
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA titanium tetrachloride-based effective methodology for the synthesis of dipeptides†
Alessandra Comandè,
Marianna Greco,
Emilia Lucia Belsito,
Angelo Liguori and
Antonella Leggio *
*
Dipartimento di Farmacia e Scienze della Salute e della Nutrizione, Università della Calabria Edificio Polifunzionale, I-87036 Arcavacata di Rende, CS, Italy. E-mail: antonella.leggio@unical.it; Fax: +39 984 493265; Tel: +39 984 493199
First published on 17th July 2019
Abstract
A series of dipeptide systems have been easily achieved through a TiCl4-assisted condensation reaction. The reaction of N-protected amino acids with amino acid methyl esters in pyridine and in the presence of TiCl4 furnished the corresponding dipeptides with high yields and diastereoselectivity. The reaction was successfully applied to amino acids protected on the α-amino function with different protecting groups. The adopted experimental conditions allowed preserving both the protecting groups on the α-amino function and on the side chain functionalities. Furthermore, the preservation of the stereochemical integrity at the amino acid chiral centres has been verified.
Introduction
Dipeptide systems, although widely used in medicinal and pharmacological fields as well as in synthetic chemistry,1 have been poorly investigated compared to single amino acids and long chain peptides.2The biological functions of dipeptides may be due to dipeptides as such or to the single amino acids deriving from them as a result of hydrolysis processes.
Several dipeptide systems show antitumor,3 antioxidant,4 and antihypertensive activity.5
Commercially available dipeptides Aspartame (L-aspartyl-L-phenylalanine methyl ester) and sustamine (L-alanyl-L-glutamine) are used as an artificial sweetener and nutritional supplement respectively.2 The dipeptide L-alanyl-L-glutamine is much more stable and soluble than the free amino acid L-glutamine (L-Gln); consequently the dipeptide system has more efficacy than the single amino acid L-glutamine. In fact, when L-Gln is supplied as L-alanyl-L-glutamine a greater transfer of L-Gln from the gut to plasma occurs compared to when the same dose is provided as the free amino acid.6
Histidine containing dipeptides, such as carnosine (β-alanyl-L-histidine) and anserine (β-alanyl-methyl-L-histidine), are easily obtained by controlled hydrolysis of animal proteins and have been proposed as food supplements able of sequestering reactive carbonyl species originating from lipid oxidative processes or from the oxidation of the sugars. These dipeptides also seem to be able to prevent the glycosylation of proteins and in particular of hemoglobin, a process that characterizes the metabolic syndrome.7 Other dipeptides are then widely used in the formulation of cosmetic products for instance cysteine-containing dipeptides,8 L-Tyr-L-Arg9 and aspartyl dipeptides, the latter possess highly effective skin and hair caring properties.10
Amide bond formation is an extremely important reaction in organic chemistry.11 Numerous synthetic strategies to synthesize organic molecules containing amide bonds and peptides have been reported.12
The development of solid-phase peptide synthesis (SPPS), also automated, has made easily available polypeptides of great interest for food, pharmaceutical and biomedical purposes.13
Solution phase peptide synthesis (commonly referred to as ‘liquid phase’) is the method of choice for preparing dipeptides and more generally small peptides. This strategy, although is not well suited for making longer peptides, is much more scalable, and allows to produce larger quantities of high-quality peptides, and at a lower cost than solid phase.
Peptide synthesis strategies in solution, but also in solid phase, require the protection of chemical functions not involved in the formation of the peptide bond. The formation of this bond is of particular importance for the purposes of stereochemistry preservation of the chiral centres of the amino acids that participate in the condensation reaction.
Therefore, the development of new methods of peptide bond formation in liquid phase is particularly attractive in order to obtain dipeptides or small peptides since this methodology can also be used with non-natural or unusual amino acids.
In this context, titanium tetrachloride could assume great importance as, due to its great affinity for the oxygen atom, it is used in organic synthesis for the transformation of various functional groups.14
In this study, we explored the applicability of titanium tetrachloride for the formation of the peptide bond. Our results have shown the successful use of titanium tetrachloride as condensing agent for the synthesis of dipeptide systems. Furthermore, the preservation of stereochemical integrity at the amino acid chiral centres has been verified.
Results and discussion
The possibility of using titanium tetrachloride (TiCl4) for the formation of the peptide bond was by no means obvious. In fact, TiCl4 could deprotect not only the amino functions of N-protected amino acids as it occurs with other Lewis acids15 but also the amino acid side chain functional groups.Accordingly, we thought to carry out the reaction in a pyridine-buffered medium. We proceeded preliminarily with the synthesis of the dipeptide Asp-Phe-OMe (Aspartame), via the Boc-based solution-phase approach.
