revealed that the SiO2@T1-RGDk NPs were distributed in the cytoplasm (red), as well as being gathered around the nucleus (blue). These results indicated that SiO2@T1-RGDk NPs have good stability in vitro and could be used for in vivo imaging potentially. The affinity assay was used to explore the tumor targeting ability of SiO2@T1-RGDk NPs (Fig. 2B), which showed that the fluorescent intensity of tumor cells exposed to SiO2@T1-RGDk NPs in the presence of RGD-Ack blocker was significantly reduced. This is consistent with RGD-Ack binding strongly to RGD receptors on the surface of cancer cells to prevent competing binding and internalization of the SiO2@T1-RGDk NPs. Quantitative analysis of cell fluorescent intensities (Fig. S8†) confirmed enhanced uptake of SiO2@T1-RGDk NPs into the cancer cells, thus demonstrating their potential use for selective in vivo tumor targeted imaging applications.
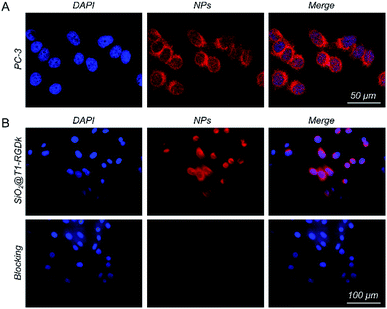 |
| | Fig. 2 (A) Confocal immunofluorescent images of SiO2@T1-RGDk NPs showing their localization in PC-3 cells. (B) Confocal immunofluorescent images of SiO2@T1-RGDk NPs showing their localization in PC-3 cells in the presence of RGD-Ack blocking agent. Cells stained with DAPI are colored blue. Cells containing SiO2@T1-RGDk NPs are colored red. | |
Toxicity assays of SiO2@T1-RGDk NPs in vitro and in vivo
The cytotoxicity of SiO2@T1-RGDk NPs, SiO2 NPs, T1, RGD-Ack were evaluated using an MTT assay. Fig. 3 reveals that the SiO2@T1-RGDk NPs exhibited no significant cytotoxicity within a certain concentration range that required for imaging applications (0.001 to 81 μg mL−1). The tail veins of healthy BALB/c mice were injected with SiO2@T1-RGDk NPs (25 mg kg−1) and their major organs (heart, liver and kidneys) harvested for histopathological analysis after 2 weeks (Fig. 4). Survival rates after 14 d were 95%, with no noticeable tissue or cellular damage being observed in the major organs of mice, when compared to control groups. These results suggest that SiO2@T1-RGDk NPs demonstrate no obvious toxicity in mice and may potentially be used as a safe fluorescent probe for tumor imaging applications.
