
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCo2+ substituted for Bi3+ in BiVO4 and its enhanced photocatalytic activity under visible LED light irradiation†
Trinh Duy Nguyen *a,
Quynh Thi Phuong Buib,
Tien Bao Leb,
T. M. Altahtamounic,
Khanh Bao Vud,
Dai-Viet N. Voa,
Nhan Thi Hong Lee,
Tuan Duy Luue,
Seong Soo Hongf and
Kwon Taek Limg
*a,
Quynh Thi Phuong Buib,
Tien Bao Leb,
T. M. Altahtamounic,
Khanh Bao Vud,
Dai-Viet N. Voa,
Nhan Thi Hong Lee,
Tuan Duy Luue,
Seong Soo Hongf and
Kwon Taek Limg
aCenter of Excellence for Green Energy and Environmental Nanomaterials (CE@GrEEN), Nguyen Tat Thanh University, Ho Chi Minh City, Vietnam. E-mail: ndtrinh@ntt.edu.vn; Fax: +84-028-39-404-759; Tel: +84-028-3941-1211
bFaculty of Chemical Technology, Ho Chi Minh City University of Food Industry, 140 LeTrong Tan, Tan Phu District, Ho Chi Minh City, Vietnam
cMaterials Science and Technology Program, College of Arts and Sciences, Qatar University, Doha 2713, Qatar
dNTT Hi-Tech Institute, Nguyen Tat Thanh University, Ho Chi Minh City, Vietnam
eDepartment of Chemical Engineering, HCMC University of Technology, VNU-HCM, Ho Chi Minh City, Vietnam
fDepartment of Chemical Engineering, Pukyong National University, Busan, Korea
gDepartment of Display Engineering, Pukyong National University, Busan, Korea
First published on 30th July 2019
Abstract
We investigated the fabrication of Co-doped BiVO4 (Bi1−xCoxVO4+δ, 0.05 < x < 0.5) by the substitution of Co ions for Bi sites in BiVO4. The X-ray diffraction (XRD), Raman, and X-ray photoelectron spectroscopy (XPS) results indicated that the substitution of Co2+ ions for Bi3+ sites in BiVO4 was successful, although a change in the crystal phase of BiVO4 did not occur. UV-vis DRS and PL results suggested that the Co-incorporation could slightly improve the visible light absorption of BiVO4 and induce the separation of photoinduced electron–hole pairs; therefore, a significant enhancement of photocatalytic performance was achieved. The Bi0.8Co0.2VO4+δ sample showed superior photocatalytic activity in comparison with other samples, achieving 96.78% methylene blue (MB) removal within 180 min. In addition, the proposed mechanism of improved photocatalytic activities and the stability of the catalyst were also investigated.
1 Introduction
Monoclinic scheelite bismuth vanadate (m-s BiVO4), a p-type semiconductor with a direct band gap (Eg) of 2.4 eV, has generated considerable interest as a visible light-responsive photocatalyst.1–4 Reportedly, different morphologies of monoclinically structured BiVO4 crystallites, such as nanospheres,5 nanorods,6,7 nanoflowers,8 nanosheets,9 nanotubes,10–12 olive,13,14 and hyperbranched,15 have been clearly created by the hydrothermal or solvothermal technique. Nevertheless, the poor charge transport and weak surface adsorption properties associated with pristine BiVO4 could cause prompt recombination of photogenerated electron–hole pairs, reducing its overall photocatalytic efficiency. Hence, various strategies aiming to enhance the photocatalytic performance of m-s BiVO4 have been explored. Such approaches could include doping BiVO4 with metals and/or non-metals,16–21 controlling the morphology and facets of BiVO4,5,13,22,23 constructing a novel p–n heterojunction photocatalyst,24–27 and controlling the monoclinic-tetragonal heterostructure of BiVO4.28Among the many methods for improving the photocatalytic activity, metal and/or non-metal doping is regarded as a simple and effective strategy. For metal-doped BiVO4, metal ions can substitute the V or Bi sites in the crystal lattice of BiVO4, exhibiting a quite different photocatalytic performance. For instance, Luo and co-workers observed that the photocurrent of the BiVO4 semiconductor could be substantially improved by Mo6+ or W6+ doping, and further demonstrated the substitution of Mo6+ or W6+ ions in V5+ positions.29 Contrarily, the incorporation of lanthanide ions CeIII,IV occurring at Bi3+ sites in the BiVO4 lattice caused a pronounced change in the crystal phase, optical properties, and the separation of photoinduced electron–hole pairs, resulting in a prominent improvement of photocatalytic activity.30 Substitution of Fe3+ ions for Bi3+ sites in BiVO4 also significantly enhanced photocatalytic performance for the decomposition of ibuprofen and greatly inactivated Escherichia coli bacteria under visible light irradiation.31 However, modification of BiVO4 by doping of cobalt ions seemed to yield contradictory results. Specifically, Regmi and co-workers found that Co substituted for V in the crystal lattice, indicated by the shift in the symmetric V–O stretching mode to lower wavenumber, was confirmed by the Raman result.32 To the contrary, another study showed that Co2+ substituted for Bi3+ in BiVO4, which was confirmed by Bo Zhang et al.33 On the other hand, it was suggested by Zhou et al. that the incorporation of cobalt as an oxide on the surface of larger BiVO catalysts caused no change in the crystal structure of the composites.34 Co-doped BiVO was also investigated by Geng et al.,35 but they did not identify the substitution of Co ions for Bi3+ or V3+ sites in BiVO4.
Therefore, the present study aimed to identify the substitution of Co ions for Bi3+ sites in BiVO4. We proposed the synthesis of Co-doped BiVO4 (Bi1−xCoxVO4+δ, 0.05 < x < 0.5) by the hydrothermal method. The effect of Co2+ ions substituted for Bi3+ sites in BiVO4 on the crystal structure, morphology, optical behavior, photoinduced charges separation, and photocatalytic performance for decomposition of methylene blue (MB) under visible LED light irradiation was carefully explored. The proposed mechanism for the improved photocatalytic activities in Co2+ doped BiVO4 is discussed. In addition, the stability and reusability of catalyst were also investigated.
