
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceClinical developments of antitumor polymer therapeutics
Shazia Parveen
 *a,
Farukh Arjmand
b and
Sartaj Tabassum
b
*a,
Farukh Arjmand
b and
Sartaj Tabassum
b
aChemistry Department, Faculty of Science, Taibah University, Yanbu Branch, 46423, Yanbu, Saudi Arabia. E-mail: sparveen@taibahu.edu.sa; Tel: +966 504522069
bDepartment of Chemistry, Aligarh Muslim University, Aligarh-202002, India
First published on 8th August 2019
Abstract
Polymer therapeutics encompasses polymer–drug conjugates that are nano-sized, multicomponent constructs already in the clinic as antitumor compounds, either as single agents or in combination with other organic drug scaffolds. Nanoparticle-based polymer-conjugated therapeutics are poised to become a leading delivery strategy for cancer treatments as they exhibit prolonged half-life, higher stability and selectivity, water solubility, longer clearance time, lower immunogenicity and antigenicity and often also specific targeting to tissues or cells. Compared to free drugs, polymer-tethered drugs preferentially accumulate in the tumor sites unlike conventional chemotherapy which does not discriminate between the cancer cells and healthy cells, thereby causing severe side-effects. It is also desirable that the drug reaches its site of action at a particular concentration and the therapeutic dose remains constant over a sufficiently long period of time. This can be achieved by opting for new formulations possessing polymeric systems of drug carriers. However, many challenges still remain unanswered in polymeric drug conjugates which need to be readdressed and therefore, can broaden the scope of this field. This review highlights some of the antitumor polymer therapeutics including polymer–drug conjugates, polymeric micelles, polymeric liposomes and other polymeric nanoparticles that are currently under investigation.
 Shazia Parveen | Dr Shazia Parveen received her Doctorate degree in Chemistry under the supervision of Prof. Farukh Arjmand from Aligarh Muslim University, Aligarh, India, in 2013. Earlier she completed her BSc (Hons.) in Chemistry in 2006 and MSc in Organic Chemistry in 2008 from the same university. During her PhD, she was awarded with Maulana Azad National Fellowship from University Grants Commission, New Delhi. Her research work includes the design and synthesis of metal based chiral chemotherapeutic agents derived from biologically active ligands viz., amino acids, benzimidazoles, peptides; and their in vitro DNA binding and cleavage studies. She has done research training in Solution Phase Peptide Synthesis (SPSS) in Laboratory of Organic Synthesis (DOSL) at Indian Institute of Chemical Technology (IICT), Hyderabad, India in 2009. She has been awarded with various awards including ‘Best Poster Presentation Award’ in “11th International Conference on Chemistry and its Role in Development” held at Mansoura University, Sharm-El-Sheikh, Egypt in 2013. After completing her postdoctoral fellowship in Jamia Hamdard, she is currently working as an Assistant Professor in Department of Chemistry, College of Sciences, Yanbu Branch, Taibah University, Madina, KSA. |
 Farukh Arjmand | Dr (Mrs) Farukh Arjmand is currently working as Full Professor of Chemistry, Aligarh Muslim University, Aligarh, India. She has completed her masters and PhD in chemistry from Aligarh Muslim University, Aligarh and later has joined her services as lecturer in 1994 in A.M.U. Dr Arjmand has research experience of 28 years in the specialization area of bioinorganic chemistry. Her research focus is on medicinal inorganic chemistry. She works on “Design and synthesis of chiral metal-based antitumor chemotherapeutic drug entities and in vitro interaction studies of metal-based compounds with biomolecules viz., DNA and RNA. She has published more than 131 research articles pertinent to her specialization area in the peer reviewed journals of international repute, has contributed 46 articles to conferences/symposium and has two patents. Dr Arjmand has 3095 citations to her credit with h-index 31 and i10-index 78. She has contributed a chapter “Antitumor activity of tin complexes” to Encyclopedia of Metalloproteins (Springer, 2012). Dr Arjmand has successfully guided 14 PhD and 4 MPhil students and has run six major research projects as PI on the design of metal-based drug candidates awarded by UGC, CSIR and DBT, Govt. of India (2001–09) and has visited many countries (China, USA, Egypt) for academic pursuits. She has joint research collaborations with national and international research institutes, IIT Kharagpur, IICT, Hyderabad and ACTREC, Mumbai (India) USTC, China, USM, Malaysia and Materials Chemistry Laboratory Oujda, Morocco and Institut de Physique de Rennes – UMR 6251, université de Rennes 1, France and The Ohio state University, USA. Prof Arjmand is presently serving as co-director of APJ Abdul Kalam STEM ER centre which is joint collaboration venture of The Ohio State University, USA and AMU, Aligarh. |
 Sartaj Tabassum | Prof. Sartaj Tabassum is working as Professor in the Department of Chemistry, Aligarh Muslim University, Aligarh. He was awarded Senior Research Fellowship (1988) and Research Associateship (1991) by CSIR New Delhi and in 1994 he was appointed as lecturer in the Department of Chemistry, Aligarh Muslim University. He has published 120 papers in the journals of international repute. He has presented his research work and delivered invited lectures in conferences/symposia. He is a life member of ICC, CRSI, ISCB, DNA Society of India and American Nano Society. He has successfully guided 17 PhD and 4 MPhil. students. He has successfully completed many research schemes granted by TWAS, Italy, CSIR, New Delhi, DBT, Govt. of India. As a distinguished Scientist, Prof. Tabassum was awarded Overseas Associateship award in 2005 by DBT, Govt. of India. He has signed several MoU and joint research collaboration with University of Camerino UNICAM, Italy, USM Malaysia and USTC Hefei, China. He has visited many countries for academic pursuit particularly, China, USA, Italy as fellow, visiting Professor and for the international conferences. He has also visited King Saud University, Riyadh, Saudi Arabia as visiting Professor for two years. Prof. Tabassum is working in the area of medicinal inorganic chemistry and his main focus is the design and synthesis of heterobimetallic complexes which have potential to act as cancer chemotherapeutics. He has been awarded three patents on cancer drugs and device He has claimed that the new drugs will be very effective with minimal side effects and it will be available at very low cost. |
1 Introduction
Cancer or the big ‘C’ is an invincible predator, not just a complex disease. Cancer is a malignant growth in which a group of cells divide uncontrollably beyond the normal limit; subsequently intrude into the near or distant tissues, a condition called metastasis, ultimately causing death. According to the World Health Organization, globally approximately 13% of all deaths worldwide are due to cancers which makes it the leading cause of man's mortality and morbidity.1 For over half a century, extensive research has been undertaken, and tremendous resources are being invested in prevention, diagnosis, and treatment of tumor. Cancer can be effectively treated with surgery, chemotherapy and radiotherapy either alone or in combination. However, the mainstay treatment regime is chemotherapy involving various natural and synthetic compounds that have the potential to kill or check the unwanted proliferation of cancerous cells.2–4The modern use of transition metal complexes as chemotherapeutic agents dates back to the serendipitous discovery of cisplatin by Rosenberg et al. in 1965.5 It is one of the most potent chemotherapeutic drug widely used for the treatment of solid malignancies.6 Following the success of cisplatin, various other Pt-based drugs7–10 have been developed, although only a small subset (e.g., carboplatin and oxaliplatin) has received US Food and Drug Administration (FDA) approval (Table 1). In spite of therapeutic success, the clinical use of platinum drugs is severely hindered by adverse side effects, systemic toxicity and intrinsic resistance and off-target reactivity.11,12 Therefore, various delivery systems have been developed to prevent the shortcomings of Pt-based chemotherapy and to increase their efficacy.13
| Complex | Structure | Clinical use | Approval | Reference |
|---|---|---|---|---|
| Cisplatin | 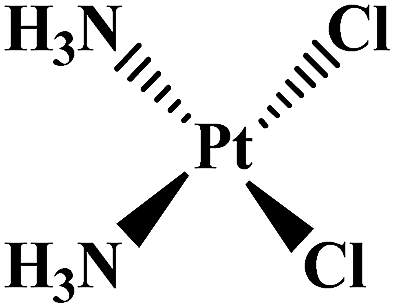 |
Metastatic, testicular, ovarian, advanced bladder tumors | Worldwide | 5 |
| Carboplatin | 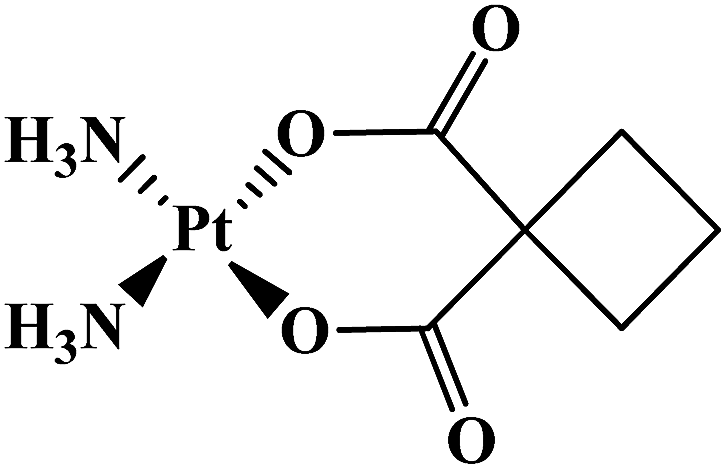 |
Advanced ovarian tumor | Worldwide | 7 |
| Oxaliplatin | 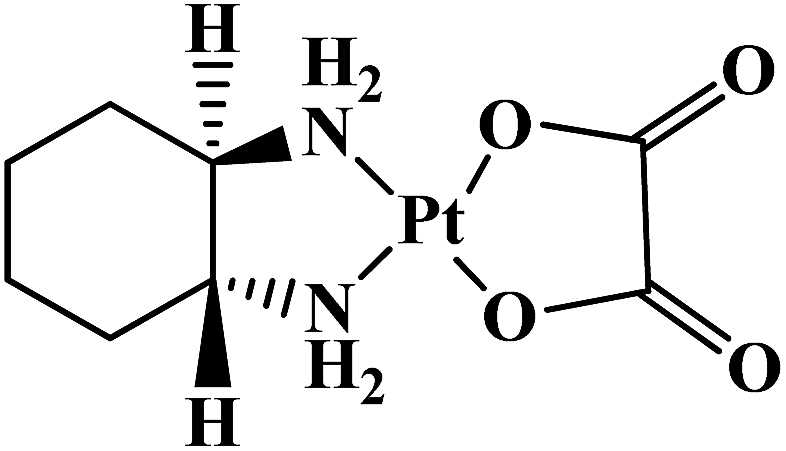 |
Metastatic colorectal tumor | Worldwide | 8 |
| Nedaplatin | 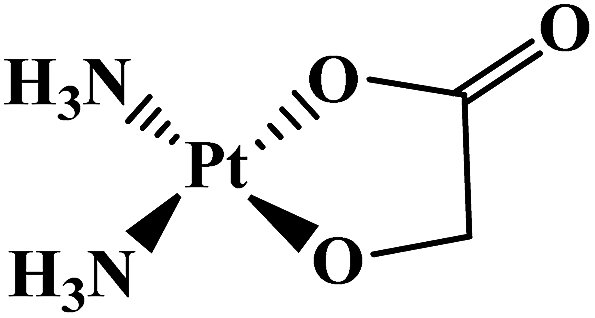 |
Head and neck tumor, small and non-small lung tumor, esophageal and bladder tumors, cervix carcinomas | Japan | 9 |
| Lobaplatin | 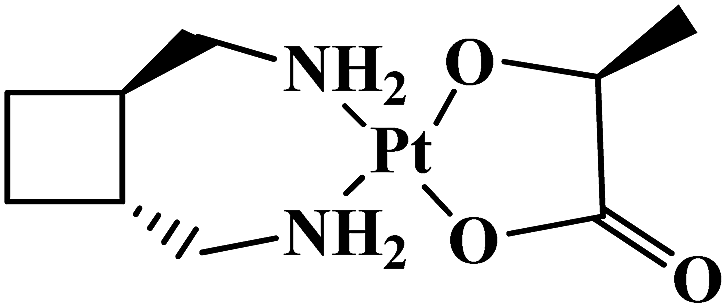 |
Breast, testicular, ovarian, small-cell lung, gastric carcinomas, chronic myeloid leukemia | China | 10 |
| Heptaplatin | 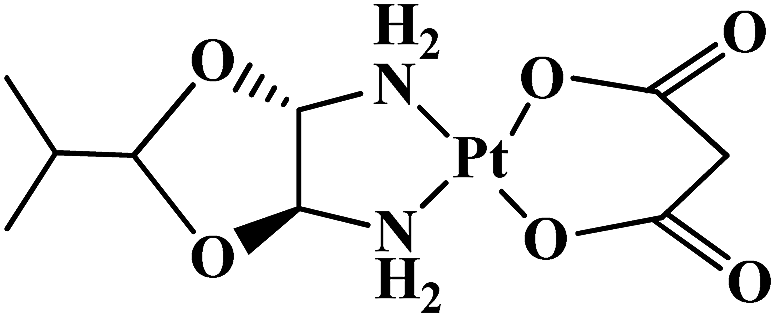 |
Gastric tumors | Korea | 6 |
Current cancer chemotherapies often fail to improve patient mortality and morbidity due to severe adverse effects on normal tissues.14 Nowadays, a number of internal triggers, including pH gradient and enzyme activity, and external stimuli such as light and magnetic field have been integrated with different inorganic and organic materials for a range of biomedical application such as diagnosis, tissue engineering and cancer therapy.15,16 Development of new drug molecule is expensive and time consuming. Improving safety efficacy ratio of “old” drugs has been attempted using different methods such as individualizing drug therapy, therapeutic drug monitoring, and targeted delivering of the drug at controlled rate. A major impetus for innovation chemotherapy is improving the therapeutic index of drugs. A vast majority of clinically used drugs in practice are low molecular-weight compounds usually less than 500 g mol−1, that exhibit a short half-life in the blood stream and a high overall clearance rate. They rapidly diffuse into healthy tissues and are distributed evenly within the body. As a result, relatively small amounts of the drug reach the target site, and therapy is associated with side effects.
However, in the last decade, much emphasis has been placed on the development of nano-medicine therapeutics by using liposomes, dendrimers, polymeric micelles, as nano-carriers for the tumor targeted delivery of anticancer agents, among which polymeric drug delivery system are of immense interest.17–19 The primary goal of drug delivery in cancer therapy is to transport sufficient drugs to target disease site(s) while minimizing their exposure to healthy tissues. Two main strategies have been extensively exploited to achieve this by altering the pharmacokinetic properties of the drug in some way. First, the use of a nanoparticle delivery vehicle, that encapsulates the drug and dictates the biodistribution through its own physicochemical properties; second, the covalent modification of the drug with some small moiety that temporarily masks or limits the drug's bioactivity and confers overall desirable pharmacokinetic properties (prodrug strategy).20
Since cancers exhibit acquired characteristics that enable their malignant phenotype, there is a need to search for more targeted cancer therapeutic drugs, which has been supported by a better understanding of malignancy at the molecular level. The traits which provide a target for drug therapy include replicative immortality, evasion of growth suppressors, angiogenesis induction, tissue invasion and metastases, resistance to cell death, deregulated cell metabolism, tumor-promoting inflammation and the ability to evade the immune system.21 Cancers interact with their local microenvironment or stroma through angiogenesis, inflammation and immune responses.22
Targeted drug delivery system is a rational solution to toxicity-related issues because it deliver the drug specifically through a carrier molecule to the site of tumor, leaving on-cancerous/healthy cells unaffected.23 An ideal drug delivery system should be capable of delivering the drug to a specific tumor site in a specific time and release pattern. Polymers have been in use to design polymer therapeutics that include linear or branched polymer chains, working either as the bioactive molecule, such as a polymeric drug or as the inert carrier to which a drug can either be covalently linked, e.g., polymer–drug conjugates, or non-covalently tethered such as dendrimers, polymeric micelles, polymeric liposomes and other nano-sized systems.24
Polymer conjugated drugs offer good opportunistic systems for antitumor drug delivery due to (i) their great versatility from the structural point of view, (ii) their nano size, (iii) prolonged plasma half-life, (iv) ability to boost stability of drugs, (v) ability to lower immunogenicity and antigenicity and specific targeting to tissues or cells, (vi) have the potential to improve pharmacological therapy of a variety of tumors, (vii) the possibilities to combine both type of drugs (hydrophobic and hydrophilic), (viii) polymer–drug interactions that offer many possibilities to design and prepare polymer–antitumor drug formulations with specific properties and functions, (ix) has potential for targeted drug delivery and (x) their biodegradable nature, they can be easily degraded into smaller non-toxic molecules in the body.25,26 Moreover, if the polymer is not biodegradable, the drug can be covalently attached to the polymeric structure by a linker which can be degraded under different conditions such as in an acidic medium or by different enzymes; while the targets can be bound covalently to the surface which will help the directionality of the vector to the site of action.27 Therefore, an ideal nano-carrier for drug delivery and cancer chemotherapy should (i) stabilize without altering the pharmacological activity of the drug, (ii) prevent premature metabolic degradation of the drug in the systemic circulation such that it arrives in a pristine state at the intended target, (iii) release the drug at the intended site/tumor, and (iv) exhibit similar or lower toxicity than that of the free drug.
Nanotechnology-based drug delivery systems are the emerging multi-disciplinary program in biomedical field and they resolve the challenges such as bioavailability due to less solubility, poor intestinal absorption mechanism by degradation, deficient delivery mechanism at target site, therapeutic potency, side effects and plasma in consistency of drugs.28
This paper reviews some of the important anticancer polymer therapeutics with emphasis on those that are either approved or are in any phase of clinical trials. These include polymer–drug conjugates, polymeric micelles, polymeric liposomes and nanoparticle-assisted antitumor agents. It gives an idea of the effects of polymer conjugation on the antitumor activity of the drug, the possible drawbacks and future perspectives that could expand this field of drug targeting via polymeric systems for the successful treatment of a wide spectrum of tumors.
