
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA restricted access molecularly imprinted polymer coating on metal–organic frameworks for solid-phase extraction of ofloxacin and enrofloxacin from bovine serum†
Zhian Sun ,
Huachun Liu,
Yanqiang Zhou,
Shanwen Zhao,
Jianmin Li,
Xiaoxiao Wang and
Bolin Gong*
,
Huachun Liu,
Yanqiang Zhou,
Shanwen Zhao,
Jianmin Li,
Xiaoxiao Wang and
Bolin Gong*
School of Chemistry and Chemical Engineering, North Minzu University, No. 204 Wenchang North Street, Xixia District, Yinchuan 750021, P. R. China. E-mail: gongbolin@163.com; Fax: +86-951-2067917; Tel: +86-951-2067917
First published on 4th September 2019
Abstract
A restricted access molecularly imprinted polymer (RAMIP) crosslinked with bovine serum albumin (BSA) was prepared on the surface of the mesoporous UiO-66-NH2 metal–organic framework (MOF). The surface morphology, imprinting behavior, and protein exclusion properties of UiO-66-NH2@RAMIP@BSA were investigated. The maximum adsorption capacity was 50.55 mg g−1 for ofloxacin, with a 99.4% protein exclusion rate. Adsorption equilibrium was reached in 9 min. Combined with RP-HPLC, a solid-phase extraction column filled with UiO-66-NH2@RAMIP@BSA was used to selectively enrich and analyze ofloxacin and enrofloxacin antibiotics from bovine serum with recoveries of 93.7–104.2% with relative standard deviations of 2.0–4.5% (n = 3). The linear range and the limit of detection were 0.1–100 μg mL−1 and 15.6 ng mL−1, respectively. These results suggest that UiO-66-NH2@RAMIP@BSA is an efficient pretreatment adsorbent for biological sample analysis.
1. Introduction
Ofloxacin (OFL) and enrofloxacin (ENRO) are common fluoroquinolone antibiotics that are widely used because of their broad spectrum and good therapeutic effects. However, overuse has caused environmental pollution and bacterial resistance. There is thus an urgent need to a develop high-performance detection technology to monitor rational applications.There are several established methods for OFL and ENRO analysis. Complex sample pretreatment is often necessary to remove interfering components (including biological macromolecules such as proteins), and to enrich the concentration of the target. Different strategies include protein precipitation, liquid–liquid extraction, solid-phase extraction, neutral salt solutions, organic solvents, and restricted-access media (RAM). Compared with other adsorbents, RAM can extract small molecules and exclude biological macromolecules,1 greatly simplifying sample pre-treatment, but it lacks target selectivity. However, molecularly imprinted polymers (MIPS) have very high selectivity for small molecules. In particular, surface imprinting overcomes the shortcomings of normal imprinting methods,2–4 with a large adsorption capacity and a fast mass transfer rate. Imprinted core materials play an important role in the adsorption properties. For example, Lv et al. prepared florfenicol Fe3O4@RAMIP (restricted access molecularly imprinted polymer) nanoparticles for the enrichment and separation of florfenicol from milk.5 Xu et al. prepared sulfamethazine silica@RAMIP for on-line detection of sulfonamides in bovine milk.6 These matrices are traditional materials, and it is necessary to find new core materials to improve the enrichment performance.
Metal–organic frameworks (MOFs) are highly crystallized porous materials of self-assembling metal ions with organic ligands.7 The incorporation of MOFs in MIPs is beginning to attract great interest for separations. For example, MOF-5@MIPs for metolcarb has a higher specific surface area and a faster mass-transfer rate relative to those of bulk MIPs.8 Nanocomposite MOF-5@MIPs for lipocalin greatly increased the analyte accessibility to the imprinted molecular cavities,9 while Fe3O4 nanoparticle HKUST-1@MIPs for gallic acid exhibited excellent extraction in real samples and a tunable porosity of HKUST-1.10 Liu et al. synthesized dummy template MIL-101@MIPs to detect pyrraline in food samples, which exhibited fast mass-transfer rates and excellent selectivity.11 UiO-66 is a MOF that has good thermal stability, chemical inertness,12 a wide pH range (1–14),13 high pressure resistance,14 and high mechanical strength.15 In particular, UiO-66-NH2 is easy to modify, and is thus an ideal imprinting composite material. However, its application in RAMIPs has not been reported.
Here, newly developed imprinted RAMIPs were synthesized using OFL as a template molecule, methacrylic acid as a functional monomer, trimethylolpropane trimethacrylate as a cross-linker, and glycidyl methacrylate as a hydrophilic monomer on the surface of UiO-66-NH2 in reversible addition–fragmentation chain transfer (RAFT) polymerization. To enhance the exclusion properties, and additional coating of cross-linked bovine serum albumin (BSA) was performed with an in-column process. The resulting RAMIPs was used to enrich OFL and ENRO from bovine serum samples.
2. Experimental
2.1. Materials and apparatus
ZrCl4 (≥99.9%, metals basis), glycidyl methacrylate (GMA, 97%), trimethylolpropane trimethacrylate (TRIM, GC, >90.0%), enrofloxacin (ENRO, 98%), chloramphenicol (CAP, 98%), sulfamerazine (SMZ, 99%), amoxicillin (AMO, 98%), methanol (HPLC, ≥99.9%), acetonitrile (HPLC, ≥99.9%), phosphoric acid (HPLC, 85–90%), triethylamine (HPLC, ≥99.5% (GC)), sodium diethyldithiocarbamate trihydrate (analytical, 99.0%), 2-chloroethyl isocyanate (97%), bovine serum albumin (BSA), glutaraldehyde (AR, 50%), sodium cyanoborohydride (NaBH3CN, 95%), 2,2′-azobis(2-methylpropionitrile) (AIBN, 99%) were all purchased from Aladdin Industrial Corporation, and AIBN was recrystallized with absolute ethanol. Methacrylic acid (MAA, 98%) was bought from Aladdin Industrial Corporation, and was removed inhibitor in a column with neutral aluminum oxide. 2-Aminoterephthalic acid (≥98%) was obtained from Shanghai D&B Biological Science and Technology Co. Ltd. Ofloxacin (OFL, 98.5%) were bought from Tianjin Sima Technology Co., Ltd. Other reagents were analytical reagents.The instruments that were used were a LC-20AT high performance liquid chromatography (HPLC) (Shimadzu, Japan), a SmartLab SE X-ray diffractometer (XRD) (Rigaku, Japan), a HT7700 transmission electron microscope (TEM) (Hitachi, Japan), FTIR-650 a Fourier transform infrared spectrometer (Gangdong, China), a SETARAM SETSYS16 thermogravimetric analyzer (TGA) (SETARAM, France), a ASAP 2020 HD88 surface area and porosity analyzer (Micromeritics, U.S.A), a Vario EL cube elemental analyzer (Elementar, Germany), and a UV2800S UV-visible spectrophotometer (Sunny Hengping, China).