2 Experimental
2.1 Synthesis of photocatalysts
The hydrothermal procedure employing Bi(NO3)3·5H2O, Co(NO3)2·6H2O, and NH4VO3 as the precursors were adopted to synthesize the Co-doped BiVO4 samples. Firstly, two clear solutions were prepared as follows: (i) Bi(NO3)3·5H2O (4 mmol) and a predetermined amount of Co(NO3)2·6H2O were dissolved in 20 mL of HNO3 2 M; (ii) 4 mmol of NH4VO3 was dissolved in 40 mL of NaOH 0.3 M. Estimation of the concentration of Co(NO3)2·6H2O followed the stoichiometric formulation of Bi1−xCoxVO4+δ (where x = 0, 0.05, 0.1, 0.2, 0.3, and 0.5; abbreviated as BVO, 0.05Co–BVO, 0.1Co–BVO, 0.2Co–BVO, 0.3Co–BVO, and 0.5Co–BVO, respectively). Then, a mixture was created by mixing the two solutions under vigorous stirring for 30 min; pH was adjusted to 10 using NaOH 4 M solution. Next, the resulting suspension was adjusted for volume to 80 mL using distilled water and then transferred to a 100 mL hydrothermal autoclave reactor with a Teflon chamber vessel. The reactor then underwent thermal treatment at 180 °C in an oven for 24 h. The obtained yellow paste was rinsed with water until the pH of the filtrate solution was around 7. Finally, the obtained powder was dried at 110 °C for 24 h, followed by calcination at 300 °C for 2 h.2.2 Characterization of photocatalysts
To characterize the crystal structure of the undoped and doped BiVO4 photocatalysts, multiple techniques including X-ray diffraction (XRD, D8 ADVANCE-Bruker), Raman spectra (HORIBA Jobin Yvon), and X-ray photoelectron spectroscopy (XPS, Thermo VG Multilab 2000) were employed. Morphological characteristics were observed by Field Emission Scanning Electron Microscopy (FE-SEM, JSM-6700F, Jeol) and transmission electron microscopy (TEM, JEM 1400, JEOL). Energy band structure was evaluated through the UV-visible diffuse reflectance spectrum (UV-Vis DRS, Shimadzu UV-2450) with BaSO4 as the standard. Photoluminescence (PL) spectroscopy was performed using an F-4500 Spectro-fluorometer (Hitachi) with an excitation wavelength of 315 nm.2.3 Photocatalytic test
The assessment of the photocatalytic performance of the bare BiVO4 and Co-doped BiVO4 was executed by the photodecomposition of MB dye in aqueous media under white LED light irradiation. Six LED lamps (XLamp XT-E White, Cree X6) with a max power of 10 W and max light output of 1040 lm acted as the white light source for this study. To perform this experiment, a mixture containing catalyst powder (0.5 g L−1) and MB aqueous solution (100 mL, 15 mg L−1) was mixed in a 250 mL double layer interbed interlayer glass beaker and vigorously stirred for 100 min to ensure the adsorption–desorption equilibrium between the surface of BiVO4 and the MB molecules. Under light exposure, aliquots of the reaction solution were withdrawn at defined time intervals, and the solid was immediately isolated by centrifugation at 8000 rpm. The concentration of MB was determined by the intensity change of absorption peak at 664 nm using a UV-visible spectrophotometer (Model Evolution 60S, Thermo Fisher Scientific). In addition, to investigate the stability of catalyst, repeated photodecomposition of MB dye over five consecutive cycles was conducted using a method similar to the above. After each reaction run, the catalyst was washed with water three times and isolated by centrifugation at 8000 rpm.2.4 Analysis of hydroxyl radical productions
We also used the PL technique with benzene-1,4-dicarboxylic acid (H2BDC) as the sensor molecule to detect the ·OH radicals produced by 0.2Co–BVO during the photocatalytic processes. H2BDC molecule reacted with the ·OH radicals to form a fluorescent molecule (2-hydroxy-1,4-benzenedicarboxylic acid, H2BDC-OH). The experimental procedures were conducted based on the above photocatalytic activity test, but MB solution was replaced by H2BDC solution ([H2BDC] = 5 × 10−4 M and [NaOH] = 2 × 10−3 M).14 After 60 min of irradiation, the reaction solution after eliminating catalyst was analyzed by a Hitachi F4500 fluorescence spectrometer at an excitation wavelength of 315 nm.3 Results and discussion
3.1 Crystal structure
Fig. 1A presents XRD patterns of the bare BiVO4 and Co-doped BiVO4 samples. The bare BiVO4 sample had a pure monoclinic scheelite phase structure (m-s BiVO4, JCPDs 14-0688) with sharp characteristic peaks at 28.9°, and 30.5°, and double peaks at 18.9°, 35°, and 47°. Similarly, all XRD peaks of the 0.05Co–BVO, 0.1Co–BVO, and 0.2Co–BVO samples were also correctly indexed to the m-s BiVO4, and no peaks of tetragonal phase (t-z BiVO4) were detected. This result indicated that the presence of Co with x = 0.05–0.2 caused no significant change in the crystal phase of BiVO4, which was consistent with previous studies.33–35 However, when further increasing the Co molar ratio to 0.3, diffraction peaks of impurities including Bi2O3 (JCPDs 41-1449) and Co2V2O7 (JCPDs 70-1189) were detected. The XRD pattern of the 0.5Co–BVO sample also showed the mixed phases of BiVO4, Bi2O3, Co2V2O7, and Co3V2O8 (JCPDs 74-1487). The presence of Bi2O3 could be due to the hydrolysis of Bi(NO3)3.36 Co2V2O7 and Co3V2O8 could be formed when the excess of Co2+ content under hydrothermal condition.37 In addition, there was no peak indexed to the cobalt oxide or cobalt hydroxide for all Co-doped BiVO4 samples, suggesting the possible doping of the cobalt ions into the lattice of BiVO4 instead of loading to the BiVO4 surface as hydroxide or oxide crystal grains. For the 0.2Co–BVO sample, we also observed that the peak at 28.9° had slightly shifted to a higher angle (Fig. S1†), implying that Co could be doped into the lattice of BiVO4 crystal.31,32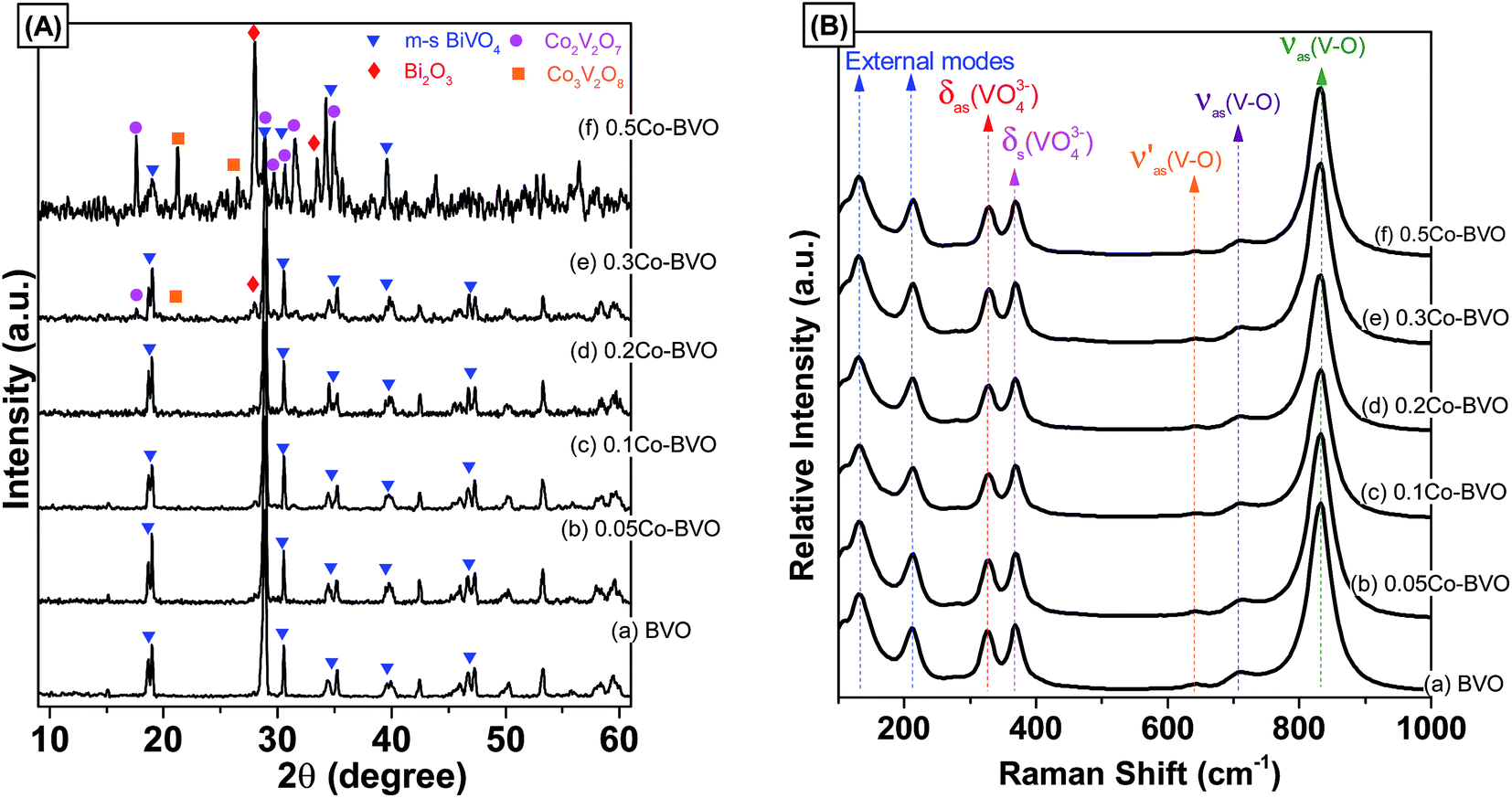 | ||
| Fig. 1 XRD patterns (A) and Raman spectra (B) of the pristine BiVO4 and Co-doped BiVO4 samples. | ||
The crystallite size (d, nm) of Bi1−xCoxVO4+δ samples were evaluated from {121} peak of XRD patterns using the Scherrer's equation.14,30 The d values of the Bi1−xCoxVO4+δ samples are presented in Table 1. As shown in Table 1, the crystallite size of Co-doped BiVO4 slightly increased compared to that of bare BiVO4 sample, implying that the presence of Co2+ could influence the formation of crystalline particles of BiVO4.