2 Targeted drug conjugates
The major limitations of prevalent Pt-based chemotherapeutic agents are the issues with solubility, formulation, biodistribution and ability to cross cell membranes. In order to overcome these shortcomings; tumor targeted delivery system employing both Pt-based and non-Pt based organic nano-antitumor drug formulations could be a viable option for improving the delivery of antitumor drugs to the target sites.23,29,30 They may be micelles, liposomes, micro- or nano-spheres, implants or simply polymer–drug conjugates.31–33 The field of drug delivery systems utilizing synthetic polymers either by covalent conjugation or by composite of micellar drugs has become a new domain for drug development program for the treatment of numerous chronic diseases viz., cancers.Targeted drug delivery involves the delivery of hydrophobic polymer drugs across the cell membranes to the specific targeted cancerous cells.34,35 Previous literature reports have demonstrated that improving the delivery of Pt-based drugs could lead to reduced side effects, greater efficacy at lower doses of drug.36,37 For a drug delivery system to be effective, it must have high plasma stability, low toxicity and immunogenicity, and protect drugs against premature metabolism. “Targeted drug delivery” is distinct from “targeted therapy” as former refers to predominant drug accumulation within a target zone that is independent of the method and route of drug administration.38 Whereas, targeted therapy means specific interaction between a drug and its receptor at the molecular level.39–41
2.1 Passive targeting and active targeting
Targeted delivery is a well-known field in which the drug carriers target tumor cells via two different processes; passive or active drug delivery.42 The passive targeting exploits the tumor vascular system through the enhanced permeability and retention (EPR) effect. Whereas, active targeting takes advantage of both the EPR effect and achieve specific cancer cell targeting by modifying the drug carrier surface through covalent attachment of drug specific for receptor molecule that is overexpressed in the cancerous cells (Fig. 1).43–47 Due to EPR effect polymer–drug conjugates can increase the therapeutic index of antitumor agents.48 In active targeting, targeting ligands are attached at the surface of the nanocarrier for binding to appropriate receptors expressed at the target site. It is the tendency for macromolecules and nanoparticles (NPs) to accumulate more in tumor than in normal tissues. Normal vessels have a tight endothelium, while tumor vessels are disorganized and leaky, allowing preferential extravasation of circulating macromolecules. In tumor tissues, the carrier-drug conjugate is cleaved to generate the active platinum species, leading to the formation of cell-lethal DNA adducts. Generally, the binding affinity of the ligands (viz., monoclonal antibodies and antibody fragments or non-antibody) influences the tumor penetration because of the “binding-site barrier” targeting ligands are either ligands.49 All polymer–drug conjugates including NPs exhibit superior antitumor efficiency owing to enhanced EPR effect.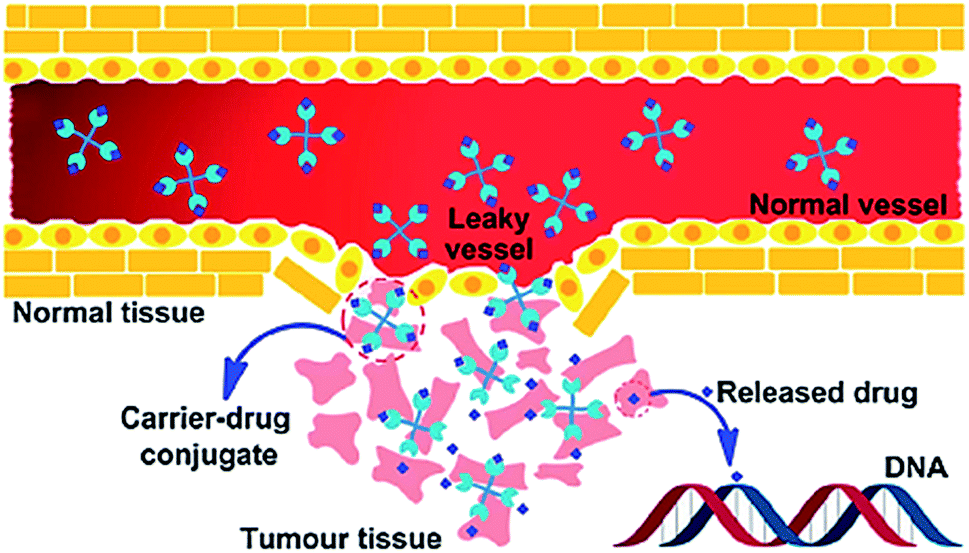 | ||
| Fig. 1 Schematic representation of the EPR effect. Reprinted with permission from ref. 42, Royal Society of Chemistry. | ||
2.2 Polymeric drug conjugates: from Ringsdorf's model to the clinic
In 1975, a rational model for pharmacologically active polymers was first proposed by Helmut Ringsdorf.50 In Ringsdorf's original model, a number of drug molecules were bound to a macromolecule through a spacer molecule, which incorporated a predetermined breaking point to ensure release of the drug at the specific target site (Fig. 2). The polymer conjugates additionally contain recognition moieties, for example, antibodies or sugar moieties, which target disease related antigens or their receptors. In addition, solubilizing groups can be attached to the polymer backbone to modify the bioavailability of the polymer–drug conjugate. These polymer–drug conjugates offer several significant advantages over traditional small molecule therapeutics. First, the aqueous solubility of a drug can be dramatically improved following conjugation to a water soluble polymer.51,52 Secondly, polymer–drug conjugates offer the potential for a drug to be delivered in a controlled manner, with drug release from the conjugate occurring over a defined time interval. In this way, the rate and duration of delivery can be custom designed to achieve the desired therapeutically effective concentration. Moreover, polymer conjugation also provides an opportunity to alter drug pharmacokinetics and biodistribution. This is particularly useful for drugs which exhibit a short blood plasma half-life due to rapid metabolism or clearance or for drugs which exhibit off target toxicities (i.e. antitumor agents). Polymer-conjugated drugs exhibit prolonged half-life, higher stability, water solubility, lower immunogenicity and antigenicity and often also specific targeting to tissues or cells.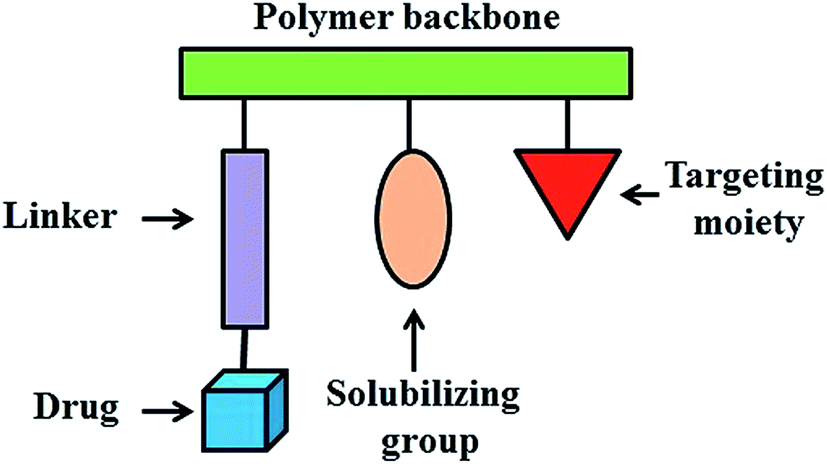 | ||
| Fig. 2 Ringsdorf's model of polymer–drug conjugate. | ||
These systems are able to selectively deliver the bioactive payload to the target tissue, cells or even sub-cellular compartments and release it there in a triggered way. In these types of polymer–drug conjugates, the goal is to improve the mechanism of cellular internalization and cell specificity to achieve optimal release of the drug at the proposed target. In contrast, the systems in which the drugs are embedded or encapsulated in the polymer seek to enhance the distribution and serum stability and to attain a decrease in drug immunogenicity through a controlled distribution at the correct site and over time.53 A large number of antitumor drug polymer conjugates have been studied in tumor therapy due to their promising clinical applications in chemotherapy.34,54–60
2.2.1.1 PEG–drug conjugates. PEG presents unique properties such as (i) lack of immunogenicity, antigenicity and toxicity; (ii) high solubility in water and in many organic solvents; (iii) high hydration and flexibility of the chain, which is at the basis of the protein rejection properties; and (iv) approval by FDA for human use. A few important polymer–drug conjugates have been summarized in Table 2.
| Name | Polymer | Drug | Function | Results | Clinical trial | Reference |
|---|---|---|---|---|---|---|
| Prothecan | PEG | Camptothecin (with glycine linker) | Gastric and gastroesophageal tumors | Lower drug loading of 1.7 wt% as compared to other polymers | Phase II | 68–70 |
| Better efficacy in animal models of human tumors than free camptothecin | ||||||
| EZN-2208 | PEG | SN38 (camptothecin analog) | Metastatic breast tumor | Prolonged blood circulation half-life | Phase II | 71 and 72 |
| Increased tumor to plasma drug concentration ratio | ||||||
| NKTR-102 | PEG | Irinotecan | second line colorectal tumor, metastatic breast tumor, platinum-resistant ovarian tumor and metastatic cervical tumor | Prolonged half-life of 15 days as compared to free irinotecan (4 h) | Phase II | 66 |
| 1.2–6.5 fold higher cumulative exposure |
Prothecan, a camptothecin conjugate, was the first PEG–drug conjugate in clinical trials in which the 20-OH position was a conjugated through camptothecin with PEG through a glycine spacer. Preclinical results with prothecan showed better efficacy in animal models of human tumors than free camptothecin.68–70 The drug loading corresponds to 1.7 wt%, which is rather low if compared with other multivalent polymers. Prothecan, currently being assessed in phase II studies administered every 3 weeks at mg m−2, displayed a promising treatment of adenocarcinoma of the stomach and gastroesophageal (GE) junction; and appeared to be well tolerated, with a low incidence of toxicities (Fig. 3a).
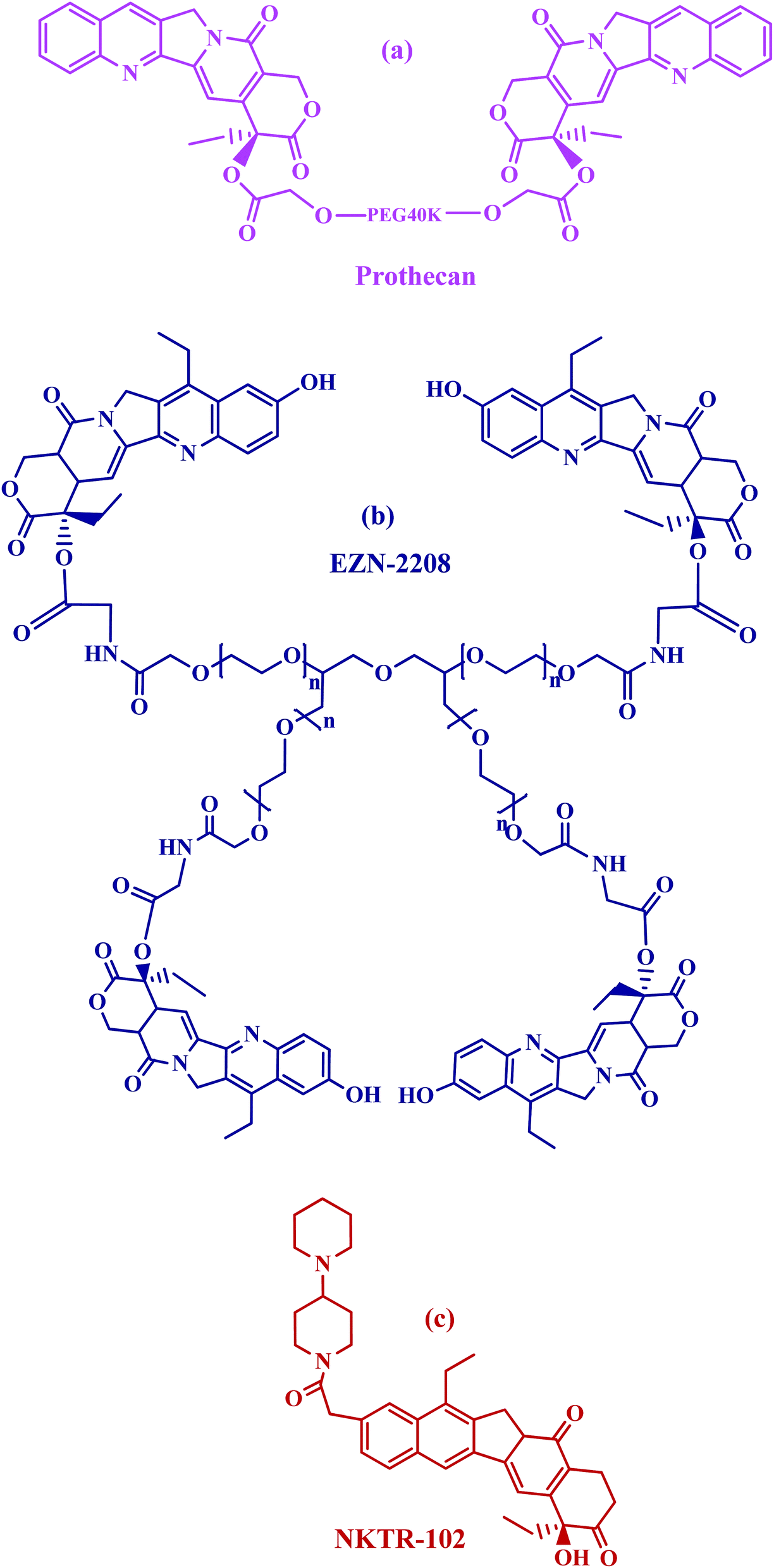 | ||
| Fig. 3 Structures of PEG–drug conjugates in clinical trials. | ||
PEG-SN38 (EZN-2208)71 is entering phase II clinical testing. This compound was obtained by coupling a 4armPEG of 40 kDa with the camptothecin derivative SN38, through a glycine spacer (Fig. 3b). SN38 is a camptothecin analog and therefore has the same function as camptothecin (topoisomerase I inhibitor) and a similar structure, containing a lactone ring. EZN-2208 showed a 207-fold higher exposure to SN38 compared to irinotecan in treated mice, longer blood circulation half-life and the a tumor to plasma drug concentration ratio increased over the time during the four days long pharmacokinetic and biodistribution studies. Antitumor efficacy was also demonstrated in xenograft models of breast, colorectal, and pancreatic tumor.72
PEG-irinotecan (NKTR-102) has been covalently bound to a four arms PEG (Fig. 3c). It is the first long-acting topoisomerase I inhibitor. In preclinical studies NKTR-102 plasma half-life was evaluated in a mouse model taking into consideration the active metabolite SN-38, released from irinotecan. The conjugate showed prolonged pharmacokinetic profiles with a half-life of 15 days when compared to 4 h with free irinotecan.66 Cumulative SN38 exposure in patients treated with NKTR-102 was 1.2 to 6.5 fold higher than that predicted for irinotecan.73–75 Multiple phase 2 studies are ongoing with NKTR-102 alone or in combination with cetuximab for the treatment of ovarian, breast, colorectal and cervical tumor.
2.2.1.2 N-(2-Hydroxypropyl)methacrylamide (HPMA) copolymers–drug conjugates. Copolymers of HPMA were first developed by Kopecek and co-workers, have been widely investigated as hydrophilic, biocompatible, polymeric drug carriers.67,76,77
HPMA copolymer doxorubicin (PK1) was the first water soluble antitumor drug conjugated with a polymer to be tested clinically.78 The structure of an HPMA copolymer contains a tetrapeptide linker degradable by thiol-dependent lysosomal proteases (glycine-phenylalanine-leucine-glycine (GFLG)). Dox was bound to the carboxy terminus of the glycine via an amide bond. PK1 in comparison with free Dox demonstrated decreased cardiotoxicity and bone marrow toxicity in animals, 17–70 fold increased tumor accumulation of doxorubicin in melanoma tumor bearing mice.79 A phase I study revealed a fivefold increased maximum tolerated dose (MTD) was 320 mg m−2 Dox equivalents, demonstrating antitumor activity in refractory cancers, no polymer related toxicity. The results of phase II trials showed antitumor activity for the treatment of breast and non-small cell lung tumor but no response in patients with colorectal tumor.80
Another HPMA copolymer-doxorubicin galactosamine (PK2) a 27 kDa HMPA copolymer derivatized with 6.5% mol/wt, <2% free doxorubicin, and 2% mol/wt galactose was synthesized having a similar structure to PK1 but having galactosamine which specifically targets liver tissues (Fig. 4).81,82 In a phase I study, 31 patients with primary or metastatic liver cancer were evaluated.83 The molecular weight and the loading ratio are very similar in both conjugates PK1 and PK2, the MTD of PK2 was 160 mg m−2 doxorubicin equivalents which is approximately half the MTD value of PK1. A dose limiting toxicity was associated with severe fatigue, neutropenia, and mucositis; therefore a dose of 120 mg m−2 doxorubicin equivalents was recommended for phase II studies.
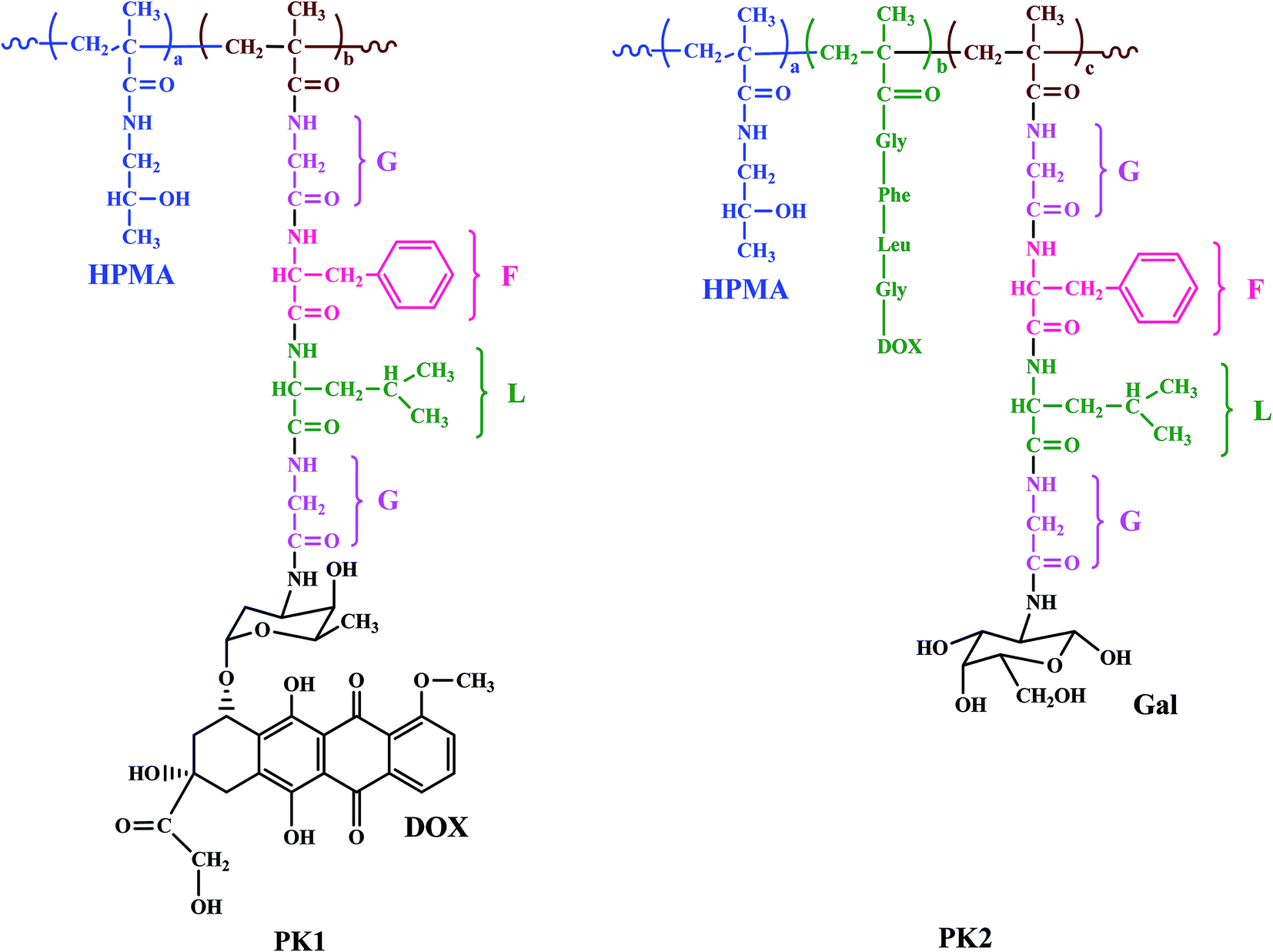 | ||
| Fig. 4 Structures of HPMA copolymer doxorubicin (PK1) and HPMA copolymer-doxorubicin galactosamine (PK2). | ||
PNU166945, HPMA copolymer-paclitaxel conjugate with same structure as HPMA copolymer-Dox but paclitaxel was conjugated to the terminal glycine via an ester bond (Fig. 5a). Since paclitaxel has poor water solubility it is pharmaceutically formulated in a mixture of ethanol and Cremophor EL (polyoxyethyleneglycerol triricinoleate 35).