2.2. Synthesis
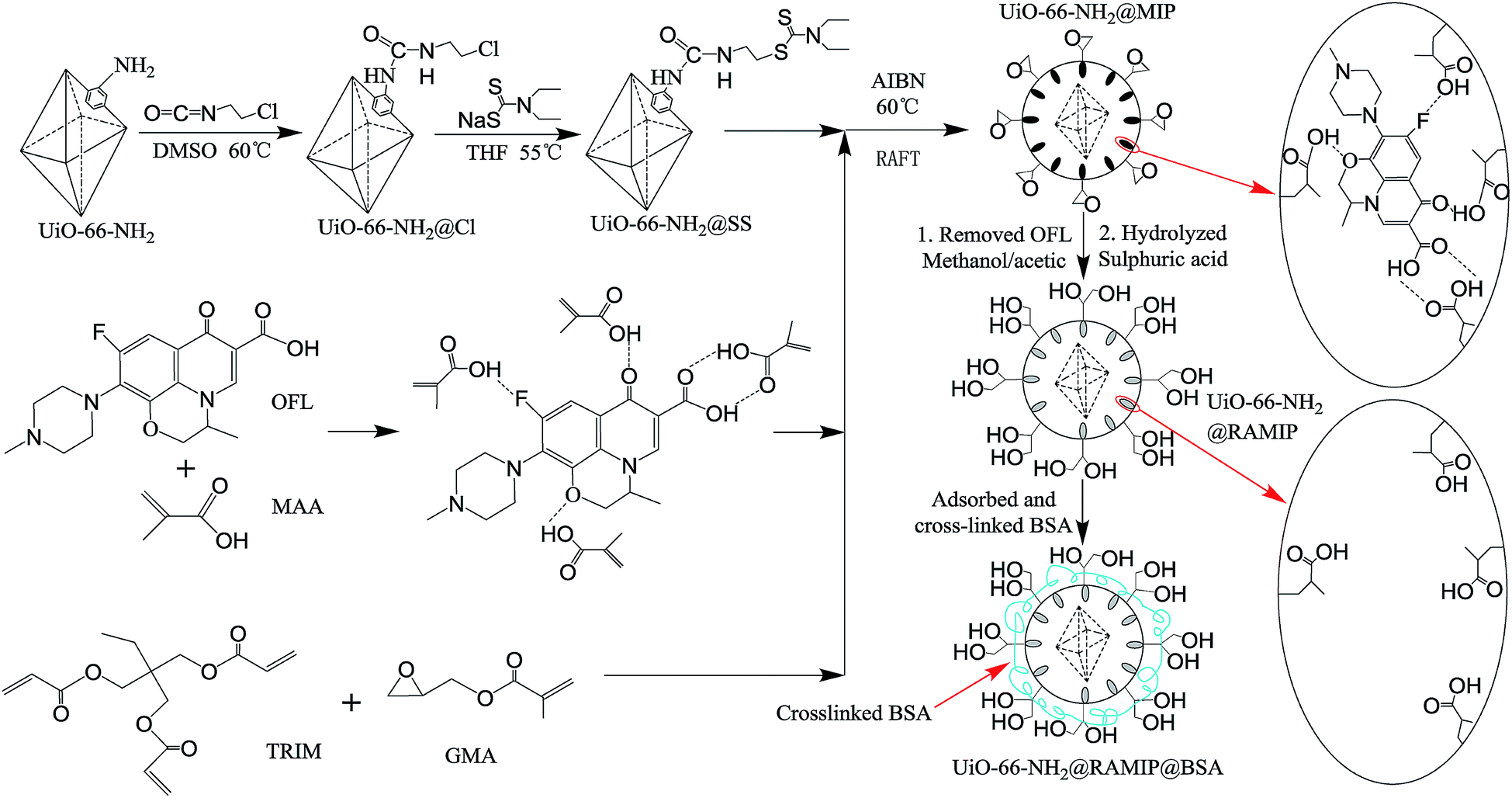 | ||
| Fig. 1 Preparation procedures of UiO-66-NH2@RAMIP@BSA. | ||
The preparation of non-imprinted polymers (UiO-66-NH2@RANIP@BSA) was the same as those of the imprinted UiO-66-NH2@RAMIP@BSA, except no template molecules were added.
2.3. Evaluation of protein exclusion efficiency
The protein exclusion efficiency of the UiO-66-NH2@RAMIP@BSA was assessed by the recoveries of BSA. A SPE column filled with UiO-66-NH2@RAMIP@BSA was activated with methanol (2 mL) and triply-distilled water (2 mL), successively. The BSA aqueous solution (1.0 mg mL−1) was flowed through the column, and the solution absorbance at 280 nm was measured. The adsorption capacity was calculated with eqn (1):| Q = (C0 − Ce)V/m | (1) |
The above procedure was the same for UiO-66-NH2@RANIP@BSA, UiO-66-NH2@RAMIP, UiO-66-NH2@RANIP, UiO-66-NH2@MIP, and UiO-66-NH2@NIP.
