
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of the intramolecular hydrogen bonding interactions and excited state proton transfer mechanism for both Br-BTN and CN-BTN systems†
Dapeng Yang *ab,
Qiaoli Zhanga,
Xiaoyan Songa and
Tianjie Zhanga
*ab,
Qiaoli Zhanga,
Xiaoyan Songa and
Tianjie Zhanga
aCollege of Physics and Electronics, North China University of Water Resources and Electric Power, Zhengzhou 450046, P. R. China. E-mail: dpyang_ncwu@163.com
bState Key Laboratory of Molecular Reaction Dynamics, Theoretical and Computational Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, P. R. China
First published on 25th July 2019
Abstract
In the present work, two novel Br-BTN and CN-BTN compounds have been investigated theoretically. We in-depth explore the excited state hydrogen bonding interactions and the excited state intramolecular proton transfer (ESIPT) behaviors for the Br-BTN and CN-BTN system. We firstly verify the formation of hydrogen bond effects of O–H⋯N based on reduced density gradient (RDG) versus sign(λ2)ρ. The simulated primary bond lengths and bond angles as well as infrared (IR) vibrational spectra reveal that the hydrogen bond O–H⋯N should be strengthened in the excited state. Combining the frontier molecular orbital (MO) investigations, we infer that the charge transfer phenomenon (from HOMO to LUMO) around hydrogen bonding moieties reveals the tendency of ESIPT reaction. Particularly, the increased electronic densities around proton acceptor atoms facilitate attracting a hydrogen proton, which plays a decisive role in opening the ESIPT reaction. Via constructing potential energy curves in both S0 and S1 states, the ultrafast ESIPT process can be verified which explains previous experimental characteristics. Furthermore, via searching the transition state (TS) structure and constructing the intrinsic reaction coordinate (IRC) reaction path, we check and confirm the ESIPT mechanism for both Br-BTN and CN-BTN systems. We sincerely hope that our theoretical work could guide novel applications based on Br-BTN and CN-BTN compounds in future.
1. Introduction
It is well known that hydrogen bonding is one of the most important weak interactions in the natural world, which plays a significant role in various branches of natural science and engineering fields.1–6 It is operative in the crystallization of materials, the formation of straightforward properties of associated liquids, and the determination of the three-dimensional structures adopted by biomolecules such as proteins and nucleic bases. Particularly, the hydrogen bond can result in relatively stable supramolecular structures. And it can also serve as an active site for initiating a plethora of chemical reactions due to the dynamic features of the proton.7–10 As one of the most fundamental processes involved in biochemical and chemical reactions along with hydrogen bonding, proton transfer (PT) deserves the attention of vast numbers of scientists.11–15 Various kinds of PT reaction have been identified and classified, such as reaction in the ground or excited states, adiabatic or non-adiabatic behaviors, and so on.16–20 It cannot be denied that people pay more attention to the excited state dynamical behaviors with the ever-accelerated experimental instruments and methods. Therefore, excited state intramolecular proton transfer (ESIPT) reactions have become the most fundamental and important research focus in recent years.As far as we know, the ESIPT reaction belongs to a kind of photo-tautomerization process that happens along with a pre-existing hydrogen bond in the excited state. Generally, the hydrogen bond links the hydrogen acceptor moiety (O or N atom) and the donor acidic parts (N–H or O–H). Based on the photo-induced excitation, the initial stable ground-state enol form could be excited to the excited states. After the de-excitation behaviors from high excited states to the first excited state as well as the thermal relaxations, the stable enol* can be formed in the S1 state with the normal emission peak. Given the energy gap between FC and enol*, the driving force of ESIPT could be provided, which further offers the relative kinetics. Subsequently, the ESIPT reactions happens via converting enol* into its photoproduct (i.e., the photo-tautomer keto*) in the first excited state. Generally, the ESIPT behavior is ultrafast with crossing a low potential energy barrier. Experimentally, this kind of ultrafast process is in the region of femtosecond to picosecond. And the fluorescence band of keto* exhibits a large Stokes shift,21–30 which could be as large as 8000–12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1. In particular, the fast charge redistribution resulting from ESIPT tautomer keto*, multiple optoelectronic applications have been designed and developed, such as fluorescence sensors, luminescent materials, UV absorbers, molecular logic gates, and so forth.31–40
000 cm−1. In particular, the fast charge redistribution resulting from ESIPT tautomer keto*, multiple optoelectronic applications have been designed and developed, such as fluorescence sensors, luminescent materials, UV absorbers, molecular logic gates, and so forth.31–40
Recently, Santra and coworkers designed and synthesized bent-shaped 1-(benzo[d]thiazol-2-yl)-6-substituted-naphthalen-2-ol (BTN) compounds.41 They characterized their photophysical and photochemical properties in both solution and solid states. Among these compounds, the Br-BTN and CN-BTN reveal the ESIPT phenomenon experimentally occurring along with the intramolecular hydrogen bond O–H⋯N in the first excited state. Santra and coworkers mainly focused on solid-state emission behavior of the new dyes, and they showed that the fluorescent nanoparticles could permeate cells and give fluorescent microscopic images. We must say that their work facilitates opening a door toward solid-state luminescent organic compounds. Nevertheless, the excited state dynamics are insufficient for Br-BTN and CN-BTN in solution. Since BTN compounds exhibit potential application prospects, the straightforward excited-state dynamic process should be very helpful for further exploitations in future. Moreover, experiments can only provide the indirect information about photochemical and photophysical properties. For clarifying detailed dynamical mechanism, the theoretical calculation should be the critical mean currently. In this present work, therefore, we mainly focus on the excited state dynamical behaviors and ESIPT mechanism for Br-BTN and CN-BTN systems based on density functional theory (DFT) and time-dependent density functional theory (TDDFT) methods (see Fig. 1). The initial stable Br-BTN and CN-BTN structures are named as Br-BTN-enol and CN-BTN-enol, and the proton-transfer configurations are named as Br-BTN-keto and CN-BTN-keto, respectively. Bond lengths and bond angles relative to hydrogen bond and homologous infrared (IR) vibrational spectra are analyzed. The hydrogen bonding interactions reveal the tendency of ESIPT behavior. Exploring photo-induced process, the charge redistribution resulting from photoexcitation facilitates the ESIPT reaction. Finally, the constructed S0 and S1 state potential energy curves as well as reaction transition state (TS) show the direct information about ESIPT mechanism for both Br-BTN and CN-BTN systems.