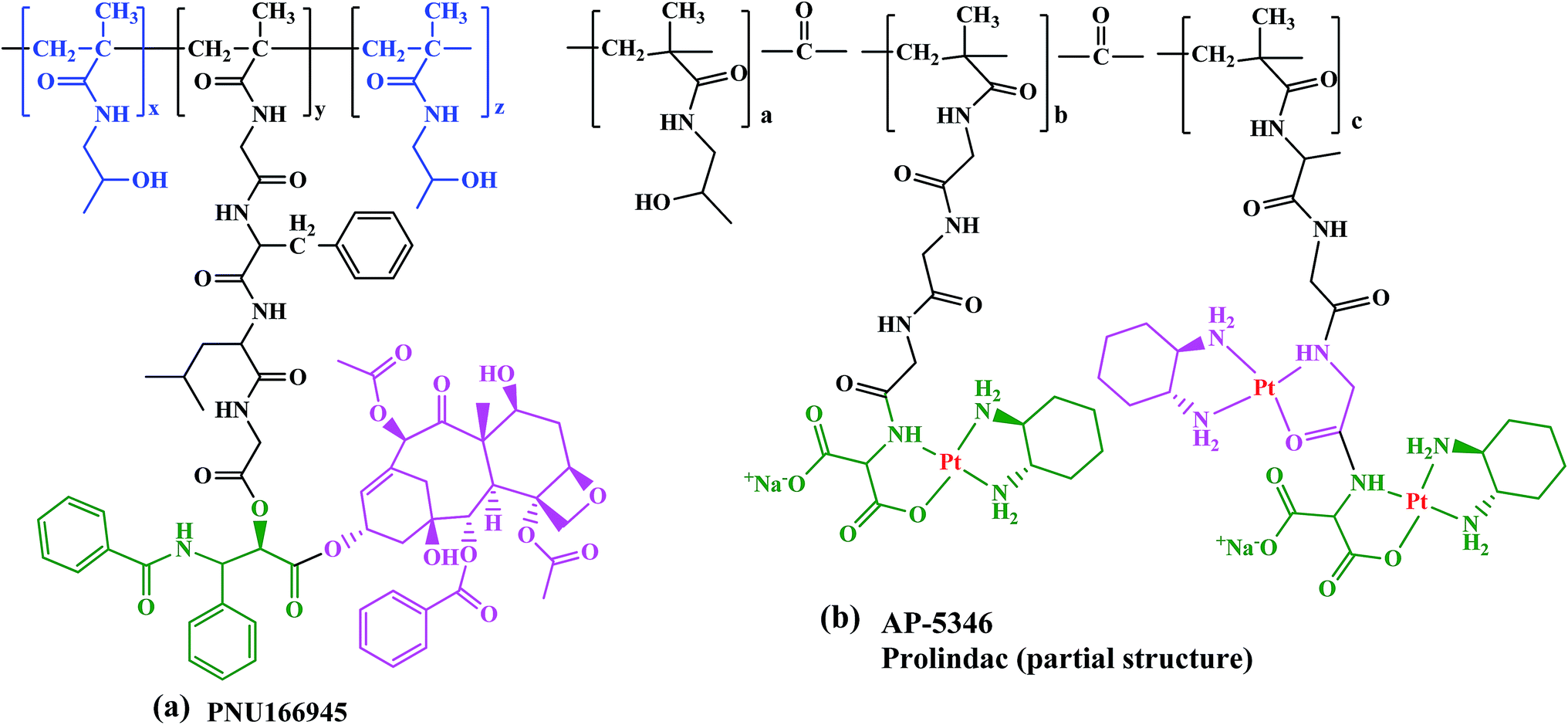 | ||
| Fig. 5 Structures of (a) PNU166945 and (b) AP 5346. | ||
Another HPMA copolymer is AP 5346 platinate that has progressed into clinical trials, which is a cytotoxic diaminocyclohexane (DACH)-platinum moiety coupled to a HPMA via a pH-sensitive linker (Fig. 5b).84–86 It is consequently released in the extracellular space of tumors and/or the intracellular lysosomal compartment. The small size of this polymer–drug conjugate (∼25 kDa), allows its renal clearance enabling it to reach tumor cells through endothelial cells. Therefore, AP 5346 has a longer half-life relative to small platinum drugs that enables it to reach tumor cells by a passive process and then release the drug. Partial responses in antitumor activity in metastatic melanoma and ovarian tumor to AP 5346 were observed in a phase I study. Thus, this drug has been shown to have a higher therapeutic index in multiple preclinical tumor models and it is entering phase II clinical testing under the new name of Prolindac.87
Although both HPMA camptothecin and paclitaxel conjugates entered phase I clinical trials, but the products showed the toxicity of the respective free drugs due to the rapid hydrolysis of the ester bond in vivo.88 Therefore, there is a need for optimization of the drug design, emphasizing on the stability of the ester linkage between polymer and drugs and drug loading.
The results have been summarized in Table 3.
| Name | Polymer–drug conjugate | Linker | Function | Results | Clinical trial | Reference |
|---|---|---|---|---|---|---|
| PK1 | HPMA-copolymer-doxorubicin | Gly-Phe-Leu-Gly | Lung and breast tumors | Decreased cardiotoxicity and bone marrow toxicity as compared to free doxorubicin | Phase II | 78 and 79 |
| 17–70% increased tumor accumulation of doxorubicin in melanoma tumor bearing mice | ||||||
| 5-Fold increased MTD of 320 mg m−2 | ||||||
| PK2 | HPMA-copolymer-doxorubicin-galactosamine | Gly-Phe-Leu-Gly | Hepatocellular carcinoma | MTD of 160 mg-m−2, but dose limiting toxicities were associated with severe fatigue, neutropenia, lower dose of 120 mg m−2 recommended for phase II | Phase I/II | 81 and 82 |
| PNU166945 | HPMA copolymer-paclitaxel | Ester | Solid tumors | Soluble in water, hence no need for cremophor El, (responsible for hypersensitivity reactions) | Phase I | 88 |
| MTD of 196 mg m−2 | ||||||
| Hematologic toxicity was mild and dose independent | ||||||
| Prolindac (AP 5346) | HPMA copolymer-platinate | A pH-sensitive linker | Solid tumors, specifically recurrent ovarian tumors | Small size allows renal clearance enabling it to reach tumor cells through endothelial cells | Phase I/II | 84–86 |
| Relatively longer half-life | ||||||
| Higher therapeutic index in multiple preclinical tumor models |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da, conjugated to paclitaxel through an ester bond reaching the impressive high drug loading of 37 wt%,89 the final conjugate molecular weight was 49000 Da. It has been studied in numerous clinical trials, including phase III trials for various solid tumors64,90 (Fig. 6a). In phase I/II clinical studies in patients with mesothelioma, renal cell carcinoma, NSCLC and in paclitaxel-resistant ovarian cancer, a significant number of partial responses stable disease were shown. Now XYOTAX is under in a phase III trial, known as the PIONEER trial, in women with advanced lung cancer to validate the earlier clinical results (Table 4).91
000 Da, conjugated to paclitaxel through an ester bond reaching the impressive high drug loading of 37 wt%,89 the final conjugate molecular weight was 49000 Da. It has been studied in numerous clinical trials, including phase III trials for various solid tumors64,90 (Fig. 6a). In phase I/II clinical studies in patients with mesothelioma, renal cell carcinoma, NSCLC and in paclitaxel-resistant ovarian cancer, a significant number of partial responses stable disease were shown. Now XYOTAX is under in a phase III trial, known as the PIONEER trial, in women with advanced lung cancer to validate the earlier clinical results (Table 4).91
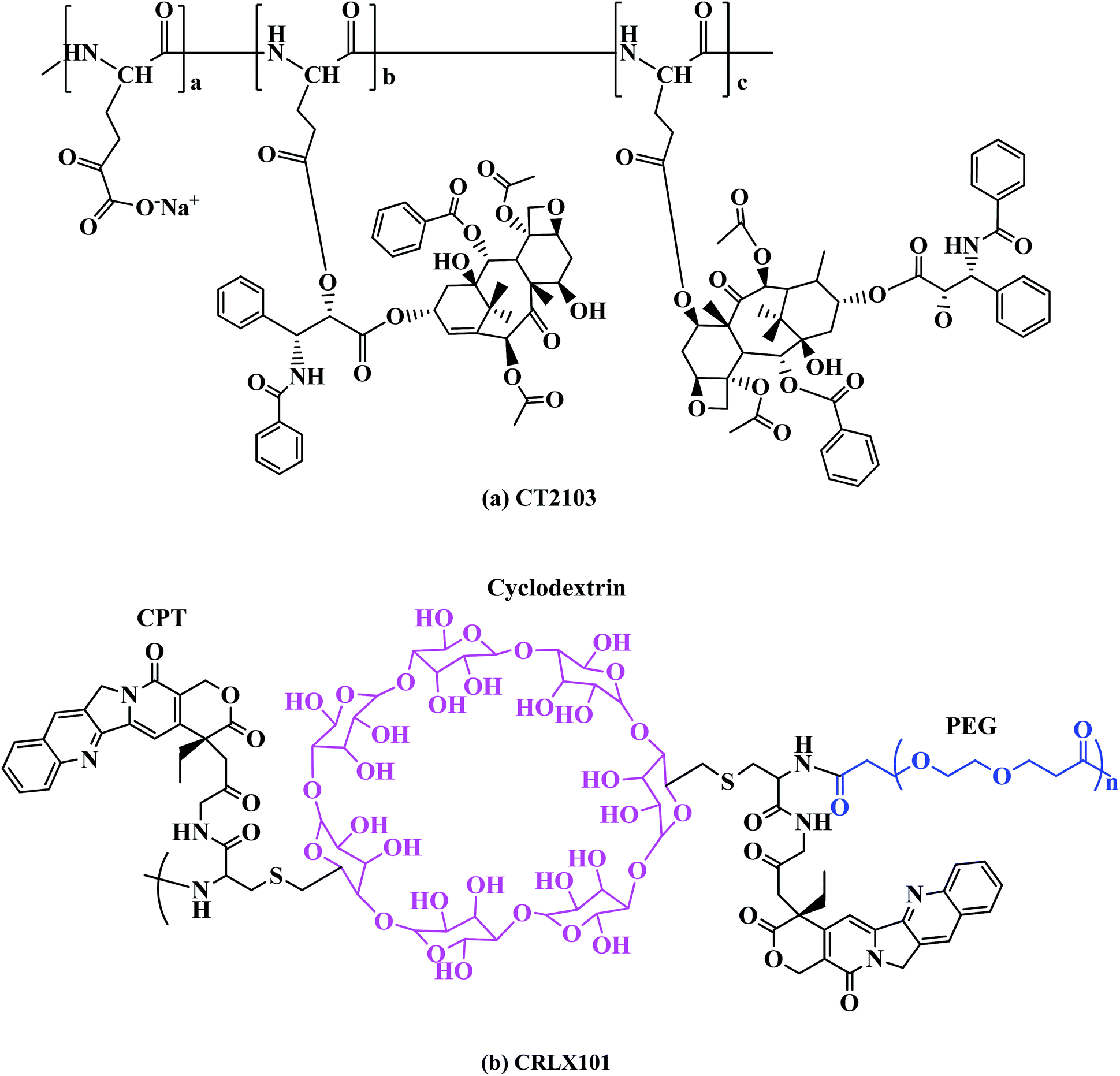 | ||
| Fig. 6 Structures of other polymer–drug conjugates. | ||
| Name | Polymer | Drug | Spacer | Function | Results | Clinical trials | Reference |
|---|---|---|---|---|---|---|---|
| XYOTAX (CT-2103) | Poly(L-glutamic acid) | Paclitaxel | Ester | Variety of solid tumors | High drug loading of 37 wt% | Phase III | 89 and 90 |
| Biodegradability of PGA backbone | |||||||
| Liberation of paclitaxel and its glutamic acid derivatives in vitro and in vivo | |||||||
| CT-2106 | Poly(L-glutamic acid) | Camptothecin | Gly-ester | Solid tumor malignancies | Increased plasma half-life | Phase I/II | 93 and 94 |
| Reduced renal clearance | |||||||
| Super aqueous solubility | |||||||
| CRLX101 | Cyclodextrin-based polymer (CDP) | Camptothecin | Advanced solid tumors | Increased plasma half-life | Phase IIa | 95 | |
| Enhanced distribution to tumor tissue as compared to free drug | |||||||
| Good tolerability against wide range of solid tumors |
CT-2106 is polymer conjugate of poly(L-glutamic acid), PG and camptothecin in which camptothecin is covalently attached to poly(L-glutamic acid) through a glycine linkage. PG was an effective solubilizing carrier of camptothecin (CPT) and stabilized the E-ring lactone structure in CPT. PG-CPT conjugate was obtained by directly coupling the hydroxy group at the C20(S)-position of CPT with the carboxylic acid of PG.70 Increasing the molecular weight of PG from 33 to 50 kDa improved the antitumor efficacy of PG-Gly-CPT, probably because of an increased plasma half-life and reduced renal clearance.92 Considering its ease of synthesis, superior aqueous solubility and stability, and marked efficacy in various in vivo tumor models, PG-Gly-CPT with 30–35% CPT was selected for further study and has entered phase I/II trials against solid tumor malignancies.93,94
CRLX101 (formerly IT-101) is a conjugate of camptothecin and a linear cyclodextrin-based polymer (CDP). The components of CDP are β-cyclodextrin and PEG (Fig. 6b). Pharmacokinetics and preclinical studies have demonstrated that this conjugate exhibited prolonged plasma half-life and enhanced distribution to the tumor tissue when compared to CPT alone.95 A phase IIa safety and pharmacokinetic study of CRLX101in the treatment of advanced solid tumors is being carried out.96,97
3 Polymeric nanoparticles for antitumor drug delivery
3.1 Polymeric micelles
Polymeric micelles are characterized by a core–shell structure. They are primarily composed of block-copolymers with hydrophilic and hydrophobic units that self-assemble into a hydrophobic core surrounded by a hydrophilic shell (Fig. 7).98 Each micellar monomer unit can be assembled in various fashions such as A–B diblock copolymers, A–B–A triblock copolymers, and grafted copolymers. Micellar encapsulation helps to solubilize, stabilize, and deliver the hydrophobic drug to the target.35 PEG due to its water-soluble nature and ability to resist uptake by the reticuloendothelial system (RES) is the most commonly employed hydrophilic polymer. However, a number of other hydrophilic polymers which include poly(N-vinyl-2-pyrrolidone) (PVP),99 poly(vinyl alcohol) (PVA),100 and poly(ethyleneimine) (PEI)101 have been used to prepare micelles. The majority of polymeric micelles investigated cannot be classified as polymer–drug conjugates, as no covalent bonds exist between the drug and the micellar carrier.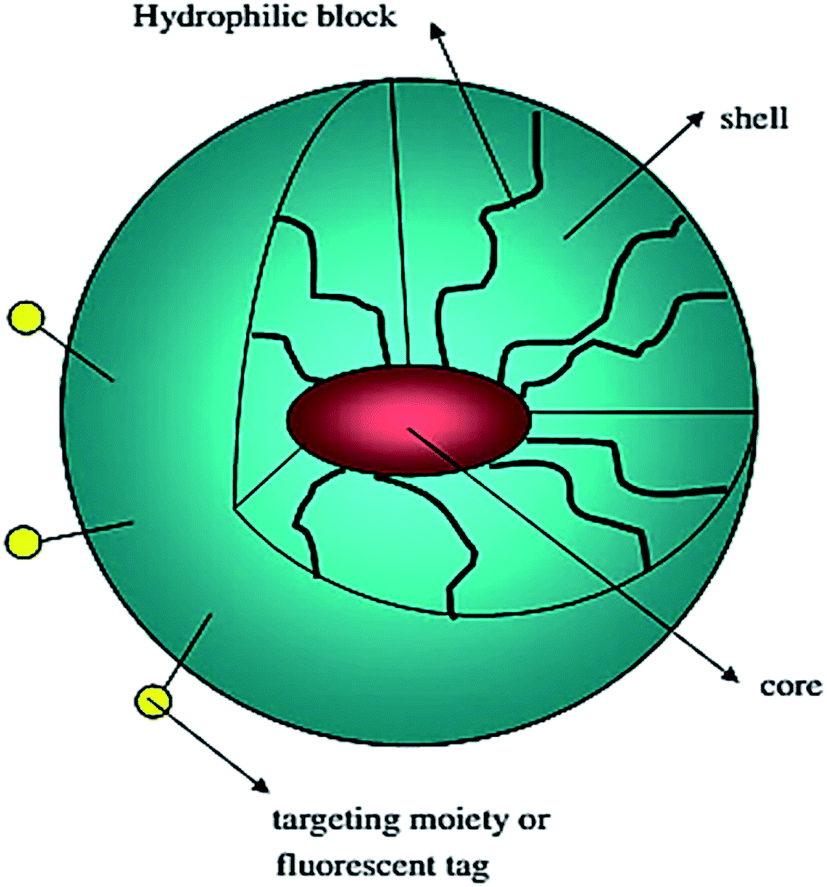 | ||
| Fig. 7 Schematic representation of a block copolymer micelle; lipophilic drug (red color) is encapsulated in the micelle core. Reprinted with permission from ref. 98, Elsevier. | ||
3.2 Polymeric micelles in clinical trials
A number of studies have been carried out to study the polymeric micelles in which the anti-tumor drug conjugated side chains form the hydrophobic micellar core, while the hydrophilic segment forms the micellar shell.57,104–110 A few polymeric micelles that are in clinical trials have been discussed here (Table 5).| Polymeric micelle | Block copolymer | Drug | Indication | Results | Clinical trial | Reference |
|---|---|---|---|---|---|---|
| NK911 | Poly(ethylene oxide)-b-poly(aspartic acid) (PEO-b-PAsp) | Doxorubicin | Pancreatic tumor | High antitumor activity than free drug | Phase I | 53 |
| MTD of 67 mg m−2 | ||||||
| NK105 | PEG-P(aspartate) | Paclitaxel | Advanced stomach tumor | Decreased hypersensitivity | Phase II | 111 and 112 |
| Can be administered without the use of antiallergic agents | ||||||
| SP1049C | Pluronic L61 and F127 | Doxorubicin | Adenocarcinoma of oesophagus, gastroesophageal junction and stomach | Increased AUC in tumor tissue | Phase III | 114 |
| NC-6004 | PEG-PGlu(cisplatin) | Cisplatin | Solid tumors | Prolonged circulation in blood | Phase I/II | 118–120 |
| Increased AUC and half-life | ||||||
| Sustained release of active Pt species | ||||||
| Low Cmax indicating low toxicity | ||||||
| Genexol-PM | PEG-P(D,L-lactide) | Paclitaxel | Breast tumor | 3-Fold increased MTD, without the occurrence of hypersensitivity reactions | Phase IV | 122 and 123 |
| Pancreatic tumor | Phase II | |||||
| Non-small-cell lung tumor in combination with carboplatin | Phase II | |||||
| Pancreatic tumor in combination with gemcitabine | Phase I/II | |||||
| Ovarian tumor in combination with carboplatin | Phase I/II |
NK911, is the first antitumor drug candidate on polymer micelles to have progressed to phase I clinical trials for antitumor therapy in Japan developed by Kataoka's group.53 It is based on poly(ethylene oxide)-b-poly(aspartic acid) (PEO-b-PAsp) block copolymers conjugated with doxorubicin. PEO is believed to form the outer shell of the micelles and the doxorubicin conjugated poly(aspartic acid) chain is hydrophobic and forms the hydrophobic core of the micelles in aqueous media. The hydrophobic core enables NK911 to entrap a sufficient amount of doxorubicin expressing higher in vivo antitumor activity than free doxorubicin because of the EPR effect. Parameters such as the maximum tolerated dose of 67 mg m−2 (grade 4 neutropenia lasting more than 5 days), dose limiting toxicities, and the recommended dose for phase II were determined.