2.4. Adsorption experiments
The saturated adsorption capacity of UiO-66-NH2@RAMIP@BSA was examined with isothermal adsorption experiments. The UiO-66-NH2@RAMIP@BSA (0.0200 g) was added to 2 mL of various OFL solutions in methanol with concentrations ranging over 100–1300 mg L−1. The adsorption process lasted for 12 h at 25 °C under oscillating conditions. The absorbance of the supernatant was measured, and the adsorption capacity was calculated from eqn (1).The adsorption model was determined by fitting the isothermal adsorption data to Langmuir, Freundlich, and Langmuir–Freundlich models. The equations are as follows:
Langmuir equation:
| Ce/Qe = Ce/Qm + 1/(KLQm) | (2) |
Freundlich equation:
ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Qe = (1/n)lnCe + lnKF Qe = (1/n)lnCe + lnKF
| (3) |
Langmuir–Freundlich equation:
| Qe = QmKLmCem/(1 + KLmCem) | (4) |
The adsorption rate was determined by kinetics experiments at 25 °C. UiO-66-NH2@RAMIP@BSA (0.1000 g) was dispersed in an OFL/methanol solution (1200 mg L−1, 10 mL). The absorbance of the supernatant was measured every 1 min and the adsorption capacity was calculated from eqn (1).
The reaction order was determined by fitting the kinetics with the following model equations:
Pseudo first-order kinetic model:
|
ln(Qe − Qt) = lnQe − k1t
| (5) |
Pseudo second-order kinetic model:
| t/Qt = 1/(k2Qe2) + t/Qe | (6) |
The procedures were the same for UiO-66-NH2@RANIP@BSA.
2.5. Adsorption selectivity
UiO-66-NH2@RAMIP@BSA (0.0200 g) was added to a 2 mL CAP/methanol solution (1200 mg L−1). The adsorption process lasted for 12 h at 25 °C under oscillating conditions. The adsorption capacity was calculated from eqn (1). The adsorption capacities of UiO-66-NH2@RAMIP@BSA for ENRO, AMO, and SMZ were measured using the same procedure. The procedures were the same for UiO-66-NH2@RANIP@BSA.The UiO-66-NH2@RAMIP@BSA selectivity was characterized by the imprinting factor IF and the selectivity coefficient SC.
| IF = QUiO-66-NH2@RAMIP@BSA/QUiO-66-NH2@RANIP@BSA | (7) |
| SC = QOFL/QCM | (8) |
2.6. Spiked recovery experiments and analysis of real samples
OFL and ENRO methanol/water (1/1, v/v) standard solutions (500 mg L−1) were prepared. To three 100 mL volumetric flasks were added 0.1 mL, 1.0 mL, and 10 mL of the OFL standard solution, and each was diluted with bovine serum to the calibration line. A SPE column filled with UiO-66-NH2@RAMIP@BSA or UiO-66-NH2@RANIP@BSA was activated with methanol (2 mL) and triply-distilled water (2 mL), successively. Without pretreatment, the bovine serum samples (10 mL) were injected directly into the SPE column, then the column was washed with 3 mL of water. The OFL and ENRO was eluted with a 3 mL mixture of methanol/acetic acid (9/1, v/v). The eluent was dried with nitrogen and dissolved again with 0.1 mL of the mobile phase. Samples (15 μL) were taken for detection. The chromatographic conditions were: C18 column (250 mm × 4.6 mm, 5 μm, Shimadzu); a mobile phase of 0.025 mol L−1 phosphoric acid aqueous solution (pH adjusted to 3 with triethylamine)/acetonitrile = 85/15 (v/v); a flow rate of 1 mL min−1; a column temperature of 40 °C; and a UV detection wavelength of 286 nm.3. Results and discussion
3.1. Synthesis of ofloxacin-imprinted UiO-66-NH2@RAMIP@BSA
The synthesis of UiO-66-NH2@RAMIP@BSA nanoparticles is illustrated in Fig. 1. The mesoporous UiO-66-NH2 substrate had excellent thermal stability, chemical inertness, stable physical and chemical properties, and a high adsorption capacity. The MIP layer was coated on the UiO-66-NH2 surface via RAFT. The first step was to prepare the initiator-functionalized UiO-66-NH2 (UiO-66-NH2@SS). In the second step, the functional monomer MAA provided hydrogen bonding with the OFL template, TRIM was the cross-linking reagent used to form the polymer network, and GMA was used to form the first hydrophilic layer. The MIP layer easily grew from the initiator according to the RAFT mechanism. After Soxhlet extraction, the surface cavities could selectively rebind template molecules because they matched in shape, size, and chemical functional groups. After undergoing hydrolysis, the UiO-66-NH2@RAMIP surface was covered with hydroxyl groups to prevent irreversible protein adsorption. Theoretically, the proportions of template molecule, functional monomer, crosslinking agent, and hydrophilic monomer could affect the adsorption performance. Hence, the ratios of OFL to MAA, MAA to TRIM, and TRIM to GAM were all optimized. The experimental range was 1:4, between 1:0.6 to 1:1.6, and between 1:1.5 to 1:3, respectively. Finally, cross-linking BSA was immobilized on the UiO-66-NH2@RAMIP surface to form a second hydrophilic layer to enhance the protein exclusion efficiency.