In this perspective, L-phenylalanine methyl ester hydrochloride (1 mmol) was preliminary treated with pyridine (5 mL), then N-Boc-L-aspartic acid-β-t-butyl ester (1 mmol) and TiCl4 (2 mmol) were added to the resulting solution (Scheme 1). The reaction was carried out in a screw-capped vial at 40 °C and was complete within about 5 h. After removal of pyridine by co-evaporation with toluene, the resulting crude product was suspended in chloroform and purified by elution through a silica gel column using chloroform as mobile phase. After evaporation of chloroform the dipeptide N-Boc-Asp-(OtBu)-Phe-OMe (1) was recovered in 84% yield and high purity as confirmed by 1H and 13C NMR analyses. Spectroscopic data analysis excluded side-chain and N-terminal amino function deprotection.
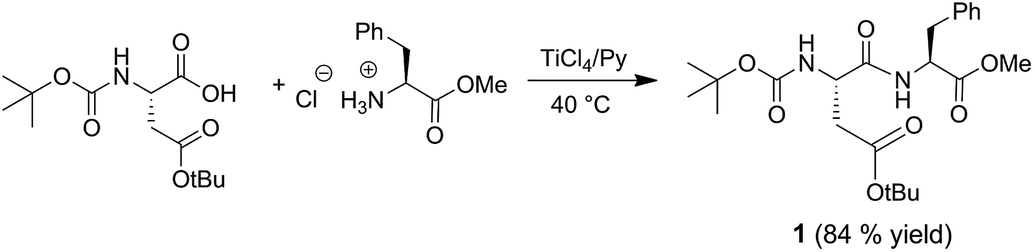 | ||
| Scheme 1 Synthesis of dipeptide N-Boc-Asp-(OtBu)-Phe-OMe (1). | ||
Afterwards Aspartame (Asp-Phe-OMe) (1a) was readily obtained in 95% yield by treating the N-Boc-protected dipeptide 1 with trifluoroacetic acid in dichloromethane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v).
:1 v/v).
The developed procedure was applied successfully for the synthesis of various N-Boc-protected dipeptides (Scheme 2 and Table 1).
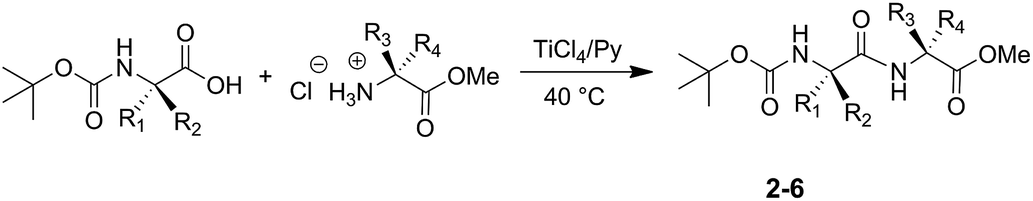 | ||
| Scheme 2 Synthesis of N-Boc protected dipeptides 2–6. | ||
All the N-Boc-dipeptides 2–6 were characterized by GC/MS (EI), 1H NMR and 13C NMR analyses. N-Boc-Ala-Cys-(SBzl)-OMe (5) and N-Boc-Ala-Lys-(Boc)-OMe (6) kept unchanged the masking groups on the amino acid side-chains.
The successful outcomes obtained with the synthesis of dipeptides 1–6 prompted us to extend the developed methodology to the preparation of dipeptide systems protected on the α-amino function with the base-labile protecting group fluorenylmethoxycarbonyl (Fmoc) (Scheme 3). N-Fmoc-dipeptide systems (7–17) were prepared in high yields without observing the formation of reaction products deprotected on the α-amino function (Table 2).