NK105 is micellar formulation consisting of PEG and modified polyaspartate, of which half of the carboxylic groups were esterified with 4-phenyl-1-butanol after treatment with the condensing agent 1,3-diisopropylcarbodiimide. This was done to increase its hydrophobicity and improve drug incorporation as hydrophobic block.111,112 Paclitaxel is physically incorporated in the core by hydrophobic interactions with the hydrophobic block. NK105 is expected to possess a clinical advantage similar to that of paclitaxel formulations and it displayed similar cytotoxicity in 12 human tumor cell lines (lung, gastric, oesophagus, colon, breast and ovarian) compared to paclitaxel112 (Fig. 8). Hamaguchi et al. has reported phase I study with NK105, less hypersensitivity reactions occurred in patients suffering from pancreatic, bile duct, gastric, and colonic tumors compared to systemic paclitaxel treatment.113 The results of this clinical trial show that NK105 can be administered safely as a short infusion (1 h) without the administration of antiallergic agents like dexamethasone and antihistamine. Currently, a phase II study in patients with advanced stomach tumor is underway.111
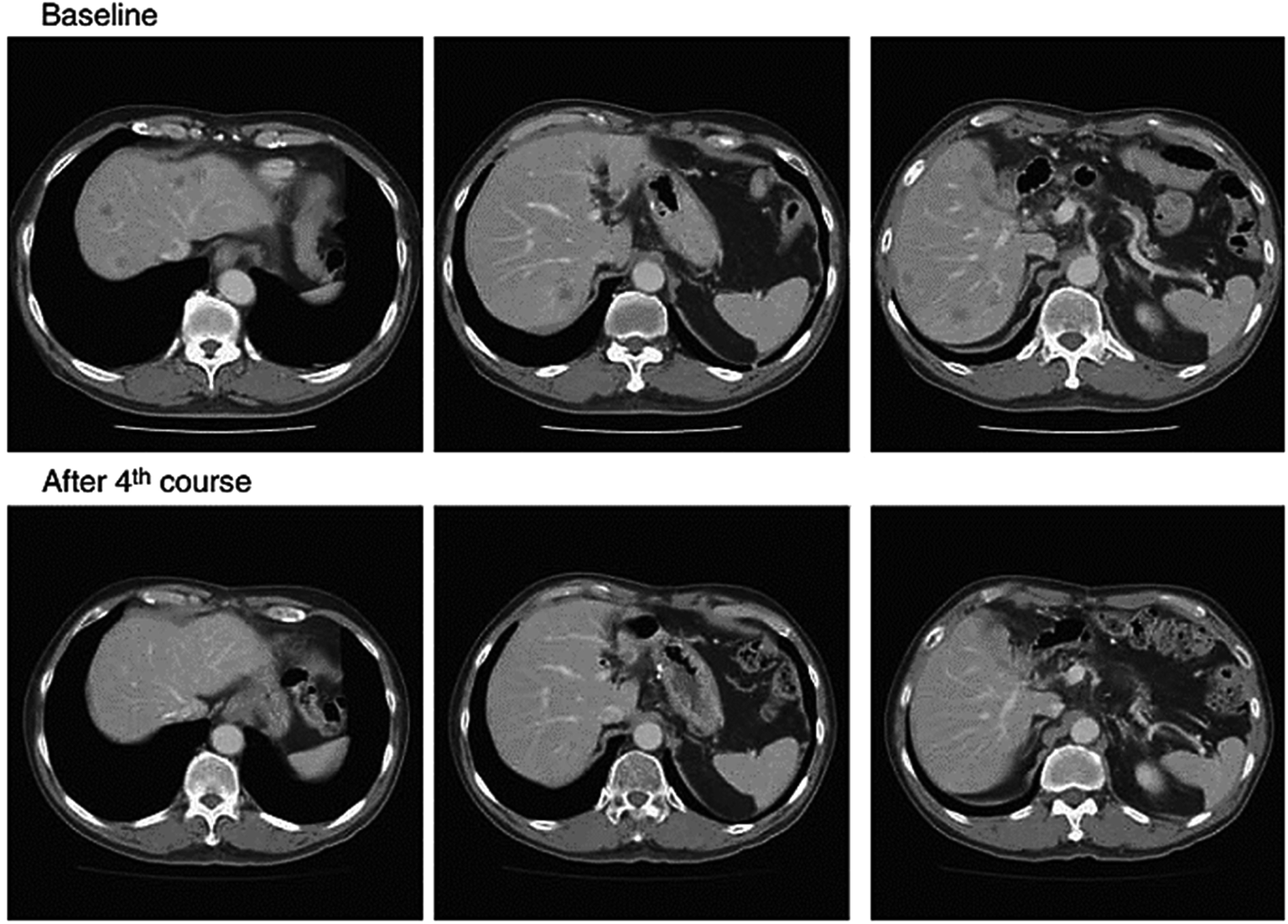 | ||
| Fig. 8 Serial CT scans of a 60 year-old male with pancreatic tumor who was treated with NK105 at a dose level of 150 mg m−2. Baseline scan (upper panels) showing multiple metastasis in the liver. Partial response, characterized by a more than 90% decrease in the size of the liver metastasis (lower panels) compared with the baseline scan. The antitumor response was maintained for nearly 1 year. Reprinted with permission from ref. 113, Springer. | ||
SP1049C is a polymeric micelle formulation of doxorubicin with Pluronic block copolymers that has shown activity in phase II clinical trials; high objective response rates of 43% in patients were observed with inoperable metastatic adenocarcinoma of the oesophagus and gastroesophageal junction.114 Pluronics are amphiphilic triblock copolymers of poly(ethylene oxide) (PEO) and poly(propylene oxide) (PPO) with PEO-PPO-PEO structure. In vitro studies revealed that SP1049C exhibited greater efficacy than doxorubicin against a variety of tumor cell lines.115 In preclinical in vivo studies, SP1049C demonstrated an increased AUC (area under plasma concentration vs. time) in tumor tissue in multiple animal tumor models and in doxorubicin resistant tumors compared to free doxorubicin.116,117 SP1049C is currently investigated in phase III in patients with metastatic adenocarcinoma of the oesophagus, gastroesophageal junction and stomach.
NC-6004 is composed of PEG and a poly(γ-benzyl L-glutamate)/cisplatin complex. In a small phase I study, it was shown that NC-6004 was well-tolerated by patients suffering from colorectal carcinoma, upper gastrointestinal tumors, non-small-cell lung carcinoma (NSCLC), melanoma and other tumor types.118,119 The pharmacokinetic analysis after the administration of NC-6004 displayed prolonged circulation of in the blood, delayed and sustained release of potentially active Pt species. A lower Cmax for ultrafilterable Pt compared with that of non-protein-bound cisplatin, might be responsible for reduction of cisplatin related toxicity. Furthermore, the higher AUC and half-life for ultrafilterable Pt compared with that of non-protein-bound cisplatin enhances the efficacy of NC-6004. Phase I study confirmed that NC-6004 exhibited pharmacokinetic profile completely different from those of cisplatin, resulting in the reduction of cisplatin-related toxicity.120 A DLT phase III study with NC-6004 in combination with gemcitabine is in progress with patients suffering from locally advanced pancreatic tumor and metastatic pancreatic tumor.121
Genexol-PM is a micellar paclitaxel formulation consisting of PEG and poly(D,L-lactic acid) (PDLLA).122 Preclinical in vivo studies with Genexol-PM demonstrated a 3-fold increase in the MTD and a significantly increased antitumor efficacy compared with free paclitaxel.123 The AUC of Genexol-PM was similar to paclitaxel, but the concentration of paclitaxel was 2–3 times higher in tissues, including liver, spleen, kidneys, lungs, heart and tumor. In phase I studies, a MTD 390 mg m−2 every 3 weeks or 120 mg m−2 every week was determined without the occurrence of hypersensitivity reactions.122,124 In phase II studies, Genexol-PM was found to be effective and safe with high response rates in patients suffering from metastatic breast cancer and advanced pancreatic cancer.125–128 It has been under clinical trials for several types of tumors including breast, ovarian,122,125,126 pancreatic,127 non-small-cell lung tumor.129
3.3 Polymeric liposomes
Liposome was first described in 1965 and is considered the most established drug-delivery vehicle.130 Liposomes are artificial, single, or multilaminar vesicles made with bilayered membrane structures, composed of natural or synthetic amphiphilic lipid molecules (Fig. 9). As drug delivery carriers, liposomes exhibit several unique properties including favorable safety profiles, long systemic circulation half-life, and ease of surface modifications.131 One major advantage of this technology is its ability to work with both hydrophobic and hydrophilic drugs; hydrophobic drugs can be enclosed within the phospholipid bilayers, while hydrophilic drugs can be entrapped in the aqueous cavity.132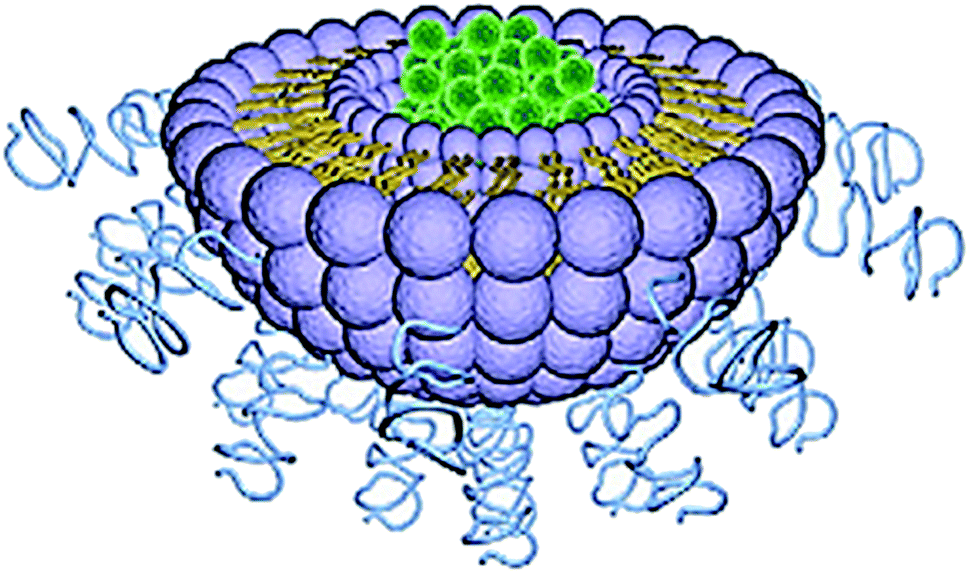 | ||
| Fig. 9 Structure of liposome. Reprinted with permission from ref. 11, Elsevier. | ||
PEGylated liposomal doxorubicin (Doxil) was the first liposomal drug formulation approved by the FDA, for the treatment of AIDS associated with Kaposi's sarcoma in 1995.133 Doxil consists of a packed PEGylated surface (2 kDa PEG chains) and is loaded with doxorubicin. The modification increases the circulatory half-life of the drug leading to its enhanced bioavailability at the tumor site.134 PEGylated liposomal doxorubicin has fewer side effects on healthy cells than regular doxorubicin.135,136 PLD has improved pharmacokinetic features, such as long circulation time of about 60–90 h for doses in the range of 35–70 mg m−2 in patients with solid tumors. After PLD administration, nearly 100% of the drug in the plasma remains in the encapsulated form. Moreover, in comparison to free doxorubicin PLD, plasma clearance is dramatically slower and its volume of distribution remains very small, which is roughly equivalent to the intravascular volume.137–139 It is currently used to treat Kaposi's sarcoma and recurrent ovarian tumor (Table 6).
| Polymeric liposome | Polymer | Drug | Function | Clinical trials | Reference |
|---|---|---|---|---|---|
| Doxil®/Caelyx® | PEG | Doxorubicin | Breast tumor, ovarian tumor, multiple myeloma, Kaposi's sarcoma | Approved | 133 |
| Thermodox® | PEG | Doxorubicin | Liver tumor, breast tumor | Phase III | 140 and 141 |
| Lipoplatin | PEG | Cisplatin | NSCLC, breast tumor, gastric tumor | Phase III | 142 and 143 |
| SPI-77 | PEG | Cisplatin | Ovarian tumor | Phase II | 150 |
| AmBisome | Amphotericin B | In tumor patients with neutropenia | Approved | 151 | |
| DaunoXome | Daunorubicin | Kaposi's sarcoma | Approved | 152 | |
| DepoCyt | Cytarabine | Leukemia | Phase III | 153 | |
| Glioblastoma | Phase I/II | ||||
| Myocet® | Doxorubicin | Breast tumor | Approved | 154 |
ThermoDox, is doxorubicin-loaded PEGylated liposome which releases encapsulated doxorubicin at elevated tissue temperature. For ThermoDox, the concentration of the drug is up to 25 times more in the treatment area than doxorubicin, and has several fold higher concentration of other liposomally encapsulated doxorubicins.140,141 Currently, it is under phase III clinical trial for hepatocellular carcinoma.
Lipoplatin is another promising PEGylated cisplatin liposomal platinum drug formulations under clinical investigation comprising ∼9% cisplatin and ∼91% lipids (w/w) corresponding to a drug-to-lipid ratio of 1:10 (ref. 142 and 143) and methoxy-PEG distearoyl phosphatidylethanolamine (mPEG2000-DSPE). Phase I human studies of Lipoplatin albeit revealed its mild hematological and gastrointestinal toxicity, did not show other side effects unlike cisplatin treatment.144 Phase II clinical studies of Lipoplatin in combination with gemcitabine demonstrated significant clinical benefit of the combination regimen in patients previously resistant to chemotherapy11 and presently it is in various phase II/III clinical studies, for NSCLC,145 HER2/neu negative metastatic breast tumor146 and advanced gastric tumor.147–149
SPI-77, a PEGylated liposomal formulation of cisplatin, is in phase II trials for patients with recurrent epithelial ovarian tumor,150 and a nano liposomal formulation of irinotecan (CPT-11) is in phase I trials for glioma. The drug loading is much lower (drug to lipid ratio ∼1:70) compared to lipoplatin. This drug did not progress to phase III because of a lack of activity in phase II trials.