3.2. Characterization of ofloxacin-imprinted UiO-66-NH2@RAMIP@BSA
The morphologies of UiO-66-NH2, UiO-66-NH2@RAMIP@BSA, and UiO-66-NH2@RANIP@BSA were imaged with TEM. As shown in Fig. 2A, UiO-66-NH2 was an octahedral crystal with a smooth surface. UiO-66-NH2@RAMIP@BSA nanoparticles were evenly encapsulated by the 21.2 nm-thick polymer, suggesting that the RAMIP layer were successfully coated on the UiO-66-NH2 surface. In addition, the thick and uniform imprinting layer could accelerate the mass-transfer rate and provide more adsorption sites. Fig. 2B and C indicates that the surface morphology of UiO-66-NH2@RANIP@BSA was similar to that of UiO-66-NH2@RAMIP@BSA. | ||
| Fig. 2 TEM images of (A) UiO-66-NH2, (B) UiO-66-NH2@RAMIP@BSA, and (C) UiO-66-NH2@RANIP@BSA. | ||
The preparation of the adsorbent was also confirmed with FTIR spectra (Fig. 3) and an elemental analysis (Table 1). In Fig. 3, the broad band at 1350–1700 cm−1 could be assigned to the COO− symmetrical and asymmetrical stretching vibration of UiO-66-NH2 in the five spectra (Fig. 3a–e). The 3353 cm−1 and 3450 cm−1 peaks in Fig. 3a were attributed to a N–H vibration from UiO-66-NH2. Comparing the a and b spectra, the absorption peaks of C–Cl (735 cm−1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1693 cm−1, redshifted by two associated N–H bonds), CH2 (2880 cm−1) and CH3 (2931 cm−1) appeared in curve Fig. 3b, which verified that 2-chloroethyl isocyanate was grafted on the UiO-66-NH2 surface. In Fig. 3c, C–S and CS peaks appeared at 615 cm−1 and 1060 cm−1, respectively, verifying the grafting of sodium diethyldithiocarbamate trihydrate. In Fig. 3d, the CO overtone peak from esters occurred at 3440 cm−1. The symmetrical and asymmetrical C–H stretching vibration (2965 cm−1) and the ester carbonyl group stretching vibration (1731 cm−1) were significantly enhanced. Two strong C–O–C peaks appeared between 1100–1300 cm−1, and the epoxy group absorption peak appeared at 908 cm−1. All of these data indicated that MAA, TRIM, and GMA were grafted onto the surface of UiO-66-NH2@SS. In Fig. 3e, the C–O stretching vibration and the O–H deformation vibration occurred at 1050 cm−1 and 1334 cm−1, respectively. Thus, the peak at 3400 cm−1 was an OH band, which verified the hydrolysis of the epoxy group.
O (1693 cm−1, redshifted by two associated N–H bonds), CH2 (2880 cm−1) and CH3 (2931 cm−1) appeared in curve Fig. 3b, which verified that 2-chloroethyl isocyanate was grafted on the UiO-66-NH2 surface. In Fig. 3c, C–S and CS peaks appeared at 615 cm−1 and 1060 cm−1, respectively, verifying the grafting of sodium diethyldithiocarbamate trihydrate. In Fig. 3d, the CO overtone peak from esters occurred at 3440 cm−1. The symmetrical and asymmetrical C–H stretching vibration (2965 cm−1) and the ester carbonyl group stretching vibration (1731 cm−1) were significantly enhanced. Two strong C–O–C peaks appeared between 1100–1300 cm−1, and the epoxy group absorption peak appeared at 908 cm−1. All of these data indicated that MAA, TRIM, and GMA were grafted onto the surface of UiO-66-NH2@SS. In Fig. 3e, the C–O stretching vibration and the O–H deformation vibration occurred at 1050 cm−1 and 1334 cm−1, respectively. Thus, the peak at 3400 cm−1 was an OH band, which verified the hydrolysis of the epoxy group.
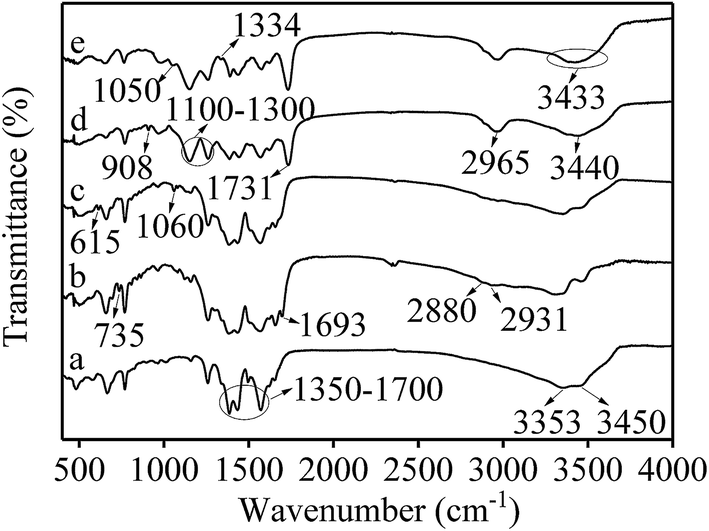 | ||
| Fig. 3 FTIR spectra of (a) UiO-66-NH2, (b) UiO-66-NH2@Cl, (c) UiO-66-NH2@SS, (d) UiO-66-NH2@MIP and (e) UiO-66-NH2@RAMIP. | ||
| Materials | N% | C% | H% | O% |
|---|---|---|---|---|
| UiO-66-NH2@RAMIP@BSA | 6.33 | 41.40 | 5.85 | 24.78 |
| UiO-66-NH2@RAMIP | 4.92 | 39.95 | 5.72 | 24.85 |
The amount of BSA bonding could be determined by the elemental analysis (Table 1). The N, C, and H content in UiO-66-NH2@RAMIP@BSA increased, which indicated that BSA had been cross-linked on the UiO-66-NH2@RAMIP surface.