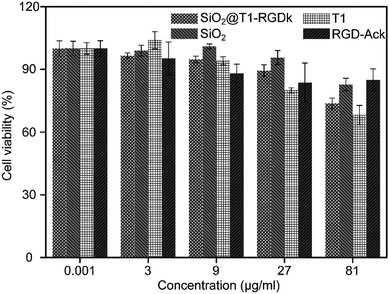 |
| | Fig. 3 Cell viability of PC-3 cells incubated with different concentrations of SiO2@T1-RGDk NPs, SiO2 NPs, T1 and RGD-Ack. | |
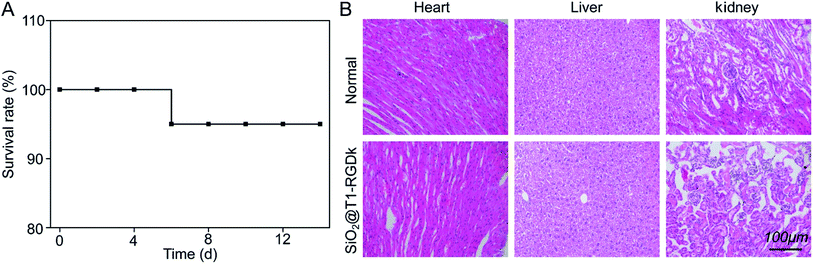 |
| | Fig. 4 Acute toxicity results of SiO2@T1-RGDk NPs in BALB/c mice; (A) survival rates of SiO2@T1-RGDk NPs in BALB/c mice; (B) H&E staining of major organs (including heart, liver and kidneys) from mice 14 days after injected with SiO2@T1-RGDk NPs. Healthy mice treated with saline were used as control. | |
In vivo fluorescence imaging and biodistribution studies
In vivo long-term monitoring of tumors is very important for accurate cancer diagnosis and treatment, so it was decided to determine whether SiO2@T1-RGDk NPs could be used for the targeted imaging of tumors in vivo. A prostate cancer subcutaneous tumor model was chosen for screening, with fluorescent images being obtained using IVIS imaging. Fig. 5 showed the in vivo fluorescence images of three groups of experimental mice within 12 hours. The excitation and emission wavelengths are 420 nm and 570 nm in the process of in vivo fluorescence imaging, which inevitably lead to the strong autofluorescence of the skin in this wavelength range, further resulting in weak fluorescence intensity of the tumor sites. In order to accurately show the changes in the fluorescence intensity of the tumor sites, the ROIs of the tumors are extracted. Subcutaneous tumors in the left and right groins of mice were used for analysis (black circle and green circle), which enabled the fluorescence intensities of ROIs to be determined accurately, with tumor ROIs being identified using white light images. The curves of fluorescence intensities of these tumors over a 12 h period were shown in Fig. 5B, with black square lines and red circle lines describing the fluorescence intensity variations of the left and right tumors for the three groups, respectively. Significant fluorescent intensity enhancement in tumors were observed after probe injection