Non-PEGylated liposomal drugs used in clinical practice today include AmBisome (amphotericin B liposomes), DaunoXome (daunorubicin liposomes), Myocet (Doxorubicin), DepoCyt (cytarabine liposomes), and Visudyne (verteporfin liposomes).151–154
3.4 Other polymeric nanoparticles
Nanoparticle drug delivery agents can improve the clinical performance of conventional chemotherapeutic agents by changing the biological disposition, reducing toxicity, enhancing the efficacy and perhaps attenuating the dose of therapy.155–158 For successful implementation of the required functional properties in the nano-delivery system, its structure should be chemically flexible. Polymer based delivery system possess a unique advantage over other nano-carriers in this regard.159–165 Structure of a polymeric nanoparticle is depicted in Fig. 10. Polymeric NPs have been used as a preferred nanoscale drug delivery vehicle especially for their excellent endocytosis efficiency, passive tumor-targeting, high encapsulation efficiency, and delivery of a wide range of therapeutic agents.166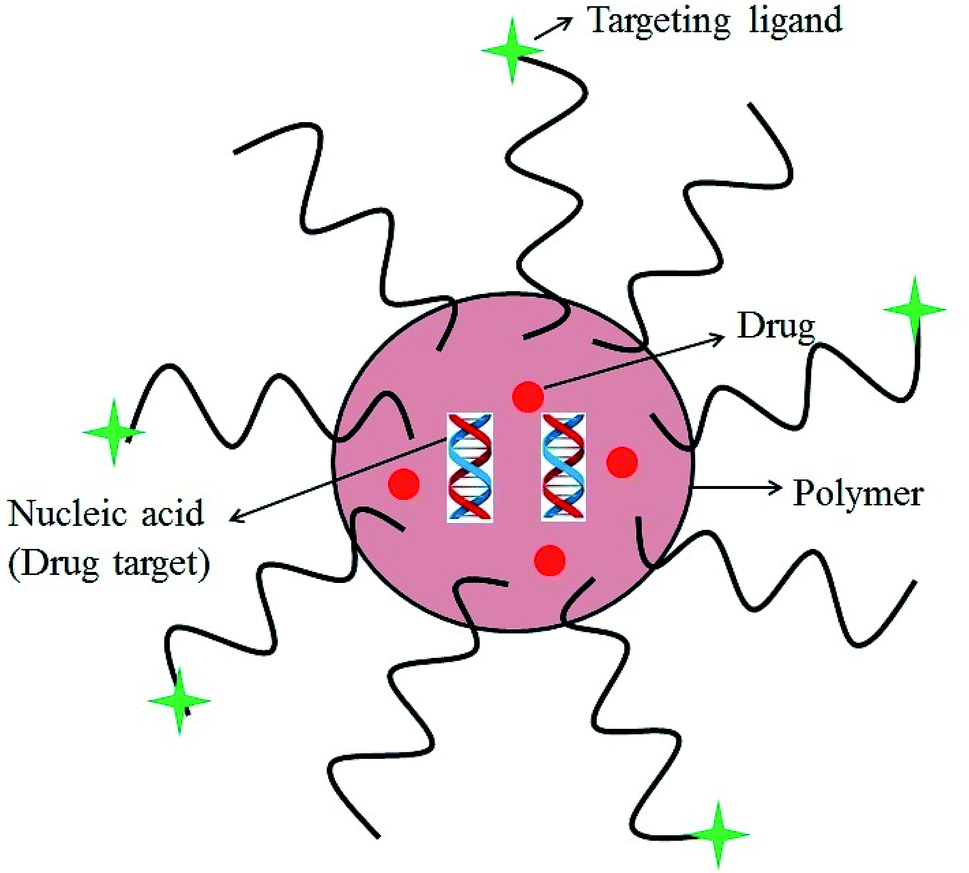 | ||
| Fig. 10 A polymeric nanoparticle. | ||
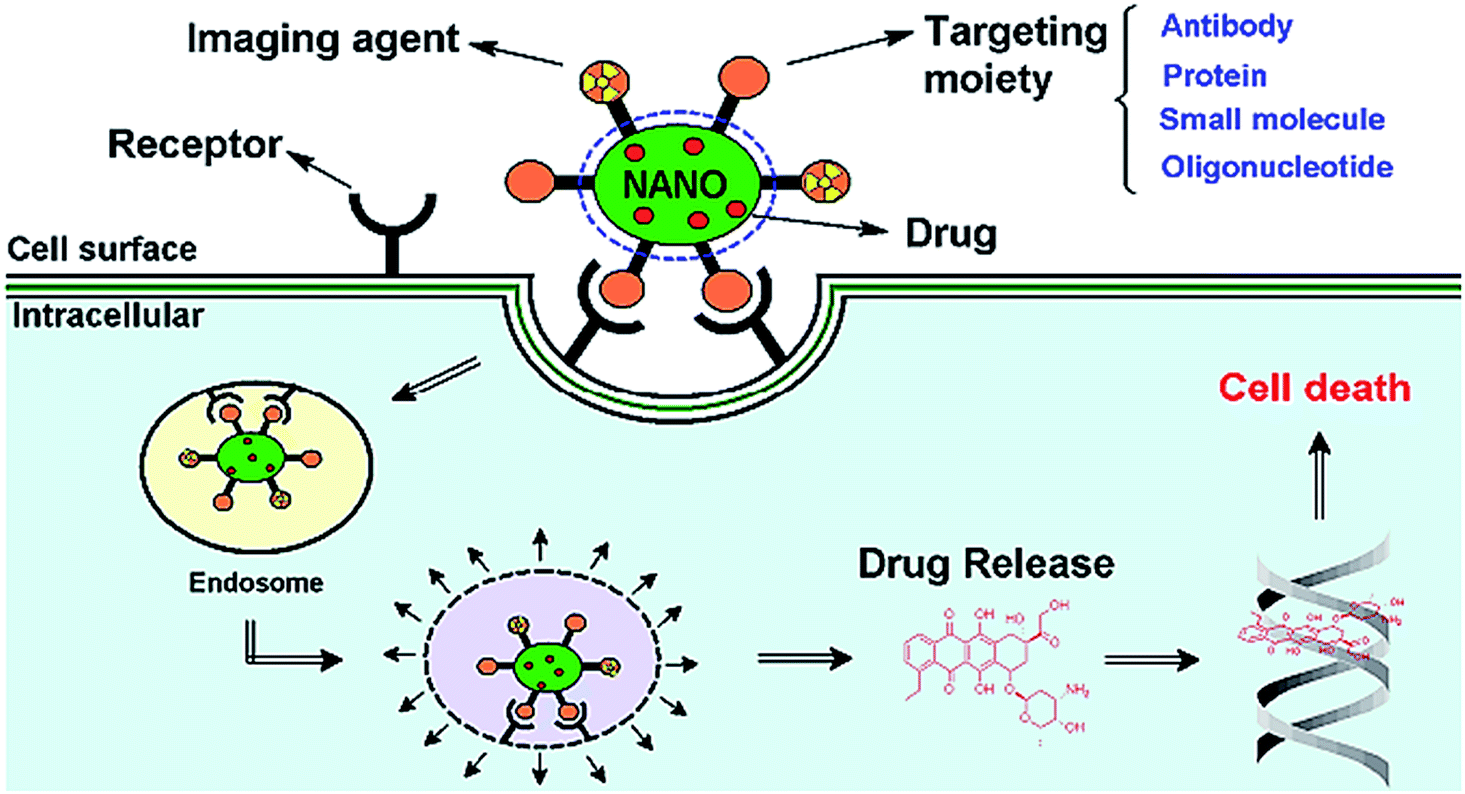 | ||
| Fig. 11 Schematic diagram representing the mode of action of targeted multifunctional nanoparticle (NP). Reprinted with permission from ref. 170. | ||
In tumor, targeted polymeric nanoparticles (NPs) can be used to deliver chemotherapies to tumor cells with greater efficacy and reduced cytotoxicity on peripheral healthy tissues. Biodegradable polymers such as poly(D,L-lactic acid) (PLA), poly(D,L-lactide-co-glycolide) (PLGA) and poly(ε-caprolactone) PCL and their copolymers diblocked or multiblocked with PEG have been commonly used to form polymeric nanoparticles (NPs) to encapsulate a variety of antitumor drugs (Fig. 12).
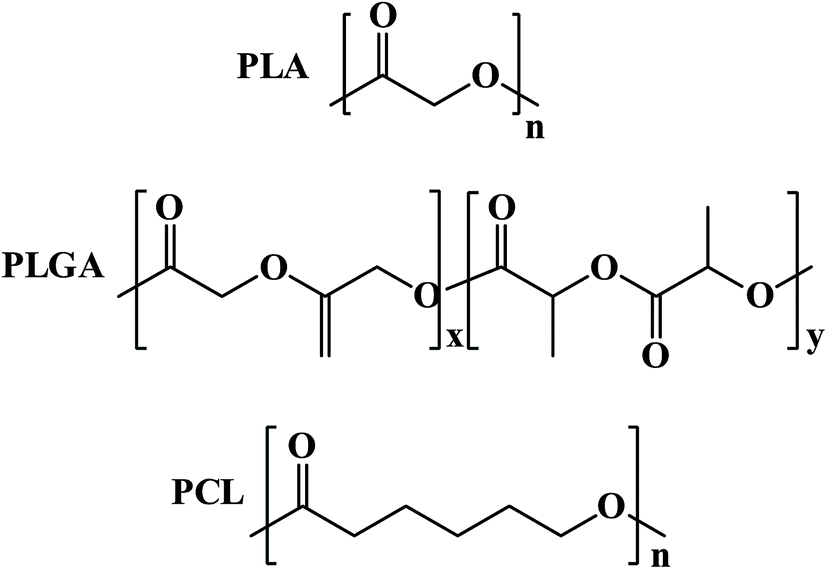 | ||
| Fig. 12 Structures of biodegradable polymers for encapsulation of antitumor drugs. | ||
9-Nitrocamptothecin (9-NC), derivative of camptothecin is a promising antitumor agent which targets topoisomerase-I enzyme. On the other hand, camptothecin derivatives lose their antitumor activity due to pH dependent rapid and reversible hydrolysis from closed lactone ring to the inactive hydroxyl carboxylated form. PLGA has been used to encapsulate 9-NC successfully by nano-precipitation methods having more than 30% encapsulation efficiency with its complete biological activity and without disturbing lactone ring.174 The in vitro drug release profile showed a sustained 9-NC release up to 160 h indicating the suitability of PLGA nanoparticles in controlled 9-NC release. The in vitro drug release profile showed a sustained 9-NC release up to 160 h indicating the suitability of PLGA nanoparticles in controlled 9-NC release.
Paclitaxel has neoplastic activity against primary ovarian carcinoma breast and colon tumors but due to its poor solubility paclitaxel has been rendered less useful for clinical administration. PLGA intermingled with vitamin E, and tocopheryl polyethylene glycol succinate (TPGS) has been used to encapsulate and in vitro controlled release of this drug has been studied.175,176 Using some additive to the PLGA nanoparticles 100% drug encapsulation efficiency was achieved with its full antitumor activity. It has also been demonstrated that incorporation of paclitaxel in the PLGA nanoparticles strongly enhances its antitumoral efficacy as compared to free drug with prolonged incubation times with cells.176
The cisplatin has been encapsulated on PLGA-mPEG nanoparticles. PLGA-methoxy(polyethylene glycol) (mPEG) nanoparticles revealed prolonged drug residence in blood upon intravenous administration.177 However, at the targeted site PLGA-mPEG nanoparticles encapsulated with cisplatin exhibited rapid degradation and sustained release. These characteristics help in preventing the tumor growth.
PEG-PCL amphiphilic block copolymeric nanospheres containing taxol with a relatively high drug loading of more than about 20% are reported to show promising antitumor activity.178 It was reported that this mPEG/PCL diblock copolymeric nanospheres system could be potentially useful as a novel delivery system for antitumor drug taxol having outer shell of mPEG and the hydrophobic inner core of PCL. In addition, considering the extremely lipophilic characteristics of taxol, this mPEG/PCL nanosphere system with high taxol loading content and suspended properties in water could be useful for the delivery of taxol.179
Abraxane® is albumin-bound nanoparticles of paclitaxel that has been successfully used to deliver paclitaxel for the treatment of metastatic breast tumor after failure of combination chemotherapy for metastatic disease or relapse within 6 months of adjuvant chemotherapy.182,183 Abraxane has entered phase III study which demonstrated the higher tumor response rates, a better safety profile and improved survival compared with conventional paclitaxel, in patients receiving second-line chemotherapy.
4 Recent developments in polymer drug conjugates
Two polymeric DOX nano-conjugates were developed, for which the design was based on the use of multi-functionalized poly(ethylene oxide)-block-poly(3-caprolactone) (PEO-b-PCL) micelles decorated with αvβ3 integrin targeting ligand (i.e. RGD4C) on the micellar surface.184 The delivery systems are based on poly(ethylene oxide)-block-poly(3-caprolactone) (PEO-b-PCL) micelles functionalized on the micellar shell (PEO) as well as the micellar core (PCL) (Fig. 13). The pH-triggered drug release, cellular uptake, intracellular distribution, and cytotoxicity against MDA-435/LCC6WT (a DOX-sensitive tumor cell line) and MDA-435/LCC6MDR (a DOX-resistant clone expressing a high level of P-glycoprotein) were evaluated. In MDA-435/LCC6WT, the best cytotoxic response was achieved using RGD4C-PEO-b-P(CL-Hyd-DOX), that correlated with the highest cellular uptake and preferential nuclear accumulation of DOX. In MDA-435/LCC6MDR, RGD4C-PEO-b-P(CL-Ami-DOX) was the most cytotoxic, and this effect correlated with the accumulation of DOX in the mitochondria.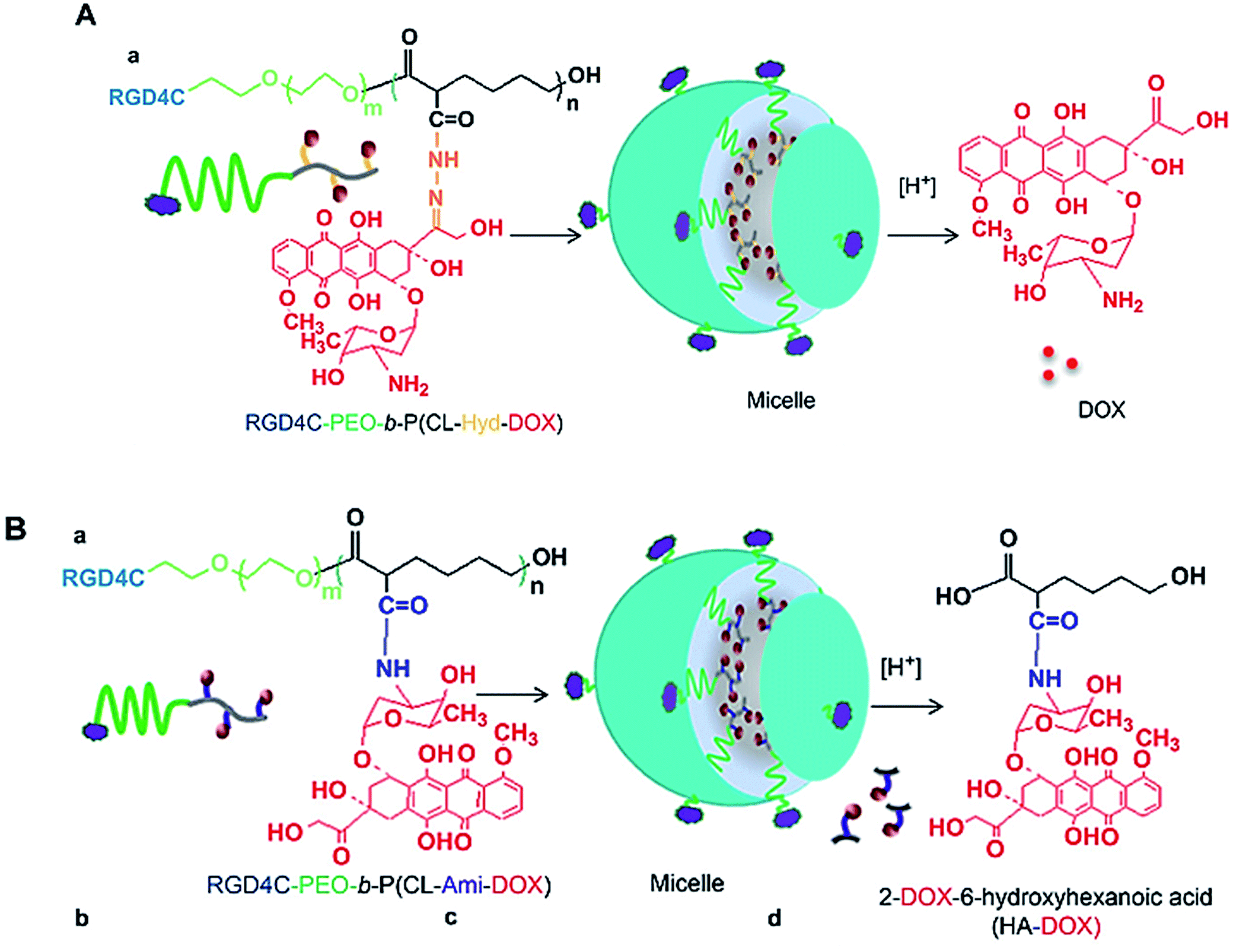 | ||
| Fig. 13 Self-assembly of (A) RGD4C-PEO-b-P(CL-Hyd-DOX) and (B) RGD4C-PEO-b-P(CL-Ami-DOX) copolymers into targeted micelles. Reprinted with permission from ref. 184, Elsevier. | ||
Chen et al. reported a nano carrier system in which Pt(II)-based drugs and catalase were incorporated into the aqueous core of PLGA which releases both Pt(II)-based drugs and O2 in response to the local H2O2 concentration which is typical of tumor environments (Fig. 14).185 Cisplatin loading capacity of the NPs and release kinetics with and without H2O2 using inductively coupled plasma optical emission spectrometry (ICP-OES) was carried out which showed negligible cisplatin release from the NPs in the absence of H2O2 within 24 h, signifying the degradation process of PLGA was rather slow. In contrast, the release rate of cisplatin from NPs in presence of H2O2 was much faster and a higher amount of cisplatin was released. The release reached a plateau after incubation with 100 mM H2O2 for about 15 h, about 80% of cisplatin originally encapsulated within the NPs was released into the medium. In vitro cytotoxicity of the cisplatin loaded PLGA NPs was compared with free cisplatin. The quantity of cisplatin released from NPs that afforded 50% cell death was significantly lower than in case of free cisplatin. However, PLGA NPs without catalase showed a much lower in vitro cytotoxicity. In these nanocarrier systems, Pt(II)-based drug exhibited the antitumor effect while O2 was utilized to overcome hypoxia-induced multidrug resistance (MDR).185
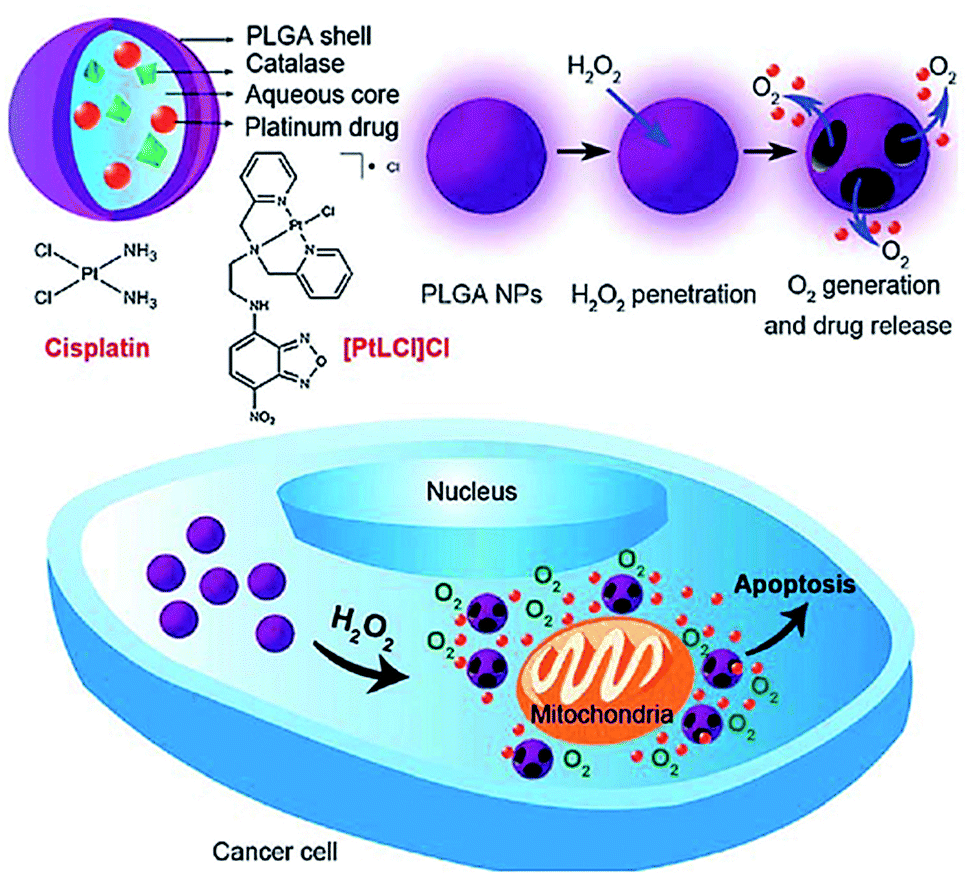 | ||
| Fig. 14 Schematic illustration of H2O2-responsive PLGA nanoparticles containing Pt(II) drugs and O2-generating catalase and the mechanism of drug release by H2O2. Reprinted with permission from ref. 185, Royal Society of Chemistry. | ||
In another recent study, Chen et.al. synthesized a novel redox-responsive polymer–drug conjugates based on hydrophilic diblock copolymer covalently linked paclitaxel (PTX) via a disulfide linker and evaluated as intracellular drug delivery.186 Utilizing the hydroxyl groups as an active reaction site, paclitaxel was covalently conjugated onto the backbone of diblock copolymer, with a disulfide linker as spacer to bridge copolymer and PTX (Scheme 1). Due to the distinguishing solubility of segments in the polymer–drug conjugate, the amphiphilic PEG-b-P(HEMA-PTX) could self-assemble into spherical micelles in aqueous solution, with hydrophobic PTX as core and hydrophilic PEG chains as a shell. The in vitro cytotoxicity results showed that the diblock copolymer was biocompatible, with no obvious cytotoxicity, whereas the PEG-b-P(HEMA-PTX) conjugate showed glutathione-dependent cytotoxicity with higher cellular proliferation inhibition against glutathione monoester pretreated HeLa cells than that of non-pretreated HeLa cells.