| Samples | d (nm) | Eg (eV) | kapp (10−3 min−1) | R2 |
|---|---|---|---|---|
| BVO | 25.45 | 2.31 | 0.775 | 0.99742 |
| 0.1Co–BVO | 29.09 | 2.30 | 1.185 | 0.95483 |
| 0.2Co–BVO | 28.65 | 2.29 | 1.882 | 0.98230 |
| 0.3Co–BVO | 31.88 | 2.26 | 0.633 | 0.98225 |
| 0.5Co–BVO | 30.31 | 2.14 | 0.333 | 0.98070 |
The as-synthesized samples then had their phase structure confirmed by Raman spectra as plotted in Fig. 1B. It was observed that all samples expressed main Raman shifts at around 131 cm−1 (external mode), 212 cm−1 (external mode), 326 cm−1 (the asymmetric bending mode δas(VO43−)), 368 cm−1 (the symmetric bending mode δs(VO43−)), 639 cm−1 (the asymmetric V–O stretching mode 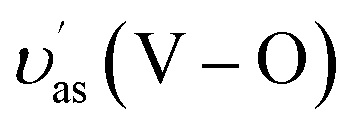 ), 709 cm−1 (the asymmetric V–O stretching mode υas(V–O)), and 832 cm−1 (the symmetric V–O stretching mode υs(V–O)), which were similar to those of the m-s BiVO4 phase. It was indicated that the existence of Co ion did not affect the crystal phase of BiVO4, which was confirmed by the XRD results. Also, a downtrend according to the increase of doped ion content in the intensity the symmetric V–O stretching mode was observed (Fig. S2†), which was caused by the weak deformation of the VO4 tetrahedron. A similar effect was reported previously in terms of Fe-doped BiVO4 (ref. 31) and Ce-doped BiVO4,30 in which this weak deformation could be due to the metal ion substitution at the Bi3+ site in the lattice of the BiVO4 crystal.
), 709 cm−1 (the asymmetric V–O stretching mode υas(V–O)), and 832 cm−1 (the symmetric V–O stretching mode υs(V–O)), which were similar to those of the m-s BiVO4 phase. It was indicated that the existence of Co ion did not affect the crystal phase of BiVO4, which was confirmed by the XRD results. Also, a downtrend according to the increase of doped ion content in the intensity the symmetric V–O stretching mode was observed (Fig. S2†), which was caused by the weak deformation of the VO4 tetrahedron. A similar effect was reported previously in terms of Fe-doped BiVO4 (ref. 31) and Ce-doped BiVO4,30 in which this weak deformation could be due to the metal ion substitution at the Bi3+ site in the lattice of the BiVO4 crystal.
The presence of substitution sites of Co in Co-doped BiVO4 was also validated by the XPS spectrum. The survey and high-resolution XPS spectrum of the pristine BiVO4 and 0.2Co–BVO photocatalysts are shown in Fig. 2A–F. Meanwhile, characteristic peaks of Bi, V, and O were present for pristine BiVO4 and 0.2Co–BVO photocatalysts exhibiting Bi, V, O, and Co peaks (Fig. 2A). In addition, according to the peak area, the molar ratios of Bi/Co in 0.2Co–BVO photocatalysts reached the value of 3.59/1, which is close to the estimated value of 4/1, indicating well-dispersion of Co in BiVO4. The C 1s peak at 284.6 eV (Fig. 2B), assigning to the signal from carbon in the instrument, was used for calibration. Co 2p could be deconvoluted into two pairs of individual peaks at 780.9 eV (Co 2p3/2) and 797.3 eV (Co 2p1/2) (Fig. 2C), which were identified as the major binding energies (BE) of Co2+.38 As also presented in Fig. 2C, the BE indexed to the cobalt metal or cobalt oxides was not detected, implying that Co could be doped into the lattice of BiVO4 crystal. Bi 4f spectrum of BVO (Fig. 2D) showed characteristic peak of Bi 4f5/2 and Bi 4f7/2 at 164.1 eV and 158.7 eV respectively, with ΔE = Bi 4f5/2 − Bi 4f7/2 of 5.4 eV. Similarly, Bi 4f5/2 (164.2 eV) and Bi 4f7/2 (158.9 eV) with ΔE of 5.3 eV were also observed in the Bi 4f spectrum of 0.2Co–BiVO4 samples (Fig. 2D). After the Gaussian–Lorentzian fitting, the asymmetric V 2p3/2 spectra can be deconvoluted into two pairs of individual components referring to the surface V4+ and V5+ states; they were 515.6 and 516.8 eV for BVO, and 515.0 and 516.7 eV for 0.2Co–BiVO4 (Fig. 2E). The V4+/V5+ molar ratios of BVO and 0.2Co–BVO samples were 0.165 and 0.214, respectively. These results showed the slight shift of Bi 4f and V 2p for the 0.2Co–BVO sample as compared with the BVO sample, implying the interaction of Co2+ with Bi3+ and V5+ ions. Moreover, considering to the ion radius of Co2+, V5+, and Bi3+, the ion radius of Co2+ (0.79 Å)32 was smaller than that of the Bi3+ (1.03 Å)30 but larger than that of V5+ (0.36 Å);29,39 thus, doping with Co2+ tended to replace some Bi3+ in the internal lattice structure of BiVO4. This outcome was also supported by Bo Zhang et al. when using periodic density functional calculations within the GGA + U approach for calculating the formation energy.33
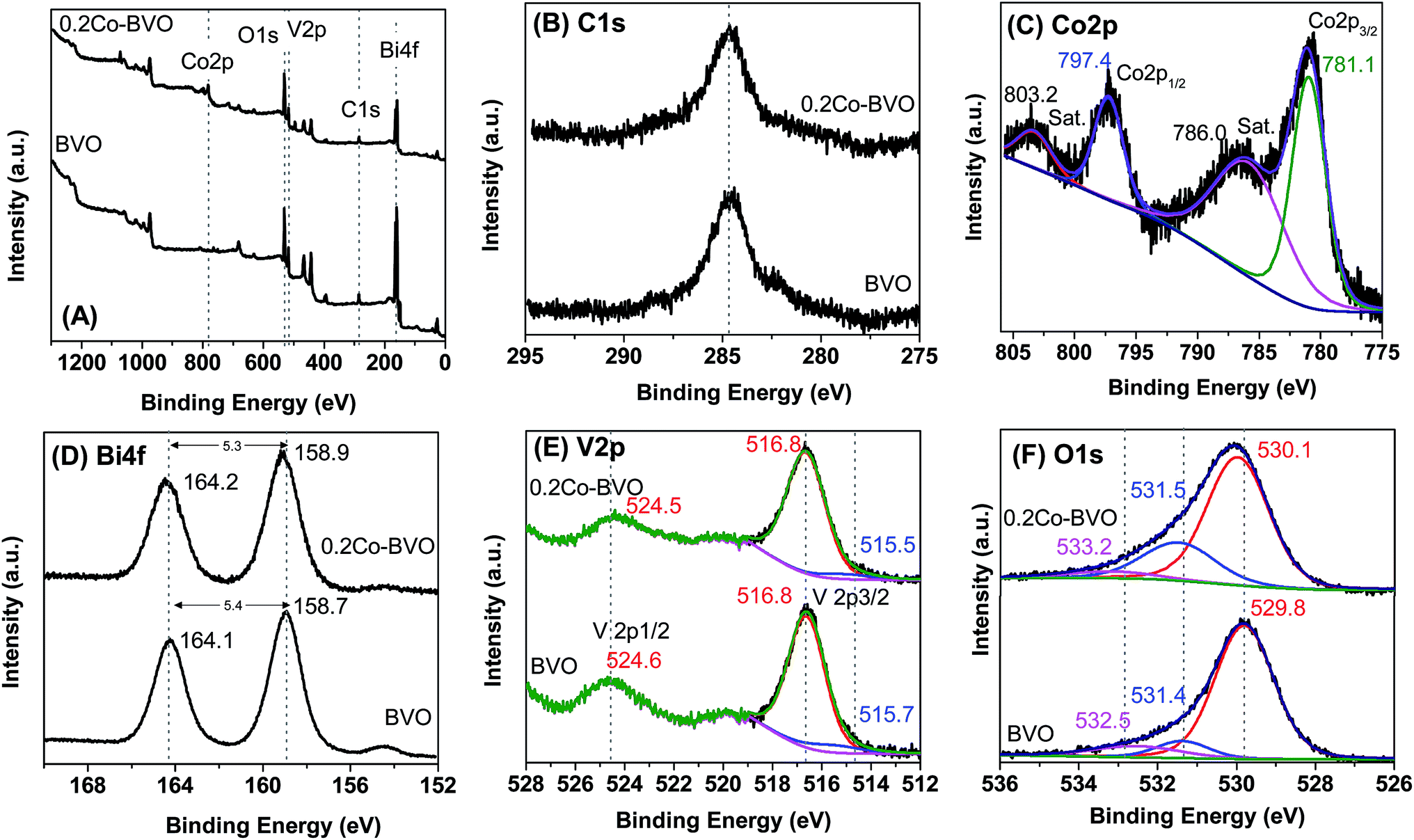 | ||
| Fig. 2 The survey and high-resolution XPS spectrum of the pristine BiVO4 and 0.2Co-BVO. | ||
Fig. 2F displays O 1s spectrum of the pure BiVO4 sample with the BE positions at 529.8 eV, 531.4 eV, and 532.5 eV, which indexed to the lattice oxide (V–O or Bi–O bonds) in BiVO4 (Olatt), the hydroxide hydrated or defective oxide (Oads) species, and the surface adsorbed water, respectively.30 As also shown in Fig. 2F, no other BE positions were observed due to the BE of V–O and Bi–O bonds being approximately the same. In terms of 0.2Co–BVO sample, after the Gaussian–Lorentzian fitting, the O 1s spectrum was also deconvoluted into three pairs of individual components: (1) the lattice oxide at BE of 530.1 eV; (2) the hydroxide hydrated or defective oxide at BE of 531.5 eV; (3) and the surface adsorbed water at BE of 533.2 eV. Notably, these peaks were slightly shifted to the high BE region in comparison to that of the bare BiVO4. This outcome could be due to the doping Co2+ replacing some Bi3+ positions in the internal lattice structure of BiVO4, implying the formation of Co–O–Bi bonds. Also, the varied Oads/Olatt molar ratio was 0.49 for 0.2Co–BVO and 0.24 for the bare BiVO4 sample. These results indicated that Co-doped BiVO4 further increases the content of Oads and oxygen vacancy, which are possibly responsible for improving the photocatalytic performance of BiVO4, as discussed below.