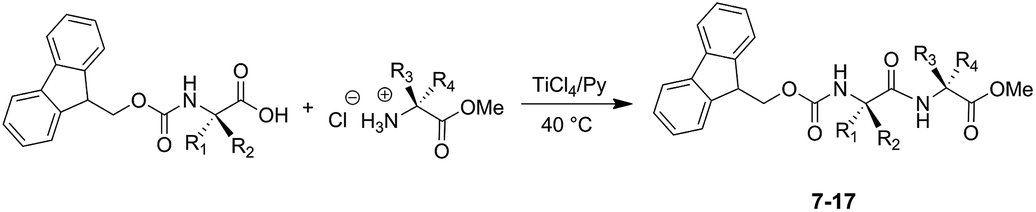 | ||
| Scheme 3 Synthesis of N-Fmoc protected dipeptides 7–17. | ||
| Dipeptide | R1 | R2 | R3 | R4 | Yielda (%) |
|---|---|---|---|---|---|
| a Isolated yield. | |||||
| 7 | (CH2)COO-(tBu) | H | CH2Ph | H | 84 |
| 8 | CH3 | H | CH3 | H | 71 |
| 9 | CH2CH(CH3)2 | H | CH3 | H | 80 |
| 10 | H | CH2CH(CH3)2 | CH3 | H | 80 |
| 11 | H | H | CH3 | H | 80 |
| 12 | CH2CH(CH3)2 | H | CH(CH3)CH2CH3 | H | 87 |
| 13 | CH2O-(tBu) | H | CH3 | H | 76 |
| 14 | CH3 | H | CH2S-(Bzl) | H | 78 |
| 15 | CH3 | H | (CH2)4NH-(Boc) | H | 60 |
| 16 | CH2S-(Bzl) | H | CH3 | H | 78 |
| 17 | H | CH2S-(Bzl) | CH3 | H | 74 |
The stability of side-chain protecting groups was also investigated by carrying out the synthesis of dipeptides N-Fmoc-Asp-(OtBu)-Phe-OMe (7), N-Fmoc-Ser-(OtBu)-Ala-OMe (13), N-Fmoc-Ala-Cys-(SBzl)-OMe (14) and N-Fmoc-Ala-Lys-(Boc)-OMe (15). In these cases, the reaction occurred without removal of the side-chain protecting groups and afforded the corresponding dipeptides in high yields (Scheme 3 and Table 2). In particular, the t-butyl group on the serine side-chain, the benzyl group on the cysteine side-chain and the Boc group on the lysine side-chain were preserved during the condensation reaction. Therefore using pyridine as reaction solvent has proved useful not only to convert the ammonium group of amino acid methyl ester hydrochloride into the free amino function able to react as nucleophile in the condensation reaction, but also to preserve the protecting groups on the masked functionalities.
The coupling reaction mediated by TiCl4 gave excellent results also when it was employed to get dipeptides protected on the amino terminal function with the benzyloxycarbonyl (Cbz) protecting group in fact, N-Cbz-L-Ala-L-Ala-OMe (18), and N-Cbz-D-Ala-L-Ala-OMe (19) were obtained in good yields (Scheme 4).
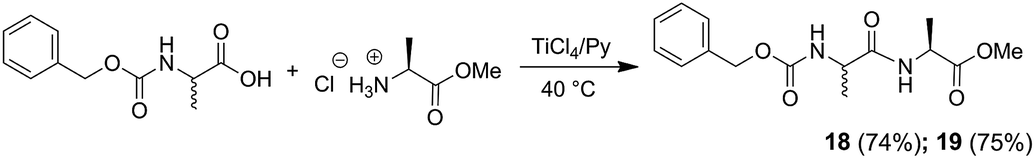 | ||
| Scheme 4 Synthesis of N-Cbz protected dipeptides 18–19. | ||
For completeness, we applied the developed procedure to the synthesis of a dipeptide system protected on the amine function with the 4-nitrobenzenesulfonyl (nosyl) group.16
Nosyl group (Ns) is an interesting protecting group as it simultaneously protects and activates the NH2 moiety of amines,17 in fact, it is widely used for the site-selective N-alkylation of amino acids and peptides both in solution and solid phase.18
The condensation reaction mediated by TiCl4 proceeded in a different way when the amino function of the N-terminal amino acid was protected with the nosyl group.
The reaction between N-Ns-L-phenylalanine and alanine methyl ester hydrochloride for obtaining the dipeptide N-Ns-L-Phe-L-Ala-OMe (20) was particularly slow.
After 7 hours, the TLC analysis (chloroform–methanol, 90:10, v/v) of reaction mixture still showed the presence of N-Ns-L-phenylalanine, then the reaction was stopped. After applying to the reaction mixture the previously described workup, the reaction product, recovered in 48% yield, was analyzed by GC/MS (EI). The resulting chromatogram gave two peaks at tr = 46.42 min and tr = 46.87 min in about 3:1 ratio respectively with identical mass spectra both characterized by the presence of the molecular ion m/z 376 consistent with that of dipeptide N-Ns-Phe-L-Ala-OMe (20) and its corresponding diastereoisomer.
The 1H NMR spectrum of the product also provided evidence of the presence of a mixture of two diastereoisomers in fact it showed, only for a few types of protons, the presence of two close signals of different intensity corresponding to the two stereoisomers.