into targeted groups, with the fluorescence intensity of the targeted group reaching a maximal level 4 h after injection. The fluorescence intensity of the non-targeted group displayed a small increase when compared to the targeted group, which is due to passive accumulation of SiO2@T1-RGDk NPs caused by the EPR effect of cells. For the blocking group, in vivo fluorescence imaging was performed by injecting RGD-Ack in advance and SiO2@T1-RGDk was injected half an hour later. RGD-Ack is able to occupy the antigenic epitopes on the surface of tumor cells, rendering the SiO2@T1-RGDk NPs unable to bind to the tumor surface antigens, thereby exhibiting a weaker fluorescent signal. Therefore, the results indicated that the SiO2@T1-RGDk NPs exhibit excellent targeting ability for tumor cells in vivo. These in vivo imaging results were verified by sacrificing the mice of all three groups which enabled their major organs (heart, liver, spleen, lungs, and kidneys) to be removed for fluorescent analysis (Fig. S9A†). Fig. S9B† reveals a strong fluorescent intensity for tumors that were exposed to targeted group, with their intensities significantly greater than for non-targeted group, and in the presence of the RGD-Ack blocking group. The ex vivo results further demonstrated the great targeting ability of the SiO2@T1-RGDk NPs for tumor cells.
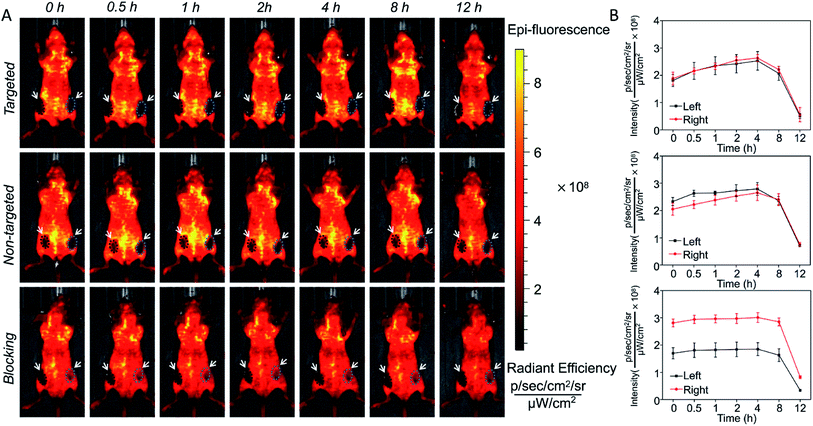 |
| | Fig. 5 (A) In vivo fluorescent imaging of SiO2@T1-RGDk NPs after 12 h. (B) Changes in fluorescent intensity of SiO2@T1-RGDk NPs that occur during in vivo imaging studies over time. | |
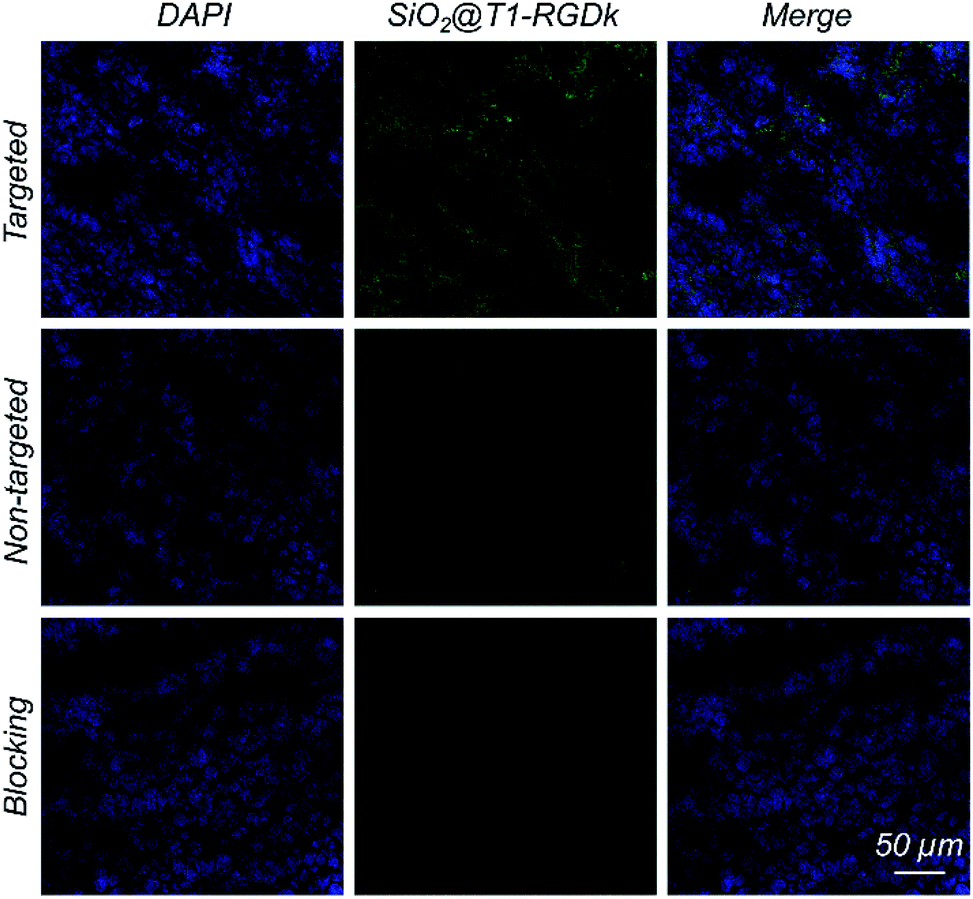
Histology
Histological studies were conducted to certify that the RGD-Ack fragment of SiO2@T1-RGDk NPs were responsible for tumor targeting. Fluorescent images of tumors were obtained using CLSM (Fig. 6), which revealed that SiO2@T1-RGDk NPs were mainly distributed throughout the cytoplasm of tumor cells as indicated by good superposition of the blue and green fluorescence signals, which correspond to the presence of DAPI and SiO2@T1-RGDk NPs, respectively.