 | ||
| Fig. 1 The structures of Br-BTN-enol, Br-BTN-keto, CN-BTN-enol and CN-BTN-keto based on DFT and TDDFT methods. | ||
2. Simulated methods
In this present work, all the quantum chemical simulations have been performed and finished using the Gaussian 09 program suite.42 The geometric optimizations of the Br-BTN and CN-BTN have been performed in the ground state based on DFT method and the electronic excited state is performed depending on TDDFT method, respectively. It is worth mentioning that the TDDFT method has become a very useful tool to research the hydrogen bond in the excited states of the hydrogen-bond system theoretically.21–30 Becke' three-parameter hybrid exchange functional with Lee–Yang–Parr gradient-corrected correlation (B3LYP functional) has been used in both the DFT and TDDFT methods.43–48 The 6-311+G(d) is chosen as basis sets throughout, which has sufficient flexibility to describe this system. There were no constraints to all the atoms, bonds, angles or dihedral angles during the geometric optimization.To evaluate the solvent effect, DMSO has been selected as solvent in the calculations depending on the model that the Polarizable Continuum Model (PCM) using the integral equation formalism variant (IEFPCM).49–51 All the local minima are confirmed by the absence of an imaginary mode in vibrational analysis calculations. The topological properties of intramolecular hydrogen bond interaction are analyzed based on the provision of Atoms-In-Molecule (AIM) method. The S0 and S1 potential energy curves have been scanned by constrained optimizations in their corresponding electronic state, and keeping the O–H distance fixed at a serious of values. Harmonic vibrational frequencies in both ground state and excited state are determined by diagonalization of the Hessian. The excited state Hessian is obtained by numerical differentiation of analytical gradients using central differences and default displacements of 0.02 bohr. The infrared intensities were determined from the gradients of the dipole moment.
3. Results and discussions
As shown in Fig. 1, based on DFT and TDDFT methods, we could locate the stable Br-BTN-enol, Br-BTN-keto (the proton-transfer Br-BTN-enol form), CN-BTN-enol and CN-BTN-keto (the proton-transfer CN-BTN-enol form) in both S0 and S1 states. Within the framework of quantum theory of atoms in molecule (QTAIM) theory, the identification of a critical point (CP) and the existence of a bond path in the equilibrium geometry are necessary and sufficient conditions for assigning an interaction between two primary atoms. The relative AIM topologic parameters involved in the optimized geometrics show that the ρ(r) at the CP moiety is close to 0.04 a.u. (the maximum threshold value to ensure the presence of hydrogen bond). Furthermore, the relative ∇2ρc value is also in the range of 0.02–0.15 a.u. Therefore, we have the reasons to believe that the hydrogen bond O–H⋯N is formed in the ground state for Br-BTN-enol and CN-BTN-enol forms. In another aspect, it is well known that the noncovalent interaction (NCI) method (i.e., reduced density gradient (RDG)) is a very popular method for investigating weak interactions. Particularly, to reveal the intra- and inter-molecular weak interactions in real space, the RDG versus sign(λ2)ρ and the lower panel low-gradient isosurfaces for both Br-BTN-enol and CN-BTN-enol systems are calculated in this work (seen in Fig. 2). The contour value is set as 0.5 a.u., and the value range of RDG isosurfaces is set form −0.04 a.u. to 0.02 a.u. According to the conclusion by Johnson and coworkers,52 the multiple weak effects could be clearly revealed, namely, positive sign(λ2)ρ refers to steric effects, negative values of sign(λ2)ρ stand for hydrogen bonding interactions, and the values near zero exhibit the van der Waals (VDW) effects.52 It should be clear that the spikes located around −0.02 to −0.01 a.u. for both Br-BTN-enol and CN-BTN-enol reveal the hydrogen bonding effects of O–H⋯N in the ground state. | ||