3.2 Morphological characteristics
The presence of Co also considerably altered the morphology of the as-synthesized samples, as exhibited in Fig. 3. The bare BiVO4 sample consisted of large quantities of corner-cut truncated bipyramid and granular like structures with smooth surfaces (Fig. 3a). The 0.05Co–BVO (Fig. S3A†), 0.1Co–BVO (Fig. S3B†), and 0.2Co–BVO (Fig. 3b) were morphologically similar to the Co-doped BiVO4 sample. However, the surface of these Co-doped BiVO4 sample (Fig. 3b) was much rougher than those of the bare BiVO4 sample and covered amorphous particles. Consistent with the results of the XRD patterns, the 0.3Co–BVO (Fig. S3C†) and 0.5Co–BVO (Fig. S3D†) samples mostly had amorphous nanoparticles because of the presence of the mixture containing BiVO4, Bi2O3, Co2V2O7, and Co3V2O8. Also, as presented in Fig. 4, the EDS-Mapping analysis of Bi, V, O, and Co in the 0.2Co–BVO was both in line with the aforementioned XPS results and indicative of uniform distribution over the BiVO4 surface. The TEM images of 0.2Co–BVO sample (Fig. S4†) also confirms that the crystal of 0.2Co–BVO sample was not homogeneous with different shapes and sizes. | ||
| Fig. 3 SEM images of the (a) BVO and (b) 0.2Co–BVO samples. | ||
 | ||
| Fig. 4 Element mapping images of 0.2Co–BVO sample: SEM image (a), Bi element (b), V element (c), O element (d) and Co element (e). | ||
3.3 Optical absorption properties
UV-vis DRS spectra of the Bi1−xCoxVO4+δ samples are displayed in Fig. 5A. All samples demonstrated intensive absorption bands in the visible light region with the absorption edges of 550–600 nm, which ascribed to the electron transfer from the valence band (VB) to the conduction band (CB). The direct band gap energy (Eg, eV) of the as-synthesized photocatalysts were derived from the tangent in the Tauc plots when plotting the (αhν)2 function with respect to hν (α, h, and ν are the absorption coefficient, Plank's constant, and incident light frequency, respectively) as shown in Fig. 5B.13,30,40,41 The Eg values of the Bi1−xCoxVO4+δ samples are presented in Table 1. Accordingly, the Eg appeared to be slightly declining with increasing Co molar ratio. This behavior indicated the slightly narrowed BiVO4 band gap, which could be due to Co incorporation and the introduction of an energy level (Ef) near the top of the VB in the forbidden band gap (Fig. 11), caused by substitution of cobalt ions to Bi3+ in BiVO4.33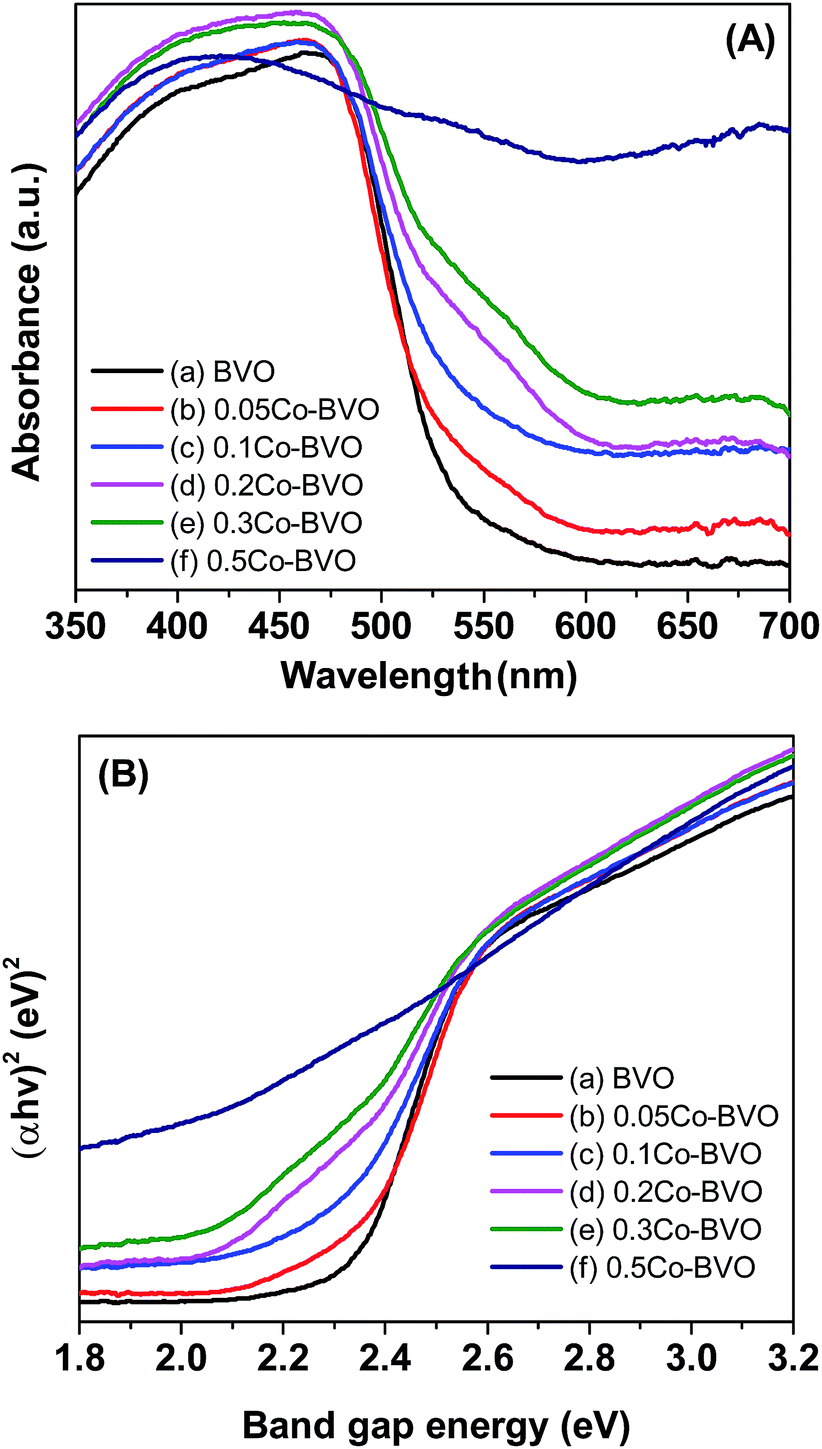 | ||
| Fig. 5 UV-vis DRS spectra (A) and band gap energy (B) of the pristine BiVO4 (a), 0.05Co–BVO (b), 0.1Co–BVO (c), 0.2Co–BVO (d), 0.3Co–BVO (e), and 0.5Co–BVO (f). | ||
The separation and transfer of photoinduced charges in Bi1−xCoxVO4+δ semiconductors could be clarified by PL spectra as presented in Fig. 6. For the undoped BiVO4 catalyst, the emission peak at an excitation wavelength of 340 nm was observed at around 478 nm. For 0.2Co–BVO, the absence of an emission peak indicated that Co incorporation, due to the increased electron mobility, could prevent photogenerated electron–hole pairs from recombining. As a result, it was apparent that the photocatalytic activity of the Bi1−xCoxVO4+δ samples was improved due to the efficient separation and transfer of photoinduced charges.