Therefore, when N-nosyl amino acids are used as N-terminal residues the reaction is incomplete, probably because of the presence of the sulfonamide function, and the stereochemical integrity of the chiral centers is not completely retained.
This result prompted us to verify, also in the cases of Boc-, Cbz- and Fmoc-protected dipeptides, the configuration preservation of the amino acids chiral centers.
To this aim, we designed and carried out the synthesis of couples of diastereoisomeric dipeptides protected on the amino function with the above-cited urethane protecting groups.
Under the typical reaction conditions by using TiCl4/pyridine reagent system, the dipeptide N-Boc-L-Ala-L-Ala-OMe (3) and its epimer N-Boc-D-Ala-L-Ala-OMe (4) were synthesized. Samples of the crude dipeptides 3 and 4 were analyzed by 1H NMR and 13C NMR and the resulting spectra were compared with that obtained analyzing a mixture, of the same two epimers.
1H NMR spectra of both single products 3 and 4 showed the presence of signals attributable to only one diastereomer and the absence of epimerized products at least within the sensibility limits of the NMR technique. The chemical shifts of the signals generated by the NH amide protons were different in the two diastereoisomers and readily resolved in the 1H NMR spectrum of the mixture of 3 and 4, prepared for this purpose, with a ratio 4:6 respectively (Fig. 1).
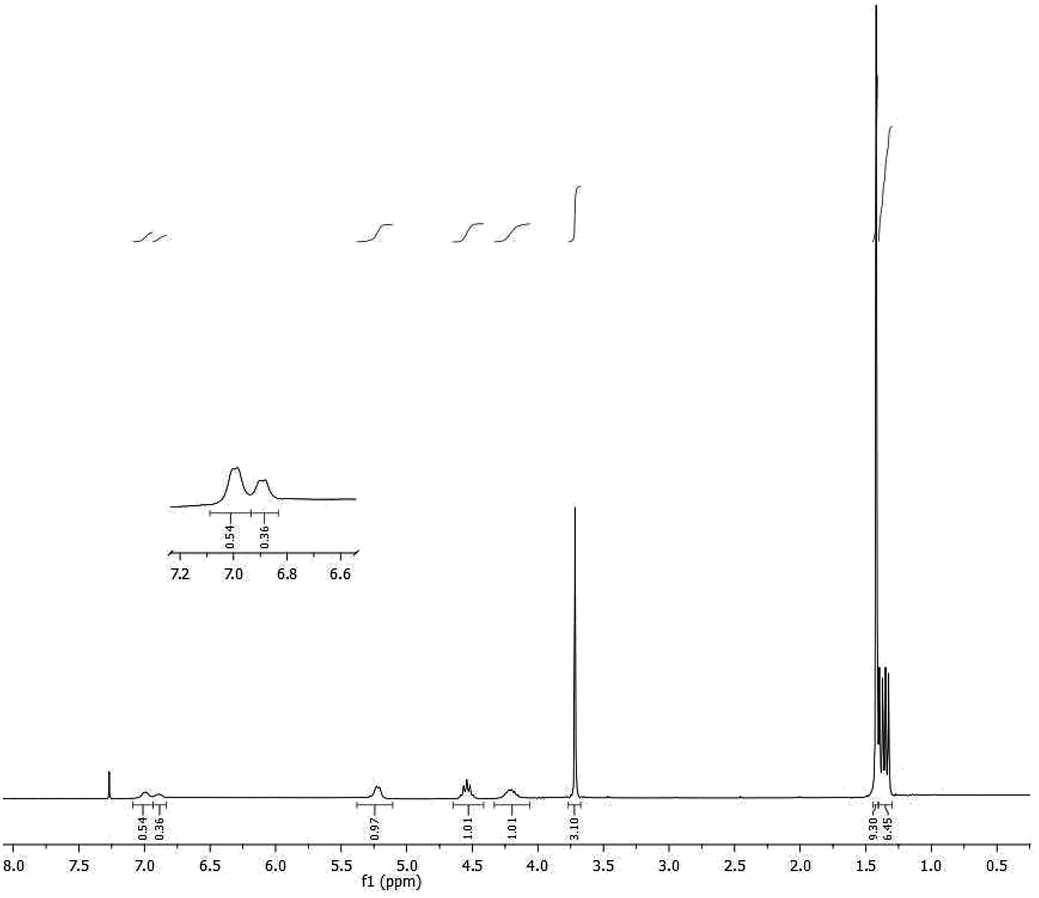 | ||
| Fig. 1 1H NMR spectrum of a mixture of 3 and 4 (approx. 4:6). | ||
The GC/MS analysis of the two epimers N-Boc-L-Ala-L-Ala-OMe dipeptides (3) and N-Boc-D-Ala-L-Ala-OMe (4) instead, indicated the presence in both chromatogram of a very small amount of the other epimer, the calculated diastereomeric excess was for both epimers satisfactory (≥95%).