The specific surface areas of UiO-66-NH2 and UiO-66-NH2@RAMIP@BSA were determined by BET (Fig. 4A) to be 332.6 m2 g−1 and 210.4 m2 g−1, respectively. These were much higher than other MIPs,17,18 which was the basis for the increase of adsorption capacity. Furthermore, as shown in Fig. 4B, UiO-66-NH2 and UiO-66-NH2@RAMIP@BSA were mostly mesoporous. Mesoporous MOFs can greatly increase the pore utilization rate and accelerate mass transfer relative to microporous MOFs.8–11
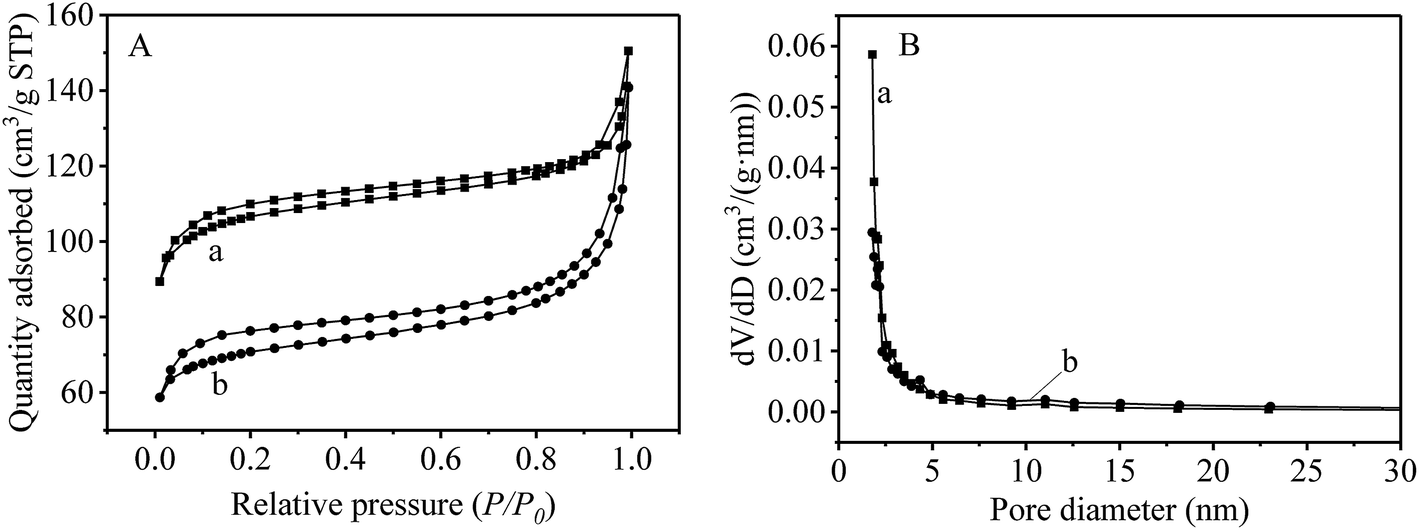 | ||
| Fig. 4 (A) The specific surface area and (B) the pore size distribution (a) UiO-66-NH2 and (b) UiO-66-NH2@RAMIP@BSA. | ||
XRD (Fig. S1 in the ESI†) and thermogravimetric analysis (Fig. S2†) revealed that UiO-66-NH2@RAMIP@BSA had good chemical and thermal stability, and could meet the needs of practical applications.
3.3. Protein exclusion efficiency of UiO-66-NH2@RAMIP@BSA
Protein adsorption on the different materials was estimated by injecting a 1 mg mL−1 BSA solution on SPE columns packed with UiO-66-NH2@MIP, UiO-66-NH2@RAMIP, UiO-66-NH2@RAMIP@BSA and the corresponding non-imprinted particles. The recovery of BSA from the columns was used to evaluate the various protein exclusion efficiencies. Table 2 shows that the BSA recoveries of UiO-66-NH2@MIP and UiO-66-NH2@NIP without grafting poly (GMA) and introducing the BSA layer were just 36.5% and 37.2%, respectively, while that of UiO-66-NH2@RAMIP@BSA and UiO-66-NH2@RANIP@BSA were 99.4% and 99.2%, respectively. The latter were much higher than those of UiO-66-NH2@RAMIP (96.0%) and UiO-66-NH2@RANIP (95.6%). Hence, further cross-linking of BSA on the UiO-66-NH2@RAMIP surface could increase the hydrophilicity of the RAMIP materials. The formation of the hydrophilic bilayer by grafting poly (GMA) chains, and after introducing BSA on the UiO-66-NH2 surface, resulted in excellent protein exclusion efficiency. Therefore, UiO-66-NH2@RAMIP@BSA was the most suitable SPE material for pre-treating biological samples.| Materials | Concentration of BSA (mg mL−1) | A (λ = 280 nm) | Binding ability (%) | Exclusion ability (%) | |
|---|---|---|---|---|---|
| Before SPE | After SPE | ||||
| UiO-66-NH2@MIP | 1 | 0.643 | 0.235 | 63.5 | 36.5 |
| UiO-66-NH2@NIP | 1 | 0.643 | 0.239 | 62.8 | 37.2 |
| UiO-66-NH2@RAMIP | 1 | 0.643 | 0.617 | 4 | 96.0 |
| UiO-66-NH2@RANIP | 1 | 0.643 | 0.615 | 4.4 | 95.6 |
| UiO-66-NH2@RAMIP@BSA | 1 | 0.643 | 0.639 | 0.6 | 99.4 |
| UiO-66-NH2@RANIP@BSA | 1 | 0.643 | 0.638 | 0.8 | 99.2 |
3.4. Absorption capacity and kinetics