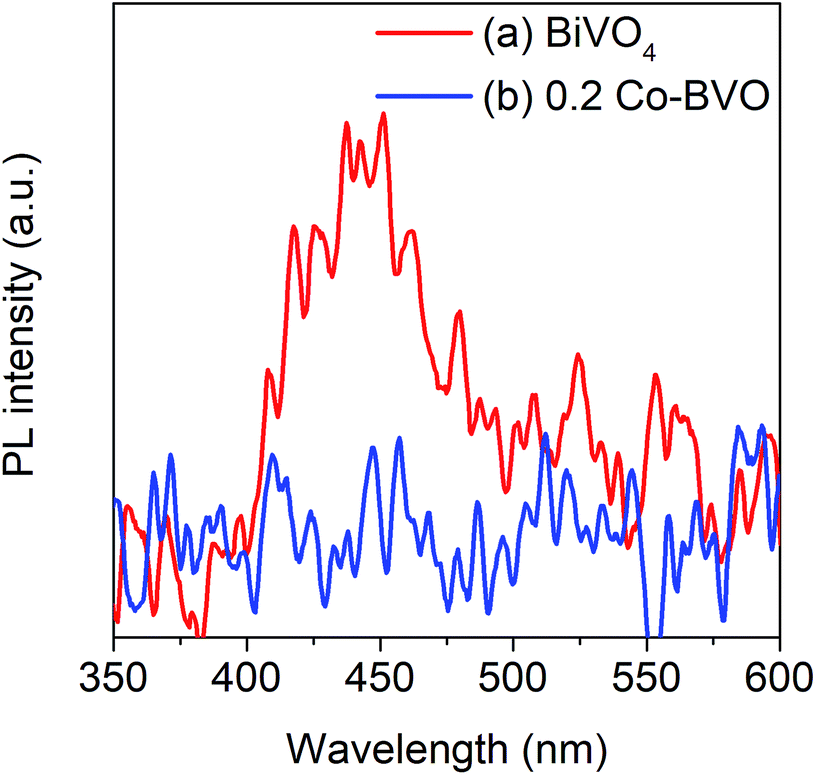 | ||
| Fig. 6 Room temperature PL spectra of the pristine BiVO4 (a) and 0.2Co–BVO (b). | ||
3.4 Photocatalytic performance
To demonstrate the improvement in photocatalytic performance of Co-doped BiVO4 catalysts in comparison with the bare BiVO4, we conducted the photodegradation of MB under white LED light irradiation using the pristine BiVO4 and Co-doped BiVO4 samples. Fig. 7 shows UV-vis absorption spectra of MB solution isolated from a reaction mixture containing 0.2Co–BVO catalyst, water, and MB. As shown in Fig. 7, the intensity of the absorption spectra decreased after the first 20 min in the dark and then remained unchanged after 60 min in the dark, indicating the adsorption–desorption equilibrium between the surface of the 0.2Co–BVO sample and the MB molecules. When visible light was provided, the intensity of the absorption peak (664 nm) dramatically declined, and the position of this peak tended to shift toward a shorter wavelength. These signs were attributed to the photodegradation of MB molecules, which was caused by 0.2Co–BVO catalyst.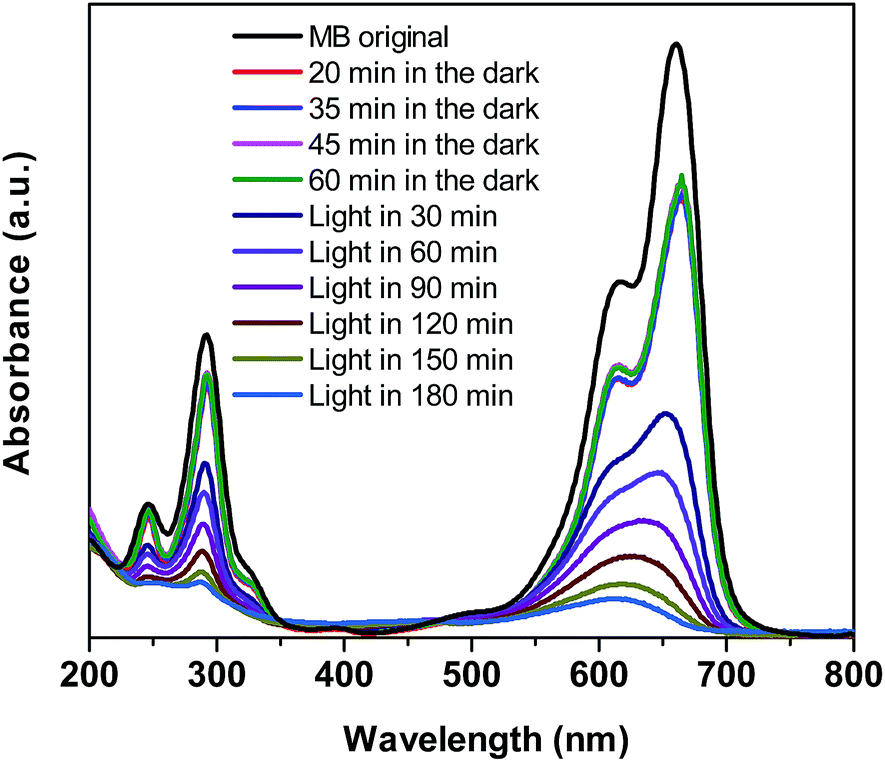 | ||
| Fig. 7 UV-vis absorption spectra of MB solution separated from catalyst suspensions during illumination using 0.2Co–BVO. | ||
Fig. 8A displays the removal efficiency of MB by the pristine BiVO4 and Co-doped BiVO4 samples. In comparison with the bare BiVO4 samples, Co-doped BiVO4 catalysts exhibited significantly improved photocatalytic activity after 180 min of irradiation in response to the incorporation of Co. The degradation rate of 0.2Co–BVO sample reached its highest value (about 98%) while the figure was 72% for the bare BiVO4 sample. However, when further increasing Co content (0.3Co–BVO and 0.5Co–BVO samples), the degradation rate witnessed a sharp decline. Such an outcome could be due to the formation of Bi2O3, Co2V2O7, and Co3V2O8 phase in 0.3Co–BVO and 0.5Co–BVO samples as described by the above XRD analysis. In addition, we employed the pseudo-first-order kinetic equation (ln(C/Co) = kt; Co and C are the concentration of MB at 0 min and t min of irradiation, respectively) to fit the degradation of MB (Fig. 8B) and obtain the reaction rate constant (k, 10−3 min−1) of the as-synthesized samples.28 The k values of Bi1−xCoxVO4+δ samples are presented in Table 1, indicating that the 0.2Co–BVO sample exhibited superior photocatalytic performance.