| Fig. 2 The reduced density gradient (RDG) (Y-axis) versus sign(λ2)ρ (X-axis) as well as the lower panel low-gradient (s = 0.5 a.u.) isosurfaces for Br-BTN-enol (a) and CN-BTN-enol (b) systems in the ground state. Herein, the interactions are shown below corresponding RDG (red: steric effects; blue: hydrogen bonding interactions; green: VDW effects). | ||
To check the changes about bond lengths and bond angles involved in hydrogen bonding moieties upon the photoexcitation process, we list the prime geometrical parameters about hydrogen bond O–H⋯N for both Br-BTN-enol and CN-BTN-enol in Table 1. It can be clearly seen that the O–H bond lengths of both Br-BTN-enol and CN-BTN-enol are elongated from S0 state to the S1 state. Meanwhile, the bond distances of hydrogen bond H⋯N of both Br-BTN-enol and CN-BTN-enol are shortened in the S1 state. In addition, comparing with the S0-state bond angle Δ(O–H⋯N), it should be noticed that Δ(O–H⋯N) in the S1 state increases for Br-BTN-enol and CN-BTN-enol. According to previous theoretical results and decision rule, we confirm the intramolecular hydrogen bond O–H⋯N of both Br-BTN-enol and CN-BTN-enol structures should be strengthening in the first excited state.53–58
| Br-BTN-enol | CN-BTN-enol | |||
|---|---|---|---|---|
| Electronic state | S0 | S1 | S0 | S1 |
| O–H | 0.999 | 1.017 | 1.002 | 1.015 |
| H⋯N | 1.664 | 1.605 | 1.654 | 1.614 |
| Δ(O–H⋯N) | 146.8° | 149.3° | 147.0° | 148.8° |
In order to verify the strengthening phenomenon of hydrogen bond in the excited state, we further perform the infrared (IR) vibrational spectra at the O–H stretching mode for Br-BTN-enol and CN-BTN-enol in both S0 and S1 states. To show the IR results clearly, we only show the O–H stretching mode from 2600–3200 cm−1 in Fig. 3. It is obvious that the O–H IR stretching vibration frequency is redshift from S0-state 3067 cm−1 to S1-state 2737 cm−1 for Br-BTN-enol and from S0-state 3027 cm−1 to S1-state 2782 cm−1 for CN-BTN-enol. That it to say, the O–H stretching vibration becomes weaker in the first excited state, which should be attributed to the strengthening hydrogen bond O–H⋯N. Consistent with the conclusion about analyzing geometrical parameters, we confirm that hydrogen bond O–H⋯N should be enhanced in the S1 state, which conduces to attractive hydrogen proton for N atom for both Br-BTN-enol and CN-BTN-enol.
 | ||
| Fig. 3 The theoretical IR vibrational spectra of O–H stretching mode for Br-BTN-enol (a) and CN-BTN-enol (b) molecules in both S0 and S1 states based on DFT and TDDFT methods and B3LYP/6-311+G(d) theoretical level in DMSO solvent. | ||
It cannot be denied that the explorations about photo-excitation behaviors could reasonability reflect the excited state dynamical process. Therefore, the vertical excitation energy calculations are carried out using the S0 state-optimized configuration based on TDDFT method with five low-lying absorbing transitions. The corresponding results about vertical excitation energies for both Br-BTN-enol and CN-BTN-enol are listed in Table 2. We find that only the first transition (S0 → S1) owns the largest oscillator strength, which demonstrates that most of S0-state Br-BTN-enol and CN-BTN-enol should be excited to the first excited state. In this work, we mainly investigate the photochemical properties in the S1 state. The calculated absorption peak (S0 → S1) for Br-BTN-enol and CN-BTN-enol is located at 388 nm and 374 nm, respectively. They are in good agreement with experimental values (400 nm for Br-BTN-enol and 373 nm for CN-BTN-enol).41 It further confirms that the theoretical level TDDFT/B3LYP/6-311+G(d) is reasonable for these two system in this work.