The adsorption capacities of UiO-66-NH2@RAMIP@BSA and UiO-66-NH2@RANIP@BSA were initially determined with adsorption isotherms. As shown in Fig. 5A, the adsorption capacities were enhanced with increasing standard solution concentration over the range 100–1300 mg L−1, and reached saturation at 50.55 mg g−1 and 16.40 mg g−1, respectively. The adsorption capacity of UiO-66-NH2@RAMIP@BSA was much larger than that of UiO-66-NH2@RANIP@BSA. Compared with reported MIP absorbents, Qmax for UiO-66-NH2@RAMIP@BSA was higher than that of absorbents using silica and polymer resin,19–27 indicating that the UiO-66-NH2@RAMIP@BSA composites exhibited efficient extraction and could have wide analysis applications. The adsorption rate of UiO-66-NH2@RAMIP@BSA was investigated. In Fig. 5B, adsorption equilibrium was reached in 9 min, which was much faster than that of MIPs prepared by traditional polymerization methods and other imprinting materials. This was because of the uses of mesoporous MOF substrate. Recently, a RAMIP was prepared using 2 μm spherical silica gel and atom-transfer radical polymerization.28 Although this material has some advantages in adsorption capacity and limits of detection, UiO-66-NH2@RAMIP@BSA has a better adsorption rate, protein exclusion efficiency, and recovery.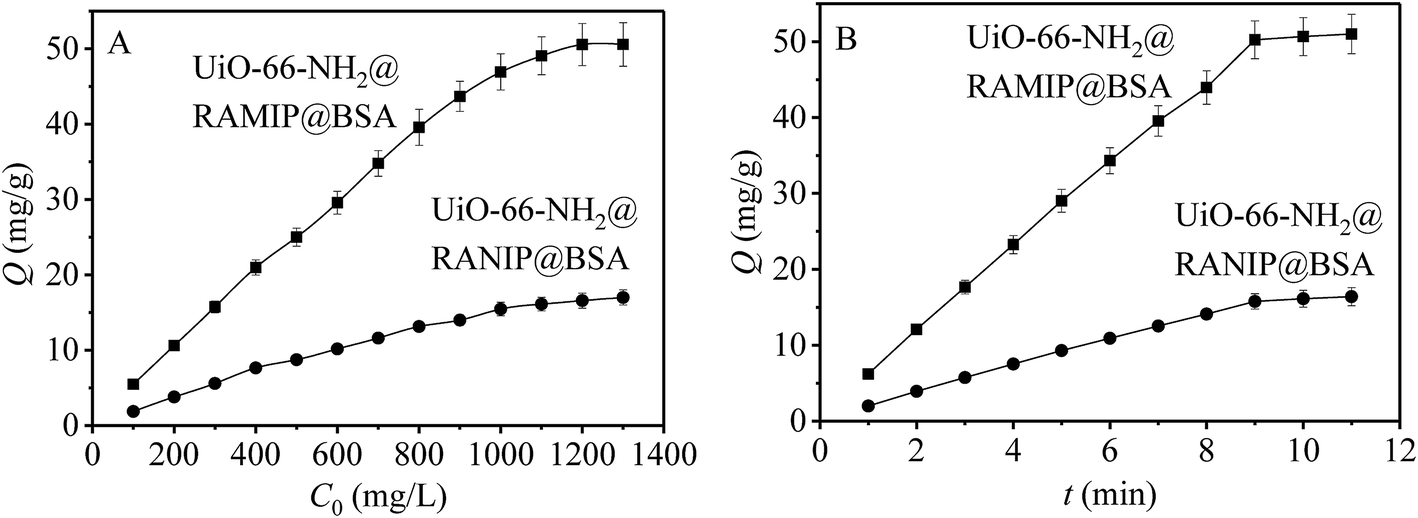 | ||
| Fig. 5 Results of (A) absorption capacity and (B) absorption kinetics. | ||
The results of the Langmuir, Freundlich, and Langmuir–Freundlich fits are shown in Table S1.† By comparing the R2 values, it could be concluded that the adsorption of OFL on UiO-66-NH2@RAMIP@BSA and UiO-66-NH2@RANIP@BSA corresponded to the Langmuir–Freundlich model. From the fitting results, the theoretical value of Qm from that model was closer to the measured value. The KL value of UiO-66-NH2@RAMIP@BSA was much larger than that of UiO-66-NH2@RANIP@BSA, which indicated that UiO-66-NH2@RAMIP@BSA was more attractive to OFL. At the same time, the m value of UiO-66-NH2@RANIP@BSA was close to 1, which indicated that its adsorption sites were more uniform because they all involved physical adsorption. The higher m value for UiO-66-NH2@RAMIP@BSA indicated nonuniform adsorption sites that involved both nonspecific physical adsorption and specific chemical adsorption.
The kinetics fits are shown in Table S2.† From the R2 values, it could be concluded that adsorption on UiO-66-NH2@RAMIP@BSA correlated with pseudo second-order kinetics. The fitting results showed that its adsorption rate was determined by the square of the number of unoccupied imprinted surface sites, and the mechanism was chemical adsorption, which involves electronic bonding between the adsorbent and the adsorbate. This result also confirmed the theoretical analysis of Section 3.1.