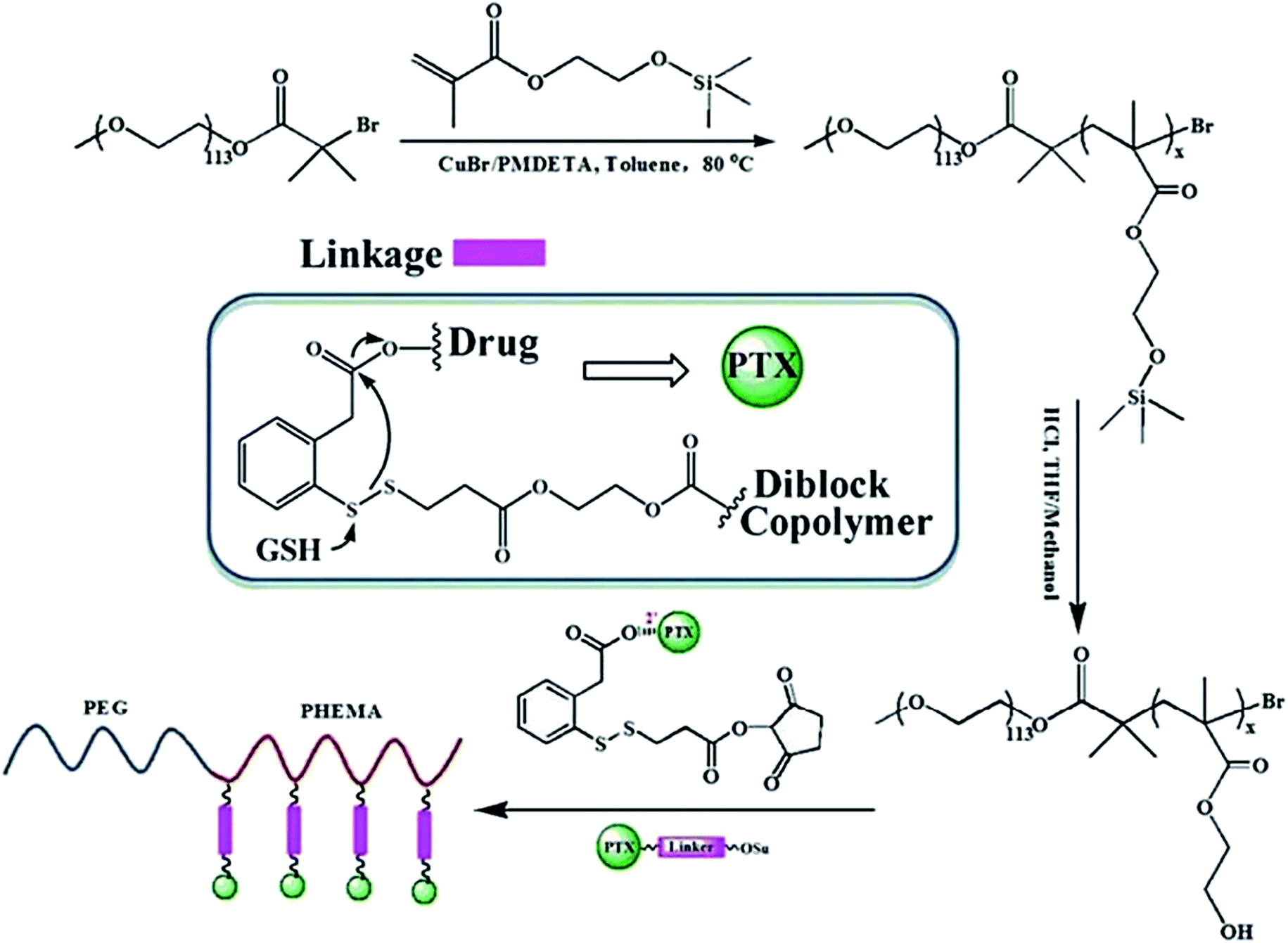 | ||
| Scheme 1 Schematic illustration of the synthesis of polymer drug conjugates of PEG-b-P(HEMA-PTX) via an disulfide linker and its drug release mechanism. Reprinted with permission from ref. 186, Royal Society of Chemistry. | ||
A CD44-targeted macromolecular conjugate of docetaxel (DTX) was prepared via a pH-sensitive linkage to hyaluronic acid by Dinarvand et. al. which were evaluated for drug release, cytotoxicity, cellular uptake, cell cycle inhibition, and subacute toxicity in mice.187 Cellular microscopic studies revealed that CD44-expressing cells including MCF-7 cancer stem cells and MDA-MB-231 metastatic breast cancer cells had internalized the conjugates via a selective receptor mediated mechanism, leading to cell cycle arrest in the G2/M phase. The toxicity of the prepared HA-DTX conjugates was tested in NIH-3T3 fibroblast cells and was compared with free DTX. Results indicated that HA-DTX had no inhibitory effect from 5.0 to 300 μM while free DTX was associated with toxicity, which was significant at 300 μM.188 Furthermore, HA-DTX conjugates showed specific toxicity only in CD44-expressing cells in vitro, along with a decreased risk of neutropenia and dose–dependent mortality in vivo.
In a recent study, Huang et. al. described a novel fluorescence method for HAase detection and tumor-targeting drug delivery and imaging, using a probe prepared by electrostatic assembly of a cationic conjugated polymer (CCP) and anionic hyaluronan (HA) conjugated with the anticancer drug, doxorubicin (Fig. 15).189 Poly{[9,9-bis(6′-(N,N,N-diethylmethylammonium)hexyl)-2,7-fluorenylene ethynylene]-alt-co-[2,5-bis(3′-(N,N,N-diethylmethylammonium)-1′-oxapropyl)-1,4-phenylene]} tetraiodide (PFEP) was used as CCP. In vitro Dox release of PFEP/HA-Dox complex in presence of HAase was carried out which demonstrated that Dox was released quickly at the first 15 min and gradually reached a plateau after 25 min. From these experiments, it could be inferred that PFEP/HA-Dox complex can be employed as a multifunctional system; tumor targeting drug delivery and cell imaging through the specific HA-CD44 binding and the degradation of HA by HAase in tumor cells.
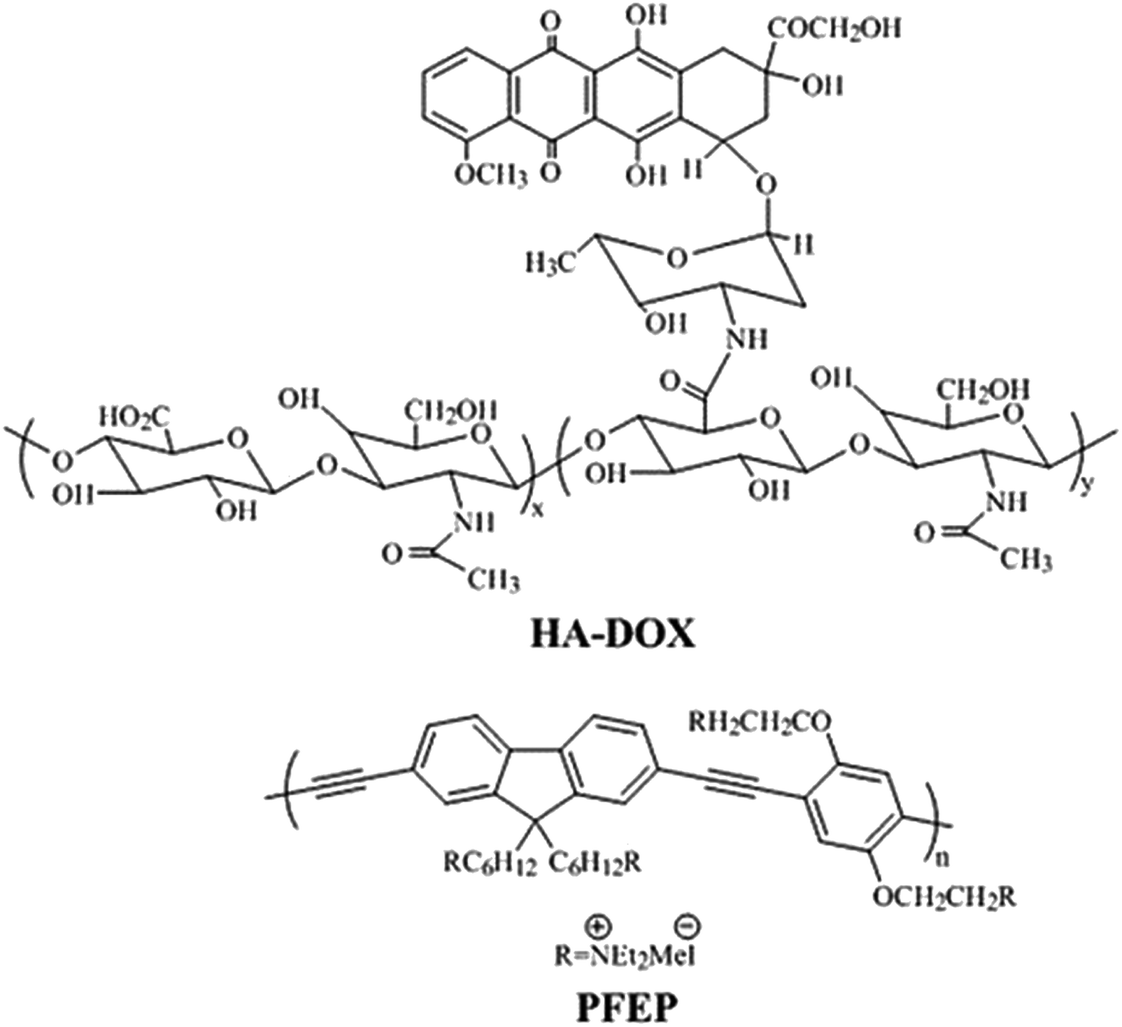 | ||
| Fig. 15 Chemical structure of HA-DOX and PEEP. Reprinted with permission from ref. 189, American Chemical Society. | ||
In another study two water soluble conjugates with an azide linker (PEG-N3-PTX) or a succinic linker (PEG-suc-PTX) were designed with 6 and 20 kDa MW PEG using ‘click chemistry’ for pulmonary application in lung cancer.190 Conjugation to polyethylene glycol improved the solubility of paclitaxel. Also, the conjugates of both molecular weights showed good stability in broncho alveolar lavage (half-life of 3 to 9 h) and in phosphate buffer saline pH 6.9 (half-life ≥ 72 h), but hydrolyzed quickly in mouse serum (half-life of 1 to 3 h). However, the conjugates showed less cytotoxicity to B16-F10 melanoma cells and LL/2 Lewis lung cancer cells than free paclitaxel or Taxol. The IC50 values of all the conjugates were 5-fold higher as compared to Taxol and 7-fold higher than free PTX which could be attributed due to the slow release of PTX from the conjugates. In vitro cytotoxicity results revealed that the formation of the prodrugs at 2′-OH on PTX increased the IC50 of all the conjugates.
Shao et. al. designed a smart prodrug of doxorubicin, Ac-Phe-Lys-PABC-ADM in which a dipeptide Phe-Lys specific for cathepsin B, and PABC (para-aminobenzyloxycarbonyl) as a self-immolative spacer was tested on gastric cancers (Fig. 16).191 In normal tissues and peripheral blood where there is little cathepsin B activity, this prodrug was found to be inactive. Since cathepsin B is activated and overexpressed on the exterior membrane of the invading cancer cells, which is responsible for the cleavage of the Phe-Lys dipeptide at the Lys-PABC bond. The resulting PABC-doxorubicin decomposes at once to para-aminobenzyl alcohol, CO2, and free doxorubicin, resulting in direct killing of the invading cancer cells.
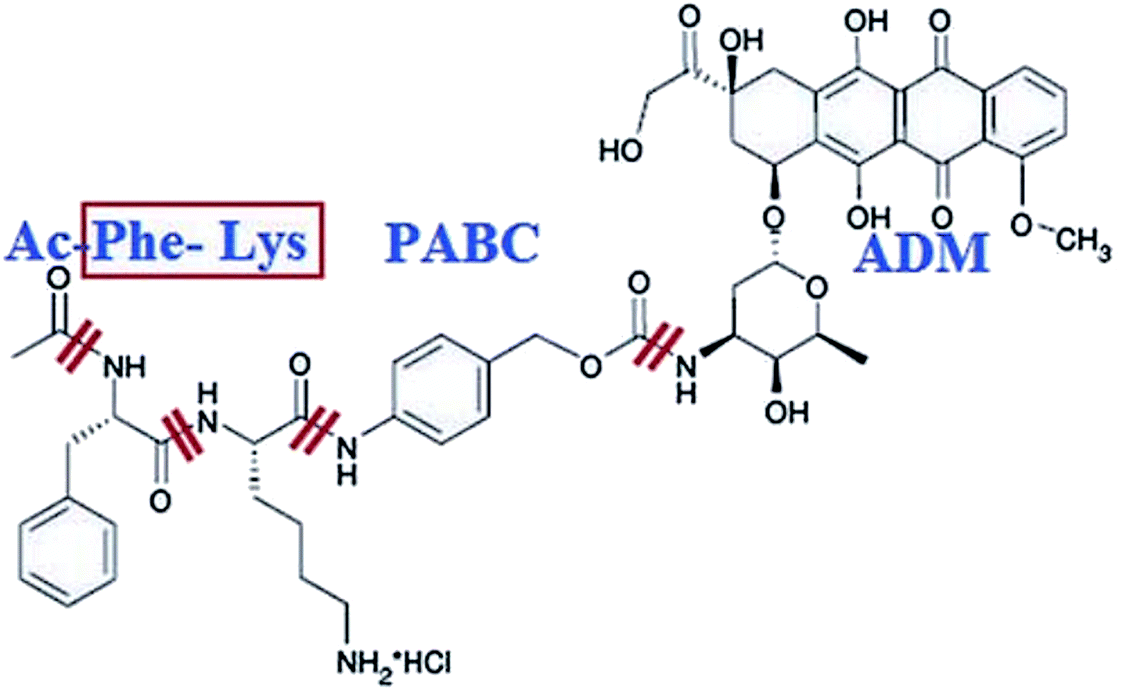 | ||
| Fig. 16 Structure of Ac-Phe-Lys-PABC-ADM hydrochloride. Reprinted with permission from ref. 191, John Wiley & Sons. | ||
Further, in vitro release study of Ac-Phe-Lys-PABC-ADM showed that the half-life of doxorubicin release at 37 °C was 16 minutes in cathepsin B solution, but no changes were observed over 6 to 7 hours period in human plasma.192 However, the prodrug has not yet been evaluated in a real life cancer model due to some limitations (i) it focused on only a single model system, limiting the antitumor spectrum of Ac-Phe-Lys-PABC-ADM and (ii) pharmacological and pharmacodynamic studies were not completely conducted.
Etrych et al. designed and synthesized two types of HPMA copolymer conjugates of doxorubicin; (i) the linear non-degradable copolymer and (ii) biodegradable high molecular weight diblock copolymer; partly bound via a pH-sensitive hydrazone and partly via enzymatically degradable amide bonds, each contributing to different mechanism of drug release of the polymer–doxorubicin conjugate.193 The results revealed that the high molecular weight diblock copolymer carrier containing a degradable disulphide bond between the polymer blocks displayed a rapid degradation in a buffer containing glutathione within the first few hours of incubation. The conjugate with the amide bond-bound DOX require the presence of lysosomal enzyme cathepsin B to release DOX, whereas the polymer–drug conjugate with the DOX bound via a hydrazone bond released DOX by pH-sensitive hydrolysis in a buffer of pH 5.0 at 37 °C but was fairly stable in buffer solutions at pH 7.4 at 37 °C, mimicking the blood stream and the endosomal environment of the target cells, respectively.
From these experimental studies, it could be inferred that the controlled release of DOX was achieved either by a simple spacer tailored for pH-controlled hydrolysis, providing the drug intracellular release, or by a more sophisticated oligopeptide spacer, enabling enzymatically controlled intracellular drug release in presence of a lysosomal enzyme.193
Recently Zhang et. al. synthesized a polymer, poly-L-lysine-lipoic acid (PLL-LA), by dimethylmaleic anhydride (DA) and applied it as a carrier which exhibited enhanced cell internalization and intracellular pH- and reduction-triggered Dox release.194 The results of MTT assay, including Dox·HCl (free drug) and Dox-micelles, displayed a dose–dependent higher cytotoxicity against A549 cell lines at pH 6.8 as compared to pH 7.4. This superior cytotoxicity of Dox-micelles at pH 6.8 was attributed to the enhanced cell internalization. These results demonstrated that the stepwise pH/reduction-responsive DA-PLL-LA micelles showed high cytotoxicity to A549 cancer cells in a pH-dependent way.
X. Chen et al. recently designed a dual sensitive dual drug backboned shattering polymer (DDBSP); starting from a PP2A inhibitor demethylcantharidin (DMC) and cisplatin, with exact composition at a fixed DMC/Pt ratio (1:2) for precise nanomedicine.195 Rapid breakdown of the polymer chain and release of the drug payload in the cancer cells is vital for the anticancer activity. This DDBSP self-assembled nanoparticle (DD-NP) was triggered intracellularly to break down in a chain-shattering manner under intracellular reduction/acidic microenvironment for optimal anticancer efficacy to release the dual drugs payload. The anticancer efficacy of DD-NP on the above PDLC model was further evaluated. The results revealed that though DD-NP were the most potent on inhibition of tumor growth, they also showed least effect on loss of mice body weight and higher survival rate. Moreover, DD-NP can be applied as contrast agent to directly track the drug itself and NP via Pt DMCT (drug-mediated computer tomography) and ICP-MS both in vitro and in vivo due to the exceptional high content of heavy metal Pt in polymer backbone. The authors claim, that DD-NP could be the first example of nanomedicine tackling the major challenges together including precise composition, direct fate monitoring of drug, drug evaluation and screening on reliable cancer models, validating the possible use of DD-NP in clinic.
In another work, X. Chen et al. reported the synthesis of photo-activatable Pt(IV) amphiphiles containing one or two hydrophilic lactose targeting ligands per hydrophobic Pt(IV) prodrug for an all-in-one precise nanomedicine.196 The self-assembly of these Pt(IV) amphiphiles resulted in either micelle or vesicle formation with a fixed Pt/targeting moiety ratio and high content of platinum. These micelles and vesicles were capable of hepatoma cell targeting, fluorescence/Pt-based CT imaging and have shown effective anticancer efficacy under laser irradiation in vitro and in vivo. The Pt biodistribution of tissues determined by average fluorescence signals and CT values corresponded well to the results detected by ICP-MS.
5 Summary and future directions
In this review, a detailed overview of some intelligent polymer based antitumor drug delivery approaches has been discussed, which include polymer–drug conjugates, polymeric micelles, polymeric liposomes and nanoparticle-assisted antitumor drug delivery agents.Polymer based drug delivery systems possess some advantages as compared to small drug molecules such as (i) improved pharmacokinetic profile with longer circulation time and (ii) potential for tissue targeting. Many PEG and HPMA-based polymer–drug conjugates have progressed into clinical development, but their market approval has been hindered by their non-biodegradable nature, reduced efficacy of therapy, instability and toxicity. Targeting moieties linked to the polymers by either chemical attachment or the physical encapsulation directs the drug to the target site of pharmacological action. However, there is a tremendous scope for the improvement of the design of polymer based delivery systems.