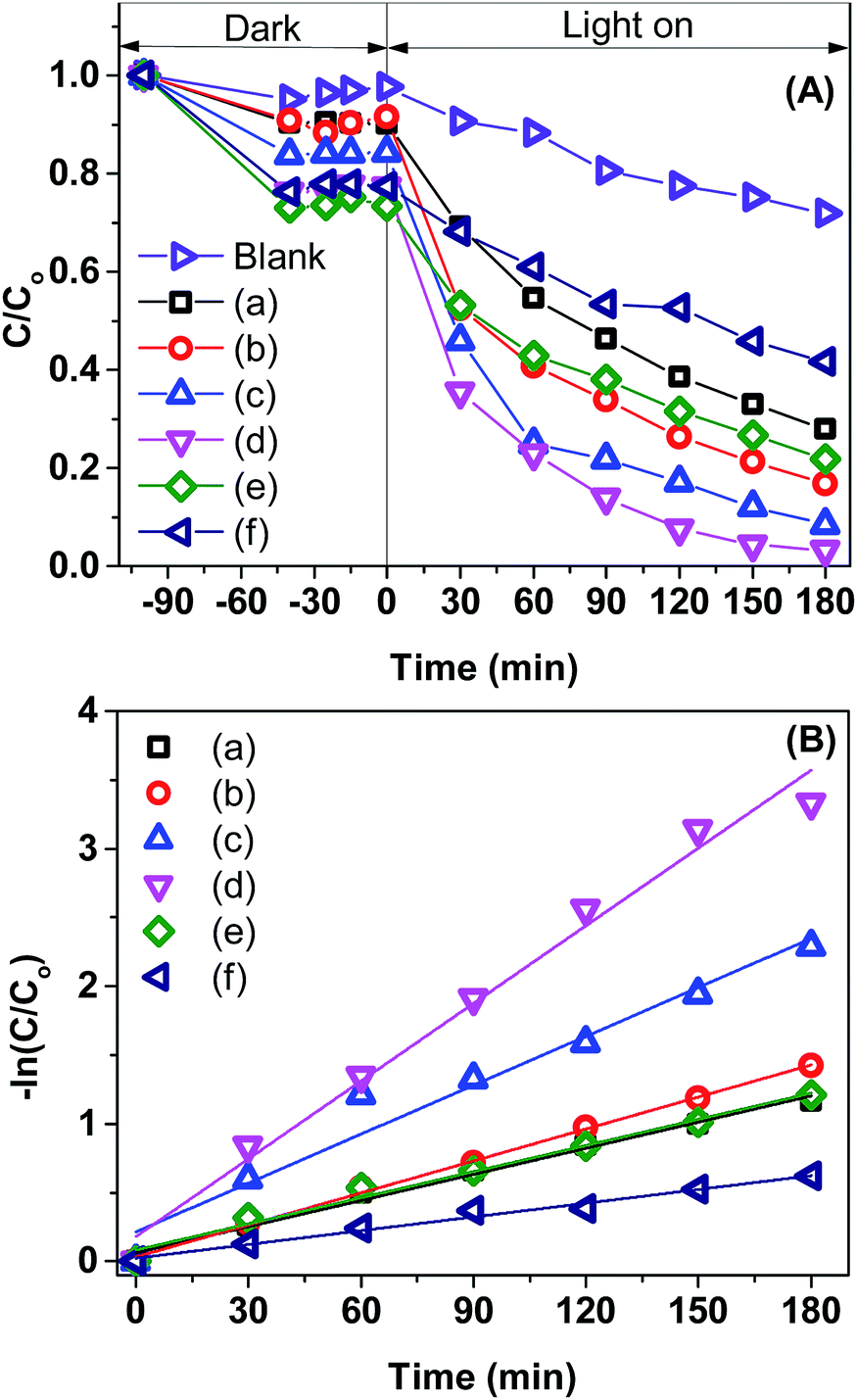 | ||
| Fig. 8 Photocatalytic degradation of MB over BiVO4 samples (A): BiVO4 (a), 0.05Co–BVO (b), 0.1Co-BV (c), 0.2Co-BV (d), 0.3Co-BV4 (e), and 0.5Co-BV (f) and plots of ln(Co/C) versus irradiation time representing the fit using a pseudo-first-order reaction rate (B). | ||
A comparison table for photocatalytic MB degradation performance of the as-synthesized Co-doped BiVO4 with other materials reported in the literature was also conducted (Table S1†). As shown in Table S1,† the degradation rate of MB over Co-doped BiVO4 samples was equal to or higher than those reported from previous studies, which further indicated that Co-doped BiVO4 could be a promising photocatalyst for the degradation of organic dyes.
To investigate the stability of the 0.2Co–BVO catalyst, five cycling photocatalytic tests were conducted by collecting and reusing the same photocatalyst. The result (Fig. S5†) exhibited an only negligible decrease in photocatalytic activity after five runs, which may have been caused by an incomplete collection of the catalyst after each runs. Moreover, no obvious differences were observed in the XRD results of the 0.2Co–BVO catalyst before/after five runs (Fig. S6†), suggesting the excellent stability and reusability of the 0.2Co–BVO catalyst.
3.5 Photodegradation mechanism
To explore the mechanism for the reactive species responsible for MB degradation over 0.2Co–BVO, radical trapping experiments in the photodegradation were performed (Fig. 9). The experimental steps were similar to the above photocatalytic activity test; however, radical scavengers were added to the reaction solution at the beginning of light irradiation. Employed radical scavengers included tert-butanol (TBA), ammonium oxalate monohydrate (AO), 1,4-benzoquinone (BQ) and potassium dichromate (K2Cr2O7) for hydroxyl radicals (·OH), hole (h+), superoxide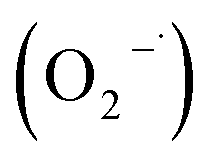 , and electron (e−). Following the addition of TBA and AO, the photodegradation rate showed a marked decline. Meanwhile, the reduction in the photodegradation rate is expected in the presence of BQ and K2Cr2O7. This observation implies that the presence of two dominant active oxidative species, namely ·OH and h+, directly oxidized MB molecules adsorbed on the surface of the 0.2Co–BVO catalyst. Similarly, trapping experiments of photocatalytic degradation of MB over the pure BiVO4 sample (Fig. S7†) also indicated that ·OH and h+ are the main active oxidative species. Therefore, the substitution of cobalt ions to Bi3+ in the BiVO4 did not influence the dominant oxidative species.
, and electron (e−). Following the addition of TBA and AO, the photodegradation rate showed a marked decline. Meanwhile, the reduction in the photodegradation rate is expected in the presence of BQ and K2Cr2O7. This observation implies that the presence of two dominant active oxidative species, namely ·OH and h+, directly oxidized MB molecules adsorbed on the surface of the 0.2Co–BVO catalyst. Similarly, trapping experiments of photocatalytic degradation of MB over the pure BiVO4 sample (Fig. S7†) also indicated that ·OH and h+ are the main active oxidative species. Therefore, the substitution of cobalt ions to Bi3+ in the BiVO4 did not influence the dominant oxidative species.
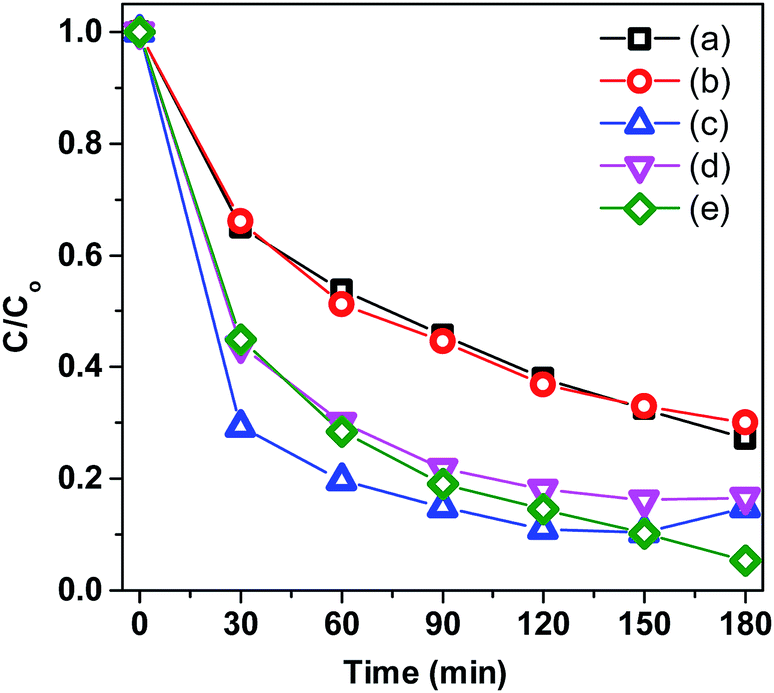 | ||
| Fig. 9 Trapping experiments of photocatalytic degradation of MB over 0.2Co–BVO with the presence of scavengers: TBA (a), AO (b), BQ (c) and K2Cr2O7 (d), and without the presence of scavenger (e). | ||
The essential role of ·OH radicals in the photocatalytic processes was further proved by the photoluminescence spectra of H2BDC-OH (Fig. 10). As displayed in Fig. 10, the intensity of H2BDC-OH caused by Co–BVO samples was enhanced as compared to the intensity of H2BDC-OH caused by the bare BiVO4 sample. The highest intensity of H2BDC-OH caused by the 0.2Co–BVO sample corresponded to the peak photocatalytic activity due to the increased formation of ·OH radicals on the surface of this sample.