3.5. Adsorption selectivity of UiO-66-NH2@RAMIP@BSA
The selectivity of UiO-66-NH2@RAMIP@BSA was compared with that of UiO-66-NH2@RANIP@BSA. The three commonly used antibiotics AMO, SMZ, and CAP, and the structural analogue ENRO of OFL, were used to investigate adsorption selectivity, as shown in Fig. 6. The IF values for OFL, ENRO, AMO, SMZ, and CAP were 3.00, 3.25, 1.14, 1.50, and 1.50, and its SC were 1.26, 33.48, 15.70, and 10.87, respectively. The molecular sizes increased in the order SMZ, CAP, OFL ≈ ENRO, and AMO. Because the molecular size and structure of the analogue ENRO were the most similar to those of OFL, its adsorption capacity was higher relative to those of SMZ, AMO, and CAP. The molecular mass of CAP was similar to that of OFL, but its structure was different, so its adsorption capacity decreased greatly. The size of SMZ was smaller than that of CAP, so its adsorption capacity was lower than that of CAP. The size of AMO was the largest; therefore, it had the lowest adsorption capacity because of less physical adsorption, which led to the minimum IF. Thus, UiO-66-NH2@RAMIP@BSA had high selectivity and could recognize OFL and ENRO in the presence of other commonly used antibiotics.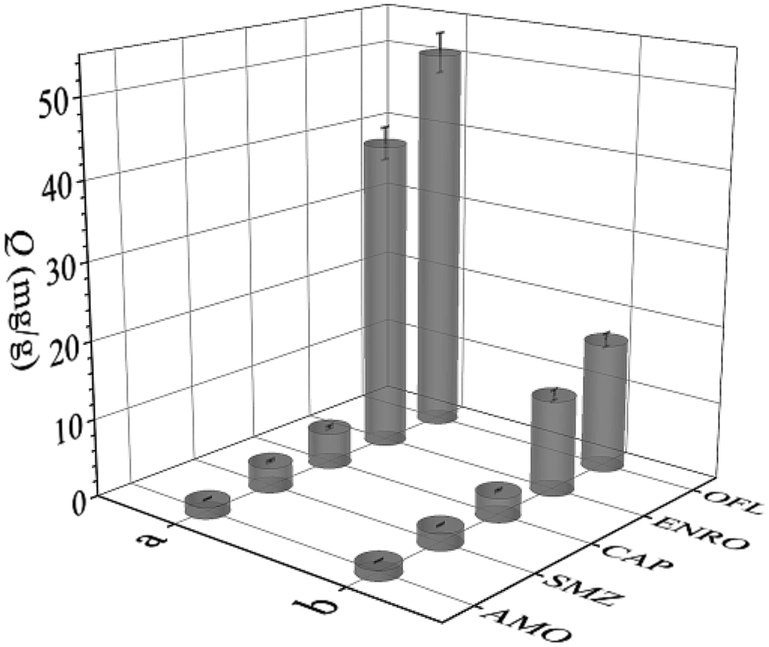 | ||
| Fig. 6 Adsorption capacity of OFL, ENRO, CAP, SMZ and AMO on (a) UiO-66-NH2@RAMIP@BSA and (b) UiO-66-NH2@RANIP@BSA. | ||
3.6. Enrichment and separation of OFL and ENRO in bovine serum by the UiO-66-NH2@RAMIP@BSA
Under the optimized conditions, UiO-66-NH2@RAMIP@BSA was used to selectively extract OFL and ENRO from bovine serum samples via RP-HPLC. Chromatograms of bovine serum samples are shown in Fig. 7. None of the target compounds were detected in the bovine serum samples (curve a), while OFL and ENRO could be detected after enrichment by using UiO-66-NH2@RAMIP@BSA (curve b) and the non-imprinted materials (curve c). The method is illustrated in Table 3. Specifically, the determinations were evaluated by using bovine serum samples spiked with OFL and ENRO at concentrations of 0.5, 5, and 50 μg mL; the recoveries of OFL and ENRO were 93.7–104.2%, and the RSDs were 2.0–4.5% (n = 3). The linear range and LOD were 0.1–100 μg mL−1 and 15.6 ng mL−1, respectively. Relative to other core materials (Table S3†), UiO-66-NH2 had advantages in addition to the slightly higher LOD. In summary, the UiO-66-NH2@RAMIP@BSA could be used for highly selective enrichment and separation of OFL and ENRO in biological samples.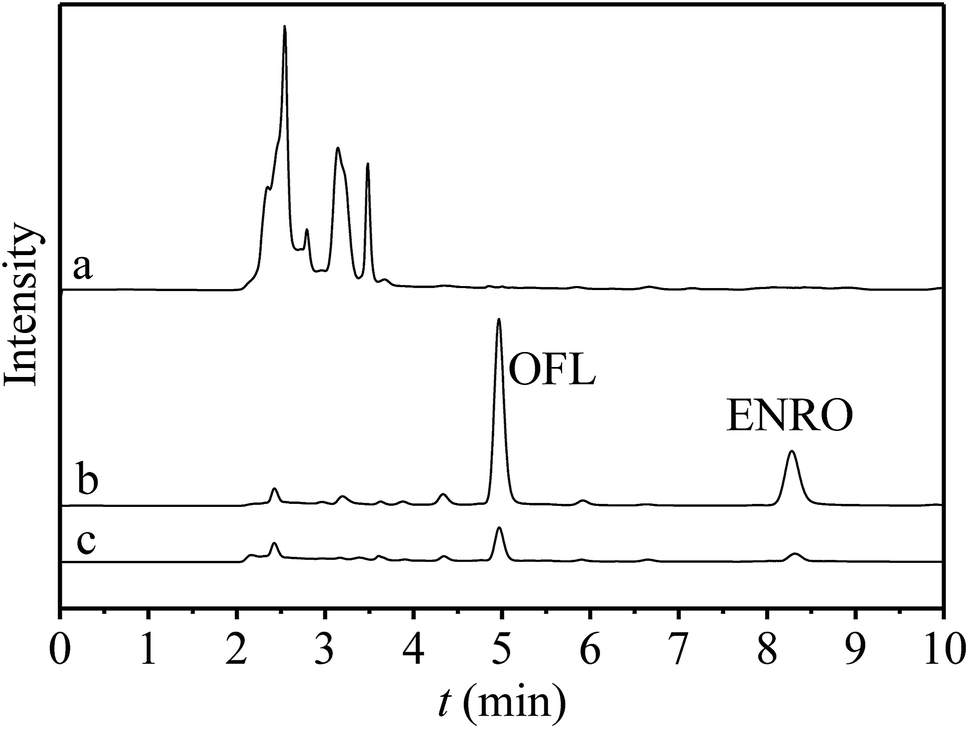 | ||
| Fig. 7 The chromatograms of (a) the blank bovine serum, (b) the elution of UiO-66-NH2@RAMIP@BSA after treatment of spiked bovine serum and (c) the elution of UiO-66-NH2@RANIP@BSA after treatment of spiked bovine serum. | ||
| Analytes | Materials | 0.5 μg mL−1 | 5 μg mL−1 | 50 μg mL−1 | |||
|---|---|---|---|---|---|---|---|
| Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | ||
| OFL | UiO-66-NH2@RAMIP@BSA | 93.7 | 3.6 | 94.9 | 4.5 | 104.2 | 2.0 |
| UiO-66-NH2@RANIP@BSA | 31.2 | 4.1 | 33.4 | 3.3 | 36.5 | 2.3 | |
| ENRO | UiO-66-NH2@RAMIP@BSA | 95.5 | 4.0 | 94.1 | 4.1 | 102.6 | 3.4 |
| UiO-66-NH2@RANIP@BSA | 30.8 | 3.8 | 32.5 | 2.9 | 35.7 | 4.4 | |
4. Conclusions
Highly hydrophilic UiO-66-NH2@RAMIP@BSA was prepared, and its protein exclusion rate was 99.4%. Its adsorption capacity was as high as 50.55 mg g−1, and the adsorption could reach equilibrium within 9 min. Combined with RP-HPLC, it was successfully used to separate and detect OFL and ENRO in bovine serum. The recoveries were 93.7–104.2% and the RSDs were 2.0–4.5%. The detection limit was 15.6 ng mL−1 and the linear range was 0.1–100 μg mL−1. Compared with previous reports, the performance of this material was much better, which greatly simplifies pretreatment steps of biological samples and improves the analysis efficiency.Conflicts of interest
The author(s) declare that they have no competing interests.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21565001) and the Key Research and Development Program of Ningxia (Special Project for Foreign Cooperation) (2019BFH02004).References
- H. D. de Faria, L. C. D. Abrao, M. G. Santos, A. F. Barbosa and E. C. Figueiredo, Anal. Chim. Acta, 2017, 959, 43–65 CrossRef PubMed.