| Transition | λ (nm) | f | Composition | CI (%) | |
|---|---|---|---|---|---|
| Br-BTN-enol | S0 → S1 | 388 | 0.4794 | H → L | 97.55 |
| S0 → S2 | 342 | 0.1371 | H−2 → L | 62.72 | |
| S0 → S3 | 312 | 0.0398 | H−1 → L | 49.71 | |
| CN-BTN-enol | S0 → S1 | 374 | 0.5087 | H → L | 95.98 |
| S0 → S2 | 326 | 0.0566 | H−1 → L | 87.12 | |
| S0 → S3 | 308 | 0.0089 | H−2 → L | 72.35 |
In order to further investigate the characteristics of the excited state, it is essential to investigate charge redistribution in electronic excited states. Therefore, the frontier molecular orbitals (MOs) of Br-BTN-enol and CN-BTN-enol have been calculated and shown in Fig. 4. Herein, we mainly focus on the S0 → S1 transition. Therefore, just the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) have been displayed in this figure since the transition HOMO → LUMO occupies more than 95% for both Br-BTN-enol and CN-BTN-enol. It can be found clearly that the HOMO orbital owns π character and the LUMO orbital has the π* character, which means the first excited state can be attributed to the π–π* transition. One thing should be noticed that the electron density on N atom increases in the LUMO orbital for both Br-BTN-enol and CN-BTN-enol, which mean the increased electron density of N atom plays an important role in enhancing the intramolecular hydrogen bonding O–H⋯N. In the same time, the electronic densities of hydroxyl O atom decrease from HOMO to LUMO orbital, which means the attraction between O and H becomes weakened upon photoexcitation, which is consistent the results via analyzing geometrical parameters and IR spectra mentioned above. Furthermore, the charge density difference (CDD) map is also calculated in this work. The relative results of CDD for both Br-BTN-enol and CN-BTN-enol systems are shown in Fig. S1, ESI.† The regions with the increasing electron density are shown in green, and those with decreasing electron density are shown in light blue. The CDD map displays that upon excitation from S0 to S1 state net charge densities shift from hydroxyl O atom to N atom. It suggests that after the excitation a driving force can be induced to facilitate the proton transfer reaction in the S1 state. This is consistent with the physical picture obtained from the HOMO–LUMO transition. Just due to this kind of charge redistribution, ESIPT tendency of both Br-BTN-enol and CN-BTN-enol can be promoted in the first excited state. Although the ESIPT behavior of Br-BTN-enol and CN-BTN-enol could be speculated via above discussions, the straightforward mechanism of ESIPT process deserves to in-depth investigation.
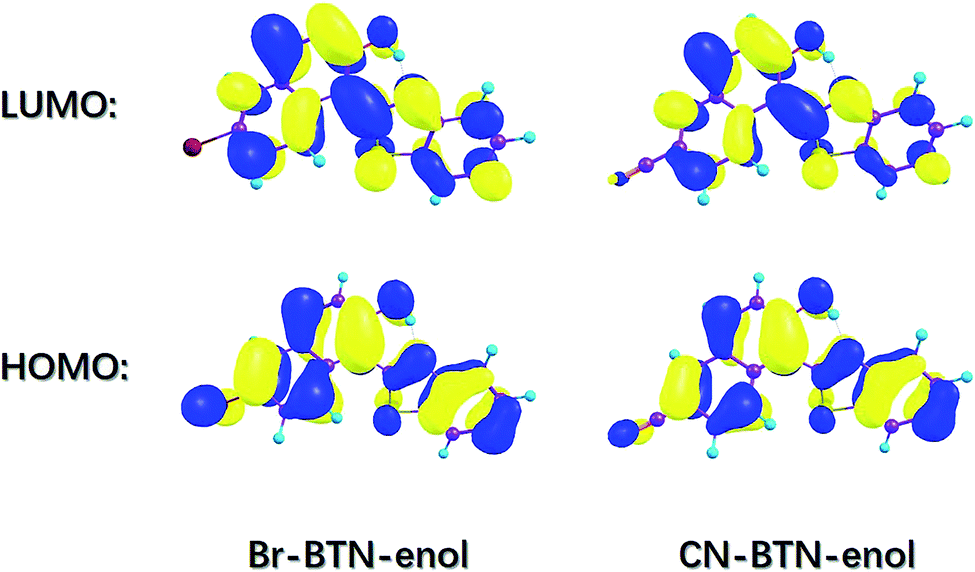 | ||
| Fig. 4 View of the simulated frontier molecular orbitals (HOMO and LUMO) for Br-BTN-enol and CN-BTN-enol systems based on TDDFT/B3LYP/6-311+G(d) theoretical level in DMSO solvent. | ||
In order to elaborate the specific ESPT mechanism for both Br-BTN-enol and CN-BTN-enol molecules, the potential energy curves of both S0 and S1 states are constructed in this work. The process of scanning potential energy curves are based on the constrained optimizations in their corresponding electronic states.59–61 We show the S0-state and S1-state potential energy curves (PECs) of both Br-BTN-enol and CN-BTN-enol molecules in Fig. 5. The PECs have been constructed via fixing the bond distance of O–H from 0.9 Å to 2.1 Å in step of 0.04 Å. Obviously, the S0-state potential energy curve increases along with the increasing of O–H bond distance for both Br-BTN-enol and CN-BTN-enol, which means the proton transfer process should be forbidden in the ground state. While in the S1 state, the potential energy curve crosses a low potential energy barrier (2.027 kcal for Br-BTN-enol and 2.184 kcal mol−1 for CN-BTN-enol) with the elongation of O–H from 1.0 Å to 1.3 Å. Then after crossing the barrier, the potential energy curve decreases along with increasing O–H bond distance. Since the potential energy barrier (2.027 kcal for Br-BTN-enol and 2.184 kcal mol−1 for CN-BTN-enol) is too low to forbid the ESIPT reaction, thus the ESIPT process is ultrafast to occur with forming Br-BTN-keto and CN-BTN-keto forms in the S1 state. Furthermore, we also consider the Cam-B3LYP and wB97XD functionals to construct the potential energy curves for Br-BTN and CN-BTN systems in both S0 and S1 states. The corresponding results are shown in Fig. S2, ESI.† Obviously, the conformations of relative potential energy curves are almost the same as the B3LYP functional. Therefore, we further confirm the rationality and correctness of B3LYP functional adopted in this work. We also consider the differences bringing from different solvents with different polarities for both Br-BTN and CN-BTN systems in this work. We show our simulated results about potential energy curves in cyclohexane and chloroform solvents in Fig. S3, ESI.† It can be found that the potential energy barriers among these three aprotic solvents (cyclohexane, chloroform and DMSO) are similar. Although the values of barriers are different, the differences are little. Therefore, we can confirm that solvent polarity plays little roles in the excited state behaviors for both Br-BTN and CN-BTN systems.