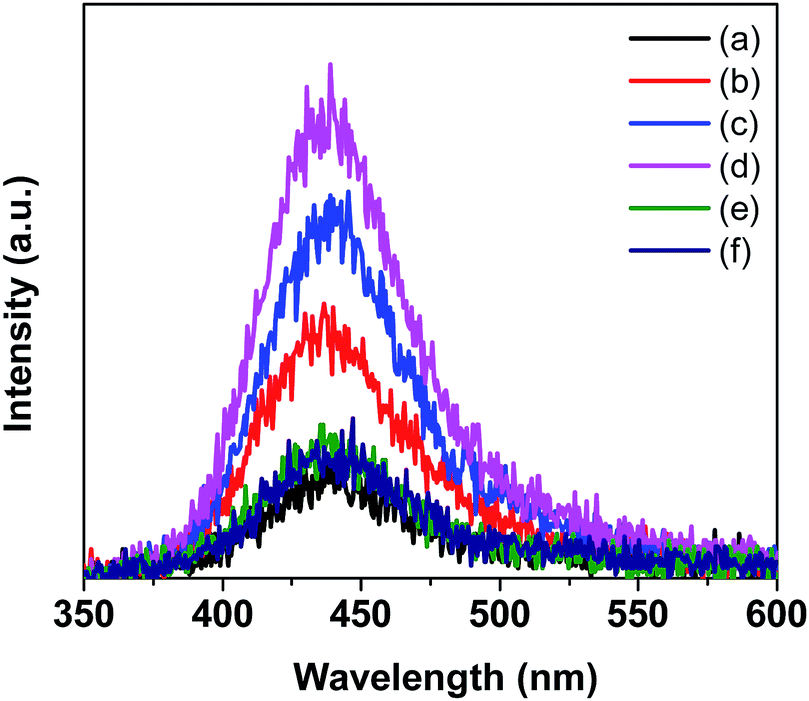 | ||
| Fig. 10 Photoluminescence spectrum of H2BDC-OH that formed upon the reaction between the ·OH radicals produced by the pristine BiVO4 (a), 0.05Co–BVO (b), 0.1Co–BVO (c), 0.2Co–BVO (d), 0.3Co–BVO (e) and 0.5Co–BVO (f) and H2BDC. | ||
The complete mechanism for photodegradation over Co-doped BiVO4 catalyst was suggested based on prior investigations3,6,42 and illustrated in Fig. 11. The process included four steps: (i) the generation of photo-excited electrons (e−) from the VB into the CB of Bi1−xCoxVO4+δ when visible LED light was provided; (ii) the corresponding holes (h+) leaving the VB; (iii) the photo-excited electrons (e−) being trapped by the acceptor defects (Co2+); (iv) the holes (h+) being transferred to the surface to oxidize the H2O and OH− to ·OH radicals which played an essential role in the photocatalytic oxidation process. In comparison with the pristine BiVO4 samples, the fabrication of Co-doped BiVO4 catalyst could encourage the efficient separation and transfer of photoinduced charges, corresponding to production of a significant amount of ·OH radicals for degradation of MB. As a result of this success, the photocatalytic performance of BiVO4 was significantly enhanced.
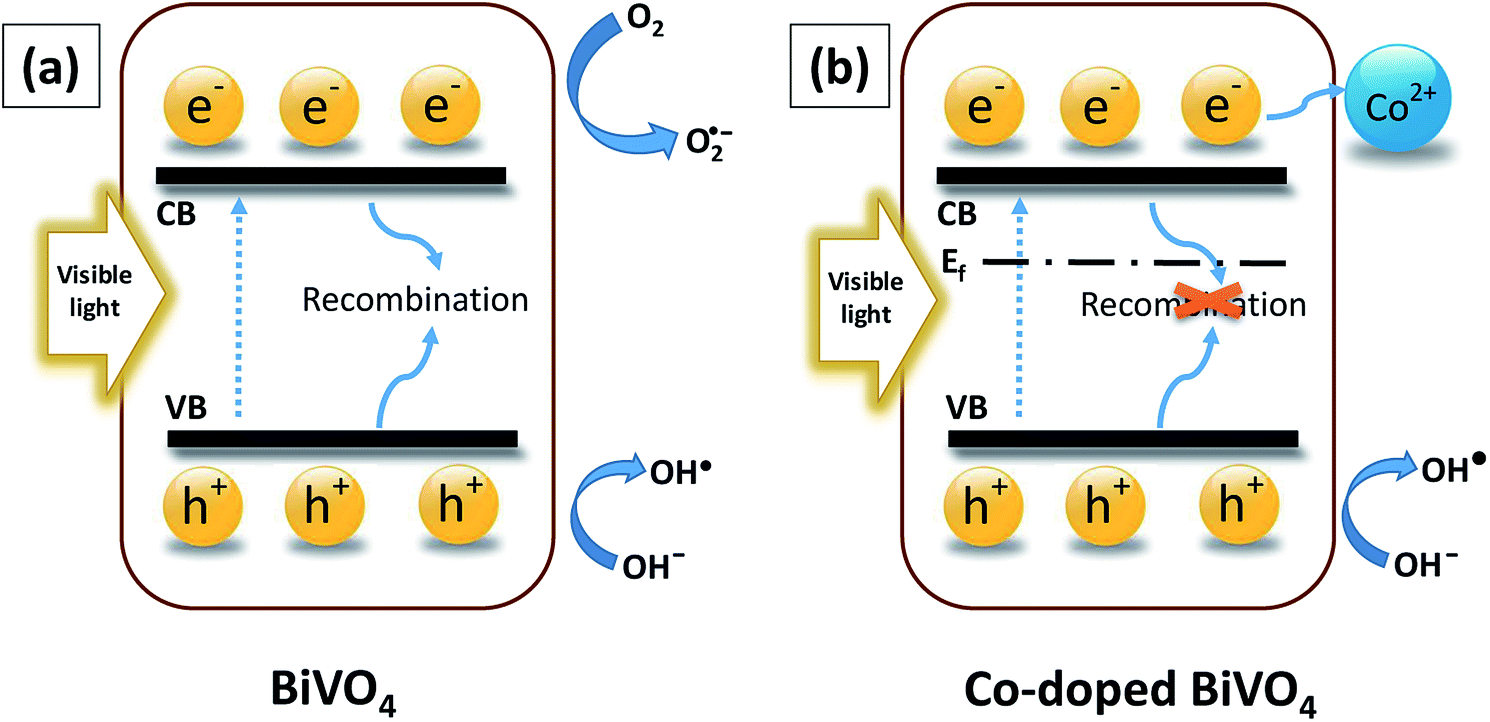 | ||
| Fig. 11 An entire mechanism for photocatalytic degradation: (a) BiVO4 and (b) Co-doped BiVO4. | ||
In addition, as described above, the photocatalytic performance of the Bi1−xCoxVO4+δ samples were not linear to the Co ion contents, but tended to increase first and then steadily decline. This downward trend could be attributed to the formation of Bi2O3, Co2V2O7, and Co3V2O8 as a result of the excessive doping Co ion content. Moreover, the excessively doped Co ion content could lead to the fast recombination of electron–hole pairs.43 Wang et al. also indicated the excessive doping metal content could occupy the space charge and hinder the penetration depth of irradiation into BiVO4 as result of fast recombination of electron–hole pairs and poor photoactivity.44
Our work first reported Co-doped BiVO4 materials synthesized according to the chemical formula Bi1−xCoxVO4+δ (0.05 < x < 0.5). Based on the XRD, Raman, and XPS results, as well as the previous studies, we could conclude that the substitution of Co2+ ions for Bi3+ sites in BiVO4, resulting in the improvement of the photocatalytic activity of BiVO4.
4 Conclusion
Monoclinic scheelite phase Bi1−xCoxVO4+δ have been successfully synthesized by the substitution of Co ions for Bi sites in BiVO4 using a hydrothermal procedure. As revealed by the crystal structural and optical absorption characterizations, the substitution of Co2+ for Bi3+ in the BiVO4 lattice was achieved; therefore, Co-doped BiVO4 exhibited the slight narrow band gap and the reduction of charge recombination. Co-doped BiVO4 catalysts show a higher removal efficiency and mineralization degree of MB than that of the pristine BiVO4 under white LED light illumination. Based on the characterization results, an entire mechanism for photocatalytic degradation of MB over Co-doped BiVO4 catalyst was proposed, in which ·OH and h+ are the predominant active oxidative species for directly oxidizing MB molecules adsorbed on the surface of the catalyst. In addition, the stability of catalyst was also investigated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 104.05-2017.315.Notes and references
- Y. Ma, H. Jiang, X. Zhang, J. Xing and Y. Guan, Ceram. Int., 2014, 40, 16485–16493 CrossRef CAS.