- M. Niu, C. Pham-Huy and H. He, Microchim. Acta, 2016, 183, 2677–2695 CrossRef CAS.
- S. Pardeshi and S. K. Singh, RSC Adv., 2016, 6, 23525–23536 RSC.
- H. Xie, W. Ji, D. Liu, W. Liu, D. Wang, R. Lv and X. Wang, RSC Adv., 2015, 5, 48885–48892 RSC.
- Y. Lv, J. Zhang, M. Li, S. Zhou, X. Ren and J. Wang, Anal. Methods, 2016, 8, 3982–3989 RSC.
- W. Xu, S. Su, P. Jiang, H. Wang, X. Dong and M. Zhang, J. Chromatogr. A, 2010, 1217, 7198–7207 CrossRef CAS PubMed.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- K. Qian, G. Fang and S. Wang, Chem. Commun., 2011, 47, 10118–10120 RSC.
- Z. Iskierko, P. S. Sharma, D. Prochowicz, K. Fronc, F. D'Souza, D. Toczydłowska, F. Stefaniak and K. Noworyta, ACS Appl. Mater. Interfaces, 2016, 8, 19860–19865 CrossRef CAS PubMed.
- A. Asfaram, M. Ghaedi and K. Dashtian, Ultrason. Sonochem., 2017, 34, 561–570 CrossRef CAS PubMed.
- H. Liu, L. Mu, X. Chen, J. Wang, S. Wang and B. Sun, J. Agric. Food Chem., 2017, 65, 986–992 CrossRef CAS PubMed.
- J. B. Decoste, G. W. Peterson, B. J. Schindler, K. L. Killops, M. A. Browe and J. J. Mahle, J. Mater. Chem. A, 2013, 1, 11922–11932 RSC.
- H. Molavi, A. Eskandari, A. Shojaei and S. A. Mousavi, Microporous Mesoporous Mater., 2018, 257, 193–201 CrossRef CAS.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- H. Wu, T. Yildirim and W. Zhou, J. Phys. Chem. Lett., 2013, 4, 925–930 CrossRef CAS PubMed.
- J. B. DeCoste, J. A. Rossin and G. W. Peterson, Chem.–Eur. J., 2015, 21, 18029–18032 CrossRef CAS PubMed.
- Y. H. Yang, L. T. Liu, M. J. Chen, S. Liu, C. B. Gong, Y. B. Wei, C. F. Chow and Q. Tang, Mater. Sci. Eng., C, 2018, 92, 365–373 CrossRef CAS PubMed.
- Z. Wang, T. Qiu, L. Guo, J. Ye, L. He and X. Li, React. Funct. Polym., 2018, 126, 1–8 CrossRef CAS.
- Z. H. Hu, Y. F. Wang, A. M. Omer and X. K. Ouyang, Int. J. Biol. Macromol., 2018, 107, 453–462 CrossRef CAS PubMed.
- Y. He, Y. Huang, Y. Jin, X. Liu, G. Liu and R. Zhao, ACS Appl. Mater. Interfaces, 2014, 6, 9634–9642 CrossRef CAS PubMed.
- J. Zhang, L. L. Wang, J. Q. Ma and Y. L. Wang, Food. Anal. Methods, 2014, 7, 721–729 CrossRef.
- H. B. He, C. Dong, B. Li, J. P. Dong, T. Y. Bo, T. L. Wang, Q. W. Yu and Y. Q. Feng, J. Chromatogr. A, 2014, 1361, 23–33 CrossRef CAS PubMed.
- X. Guan, T. Cheng, S. Wang, X. Liu and H. Zhang, Anal. Bioanal. Chem., 2017, 409, 3127–3133 CrossRef CAS PubMed.
- Y. F. Wang, H. X. Jin, Y. G. Wang, L. Y. Yang, X. K. OuYang and W. J. Wu, Molecules, 2016, 21, 915 CrossRef PubMed.
- T. Zhao, X. Guan, W. Tang, Y. Ma and H. Zhang, Anal. Chim. Acta, 2015, 853, 668–675 CrossRef CAS PubMed.
- H. Yan and F. Qiao, J. Liq. Chromatogr. Relat. Technol., 2014, 37, 1237–1248 CrossRef CAS.
- H. Wang, R. Wang and Y. Han, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 949–950, 24–29 CAS.
- J. Li, L. Zhao, C. Wei, Z. Sun, S. Zhao, T. Cai and B. Gong, J. Sep. Sci., 2019, 42, 2491–2499 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04143e |
| This journal is © The Royal Society of Chemistry 2019 |