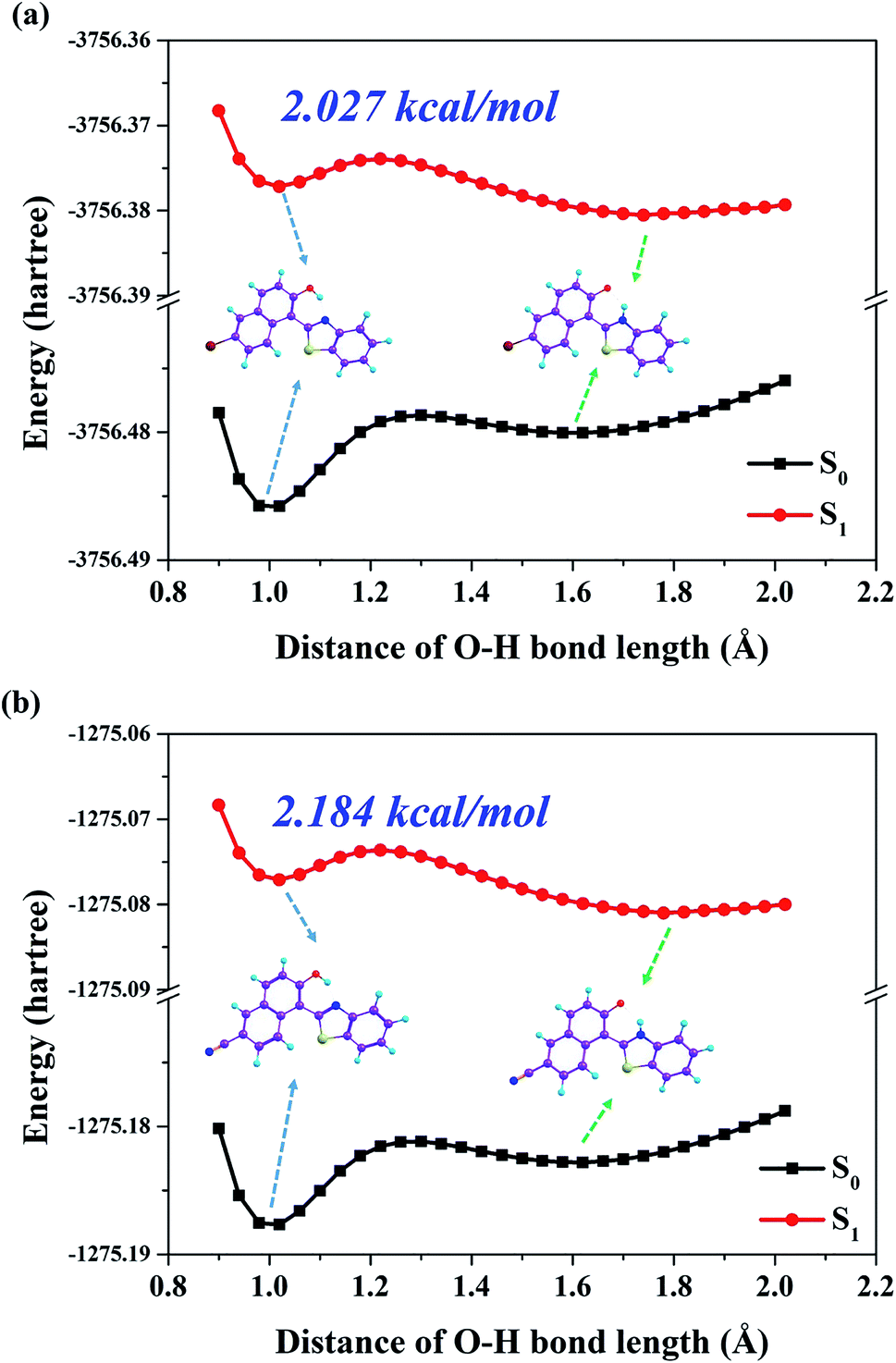 | ||
| Fig. 5 The constructed potential energy curves for both Br-BTN (a) and CN-BTN (b) based on fixing O–H bond lengths along with hydrogen bond O–H⋯N in both S0 and S1 states. | ||
In addition, based on the Berny optimization method coupling with the TDDFT/B3LYP/6-311+G(d) theoretical level, we search the transition state (TS) structure on the S1-state potential energy curve (shown in Fig. 6). Our theoretical TS form has been confirmed to be only one imaginary frequency, and its vibrational eigenvector points to the correct direction. The zero-point energy corrections have been also performed according to the harmonic vibrational frequencies. The imaginary frequencies of Fig. 6 are calculated to be −1687.3 cm−1 for Br-BTN-enol and −1703.6 cm−1. Furthermore, to the best of our knowledge, the excited state dynamical reaction path could be constructed via integrating the intrinsic reaction coordinate (IRC) at the same level of theory. And this scheme of integration over the S1-state potential energy curve can be obtained via combining the first-order Euler predictor approach with the modified Bulirsch–Stoer integrator for the corrector algorithm. The corresponding IRC curves of Br-BTN-enol and CN-BTN-enol systems have been shown in Fig. 7. Beginning from the TS structure, two minima (both reactant and product) could be searched according to the direction of energy decrease, which further verifies the ESIPT mechanism for Br-BTN-enol and CN-BTN-enol in the S1 state. Furthermore, beginning from the TS structures for both Br-BTN and CN-BTN system, the classical trajectory calculations are performed to explore the dynamical behavior with adopting the Born–Oppenheimer molecular dynamics (BOMD) method. The time evolution of the important structural parameters are shown in Fig. S4, ESI.