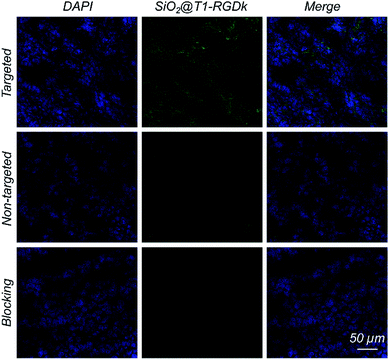 |
| | Fig. 6 Fluorescent images of frozen tumor cross-sections, shown by good superposition of blue and green fluorescence signals that correspond to DAPI and dSiO2@T1-RGDk NPs, respectively. | |
Conclusions
In summary, photo click methodology has been used to prepare stable SiO2@T1-RGDk NPs as a fluorescent tumor imaging agent that exhibits low toxicity and good tumor targeting ability. Fluorescence imaging studies in mice reveal that these SiO2@T1-RGDk NPs exhibit excellent cancer cell targeting ability which results in their accumulation at tumor sites. Their NP character was found to protect their RGD-Ack fragment from degradation in vivo, which resulted in lengthy circulation times. These results suggest that these SiO2@T1-RGDk NPs are highly promising fluorescence imaging agents for carrying out targeted tumor imaging studies for precise diagnosis and treatment. The excitation and emission wavelengths of these SiO2@T1-RGDk NPs are 410 and 550 nm, respectively, which means that the background in vivo fluorescence imaging is relatively high, which can result in relatively poor contrast levels between tumors and the epidermis. Therefore, we are currently exploring the use of longer-wavelength nanoprobes for the fluorescent imaging of tumors that afford higher levels of resolution, the properties and performance of which will be reported in due course.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported, in part, by the National Key R&D Program of China under Grant No. 2018YFC0910602, the National Natural Science Foundation of China under Grant No. 81627807, 11727813, 81571725, 91859109, 81871397, 81701853, 81660505, the Fok Ying-Tong Education Foundation of China under Grant 161104, the Program for the Young Top-notch Talent of Shaanxi Province, the Research Fund for Young Star of Science and Technology in Shaanxi Province under Grant No. 2018KJXX-018, the Natural Science Basic Research Plan in Shaanxi Province of China under Grant No. 2018JM7072, 2019JQ-519, 2019JQ-045, and the Fundamental Research Funds for the Central Universities (JB181203, JB191201).
References
- L. A. Torre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortet-Tieulent and A. Jemal, Ca–Cancer J. Clin., 2015, 65, 87–108 CrossRef PubMed.
- R. Weissleder and M. J. Pittet, Nature, 2008, 452, 580–589 CrossRef CAS PubMed.
- H. Li, K. Li, Y. Dai, X. Xu, X. Cao, Q. Zeng, H. He, L. Pang, J. Liang, X. Chen and Y. Zhan, Nanomed. Nanotechnol. Biol. Med., 2018, 14, 1867–1877 CrossRef CAS PubMed.
- E. A. Owens, M. Henary, G. El Fakhri and H. S. Choi, Acc. Chem. Res., 2016, 49, 1731–1740 CrossRef CAS PubMed.
- Y. Miyata, T. Ishizawa, M. Kamiya, S. Yamashita, K. Hasegawa, A. Ushiku, J. Shibahara, M. Fukayama, Y. Urano and N. Kokudo, Sci. Rep., 2017, 7, 1–10 CrossRef CAS PubMed.
- Y. Dai, J. Yin, Y. Huang, X. Chen, G. Wang, Y. Liu, X. Zhang, Y. Nie, K. Wu and J. Liang, Biomed. Opt. Express, 2016, 7, 1149–1159 CrossRef CAS PubMed.
- B. Bahmani, Y. Guerrero, D. Bacon, V. Kundra, V. I. Vullev and B. Anvari, Lasers Surg. Med., 2014, 46, 582–592 CrossRef PubMed.
- A. van der Ende, T. Croce, S. Hamilton, V. Sathiyakumar and E. Harth, Soft Matter, 2009, 5, 1417–1425 RSC.
- W. Cai, X. Zhang, Y. Wu and X. Chen, J. Nucl. Med., 2006, 47, 1172–1180 CAS.
- Z. Y. Li, J. J. Hu, Q. Xu, S. Chen, H. Z. Jia, Y. X. Sun, R. X. Zhuo and X. Z. Zhang, J. Mater. Chem. B, 2015, 3, 39–44 RSC.
- Q. Lei, W. X. Qiu, J. J. Hu, P. X. Cao, C. H. Zhu, H. Cheng and X. Z. Zhang, Small, 2016, 12, 4286–4298 CrossRef CAS PubMed.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- V. D. Bock, H. Hiemstra and J. H. van Maarseveen, Eur. J. Org. Chem., 2006, 1, 51–68 CrossRef.
- S. G. Agalave, S. R. Maujan and V. S. Pore, Chem. Asian J., 2011, 6, 2696–2718 CrossRef CAS PubMed.
- E. Lallana, F. Fernandez-Trillo, A. Sousa-Herves, R. Riguera and E. Fernandez-Megia, Pharm. Res., 2012, 29, 902–921 CrossRef CAS PubMed.