A diastereomeric excess ≥ 96% was measured by analyzing by GC/MS the single dipeptides N-Cbz-L-Ala-L-Ala-OMe (18) and its epimer N-Cbz-D-Ala-L-Ala-OMe (19).
The 1H NMR spectra of the single products 18 and 19 did not show residual signals attributable to the other diastereomer while, in the 1H NMR analysis of a mixture of 18 and 19, approximately 4.2:5.8 respectively, the signals corresponding to the amide proton (6.89 ppm, 6.83 ppm) and to the methyl ester protons (3.72 ppm, 3.73 ppm) of the two epimers resulted separated.
Finally, also the dipeptides N-Fmoc-L-Leu-L-Ala-OMe (9) and N-Fmoc-D-Leu-L-Ala-OMe (10), obtained through the developed methodology, showed no detectable loss of stereo integrity. In fact 1H NMR spectra of both single products 9 and 10 indicated the presence of signals corresponding to only one diastereomer while in the 1H NMR spectrum of a mixture of both epimers 9 and 10 the chemical shifts of amide and urethanic protons were resolved.
Additional experiments have been performed in order to test the epimerization process when sensitive Cys(Bzl)OH is used as N-terminal residue. To this aim we synthesized a couple of diastereomeric dipeptide systems N-Fmoc-L-Cys(Bzl)-L-Ala-OMe (16) and N-Fmoc-D-Cys(Bzl)-L-Ala-OMe (17) (Scheme 3 and Table 2). In the crude 1H NMR spectra of both systems (16 and 17) the presence of about 10% of the other diastereoisomer has been detected indicating in this case a higher epimerization level. In particular, we observed in both spectra two different signals corresponding to the NH amide proton in a ratio of about 0.9:0.1.
The formation of peptide bond under the adopted reaction conditions could occur through the mechanism depicted in Scheme 5. We hypothesized that TiCl4 interacts with the N-protected amino acid pyridinium salt by forming the intermediate A. This could react with the amino acid methyl ester directly or after conversion into the corresponding chloride (C) or acyl pyridinium ion (B) to form the dipeptide system.
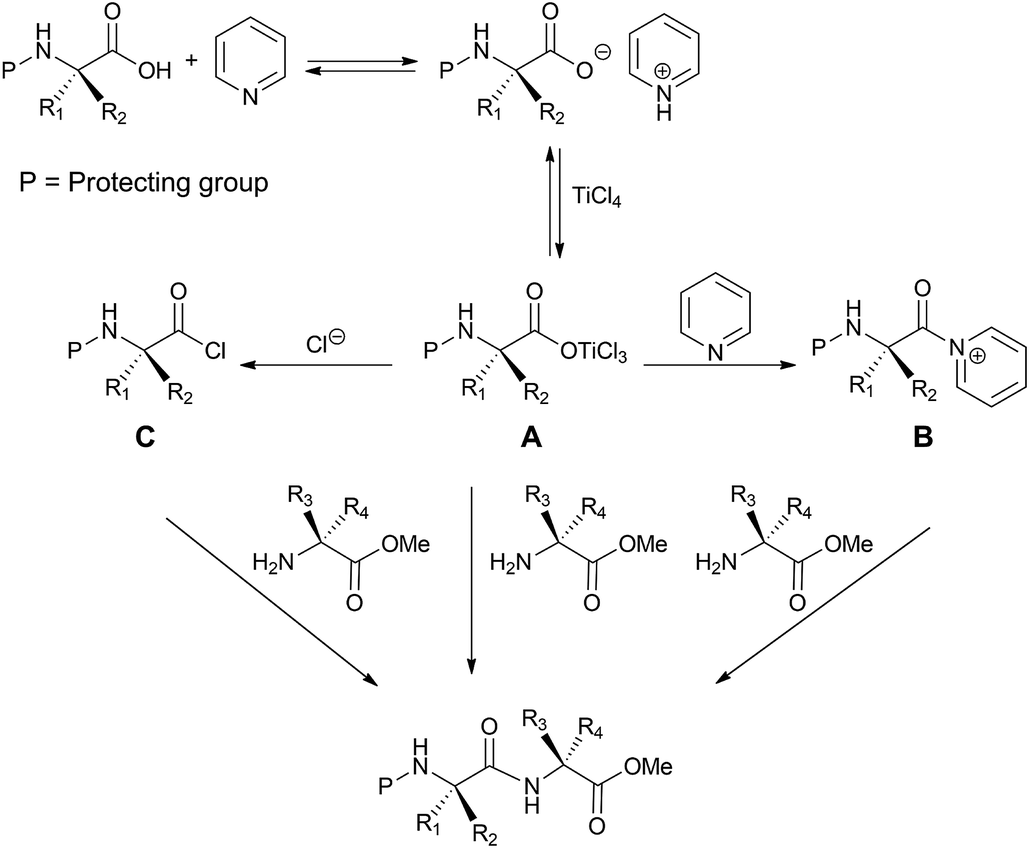 | ||
| Scheme 5 Proposed mechanism for the TiCl4-mediated synthesis of dipeptides. | ||
Conclusions
Here we developed a synthetic process mediated by TiCl4 that yields dipeptide systems having high stereochemical and chemical purity. TiCl4/pyridine reagent system represents a valuable tool for synthesizing peptides. Dipeptide systems were synthesized easily and in high yields through a TiCl4-assisted condensation reaction between N-protected amino acids and amino acid methyl esters in pyridine.The reaction was applied successfully to amino acids protected on the α-amino function with different protecting groups. The adopted experimental conditions allowed preserving not only the α-amino protecting groups but also the acid-labile side chain protecting groups.