† In fact, it could be noticed that the bond distance of H–N tends to become stable (1.05 Å) for both Br-BTN and CN-BTN molecules after evolving about 25 fs. The stable 1.05 Å is consistent with the optimized S1-state bond distance of H–N for Br-BTN-keto and CN-BTN-keto systems. In addition, Given the O–H bond distance, along with time evolution more than 300 fs, it can be found bond distance of O–H tends to become 1.65–1.70 Å. They are also close to the S1-state stable O⋯H for Br-BTN-keto and CN-BTN-keto structures. Therefore, from the view of kinetic point, we further confirm the ESIPT mechanism for both Br-BTN and CN-BTN systems.
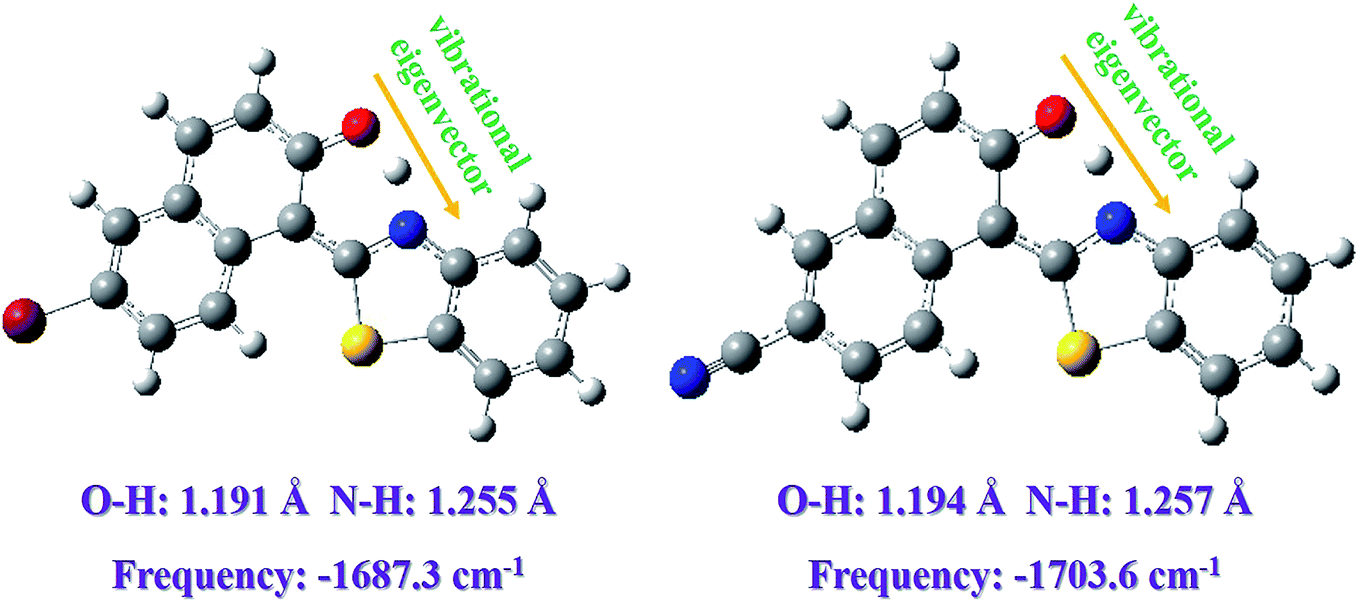 | ||
| Fig. 6 The searched transition state (TS) structure for both Br-BTN and CN-BTN systems along with the ESIPT reaction path in the S1 state. Herein, the bond length and corresponding imaginary frequency of TS structures are also listed. | ||
 | ||
| Fig. 7 The energy profile along with the first excited state IRC for Br-BTN (a) and CN-BTN (b) based on TDDFT/B3LYP/6-311+G(d) theoretical level in DMSO solvent. | ||
4. Conclusion
In this work, we theoretically investigate the excited state hydrogen bonding interactions and ESIPT behaviors for Br-BTN-enol and CN-BTN-enol systems. Firstly, we check and verify the formation of intramolecular hydrogen bond O–H⋯N of Br-BTN-enol and CN-BTN-enol in the ground state. Then, via comparing bond lengths and bond angles involved in hydrogen bonding moieties, we confirm that hydrogen bond should be strengthening in the first excited state. The analyses about IR vibrational spectra further verify the strengthening phenomenon. When it comes to the photoexcitation process, it should be mentioned that the charge redistribution plays important roles in excited state dynamical tendency. The increased electronic densities around proton acceptor atom facilitate attracting hydrogen proton, which plays a decisive role in opening ESIPT reaction. Given the specific ESIPT mechanism, we construct the potential energy curves and confirm the ultrafast ESIPT behavior due to low potential energy barrier. Further, we search the TS structure and calculate the IRC path, based on which we further verifies the ultrafast ESIPT mechanism. We sincerely wish this work could promote the corresponding novel development and applications based on Br-BTN and CN-BTN systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Key Scientific Research Project of Colleges and Universities of Henan Province of China (grant no. 18A140023).References