Our current understanding of drug targeting to tumor cells is based on the combination of a few independent phenomena involving EPR effect, properties of nanoparticles, increased retention in the blood circulation, and the type of ligand–receptor interactions. Nanoparticles have emerged as a promising scaffold for drug delivery because of low inherent toxicity, high surface area and tunable stability that provide a useful complement to more traditional delivery vehicles. NP-mediated drug delivery systems seem to be a promising strategy as they have advantages over conventional drug delivery systems. They can increase the bioavailability, solubility and permeability of many potent drugs which are otherwise difficult to deliver orally. There is a need to develop non-toxic, stable, and controlled drug delivery system for cancer therapy.
To address these issues, biodegradable conjugates could offer advantages in terms of biocompatibility, efficacy, ability to optimize pharmacokinetics via increase in molecular weight, and safety, due to their biodegradation and elimination post-treatment. Biodegradable polymeric systems can offer immense flexibility in customization and optimization of nano-carriers to efficiently deliver new therapeutics and provide an integral step in aiding their progression to clinical practice.
There is a need to synthesize more conjugates using numerous lower molecular weight drugs with hydrophilic polymers and to evaluate them on various cancer models. There is a need for more clinical data to fully understand the advantages and disadvantages of polymer therapeutics. This new generation of polymer platforms could be more promising to improve cancer chemotherapy. Another aspect in this regard could be time-controlled targeted drug delivery which can be achieved depending on the features of the polymer by modulating the structure of the polymer which can grant either of these; an extended release formulation (either by swelling of polymeric matrix or degradation of polymeric capsules), a sustained-release formulation, a pulsatile-release formulation (in which drugs are released only when required by the body) and a delayed-release formulation, in which the active ingredient is released at a time other than at the time of administration.
Conflicts of interest
The authors declare that they have no conflicts of interest to disclose.Acknowledgements
The author Dr Shazia Parveen acknowledge Taibah University, Yanbu Branch, Saudi Arabia for the support.References
- M. Shaha, V. Pandian, M. A. Choti, E. Stotsky, J. M. Herman, Y. Khan, C. Libonati, T. M. Pawlik, R. D. Schulick and A. E. Belcher, Support. Care Cancer, 2011, 19(2), 271 CrossRef PubMed.
- N. H. Rekers, E. G. C. Troost, C. M. L. Zegers, W. T. V. Germeraad, L. J. Dubois and P. Lambin, Cancer Radiother., 2011, 18(5–6), 391 Search PubMed.
- L. L. Lazdunski, Lung Cancer, 2014, 84(2), 103 CrossRef PubMed.
- S. B. Baylin and J. E. Ohm, Nat. Rev. Cancer, 2006, 6(2), 107 CrossRef CAS PubMed.
- B. Rosenberg, L. V. Camp and T. Krigas, Nature, 1965, 205, 698 CrossRef CAS PubMed.
- T. C. Johnstone, S. Kogularamanan and S. J. Lippard, Chem. Rev., 2016, 116(5), 3436 CrossRef CAS PubMed.
- T. C. Johnstone, S. M. Alexander, J. J. Wilson and S. J. Lippard, Dalton Trans., 2015, 44, 119 RSC.
- J. Graham, M. Muhsin and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2004, 3, 11 CrossRef CAS PubMed.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113 RSC.
- Lobaplatin, Drugs R&D, 2003, 4, 369 Search PubMed.
- A. A. Argyriou, P. Polychronopoulos, G. Iconomou, E. Chroni and H. P. Kalofonos, Cancer Treat Rev., 2008, 34, 368 CrossRef CAS PubMed.
- S. R. McWhinney, R. M. Goldberg and H. L. McLeod, Mol. Cancer Ther., 2009, 8, 10 CrossRef CAS PubMed.
- H. S. Oberoi, N. V. Nukolova, A. V. Kabanov and T. K. Bronich, Adv. Drug Deliv. Rev., 2013, 65, 1667 CrossRef CAS PubMed.
- V. Patil, K. Gada, R. Panwar, S. Majewski, Y. Tekabe, A. Varvarigou and B. A. Khaw, J. Drug Target., 2013, 21(10), 1012 CrossRef CAS PubMed.
- C. Qian, P. Feng, J. Yu, Y. Chen, Q. Hu, W. Sun, X. Xiao, X. Hu, A. Bellotti, Q. D. Shen and Z. Gu, Angew. Chem., Int. Ed., 2017, 56, 2588 CrossRef CAS PubMed.
- Q. Hu, Q. Chen and Z. Gu, Biomaterials, 2018, 178, 546 CrossRef CAS PubMed.
- L. Mei, Z. P. Zhang, L. Y. Zhao, L. Q. Huang, X. L. Yang, J. T. Tang and S. S. Feng, Adv. Drug Deliv. Rev., 2013, 65(6), 880 CrossRef CAS PubMed.
- S. Gao, G. Tang, D. Hua, R. Xiong, J. Han, S. Jiang, Q. Zhang and C. Huang, J. Mater. Chem. B, 2019, 7, 709 RSC.
- H. Xiao, L. Yan, E. M. Dempsey, W. Song, R. Qi, W. Li, Y. Huang, X. Jing, D. Zhou, J. Ding and X. Chen, Prog. Polym. Sci., 2018, 87, 70 CrossRef CAS.
- Y. Wang, A. G. Cheetham, G. Angacian, H. Su, L. Xie and H. Cui, Adv. Drug Deliv. Rev., 2017, 110–111, 112 CrossRef CAS PubMed.
- Y. F. Fouad and C. Aanei, Am. J. Cancer Res., 2017, 7(5), 1016 CAS.
- R. J. Seager, C. Hajal, F. Spill, R. D. Kamm and M. H. Zaman, Convergent Sci. Phys. Oncol., 2017, 3, 034002 CrossRef PubMed.
- M. J. Akhtar, M. Ahamed, H. A. Alhadlaq, S. A. Alrokayan and S. Kumar, Clin. Chim. Acta, 2014, 436, 78 CrossRef CAS PubMed.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347 CrossRef CAS PubMed.
- J. P. Patil and H. S. Mahajan, Int. J. Pharm. Sci. Res., 2015, 6(11), 4611 CAS.
- S. Hobel and A. Aigner, Methods Mol. Biol., 2010, 623, 283 CrossRef PubMed.
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1 CrossRef CAS PubMed.
- J. Jacob, J. T. Haponiuk, S. Thomas and S. Gopi, Mater. Today Chem., 2018, 9, 43 CrossRef CAS.
- R. Tong and J. J. Cheng, Polym. Rev., 2007, 47, 345 CrossRef CAS.
- M. Galanski and B. K. Keppler, Anti Cancer Agents Med. Chem., 2007, 7, 55 CrossRef CAS PubMed.
- K. Letchford and H. Burt, Eur. J. Pharm. Biopharm., 2007, 65, 259 CrossRef CAS PubMed.
- O. Pillai and R. Panchagnula, Curr. Opin. Chem. Biol., 2001, 5, 447 CrossRef CAS PubMed.
- L. Qiu and Y. Bae, Pharm. Res., 2006, 23, 1 CrossRef CAS PubMed.
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688 CrossRef CAS PubMed.
- R. Haag and F. Kratz, Angew. Chem., Int. Ed., 2006, 45, 1198 CrossRef CAS PubMed.
- F. Kratz and M. T. Schutte, Cancer J., 1998, 11, 60 CAS.
- S. van Zutphen and J. Reedijk, Coord. Chem. Rev., 2005, 249, 2845 CrossRef CAS.
- V. P. Torchilin, Eur. J. Pharm. Sci., 2000, 11, S81 CrossRef CAS PubMed.
- D. E. Gerber, Am. Fam. Physician, 2008, 77, 311 Search PubMed.
- M. Mimeault, R. Hauke and S. K. Batra, Clin. Pharmacol. Ther., 2008, 83, 673 CrossRef CAS PubMed.
- K. Strebhardt and A. Ullrich, Nat. Rev. Cancer, 2008, 8, 473 CrossRef CAS PubMed.
- X. Wang and Z. Guo, Chem. Soc. Rev., 2013, 42, 202 RSC.
- A. Beduneau, P. Saulnier, F. Hindre, A. Clavreul, J. C. Leroux and J. P. Benoit, Biomaterials, 2007, 28, 4978 CrossRef CAS PubMed.
- P. M. Deckert, Curr. Drug Targets, 2009, 10, 158 CrossRef CAS PubMed.
- M. Hong, S. Zhu, Y. Jiang, G. Tang and Y. Pei, J. Controlled Release, 2009, 133, 96 CrossRef CAS PubMed.
- A. Zensi, D. Begley, C. Pontikis, C. Legros, L. Mihoreanu, S. Wagner, C. Büchel, H. V. Briesen and J. Kreuter, J. Controlled Release, 2009, 137, 78 CrossRef CAS PubMed.
- F. Canal, M. J. Vicent, G. Pasut and O. Schiavon, J. Controlled Release, 2010, 146, 388 CrossRef CAS PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46(12 pt 1), 6387 CAS.
- F. Danhier, O. Feron and V. Préat, J. Controlled Release, 2010, 48, 135 CrossRef PubMed.
- L. Gros, H. Ringsdorf and H. Schupp, Angew. Chem., Int. Ed. Engl., 1981, 20, 305 CrossRef.
- S. Van, S. K. Das, X. Wang, Z. Feng, Y. Jin, Z. Hou, F. Chen, A. Pham, N. Jiang, S. B. Howell and L. Yu, Int. J. Nanomed., 2010, 5, 825 CrossRef CAS PubMed.
- P. Zhou, Z. Li and Y. Chau, Eur. J. Pharm. Sci., 2010, 41(3–4), 464 CrossRef CAS PubMed.
- G. Vilar, J. T. Puche and F. Albericio, Curr. Drug Deliv., 2012, 9(4), 367 CrossRef CAS PubMed.
- B. Jung, Y. C. Jeong, J. H. Min, J. E. Kim, Y. J. Song, J. K. Park, J. H. Park and J. D. Kim, J. Mater. Chem., 2012, 22, 9385 RSC.
- F. Kratz, I. A. Muller, C. Ryppa and A. Warnecke, Curr. Med. Chem., 2008, 3(1), 20 CAS.
- F. Greco and M. J. Vincent, Adv. Drug Deliv. Rev., 2009, 61, 1203 CrossRef CAS PubMed.
- N. Larson and H. Ghandehari, Chem. Mater., 2012, 24, 840 CrossRef CAS PubMed.
- C. Li and S. Wallace, Adv. Drug Deliv. Rev., 2008, 60, 886 CrossRef CAS PubMed.
- S. A. Senevirathne, S. Boonsith, D. Oupicky, M. C. Biewer and M. C. Stefan, Polym. Chem., 2015, 6, 2386 RSC.
- R. Duncan, M. J. Vicent, F. Greco and R. I. Nicholson, Endocr. Relat. Cancer, 2005, 12, S189 CAS.
- Y. Kaneda, Y. Tsutsumi, Y. Yoshioka, H. Kamada, Y. Yamamoto, H. Kodaira, S. Tsunoda, T. Okamoto, Y. Mukai, H. Shibata, S. Nakagawa and T. Mayumi, Biomaterials, 2004, 25(16), 3259 CrossRef CAS PubMed.
- H. Kamada, Y. Tsutsumi, Y. Yamamoto, T. Kihira, Y. Kaneda, Y. Mu, H. Kodaira, S. I. Tsunoda, S. Nakagawa and T. Mayumi, Cancer Res., 2000, 60(22), 6416 CAS.
- T. Yasukawa, H. Kimura, Y. Tabata, H. Miyamoto, Y. Honda, Y. Ikada and Y. Ogura, Invest. Ophthalmol. Visual Sci., 1999, 40(11), 2690 CAS.
- S. D. Chipman, F. B. Oldham, G. Pezzoni and J. W. Singer, Int. J. Nanomed., 2006, 1(4), 375 CrossRef CAS PubMed.
- J. Y. Ljubimova, M. Fujita, A. V. Ljubimov, V. P. Torchilin, K. L. Black and E. Holler, Nanomedicine, 2008, 3(2), 247 CrossRef CAS PubMed.
- G. Pasut and F. M. Veronese, Adv. Drug Deliv. Rev., 2009, 61(13), 1177 CrossRef CAS PubMed.
- J. Kopecek and P. Kopeckova, Adv. Drug Deliv. Rev., 2010, 62(2), 122 CrossRef CAS PubMed.
- R. B. Greenwald, A. Pendri, C. D. Conover, C. Lee, Y. H. Choe, C. Gilbert, A. Martinez, Y. Xia, D. Wu and M. Hsue, Bioorg. Med. Chem., 1998, 6, 551 CrossRef CAS PubMed.
- D. Fraier, E. Frigerio, G. Brianceschi, M. Casati, A. Benecchi and C. James, J. Pharm. Biomed. Anal., 2000, 19, 505 CrossRef.
- J. W. Singer, R. Bhatt, J. Tulinsky, K. R. Buhler, E. Heasley, P. Klein and P. de Vries, J. Controlled Release, 2001, 74, 243 CrossRef CAS PubMed.
- F. Pastorino, M. Loi, P. Sapra, P. Becherini, M. Cilli, L. Emionite, D. Ribatti, L. M. Greenberger, I. D. Horak and M. Ponzoni, Clin. Cancer Res., 2010, 16, 4809 CrossRef CAS PubMed.
- P. Sapra, H. Zhao, M. Mehlig, J. Malaby, P. Kraft, C. Longley, L. M. Greenberger and I. D. Horak, Clin. Cancer Res., 2008, 14, 1888 CrossRef CAS PubMed.
- G. S. Jameson, J. T. Hamm, G. J. Weiss, C. Alemany, S. Anthony, M. Basche, R. K. Ramanathan, M. J. Borad, R. Tibes, A. Cohn, I. Hinshaw, R. Jotte, L. S. Rosen, U. Hoch, M. A. Eldon, R. Medve, K. Schroeder, E. White and D. D. Von Hoff, Clin. Cancer Res., 2013, 19, 268 CrossRef CAS PubMed.
- I. B. Vergote, A. Garcia, J. Micha, C. Pippitt, J. Bendell, D. Spitz, N. Reed, G. Dark, P. M. Fracasso, E. N. Ibrahim, V. A. Armenio, L. Duska, C. Poole, C. Gennigens, L. Y. Dirix, A. C. Leung, C. Zhao, R. S. Mahjoubi and G. J. Rustin, J. Clin. Oncol., 2013, 31, 4060 CrossRef CAS PubMed.
- A. Awada, A. A. Garcia, S. Chan, G. H. Jerusalem, R. E. Coleman, M. T. Huizing, A. Mehdi, S. M. O'Reilly, J. T. Hamm, P. J. Barrett–Lee, V. Cocquyt, K. Sideras, D. E. Young, C. Zhao, Y. L. Chia, U. Hoch, A. L. Hannah and E. A. Perez, Lancet Oncol., 2013, 14, 1216 CrossRef CAS PubMed.
- T. Lammers, Adv. Drug Deliv. Rev., 2010, 62(2), 203 CrossRef CAS PubMed.
- R. Duncan, Adv. Drug Deliv. Rev., 2009, 61(13), 1131 CrossRef CAS PubMed.
- R. Duncan and M. J. Vicent, Adv. Drug Deliv. Rev., 2010, 62(2), 272 CrossRef CAS PubMed.
- P. A. Vasey, S. B. Kaye, R. Morrison, C. Twelves, P. Wilson, R. Duncan, A. H. Thomson, L. S. Murray, T. E. Hilditch, T. Murray, S. Burtles, D. Fraier, E. Frigerio and J. Cassidy, Clin. Cancer Res., 1999, 5, 83 CAS.
- L. W. Seymour, D. R. Ferry, D. J. Kerr, D. Rea, M. Whitlock, R. Poyner, C. Boivin, S. Hesslewood, C. Twelves, R. Blackie, A. Schatzlein, D. Jodrell, D. Bisset, H. Calvert, M. Lind, A. Robbins, S. Burtles, R. Duncan and J. Cassidy, Int. J. Oncol., 2009, 34, 1629 CrossRef CAS PubMed.
- J. W. Hopewel, R. Duncan, D. Wilding and K. Chakrabarti, Hum. Exp. Toxicol., 2001, 20, 461 CrossRef CAS PubMed.
- P. J. Julyan, L. W. Seymour, D. R. Ferry, S. Daryani, C. M. Boivin, J. Doran, M. David, D. Anderson, C. Christodoulou, A. M. Young, S. Hesslewood and D. J. Kerr, J. Controlled Release, 1999, 57, 281 CrossRef CAS PubMed.
- L. W. Seymour, D. R. Ferry, D. Anderson, S. Hesslewood, P. J. Julyan, R. Poyner, J. Doran, A. M. Young, S. Burtles and D. J. Kerr, J. Clin. Oncol., 2002, 20, 1668 CrossRef CAS PubMed.
- M. Campone, J. M. Rademaker-Lakhai, J. Bennouna, S. B. Howell, D. P. Nowotnik, J. H. Beijnen and J. H. Schellens, Cancer Chemother. Pharmacol., 2007, 60, 523 CrossRef CAS PubMed.
- J. R. Rice and S. B. Howell, Drugs Future, 2004, 29, 561 CrossRef CAS.
- L. Kelland, Expert Opin. Investig. Drugs, 2007, 16(7), 1009 CrossRef CAS PubMed.
- D. P. Nowotnik and E. Cvitkovic, Adv. Drug Deliv. Rev., 2009, 61, 1214 CrossRef CAS PubMed.
- J. M. M. Terwogt, W. W. ten Bokkel Huinink, J. H. M. Schellens, M. Schot, I. Mandjes, M. Zurlo, M. Rocchetti, H. Rosing, F. Koopman and J. H. Beijnen, Anticancer Drugs, 2001, 12, 315 CrossRef CAS.
- J. W. Singer, B. Baker, P. de Vries, A. Kumar, S. Shaffer, E. Vawter, M. Bolton and P. Garzone, Adv. Exp. Med. Biol., 2003, 519, 81 CrossRef CAS PubMed.
- S. A. Shaffer, C. B. Lee, A. Kumar and J. W. Singer, Eur. J. Cancer, 2002, 38(suppl. 7), s129 Search PubMed.
- E. Auzenne, N. J. Donato, C. Li, E. Leroux, R. E. Price, D. Farquhar and J. Klostergaard, Clin. Cancer Res., 2002, 8, 573 CAS.
- J. W. Singer, P. De Vries, R. Bhatt, J. Tulinsky, P. Klein, C. Li, L. Milas, R. A. Lewis and S. Wallace, Ann. N. Y. Acad. Sci., 2000, 922, 136 CrossRef CAS PubMed.