- D. Wang, H. Jiang, X. Zong, Q. Xu, Y. Ma, G. Li and C. Li, Chem.–Eur. J., 2011, 17, 1275–1282 CrossRef CAS PubMed.
- Y. Hu, D. Li, F. Sun, H. Wang, Y. Weng, W. Xiong and Y. Shao, RSC Adv., 2015, 5, 54882–54889 RSC.
- M. Hofmann, M. Rainer, S. Schulze, M. Hietschold and M. Mehring, ChemCatChem, 2015, 7, 1357–1365 CrossRef CAS.
- M. Hojamberdiev, G. Zhu, Z. C. Kadirova, J. Han, J. Liang, J. Zhou, X. Wei and P. Liu, Mater. Chem. Phys., 2015, 165, 188–195 CrossRef CAS.
- Y. Zhang, Y. Guo, H. Duan, H. Li, C. Sun and H. Liu, Phys. Chem. Chem. Phys., 2014, 16, 24519–24526 RSC.
- G. Xi and J. Ye, Chem. Commun., 2010, 46, 1893 RSC.
- H. Li, K. Yu, X. Lei, B. Guo, H. Fu and Z. Zhu, J. Phys. Chem. C, 2015, 119, 22681–22689 CrossRef CAS.
- L. Zhang, D. Chen and X. Jiao, J. Phys. Chem. B, 2006, 110, 2668–2673 CrossRef CAS PubMed.
- L. Ren, L. Jin, J.-B. Wang, F. Yang, M.-Q. Qiu and Y. Yu, Nanotechnology, 2009, 20, 115603 CrossRef PubMed.
- L. Zhou, W. Wang, L. Zhang, H. Xu and W. Zhu, J. Phys. Chem. C, 2007, 111, 13659–13664 CrossRef CAS.
- L. Ren, L. Ma, L. Jin, J.-B. Wang, M. Qiu and Y. Yu, Nanotechnology, 2009, 20, 405602 CrossRef PubMed.
- B. Wang, L. Guo and T. He, RSC Adv., 2016, 6, 30115–30124 RSC.
- D. T. Nguyen and S.-S. Hong, J. Nanosci. Nanotechnol., 2017, 17, 2690–2694 CrossRef CAS PubMed.
- M. Y. A. Khan, M. Zahoor, A. Shaheen, N. Jamil, M. I. Arshad, S. Z. Bajwa, N. A. Shad, R. Butt, I. Ali, M. Z. Iqbal, A. Wu, G. Nabi, S. Hussain, T. Mahmood, I. Aslam and W. S. Khan, Mater. Res. Bull., 2018, 104, 38–43 CrossRef.
- M. Wang, H. Zheng, J. Liu, D. Dong, Y. Che and C. Yang, Mater. Sci. Semicond. Process., 2015, 30, 307–313 CrossRef CAS.
- J. Q. Li, Z. Y. Guo, H. Liu, J. Du and Z. F. Zhu, J. Alloys Compd., 2013, 581, 40–45 CrossRef CAS.
- W. Yang, G. Tan, H. Ren, L. Zhang, C. Zhao and A. Xia, RSC Adv., 2015, 5, 7324–7329 RSC.
- B. Zhou, X. Zhao, H. Liu, J. Qu and C. P. Huang, Sep. Purif. Technol., 2011, 77, 275–282 CrossRef CAS.
- Z. Jiang, Y. Liu, T. Jing, B. Huang, X. Zhang, X. Qin, Y. Dai and M.-H. Whangbo, J. Phys. Chem. C, 2016, 120, 2058–2063 CrossRef CAS.
- T. Liu, G. Tan, C. Zhao, C. Xu, Y. Su, Y. Wang, H. Ren, A. Xia, D. Shao and S. Yan, Appl. Catal., B, 2017, 213, 87–96 CrossRef CAS.
- L. Chen, J. Wang, D. Meng, Y. Xing, C. Wang, F. Li, Y. Wang and X. Wu, Mater. Lett., 2015, 147, 1–3 CrossRef CAS.
- L. Ma, W. H. Li and J. H. Luo, Mater. Lett., 2013, 102–103, 65–67 CrossRef CAS.
- S. Y. Chae, C. S. Lee, H. Jung, O.-S. Joo, B. K. Min, J. H. Kim and Y. J. Hwang, ACS Appl. Mater. Interfaces, 2017, 9, 19780–19790 CrossRef CAS PubMed.
- H. Yuan, J. Liu, J. Li, Y. Li, X. Wang, Y. Zhang, J. Jiang, S. Chen, C. Zhao and D. Qian, J. Colloid Interface Sci., 2015, 444, 58–66 CrossRef CAS PubMed.
- M. Xie, Y. Feng, X. Fu, P. Luan and L. Jing, J. Alloys Compd., 2015, 631, 120–124 CrossRef CAS.
- A. Petala, R. Bontemps, A. Spartatouille, Z. Frontistis, M. Antonopoulou, I. Konstantinou, D. I. Kondarides and D. Mantzavinos, Catal. Today, 2017, 280, 122–131 CrossRef CAS.
- D. T. Nguyen and S. S. Hong, Top. Catal., 2017, 60, 782–788 CrossRef CAS.
- W. Luo, J. Wang, X. Zhao, Z. Zhao, Z. Li and Z. Zou, Phys. Chem. Chem. Phys., 2013, 15, 1006–1013 RSC.
- S. Gu, W. Li, F. Wang, H. Li and H. Zhou, Catal. Sci. Technol., 2016, 6, 1870–1881 RSC.
- C. Regmi, Y. K. Kshetri, T.-H. Kim, R. P. Pandey and S. W. Lee, Mol. Catal., 2017, 432, 220–231 CrossRef CAS.
- C. Regmi, T.-H. Kim, S. K. Ray, T. Yamaguchi and S. W. Lee, Res. Chem. Intermed., 2017, 43, 5203–5216 CrossRef CAS.
- B. Zhang, H. Zhang, Z. Wang, X. Zhang, X. Qin, Y. Dai, Y. Liu, P. Wang, Y. Li and B. Huang, Appl. Catal., B, 2017, 211, 258–265 CrossRef CAS.
- B. Zhou, X. Zhao, H. Liu, J. Qu and C. P. Huang, Appl. Catal., B, 2010, 99, 214–221 CrossRef CAS.
- Y. Geng, P. Zhang, N. Li and Z. Sun, J. Alloys Compd., 2015, 651, 744–748 CrossRef CAS.
- Y. Cheng, H. Wang, Y. Zhu, F. Liao, Z. Li and J. Li, J. Mater. Sci.: Mater. Electron., 2014, 26, 1268–1274 CrossRef.
- G. Yang, H. Cui, G. Yang and C. Wang, ACS Nano, 2014, 8, 4474–4487 CrossRef CAS PubMed.
- J. Yang, H. Liu, W. N. Martens and R. L. Frost, J. Phys. Chem. C, 2010, 114, 111–119 CrossRef CAS.
- R. Liu, J. Ren, D. Zhao, J. Ning, Z. Zhang, Y. Wang, Y. Zhong, C. Zheng and Y. Hu, Inorg. Chem. Front., 2017, 4, 2045–2054 RSC.
- M. Yan, Y. Yan, Y. Wu, W. Shi and Y. Hua, RSC Adv., 2015, 5, 90255–90264 RSC.
- M. Guo, Y. Wang, Q. He, W. Wang, W. Wang, Z. Fu and H. Wang, RSC Adv., 2015, 5, 58633–58639 RSC.
- S. Nikam and S. Joshi, RSC Adv., 2016, 6, 107463–107474 RSC.
- M. Wang, P. Guo, T. Chai, Y. Xie, J. Han, M. You, Y. Wang and T. Zhu, J. Alloys Compd., 2017, 691, 8–14 CrossRef CAS.
- Y. Wang, F. Liu, Y. Hua, C. Wang, X. Zhao, X. Liu and H. Li, J. Colloid Interface Sci., 2016, 483, 307–313 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: XRD analysis, Raman spectra, SEM images, photo-stability tests, trapping experiments, and a comparison of photocatalytic activity of photocatalysts. See DOI: 10.1039/c9ra04188e |
| This journal is © The Royal Society of Chemistry 2019 |