- Z. Guo, L. Liang, J. J. Liang, Y. F. Ma, X. Y. Yang, D. M. Ren, Y. S. Chen and J. Y. Zheng, J. Nanoparticle Res., 2008, 10, 1077–1083 CrossRef CAS.
- P. Wu and V. V. Fokin, Aldrichim Acta, 2007, 40, 7–17 CAS.
- M. van Dijk, D. T. S. Rijkers, R. M. J. Liskamp, C. F. van Nostrum and W. E. Hennink, Bioconjugate Chem., 2009, 20, 2001–2016 CrossRef CAS PubMed.
- Y. Wang, C. I. R. Vera and Q. Lin, Org. Lett., 2007, 9, 4155–4158 CrossRef CAS PubMed.
- Y. Wang, W. J. Hu, W. Song, R. K. V. Lint and Q. Lin, Org. Lett., 2008, 10, 3725–3728 CrossRef CAS PubMed.
- Z. Li, L. Qian, L. Li, J. C. Bernhammer, H. V. Huynh, J.-S. Lee and S. Q. Yao, Angew. Chem., Int. Ed., 2016, 55, 2002–2006 CrossRef CAS PubMed.
- M. Zhou, J. Hu, M. M. Zheng, Q. H. Song, J. B. Li and Y. Zhang, Chem. Commun., 2016, 52, 2342–2345 RSC.
- Y. Gai, G. Xiang, X. Ma, W. Hui, Q. Ouyang, L. Sun, J. Ding, J. Sheng and D. Zeng, Bioconjugate Chem., 2016, 27, 515–520 CrossRef CAS PubMed.
- X. Y. Xu, H. R. Li, K. Li, Q. Zeng, Y. Liu, Y. Zeng, D. Chen, J. M. Liang, X. L. Chen and Y. H. Zhan, J. Nanomed. Nanotechnol., 2019, 19, 136–144 CrossRef CAS PubMed.
- X. Wu, M. Wu and J. X. Zhao, Nanomed. Nanotechnol. Biol. Med., 2014, 10, 297–312 CrossRef CAS PubMed.
- A. Liberman, N. Mendez, W. C. Trogler and A. C. Kummel, Surf. Sci. Rep., 2014, 69, 132–158 CrossRef CAS PubMed.
- M.-H. Chan and H.-M. Lin, Biomaterials, 2015, 46, 149–158 CrossRef CAS PubMed.
- L. Kong, C. S. Alves, W. Hou, J. Qiu, H. Moehwald, H. Tomas and X. Shi, ACS Appl. Mater. Interfaces, 2015, 7, 4833–4843 CrossRef CAS PubMed.
- H. Gao, Y. Xiong, S. Zhang, Z. Yang, S. Cao and X. Jiang, Mol. Pharm., 2014, 11, 1042–1052 CrossRef CAS PubMed.
- Q. Chen, H. Wang, H. Liu, S. Wen, C. Peng, M. Shen, G. Zhang and X. Shi, Anal. Chem., 2015, 87, 3949–3956 CrossRef CAS PubMed.
- P. Padmanabhan, A. Kumar, S. Kumar, R. K. Chaudhary and B. Gulyas, Acta Biomater., 2016, 41, 1–16 CrossRef CAS PubMed.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 1–12 Search PubMed.
- G. Chen, I. Roy, C. Yang and P. N. Prasad, Chem. Rev., 2016, 116, 2826–2885 CrossRef CAS PubMed.
- F. Tang, L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
- Q. He and J. Shi, Adv. Mater., 2014, 26, 391–411 CrossRef CAS PubMed.
- W.-H. Chen, G.-F. Luo, Q. Lei, F.-Y. Cao, J.-X. Fan, W.-X. Qiu, H.-Z. Jia, S. Hong, F. Fang, X. Zeng, R.-X. Zhuo and X.-Z. Zhang, Biomaterials, 2016, 76, 87–101 CrossRef CAS PubMed.
- F. Chen, H. Hong, S. Shi, S. Goel, H. F. Valdovinos, R. Hernandez, C. P. Theuer, T. E. Barnhart and W. Cai, Sci. Rep., 2014, 4, 1–10 Search PubMed.
- S. Ito, Y. Tanaka, A. Kakehi and K. I. Kondo, Bull. Chem. Soc. Jpn., 1976, 49, 1920–1923 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04513a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a