The recovery of the dipeptide products has been achieved by simple filtration through a silica gel column, which greatly simplifies the reaction work-up and avoids the formation of emulsions and product losses.
The maintenance of amino acids stereochemical integrity is almost complete for dipeptides protected on the amino function with urethane protecting groups (Fmoc, Z, Boc). A non-negligible loss of stereochemical integrity was instead observed in N-nosyl-protected dipeptides.
Experimental
General experimental details
Reagents were commercially available with analytical grade and used as purchased without further purification. Solvents were purified according to well-known laboratory methods and freshly distilled prior to use. Reaction were carried out in a tightly sealed screw-capped vial. Reactions were magnetically stirred and monitored by thin layer chromatography using Merck-Kieselgel 60 F254 plates. Spots on the TLC plates were visualized with a UV lamp (254 nm) and by spraying with 0.2% ninhydrin in ethanol and charring after elution. 1H and 13C NMR spectra were recorded on a Bruker Avance 300 instrument at 300 MHz and 75 MHz, respectively. Spectroscopic analysis was performed at 293 K on diluted solutions of each compound by using CDCl3 or DMSO-d6 as solvents. Chemical shifts (δ) are reported in ppm. Coupling constants (J) are reported in Hertz (Hz). GC-MS analyses were performed with a DB-35MS (20 m × 0.18 mm, 35% phenyl 65% dimethylpolysiloxane) capillary column. The mass detector was operated in the electron impact ionization mode (EI/MS) with an electron energy of 70 eV. The injection port was heated to 250 °C. The oven temperature program was initially set at 70 °C with a hold of 2 min and ramped to 280 °C at 20 °C min−1 with a hold of 10 min. In order to obtain a good separation of diastereisomers the oven temperature program was initially set at 40 °C with a hold of 2 min and ramped to 280 °C at 5 °C min−1 with a hold of 10 min.General procedure for the synthesis of dipeptides 1–18
α-Amino acid methyl ester hydrochloride (1 mmol) is first solubilized in 5 mL of anhydrous pyridine by magnetic stirring. After about 5 minutes the N-protected-α-amino acid (1 mmol) and TiCl4 (2 mmol) are added. The tightly sealed screw-capped vial containing the reaction mixture is left under magnetic stirring at 40 °C, maintaining the pH at values as close as possible to neutrality by adding further amounts of pyridine. The reaction, monitored by TLC analysis (chloroform–methanol, 90:10, v/v), is completed after about 5 hours. After removal of pyridine by co-evaporation with toluene, the resulting crude product is suspended in chloroform and purified by elution through a silica gel column also containing a layer of NaHCO3 and one of NaHSO4 separated by a silica gel one and using chloroform as mobile phase. The evaporation of chloroform under reduced pressure affords the corresponding N-protected dipeptide 1–18 with yields ranging from 60 to 87%.
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) COOCH3), 4.40 (m, 1H, CCONH), 3.70 (s, 3H, OCH3), 3.05 (d, J = 6.2 Hz, 2H, CH2Ph), 1.39 (s, 9H, (CH3)3C), 1.33 (d, J = 7.2 Hz, 3H, C3CH); 13C NMR (75 MHz, CDCl3) δ: 172.82, 170.82, 155.38, 136.58, 129.35, 128.56, 126.88, 80.14, 55.53, 52.33, 48.08, 38.36, 28.22, 18.25; GC/MS (EI, 70 eV) m/z (% rel.): 294 (11), 277 (7), 263 (4), 248 (1) 233 (18), 220 (10), 191 (3.7), 178 (15), 164 (41.5), 159 (27), 146 (9), 120 (100), 91 (52), 57 (94). Found: C, 61.49; H, 7.45; N, 7.96. C18H26N2O5 requires C, 61.70; H, 7.48; N, 7.99%.
COOCH3), 4.40 (m, 1H, CCONH), 3.70 (s, 3H, OCH3), 3.05 (d, J = 6.2 Hz, 2H, CH2Ph), 1.39 (s, 9H, (CH3)3C), 1.33 (d, J = 7.2 Hz, 3H, C3CH); 13C NMR (75 MHz, CDCl3) δ: 172.82, 170.82, 155.38, 136.58, 129.35, 128.56, 126.88, 80.14, 55.53, 52.33, 48.08, 38.36, 28.22, 18.25; GC/MS (EI, 70 eV) m/z (% rel.): 294 (11), 277 (7), 263 (4), 248 (1) 233 (18), 220 (10), 191 (3.7), 178 (15), 164 (41.5), 159 (27), 146 (9), 120 (100), 91 (52), 57 (94). Found: C, 61.49; H, 7.45; N, 7.96. C18H26N2O5 requires C, 61.70; H, 7.48; N, 7.99%.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from MIUR (Ministero Italiano dell’Università e della Ricerca, ex-60% funds).Notes and references
- (a) M. S. Iyer, K. M. Gigstad, N. D. Namdev and M. Lipton, J. Am. Chem. Soc., 1996, 118, 4910–4911 CrossRef CAS; (b) J. Oku, N. Ito and S. Inoue, Makromol. Chem., 1979, 180, 1089–1091 CrossRef CAS.
- (a) M. Yagasaki and S. Hashimoto, Appl. Microbiol. Biotechnol., 2008, 81, 13–22 CrossRef CAS; (b) J. Arima, Y. Uesugi, M. Uraji, M. Iwabuchi and T. Hatanaka, Appl. Environ. Microbiol., 2006, 72, 4225–4231 CrossRef CAS.
- (a) V. K. Khavinson and V. N. Anisimov, Dokl. Akad. Nauk, 2000, 372, 421–423 CAS; (b) M. Nath, S. Pokharia, G. Eng, X. Q. Song and A. Kumar, Spectrochim. Acta, Part A, 2006, 63, 66–75 CrossRef PubMed.
- A. Guiotto, A. Calderan, P. Ruzza and G. Borin, Curr. Med. Chem., 2005, 12, 2293–2315 CrossRef CAS.
- D. D. Kitts and K. Weiler, Curr. Pharm. Des., 2003, 9, 1309–1323 CrossRef CAS.
- R. C. Harris, J. R. Hoffmanb, A. Allsoppc and N. B. H. Routledge, Nutr. Res., 2012, 32, 272–277 CrossRef CAS.
- B. C. Song, N.-S. Joo, G. Aldini and K.-J. Yeum, Nutr. Res. Pract., 2014, 8, 3–10 CrossRef CAS.
- T.-S. Tseng, K.-C. Tsai, W.-C. Chen, Y.-T. Wang, Y.-C. Lee, C.-K. Lu, M.-J. Don, C.-Y. Chang, C.-H. Lee, H.-H. Lin, H.-J. Hsu and N.-W. Hsiao, J. Agric. Food Chem., 2015, 63, 6181–6188 CrossRef CAS.
- K. Lintner and O. Peschard, Int. J. Cosmet. Sci., 2000, 22, 207–218 CrossRef CAS.
- A. Sallam, M. Krehenbrink and D. Kalkandzhiev,WO 2017/162879 A1, 2017.
- (a) F. Albericio, Curr. Opin. Chem. Biol., 2004, 8, 211–221 CrossRef CAS PubMed; (b) T. Kimmerlin and D. Seebach, J. Pept. Res., 2005, 65, 229–260 CrossRef CAS PubMed.
- (a) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS; (b) A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS; (c) R. De Marco, M. Spinella, A. De Lorenzo, A. Leggio and A. Liguori, Org. Biomol. Chem., 2013, 11, 3786–3796 RSC; (d) A. Leggio, E. L. Belsito, G. De Luca, M. L. Di Gioia, V. Leotta, E. Romio, C. Siciliano and A. Liguori, RSC Adv., 2016, 6, 34468–34475 RSC; (e) A. Leggio, A. Comandè, E. L. Belsito, M. Greco, L. Lo Feudo and A. Liguori, Org. Biomol. Chem., 2018, 16, 5677–5683 RSC.
- (a) R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS; (b) M. L. Di Gioia, A. Leggio, A. Liguori and F. Perri, J. Org. Chem., 2007, 72, 3723–3728 CrossRef CAS; (c) D. M. M. Jaradat, Amino Acids, 2018, 50, 39–68 CrossRef CAS.
- (a) M. T. Reetz, Organotitanium Reagents in Organic Synthesis, Springer-Verlag, Berlin, 1986 CrossRef; (b) M. L. Di Gioia, A. Leggio, A. Le Pera, A. Liguori, A. F. Pitrelli and C. Siciliano, Protein Pept. Lett., 2005, 12, 357–362 CrossRef CAS; (c) A. Leggio, E. L. Belsito, M. L. Di Gioia, V. Leotta, E. Romio, C. Siciliano and A. Liguori, Tetrahedron Lett., 2015, 56, 2062–2066 CrossRef; (d) M. L. Di Gioia, A. Leggio, I. F. Guarino, V. Leotta, E. Romio and A. Liguori, Tetrahedron Lett., 2015, 56, 5341–5344 CrossRef CAS; (e) A. Leggio, E. L. Belsito, S. Gallo and A. Liguori, Tetrahedron Lett., 2017, 58, 1512–1514 CrossRef CAS; (f) A. Leggio, J. Bagalà, E. L. Belsito, A. Comandè, M. Greco and A. Liguori, Chem. Cent. J., 2017, 11(87), 1–12 Search PubMed.
- (a) A. Leggio, A. Liguori, A. Napoli, C. Siciliano and G. Sindona, Eur. J. Org. Chem., 2000, 5732575 Search PubMed; (b) M. L. Di Gioia, A. Leggio, A. Le Pera, A. Liguori, F. Perri and C. Siciliano, Eur. J. Org. Chem., 2004, 4437–4441 CrossRef CAS; (c) M. L. Di Gioia, A. Leggio, A. Le Pera, C. Siciliano, G. Sindona and A. Liguori, J. Pept. Res., 2004, 63, 383–387 CrossRef CAS.
- (a) A. Leggio, M. L. Di Gioia, F. Perri and A. Liguori, Tetrahedron, 2007, 63, 8164–8173 CrossRef CAS; (b) R. De Marco, M. L. Di Gioia, A. Leggio, A. Liguori and M. C. Viscomi, Eur. J. Org. Chem., 2009, 3795–3800 CrossRef CAS.
- (a) T. Fukuyama, C.-K. Jow and M. Cheung, Tetrahedron Lett., 1995, 36, 6373–6374 CrossRef CAS; (b) A. Le Pera, A. Leggio and A. Liguori, Tetrahedron, 2006, 62, 6100–6106 CrossRef CAS.
- (a) S. C. Miller and T. S. Scanlan, J. Am. Chem. Soc., 1997, 119, 2301–2302 CrossRef CAS; (b) M. L. Di Gioia, A. Leggio, A. Liguori, F. Perri, C. Siciliano and M. C. Viscomi, Amino Acids, 2010, 38, 133–143 CrossRef CAS; (c) E. Biron and H. Kessler, J. Org. Chem., 2005, 70, 5183–5189 CrossRef CAS; (d) C. Aroulanda, G. Celebre, G. De Luca and M. Longeri, J. Phys. Chem. B, 2006, 110, 10485–10496 CrossRef CAS; (e) E. L. Belsito, M. L. Di Gioia, A. Greco, A. Leggio, A. Liguori, F. Perri, C. Siciliano and M. C. Viscomi, J. Org. Chem., 2007, 72, 4798–4802 CrossRef CAS; (f) A. Leggio, E. L. Belsito, R. De Marco, A. Liguori, F. Perri and M. C. Viscomi, J. Org. Chem., 2010, 75, 1386–1392 CrossRef CAS; (g) R. De Marco, A. Leggio, A. Liguori, T. Marino, F. Perri and N. Russo, J. Org. Chem., 2010, 75, 3381–3386 CrossRef CAS; (h) M. L. Di Gioia, A. Leggio and A. Liguori, J. Org. Chem., 2005, 70, 3892–3897 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data and copies of the 1H NMR and 13C-NMR spectra of dipeptides 1–20 and MS (EI) spectra of dipeptides 1–6 and 18–20; GC/MS (EI) analyses of dipeptides 3–4, 18–19 and 20. See DOI: 10.1039/c9ra04058g |
| This journal is © The Royal Society of Chemistry 2019 |