- D. Ray, A. Pramanik and N. Guchhait, J. Phys. Chem. B, 2015, 119, 10114 CrossRef CAS PubMed.
- T. Bruhn and C. Bruckner, Phys. Chem. Chem. Phys., 2015, 17, 3560 RSC.
- J. Li, M. Zhang, C. Du, P. Song and X. Li, J. Phys. Org. Chem., 2018, 31, e3867 CrossRef.
- Y. Cui, H. Zhao, J. Zhao, P. Li, P. Song and L. Xia, New J. Chem., 2015, 39, 9910 RSC.
- S. Sahu, M. Das, A. Bharti and G. Krishnamoorthy, Phys. Chem. Chem. Phys., 2018, 20, 27131 RSC.
- G. Fan, K. Han and G. He, Chin. J. Chem. Phys., 2013, 26, 635 CrossRef CAS.
- N. Basaric, S. Thomas, V. Bregovic, N. Cindro and C. Bohne, J. Org. Chem., 2015, 80, 4430 CrossRef CAS PubMed.
- Z. Vealey, L. Foguel and P. Vaccaro, J. Phys. Chem. Lett., 2018, 9, 4949 CrossRef CAS PubMed.
- J. Zhao, H. Dong and Y. Zheng, J. Lumin., 2018, 195, 228 CrossRef CAS.
- S. Aoi, K. Mase, K. Ohkubo and S. Fukuzumi, Chem. Commun., 2015, 51, 15145 RSC.
- G. Li and K. Han, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1351 Search PubMed.
- D. Presti, A. Pedone, I. Ciofini, F. Labat, M. Menziani and C. Adamo, Theor. Chem. Acc., 2016, 135, 86 Search PubMed.
- Q. Xiao, L. Tian, R. Tan, Y. Xia, D. Qiu, Y. Zhang and J. Wang, Org. Lett., 2012, 14, 4230 CrossRef CAS PubMed.
- Q. Zhu, B. An, H. Yuan, Y. Li, X. Guo and J. Zhang, Mol. Phys., 2017, 115, 288 CrossRef CAS.
- H. Yin, Y. Zhang, H. Zhao, G. Yang, Y. Shi, S. Zhang and D. Ding, Dyes Pigm., 2018, 159, 506 CrossRef CAS.
- J. Zhao, H. Dong and Y. Zheng, J. Phys. Chem. A, 2018, 122, 1200 CrossRef CAS PubMed.
- J. Huang, K. Yu, H. Ma, S. Chai and B. Dong, Dyes Pigm., 2017, 141, 441 CrossRef CAS.
- J. Zhao, P. Song and F. Ma, Communications in Computational Chemistry, 2014, 2, 117 CrossRef.
- J. Huang, J. Wu, H. Dong, P. Song and J. Zhao, Communications in Computational Chemistry, 2017, 5, 27 CrossRef.
- O. Mukhina and A. Kutateladze, J. Am. Chem. Soc., 2016, 138, 2110 CrossRef CAS PubMed.
- J. Huang, W. Teng, D. Chen and H. Ma, J. Mol. Liq., 2018, 249, 957 CrossRef CAS.
- J. Li, X. Li, S. Cheng, P. Song and J. Zhao, Journal of Atomic and Molecular Sciences, 2018, 9, 1 CrossRef.
- C. Du, Q. Zhou, M. Zhang, P. Song and F. Ma, J. Phys. Org. Chem., 2019, 32, e3901 CrossRef.
- J. Zhao, J. Chen, Y. Cui, J. Wang, L. Xia, Y. Dai, P. Song and F. Ma, Phys. Chem. Chem. Phys., 2015, 17, 1142 RSC.
- J. Zhao, H. Dong, H. Yang and Y. Zheng, Org. Chem. Front., 2018, 5, 2710 RSC.
- Z. Liu, L. Wang, H. Tan, S. Zhou, T. Fu, Y. Xia, Y. Zhang and J. Wang, Chem. Commun., 2014, 50, 5061 RSC.
- C. Li, C. Ma, D. Li and Y. Liu, J. Lumin., 2016, 172, 29 CrossRef CAS.
- C. Hsieh, C. Jiang and P. Chou, Acc. Chem. Res., 2010, 43, 1364 CrossRef CAS PubMed.
- M. Nayak, J. Photochem. Photobiol., A, 2012, 241, 26 CrossRef CAS.
- J. Han, X. Liu, C. Sun, Y. Li, H. Yin and Y. Shi, RSC Adv., 2018, 8, 29589 RSC.
- H. Tseng, J. Liu, Y. Chen, C. Chao, K. Liu, C. Chen, T. Lin, C. Hung, Y. Chou, T. Lin, T. Wang and P. Chou, J. Phys. Chem. Lett., 2015, 6, 1477 CrossRef CAS PubMed.
- F. Yu, P. Li, G. Li, G. Zhao, T. Chu and K. Han, J. Am. Chem. Soc., 2011, 133, 11030 CrossRef CAS PubMed.
- A. Demchenko, K. Tang and P. Chou, Chem. Soc. Rev., 2013, 42, 1379 RSC.
- Y. Liu, J. Zhao, Y. Wang, J. Tian, X. Fei and H. Wang, J. Mol. Liq., 2017, 233, 303 CrossRef CAS.
- J. Zhao, X. Liu and Y. Zheng, J. Lumin., 2017, 188, 1 CrossRef CAS.
- M. Zhang, Q. Zhou, C. Du, Y. Ding and P. Song, RSC Adv., 2016, 6, 59389 RSC.
- J. Zhao, H. Dong, H. Yang and Y. Zheng, ACS Appl. Bio Mater., 2019, 2, 2060 CrossRef CAS.
- M. Zhang, Q. Zhou, M. Zhang, Y. Dai, P. Song and Y. Jiang, J. Cluster Sci., 2017, 28, 1191 CrossRef CAS.
- G. Li, G. Zhao, K. Han and G. He, J. Comput. Chem., 2011, 32, 668 CrossRef CAS PubMed.
- J. Zhao and P. Li, RSC Adv., 2015, 5, 73619 RSC.
- M. Santra, Y. Jun, Y. Reo, S. Sarkar, W. Choi, J. Kwon, S. Part and K. Ahn, ACS Appl. Bio Mater., 2018, 1, 136 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Scalmani, G. Cheeseman, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200 CrossRef CAS.
- W. Kolth, A. Becke and R. Parr, J. Phys. Chem., 1996, 100, 12974 CrossRef.
- O. Treutler and R. Ahlrichs, J. Chem. Phys., 1995, 102, 346 CrossRef CAS.
- F. Furche and R. Ahlrichs, J. Chem. Phys., 2002, 117, 7433 CrossRef CAS.
- B. Mennucci, E. Cances and J. Tomasi, J. Phys. Chem. B, 1997, 101, 10506 CrossRef CAS.
- E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef CAS.
- R. Cammi and J. Tomasi, J. Comput. Chem., 1995, 16, 1449 CrossRef CAS.
- E. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498 CrossRef CAS PubMed.
- G. Zhao and K. Han, J. Phys. Chem. A, 2007, 111, 9218 CrossRef CAS PubMed.
- G. Zhao and K. Han, J. Phys. Chem. A, 2007, 111, 2469 CrossRef CAS PubMed.
- J. Zhao, J. Chen, J. Liu and M. Hoffmann, Phys. Chem. Chem. Phys., 2015, 17, 11990 RSC.
- J. Zhao, H. Yao, J. Liu and M. Hoffmann, J. Phys. Chem. A, 2015, 119, 681 CrossRef CAS PubMed.
- G. Zhao and K. Han, Acc. Chem. Res., 2012, 45, 404 CrossRef CAS PubMed.
- J. Zhao, X. Liu and Y. Zheng, J. Phys. Chem. A, 2017, 121, 4002 CrossRef CAS PubMed.
- Y. Saga, Y. Shibata and H. Tamiaki, J. Photochem. Photobiol., C, 2010, 11, 15 CrossRef CAS.
- J. Zhao and Y. Zheng, Sci. Rep., 2017, 7, 44897 CrossRef CAS PubMed.
- V. Tomin, A. Demchenko and P. Chou, J. Photochem. Photobiol., C, 2015, 22, 1 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04258j |
| This journal is © The Royal Society of Chemistry 2019 |