- J. Homsi, G. R. Simon, C. R. Garrett, G. Springett, R. De Conti, A. A. Chiappori, P. N. Munster, M. K. Burton, S. Stromatt, C. Allievi, P. Angiuli, A. Eisenfeld, D. M. Sullivan and A. I. Daud, Clin. Cancer Res., 2007, 13, 5855 CrossRef CAS PubMed.
- G. M. Springett, C. Takimoto, M. McNamara, J. H. Doroshow, S. Syed, E. Eastham, D. Spriggs, S. Pezzulli and G. Michelson, J. Clin. Oncol., 2004, 22, 3127 Search PubMed , ASCO Annual Meeting Proceedings.
- T. Schluep, J. Cheng, K. Khin and M. Davis, Cancer Chemother. Pharmacol., 2006, 57, 654 CrossRef CAS PubMed.
- T. Schluep, J. Hwang, J. Cheng, J. D. Heidel, D. W. Bartlett, B. Hollister and M. E. Davis, Clin. Cancer Res., 2006, 12, 1606 CrossRef CAS PubMed.
- C. Young, T. Schluep, J. Hwang and S. Eliasof, Curr. Bioact. Compd., 2011, 7, 8 CrossRef CAS PubMed.
- N. Rapoport, Prog. Polym. Sci., 2007, 32, 962 CrossRef CAS.
- O. Soga, C. F. van Nostrum, M. Fens, C. J. Rijcken, R. M. Schiffelers, G. Storm and W. E. Hennink, J. Controlled Release, 2005, 103, 341 CrossRef CAS PubMed.
- P. Opanasopit, M. Yokoyama, M. Watanabe, K. Kawano, Y. Maitani and T. Okano, J. Controlled Release, 2005, 104, 313 CrossRef CAS PubMed.
- J. B. Liu, F. Q. Zeng and C. Allen, J. Controlled Release, 2005, 103, 481 CrossRef CAS PubMed.
- V. P. Torchilin, Pharm. Res., 2007, 24, 1 CAS.
- T. C. Lai, H. Cho and G. S. Kwon, Polym. Chem., 2014, 5, 1650 RSC.
- M. Shahin and A. Lavasanifar, Int. J. Pharm., 2010, 389, 213 CrossRef CAS PubMed.
- X. Xu, X. Zhang, X. Wang, Y. Li and X. Jing, Polym. Adv. Technol., 2009, 20, 843 CrossRef CAS.
- Y. Bae, N. Nishiyama, S. Fukushima, H. Koyama, M. Yasuhiro and K. Kataoka, Bioconjugate Chem., 2005, 16, 122 CrossRef CAS PubMed.
- M. Yokoyama, T. Sugiyama, T. Okano, Y. Sakurai, M. Naito and K. Kataoka, Pharm. Res., 1993, 10, 895 CrossRef CAS.
- M. L. Forrest, J. Yanez, C. Remsberg, Y. Ohgami, G. Kwon and N. Davies, Pharm. Res., 2008, 25, 194 CrossRef CAS PubMed.
- A. Rapisarda, B. Uranchimeg, D. A. Scudiero, M. Selby, E. A. Sausville, R. H. Shoemaker and G. Melillo, Cancer Res., 2002, 62, 4316 CAS.
- J. Liu, W. Liu, I. Weitzhandler, J. Bhattacharyya, X. Li, J. Wang, Y. Qi, S. Bhattacharjee and A. Chilkoti, Angew. Chem., Int. Ed., 2015, 54, 1002 CrossRef CAS PubMed.
- Y. Matsumura, Adv. Drug Deliv. Rev., 2008, 60, 899 CrossRef CAS PubMed.
- T. Hamaguchi, Y. Matsumura, M. Suzuki, K. Shimizu, R. Goda, I. Nakamura, I. Nakatomi, M. Yokoyama, K. Kataoka and T. Kakizoe, Br. J. Cancer, 2005, 92, 1240 CrossRef CAS PubMed.
- T. Hamaguchi, K. Kato, H. Yasui, C. Morizane, M. Ikeda, H. Ueno, K. Muro, Y. Yamada, T. Okusaka, K. Shirao, Y. Shimada, H. Nakahama and Y. Matsumura, Br. J. Cancer, 2007, 97, 170 CrossRef CAS PubMed.
- J. W. Valle, A. Armstrong, C. Newman, V. Alakhov, G. Pietrzynski, J. Brewer, S. Campbell, P. Corrie, E. K. Rowinsky and M. Ranson, Invest. New Drugs, 2010, 29(5), 1029 CrossRef PubMed.
- A. Venne, S. Li, R. Mandeville, A. Kabanov and V. Alakhov, Cancer Res., 1996, 56, 3626 CAS.
- E. V. Batrakova, T. Y. Dorodnych, E. Y. Klinskii, E. N. Kliushnenkova, O. B. Shemchukova, O. N. Goncharova, S. A. Arjakov, V. Y. Alakhov and A. V. Kabanov, Br. J. Cancer, 1996, 74, 1545 CrossRef CAS PubMed.
- V. Alakhov, E. Klinski, S. M. Li, G. Pietrzynski, A. Venne, E. V. Batrakova, T. Bronitch and A. Kabanov, Colloids Surf., B, 1999, 16, 113 CrossRef CAS.
- Y. Matsumura, Jpn. J. Clin. Oncol., 2008, 38, 793 CrossRef PubMed.
- R. H. Wilson, R. Plummer, J. Adam, M. Eatock, A. V. Boddy, M. Griffin, R. Miller, Y. Matsumura, T. Shimizu and H. Calvertet, J. Clin. Oncol., 2008, 26(20 suppl.), 2573 Search PubMed.
- R. Plummer, R. H. Wilson, H. Calvert, A. V. Boddy, M. Griffin, J. Sludden, M. J. Tilby, M. Eatock, D. G. Pearson, C. J. Ottley, Y. Matsumura, K. Kataoka and T. Nishiya, Br. J. Cancer, 2011, 104, 593 CrossRef CAS PubMed.
- United States National Library of Medicine, overview of clinical trials available via www.clinicaltrials.org, accessed 01-27-10.
- T. Y. Kim, D. W. Kim, J. Y. Chung, S. G. Shin, S. C. Kim, D. S. Heo, N. K. Kim and Y. J. Bang, Clin. Cancer Res., 2004, 10, 3708 CrossRef CAS PubMed.
- S. C. Kim, D. W. Kim, Y. H. Shim, J. S. Bang, H. S. Oh, K. S. Wan and M. H. Seo, J. Controlled Release, 2001, 72, 191 CrossRef CAS PubMed.
- E. Tan, S. S. Leong, W. T. Lim, C. K. Toh, and B. Chowbay, Gastrointestinal Cancers Symposium, 2007 Search PubMed.
- K. S. Lee, H. C. Chung, S. A. Im, Y. H. Park, C. S. Kim, S. B. Kim, S. Y. Rha, M. Y. Lee and J. Ro, Breast Canc. Res. Treat., 2008, 108, 241 CrossRef CAS PubMed.
- N. A. Podoltsev, M. Rubin, J. Figueroa, M. Y. Lee, J. Kwonj, J. Yu, R. O. Kerr and M. W. Saif, J. Clin. Oncol., 2008, 26(20 suppl.), 4627 Search PubMed.
- M. W. Saif, M. Rubin, J. Figueroa and R. O. Kerr,Gastrointestinal Cancers Symposium, 2008 Search PubMed.
- C. Kim, S. Bae, N. Lee, K. Lee, S. Park, D. Kim, J. Won, D. Hong and H. Park, J. Clin. Oncol., 2006, 24(20 suppl.), 17049 Search PubMed.
- D. W. Kim, S. Y. Kim, H. K. Kim, S. W. Kim, S. W. Shin, J. S. Kim, K. Park, M. Y. Lee and D. S. Heo, Ann. Oncol., 2007, 18, 2009 CrossRef PubMed.
- A. D. Bangham, M. M. Standish and J. C. Watkins, J. Mol. Biol., 1965, 13(1), 238 CrossRef CAS PubMed.
- S. G. Antimisiaris, P. Kallinteri and D. G. Fatouros, Frontiers in anticancer drug discovery, Wiley, New York, 2007, p. 443 Search PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discov., 2005, 4, 145 CrossRef CAS PubMed.
- P. Caliceti and F. M. Veronese, Adv. Drug Deliv. Rev., 2003, 55, 1261 CrossRef CAS PubMed.
- S. E. Krown, D. W. Northfelt, D. Osoba and J. S. Stewart, Semin. Oncol., 2004, 31, 36 CrossRef CAS PubMed.
- A. M. Rahman, S. W. Yusuf and M. S. Ewer, Int. J. Nanomed., 2007, 2, 567 CAS.
- M. E. O'Brian, N. Wigler, M. Inbar, R. Rosso, E. Grischke, A. Santoro, R. Catane, D. G. Kieback, P. Tomczak, S. P. Ackland, F. Orlandi, L. Mellars, L. Alland and C. Tendler, Ann. Oncol., 2004, 15, 440 CrossRef PubMed.
- A. Gabizon, Applications of liposomal drug delivery systems to cancer therapy, in Nanotechnology for cancer therapy, New York, CRC Press, 2006, p. 595 Search PubMed.
- A. Gabizon, R. Catane, B. Uziely, B. Kaufman, T. Safra, R. Cohen, F. Martin, A. Huang and Y. Barenholz, Cancer Res., 1994, 54, 987 CAS.
- M. A. Amantea, A. Forrest, D. W. Northfelt and R. Mamelok, Clin. Pharmacol. Ther., 1997, 61, 301 CrossRef CAS PubMed.
- M. Slingerland, H. J. Guchelaar and H. Gelderblom, Drug Discovery Today, 2012, 17, 160 CrossRef CAS PubMed.
- P. S. Yarmolenko, Y. Zhao, C. Landon, I. Spasojevic, F. Yuan, D. Needham, B. L. Viglianti and M. W. Dewhirst, Int. J. Hyperthermia, 2010, 26, 485 CrossRef CAS PubMed.
- G. P. Stathopoulos and T. Boulikas, J. Drug Delivery, 2012, 2012, 581363 CAS.
- T. Boulikas, Oncol. Rep., 2004, 12, 3 CAS.
- G. P. Stathopoulos, T. Boulikas, M. Vougiouka, G. Deliconstantinos, S. Rigatos, E. Darli, V. Viliotou and J. G. Stathopoulos, Oncol. Rep., 2005, 13, 589 CAS.
- C. Kosmas, J. Angel, A. Athanasiou, A. Rapti, C. Karanikas, S. Lambaki, N. Politis and N. Mylonakis, Eur. J. Cancer Suppl., 2009, 7, 531 CrossRef.
- F. Farhat, J. Kattan, K. Ibrahim, N. Bitar, N. Haddad, S. Tamraz, H. Hatoum and A. Shamseddine, Eur. J. Cancer Suppl., 2010, 8, 192 CrossRef.
- M. I. Koukourakis, A. Giatromanolaki, M. Pitiakoudis, G. Kouklakis, P. Tsoutsou, I. Abatzoglou, M. Panteliadou, K. Sismanidou, E. Sivridis and T. Boulikas, Int. J. Radiat. Oncol., Biol., Phys., 2010, 78, 150 CrossRef CAS PubMed.
- T. Boulikas, G. P. Stathopoulos, N. Volakakis and M. Vougiouka, Anticancer Res., 2005, 25, 3031 CAS.
- T. Boulikas, Expert Opin. Investig. Drugs, 2009, 18, 1197 CrossRef CAS PubMed.
- N. Seetharamu, E. Kim, H. Hochster, F. Martin and F. Muggia, Anticancer Res., 2010, 30(2), 541 CAS.
- P. Tardi, S. Johnstone, N. Harasym, S. Xie, T. Harasym, N. Zisman, P. Harvie, D. Bermudes and L. Mayer, Leuk. Res., 2009, 33(1), 129 CrossRef CAS PubMed.
- T. O. Harasym, P. G. Tardi, N. L. Harasym, P. Harvie, S. A. Johnstone and L. D. Mayer, Oncol. Res., 2007, 16(8), 361 CrossRef PubMed.
- G. Batist, K. A. Gelmon, K. N. Chi, W. H. Miller Jr, S. K. Chia, L. D. Mayer, C. E. Swenson, A. S. Janoff and A. C. Louie, Clin. Cancer Res., 2009, 15(2), 692 CrossRef CAS PubMed.
- V. Sanna, N. Pala and M. Sechi, Int. J. Nanomed., 2014, 9, 467 CAS.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818 CrossRef CAS PubMed.
- R. Langer, Nature, 1998, 392, 5 CAS.
- L. E. van Vlerken, Z. Duan, M. V. Seiden and M. M. Amiji, Cancer Res., 2007, 67, 4843 CrossRef CAS PubMed.
- K. Riehemann, S. W. Schneider, T. A. Luger, B. Godin, M. Ferrari and H. Fuchs, Angew. Chem., Int. Ed., 2009, 48, 872 CrossRef CAS PubMed.
- Y. Bae and K. Kataoka, J. Controlled Release, 2006, 116, 49 CrossRef PubMed.
- X. B. Xiong, A. Mahmud, H. Uludag and A. Lavasanifar, Pharm. Res., 2008, 25, 2555 CrossRef CAS PubMed.
- A. Lavasanifar, J. Samuel and G. S. Kwon, Adv. Drug Deliv. Rev., 2002, 54, 169 CrossRef CAS PubMed.
- V. P. Torchilin, A. N. Lukyanov, Z. Gao and B. P. Sternberg, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6039 CrossRef CAS PubMed.
- A. Harada and K. Kataoka, Prog. Polym. Sci., 2006, 31, 949 CrossRef CAS.
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., Int. Ed. Engl., 2003, 42, 4640 CrossRef CAS PubMed.
- Y. Lee, S. Fukushima, Y. Bae, S. Hiki, T. Ishii and K. Kataoka, J. Am. Chem. Soc., 2007, 129, 5362 CrossRef CAS PubMed.
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Biomaterials, 2010, 31, 3657 CrossRef CAS PubMed.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, Pharmacol. Rev., 2001, 53, 283 CAS.
- A. Lamprecht, N. Ubrich, H. Yamamoto, U. Schäfer, H. Takeuchi, P. Maincent, Y. Kawashima and C. M. Lehr, J. Pharmacol. Exp. Ther., 2001, 299, 775 CAS.
- P. T. Wong and S. K. Choi, Chem. Rev., 2015, 115, 3388 CrossRef CAS PubMed.
- M. Srinivasan, M. Rajabi and S. A. Mousa, Nanomaterials, 2015, 5, 1690 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271 CrossRef CAS PubMed.
- Y. Cheng, A. C. Samia, J. D. Meyers, I. Panagopoulos, B. Fei and C. Burda, J. Am. Chem. Soc., 2008, 130, 10643 CrossRef CAS PubMed.
- H. S. Choi, B. I. Ipe, P. Misra, J. H. Lee, M. G. Bawendi and J. V. Frangioni, Nano Lett., 2009, 9, 2354 CrossRef CAS PubMed.
- K. Derakhshandeh, M. Erfan and S. Dadashzadeh, Eur. J. Pharm. Biopharm., 2007, 66(1), 34 CrossRef CAS PubMed.
- Y. M. Wang, H. Sato, I. Adachi and I. Horikoshi, Chem. Pharm. Bull., 1996, 44(10), 1935 CrossRef CAS PubMed.
- C. Fonseca, S. Simoes and R. Gaspar, J. Controlled Release, 2002, 83(2), 273 CrossRef CAS PubMed.
- K. Avgoustakis, A. Beletsi, Z. Panagi, P. Klepetsanis, A. G. Karydas and D. S. Ithakissios, J. Controlled Release, 2002, 79(1–3), 123 CrossRef CAS PubMed.
- S. Y. Kim and Y. M. Lee YM, Biomaterials, 2001, 22(13), 1697 CrossRef CAS PubMed.
- H. Gao, Y. N. Wang, Y. G. Fan and J. B. Ma, J. Controlled Release, 2005, 107(1), 158 CrossRef CAS PubMed.
- P. N. Cheng, Y. C. Leung, W. H. Lo, S. M. Tsui and K. C. Lam, Cancer Lett., 2005, 224, 67 CrossRef CAS PubMed.
- J. M. Lee, T. J. Yoon and Y. S. Cho, BioMed Res. Int., 2013, 2013, 782041 Search PubMed.
- W. J. Gradishar, S. Tjulandin, N. Davidson, H. Shaw, N. Desai, P. Bhar, M. Hawkins and J. O'Shaughnessy, J. Clin. Oncol., 2005, 23(31), 7794 CrossRef CAS PubMed.
- W. Gradishar, T. Vishalpura, M. Franklin and T. Bramley, Breast Canc. Res. Treat., 2005, 94, S220 Search PubMed.
- X. B. Xiong, Z. Ma, R. Lai and A. Lavasanifar, Biomaterials, 2010, 31, 757 CrossRef CAS PubMed.
- H. Chen, W. He and Z. Guo, Chem. Commun., 2014, 50, 9714 RSC.
- W. Chen, L. A. Shah, L. Yuan, M. Siddiq, J. Hu and D. Yang, RSC Adv., 2015, 5, 7559 RSC.
- N. Goodarzi, M. H. Ghahremani, M. Amini, F. Atyabi, S. N. Ostad, N. S. Ravari, N. Nateghian and R. Dinarvand, Chem. Biol. Drug Des., 2014, 83(6), 741 CrossRef CAS PubMed.
- G. Tzircotis, R. F. Thorne and C. M. Isacke, J. Cell Sci., 2005, 118, 5119 CrossRef CAS PubMed.
- Y. Huang, C. Song, H. Li, R. Zhang, R. Jiang, X. Liu, G. Zhang, Q. Fan, L. Wang and W. Huang, ACS Appl. Mater. Interfaces, 2015, 7, 21529 CrossRef CAS PubMed.
- T. Luo, J. Magnusson, V. Préat, R. Frédérick, C. Alexander, C. Bosquillon and R. Vanbever, Pharm. Res., 2016, 33(7), 1671 CrossRef CAS PubMed.
- L. H. Shao, S. P. Liu, J. H. Hou, Y. H. Zhang, C. W. Peng, Y. J. Zhong, X. Liu, X. L. Liu, Y. P. Hong, R. A. Firestone and Y. Li, Cancer, 2012, 118(11), 2986 CrossRef CAS PubMed.
- G. M. Dubowchik and R. A. Firestone, Bioorg. Med. Chem. Lett., 1998, 3341 CrossRef CAS.
- T. Etrych, V. Šubr, R. Laga, B. Ríhová and K. Ulbrich, Eur. J. Pharm. Sci., 2014, 58, 1 CrossRef CAS PubMed.
- S. Yang, Y. Wang, Z. Ren, M. Chen, W. Chen and X. Zhang, Mater. Sci. Eng., C, 2018, 82, 234 CrossRef CAS PubMed.
- Y. Cong, H. Xiao, H. Xiong, Z. Wang, J. Ding, C. Li, X. Chen, X. J. Liang, D. Zhou and Y. Huang, Adv. Mater., 2018, 30, 1706220 CrossRef PubMed.
- S. He, C. Li, Q. Zhang, J. Ding, X. J. Liang, X. Chen, H. Xiao, X. Chen, D. Zhou and Y. Huang, ACS Nano, 2018, 12, 7272 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |