
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLonimacranaldes A–C, three iridoids with novel skeletons from Lonicera macranthoides†
Yu-Dan Mei‡
ab,
Hai-Bo Li‡c,
Qian-Qian Panga,
Ting Lia,
Da-Bo Pana,
Yi Daia,
Da-Peng Qina,
He Mengb,
Xin-Sheng Yao *a and
Yang Yu*a
*a and
Yang Yu*a
aInstitute of Traditional Chinese Medicine & Natural Products, College of Pharmacy, Jinan University, Guangzhou 510632, P. R. China. E-mail: 1018yuyang@163.com; tyaoxs@jnu.edu.cn; Fax: +86-20-85221559; Tel: +86-20-85221559
bXiangxue Academician Workstation, Xiangxue Pharmaceutical Co. Ltd., Guangzhou, 510663, P. R. China
cJiangsu Kanion Pharmaceutical Co., Ltd., State Key Laboratory of New-tech for Chinese Medicine Pharmaceutical Process, Jiangsu, Lianyungang, 222001, China
First published on 16th July 2019
Abstract
Lonimacranaldes A and B (1 and 2), along with one biogenetically related intermediate, lonimacranalde C (3), were isolated from the flower buds of Lonicera macranthoides. Characterized by an iridoid structure and an additional C-6 unit with an aldehyde group, compounds 1 and 2 are the first examples of hybrid iridoids possessing an unexpected 6/5/6 fused tricyclic ring system, while compound 3 serves as an important precursor for their generation. The structures of lonimacranaldes A–C (1–3) were revealed by extensive spectroscopic and X-ray diffraction analyses. A plausible biogenetic pathway for them was proposed. Compound 3 showed anti-inflammatory activities by inhibiting the production of IL-6 on LPS-induced RAW 264.7 cells with an IC50 value of 6.33 μM.
Introduction
Naturally occurring iridoids with a cyclopenta[c]pyran skeleton are monoterpenoids which usually exist as glycosides with a monosaccharide group at C-1.1 Cleavage of the C-7 and C-8 bonds of the cyclopentane ring of iridoids affords a subclass known as secoiridoids. Iridoids possess a broad range of biological activities such as hepatoprotective,2,3 anti-inflammatory,4,5 antitumor,6,7 antimicrobial,8 allelopathic,9 hypoglycemic,10 cardiovascular,11 choleretic, antispasmodic, antiviral, and immunomodulatory activity.12,13Lonicera macranthoids Hand.-Mazz., an important source of Lonicerae Flos (also Shan Yin-Hua in Chinese), are commonly used in traditional Chinese medicine (TCM) for its similar heat-clearing and detoxicating effects as those of Lonicerae japonicae Flos (Jin Yin-Hua). Our previous studies on the 70% EtOH extract of the flower buds of L. macranthoides resulted in the isolation of 11 chlorogenic acids and 12 triterpenoids, including three new chlorogenic acids with a monoterpene unit and two anti-inflammatory triterpenoids.14,15
In our continuous efforts to discover new bioactive iridoids from the flower buds of Lonicera macranthoids, three iridoids with novel skeletons (lonimacranaldes A–C, 1–3) were isolated unexpectedly. The common structural feature among 1–3 was biogenetically derived from two precursors, an iridoid (secologanic acid, 4) and a volatile component with C-6 unit (hexanal, 5), both of which had been reported in Lonicera genus before.16,17 Obviously, the aldol condensation of the two precursors led to 3, while 1 and 2 might be derived from 3 by an oxidative reaction and an intramolecular Diels–Alder reaction to form an unexpected 6/5/6 fused tricyclic ring skeleton system (Fig. 1, rings A/B/C). Herein, we reported the isolation, structure elucidation, and plausible biosynthetic pathway of 1–3, and their potential anti-inflammatory activities.
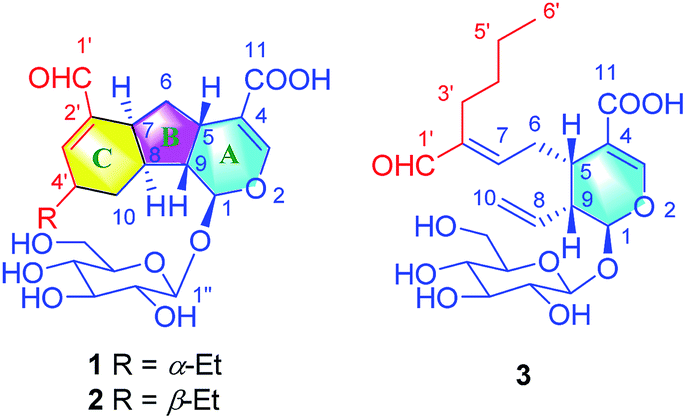 | ||
| Fig. 1 Chemical structures of compounds 1–3. | ||
Results and discussion
Lonimacranalde A (1) was obtained as white powder and subsequently crystallized from EtOH/H2O as colorless needles with negative rotation ([α]28D – 18.7, c = 0.6, in CH3OH). The UV spectrum showed a characteristic absorption maximum of iridoid at 232 nm (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε 3.9), while the IR spectrum showed an absorption band of hydroxyl (3366 cm−1) and carbonyl (1700 cm−1). Its molecular formula was determined as C22H30O10 based on HR-ESI-Q-TOF-MS (m/z 477.1747 [M + Na]+, calcd for C22H30O10Na, 477.1737). The 1H NMR spectrum of 1 exhibited characteristic signals of iridoid glycoside, including an acetal proton [δH 5.21 (1H, d, J = 7.1 Hz, H-1)], a tri-substituted olefinic proton [δH 7.48 (1H, d, J = 1.2 Hz, H-3)], and an anomeric proton [δH 4.72 (1H, d, J = 7.9 Hz, H-1′′)]. In addition, a proton signal assignable to an aldehyde group [δH 9.38 (1H, s, H-1′)] was also observed. The sugar moiety of 1 was determined to be D-glucose by HPLC analysis after acid hydrolysis and glycosyl derivatization. Combined with the J value of the anomeric proton mentioned above, a β-D-glucosyl residue was deduced.
ε 3.9), while the IR spectrum showed an absorption band of hydroxyl (3366 cm−1) and carbonyl (1700 cm−1). Its molecular formula was determined as C22H30O10 based on HR-ESI-Q-TOF-MS (m/z 477.1747 [M + Na]+, calcd for C22H30O10Na, 477.1737). The 1H NMR spectrum of 1 exhibited characteristic signals of iridoid glycoside, including an acetal proton [δH 5.21 (1H, d, J = 7.1 Hz, H-1)], a tri-substituted olefinic proton [δH 7.48 (1H, d, J = 1.2 Hz, H-3)], and an anomeric proton [δH 4.72 (1H, d, J = 7.9 Hz, H-1′′)]. In addition, a proton signal assignable to an aldehyde group [δH 9.38 (1H, s, H-1′)] was also observed. The sugar moiety of 1 was determined to be D-glucose by HPLC analysis after acid hydrolysis and glycosyl derivatization. Combined with the J value of the anomeric proton mentioned above, a β-D-glucosyl residue was deduced.
Besides signals attributable to a glucosyl residue, the rest carbons were revealed as two carbonyls (δC 196.4, 170.9), two tri-substituted double bonds (δC 158.1, 153.5, 145.1, 112.2), and ten sp3 carbons, which comprised six methines (δC 98.6, 46.1, 38.4, 36.1, 35.7, 35.2), three methylenes (δC 38.9, 32.1, 28.7), and a methyl (δC 11.8) according to 13C NMR and DEPT 135 spectra. All of the proton and carbon signals were assignable by COSY, HSQC, and HMBC spectra (Table S1†).
The aglucone of compound 1 was determined by the COSY correlations of H-1/H-9(H-8)/H-5/H2-6/H-7/H-8/H2-10/H-4′(H-3′)/H2-5′/H3-6′, together with the HMBC correlations of H-3/C-1, 4, 5, 11, H-1/C-3, 5, 8, 9 and H-1′/C-7, 2′, 3′, (Fig. 2). Moreover, other HMBC correlations of H-1′′/C-1 and H-1/C-1′′ revealed that the β-D-glucosyl was located at C-1. Hence, the planar structure of compound 1 was established.
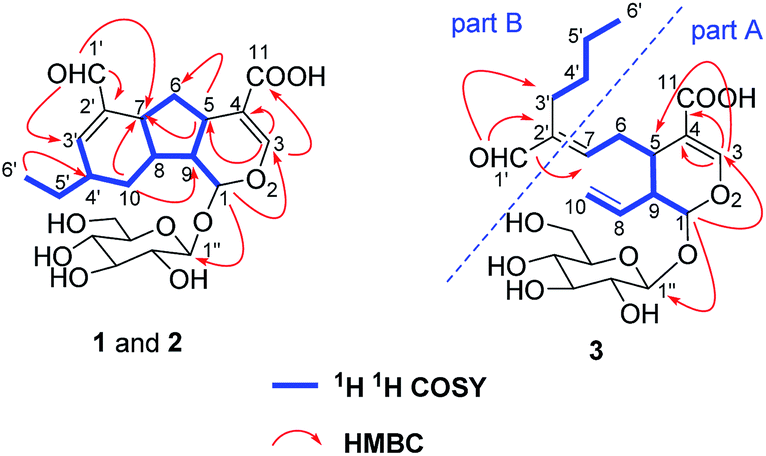 | ||
| Fig. 2 Key 1H–1H COSY, HMBC, and ROESY correlations of 1–3. | ||
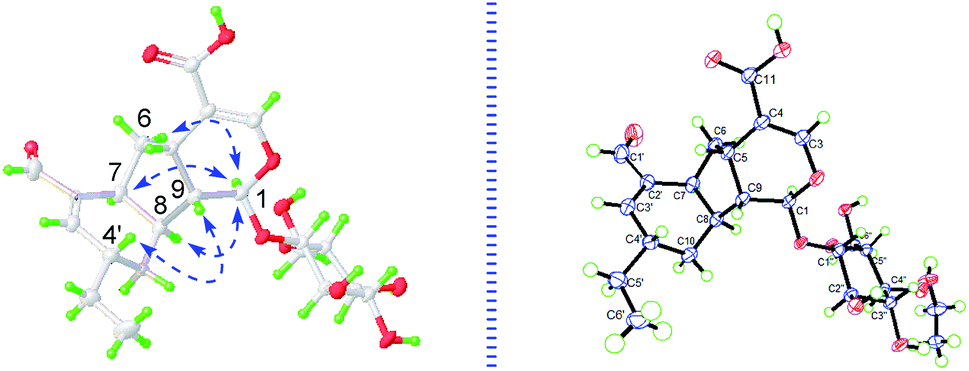 | ||
| Fig. 3 Key NOESY correlations and X-ray crystallographic analysis of 1. | ||
The cyclopentanopyran ring in iridoids was commonly H-5/H-9 β,β-cis-fused, while H-1 α-oriented. Since the C-1 resonance was at δ 98.6 (compared to δ 101–103 for trans-fused iridoids) and the allylic coupling of the H-3/H-5 resonances was 1.2 Hz (compared to 2.0–2.5 Hz for trans-fused iridoids),14,18 the H-5/H-9 in 1 could be preliminarily assumed as β,β-cis-fused. Then, based on the NOE correlations of H2-6/H-1, H-1/H-7 and H-1/H-8, A/B and B/C rings in 1 were determined as H-5/H-9 β,β-cis-fused, and H-7/H-8 α,α-cis-fused, respectively. Furthermore, the NOE correlations of H-4′/H-9 indicated that H-4′ was β-oriented. For the deductions above, the relative configurations in 1 were all consistent with the absolute configurations determined by X-ray (Fig. 3). Consequently, the structure of 1 was unmistakably and completely confirmed and named lonimacranalde A.
Lonimacranalde B (2), isolated as colorless needle ([α]28D – 9.7, c = 0.6, in CH3OH), has the same molecular formula with 1 based on the HR-ESI-MS data. The 1H NMR and 13C NMR data of 2 were assignable by COSY, HSQC, and HMBC spectra (Table S1†), which were highly similar to 1, suggesting that they were two isomers.
There is no single crystal data of 2, so its configurations were solved by analyzing the coupling constants and NOESY data, as well as the CD spectrum. Similarly, for the NOE correlations of H-6 (δH 2.23)/H-1, H-1/H-7 and H-1/H-8, A/B and B/C rings in 2 were determined as β,β-cis-fused and α,α-cis-fused, respectively, as well. The coupling constants of H-10a, H-4′, and H-8 (J10a,4′ = 11.0 Hz, J10a,8 = 11.0 Hz) indicated that H-10a/H-4′ and H-10a/H-8 were aa coupling. Since that, H-4′ was β-oriented as H-8. It also meant that the dominant conformation of ring C in compound 2 was the same as 1 with 4′-ethyl in equatorial bond. The NOE correlations of H-9/H2-10 and H-10b (δH 1.88)/H3-6 further provided more evidences for such deduction (Fig. 4). Obviously, 2 was a C-4′ epimer of 1, which can further be validated by their experimental CD spectra with the same Cotton effects (Fig. 5). In conclusion, compound 2 was determined and named lonimacranalde B.
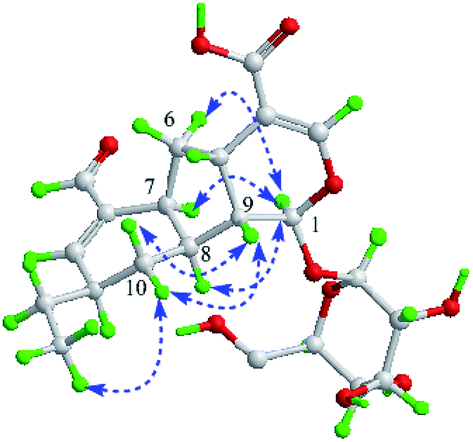 | ||
| Fig. 4 Key NOESY correlations of 2. | ||
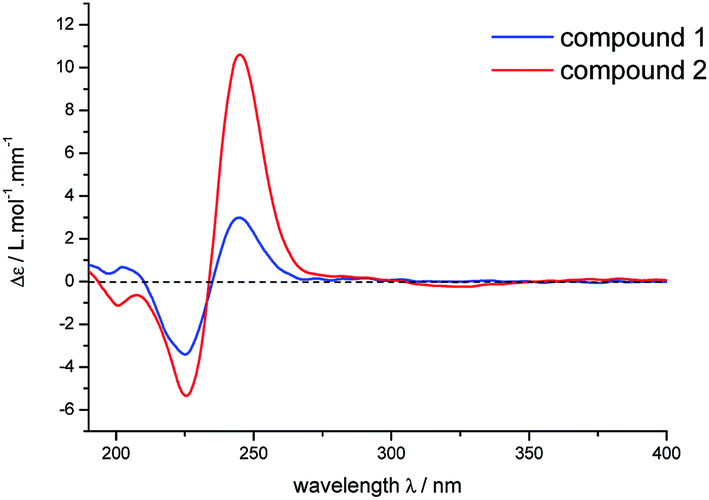 | ||
| Fig. 5 Experimental CD spectra of 1 and 2. | ||
Lonimacranalde C (3), obtained as light yellow powder ([α]28D – 150.4, c = 0.3, in CH3OH), its molecular formula was determined as C22H32O10 on the basis of the HR-ESI-MS data (m/z 479.1895 [M + Na]+, calcd for C22H32O10Na: 479.1893). Compound 3 was considered as a secoiridoid-type glycoside for the typical protons attributed to an acetal [δH 5.55 (1H, d, J = 5.6 Hz, H-1)], a tri-substituted olefinic proton [δH 7.52 (1H, d, J = 1.1 Hz, H-3)], and a mono-substituted vinyl group [δH 5.77 (1H, ddd, J = 17.1, 10.4, 8.9 Hz, H-8), 5.27 (2H, m, H2-10)], together with an anomeric proton [δH 4.69 (1H, d, J = 7.9 Hz, H-1′)]. Additionally, the sugar moiety in 3 was identified as β-D-glucosyl by comparing the NMR data with 1.
According to the COSY correlations of H-1/H-9/H-5/H2-6/H-7 and H-9/H-8/H2-10, as well as the HMBC correlations of H-1/C-1′′, 3, and H-3/C-4, 5, 11, a 7-substituted secologanic acid moiety in 3 was established (part A, Fig. 2). Except for the structure of part A, the rest six carbon signals included four sp3 carbons, an olefinic quaternary carbon, and an aldehyde. Thereafter, a hexanal moiety was further confirmed by the COSY correlations of H2-3′/H2-4′/H2-5/′H3-6′ and the HMBC correlations of H-1′/C-2′, 3′ (part B, Fig. 2). Moreover, part A and part B in 3 were linked through the C-7/C-2′ double bond, which was verified by the HMBC correlations of H-1′/C-7. Thus, the planar structure of 3 was established and data assignment was shown in Table 1.
| No. | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| a Recorded at 600 (1H) and 150 MHz (13C) in CD3OD. Multiplets or overlapped signals are reported without designating multiplicity. | ||||||
| 1 | 98.6 | 5.21, d (7.1) | 97.2 | 5.35, d (5.9) | 97.7 | 5.55, d, (5.6) |
| 3 | 153.5 | 7.48, d (1.2) | 153.4 | 7.49, d (1.1) | 153.9 | 7.52, d (1.1) |
| 4 | 112.2 | 113.0 | 110.8 | |||
| 5 | 35.2 | 2.68, q (7.8) | 33.6 | 3.06, ddd (8.8, 8.0, 4.3) | 33.5 | 3.04 |
| 6 | 38.9 | 2.00, ddd (13.0, 6.7, 4.4) | 37.8 | 2.23 | 30.2 | 2.50, ddd (15.2, 8.0, 7.0) |
| 1.85 | 1.68, ddd (13.3, 10.5, 8.8) | 2.99 ddd (15.2, 7.0, 7.0) | ||||
| 7 | 35.7 | 2.89 | 34.7 | 2.79, q (9.0) | 155.5 | 6.63, t (7.0) |
| 8 | 38.4 | 2.47 | 40.4 | 2.25 | 135.4 | 5.77, ddd (17.1, 10.4, 9.0) |
| 9 | 46.1 | 1.94, td (7.8, 4.8) | 48.8 | 2.09, ddd (8.0, 5.9, 2.5) | 45.4 | 2.70, dt (9.0, 5.6) |
| 10 | 32.1 | 1.88 | 33.7 | 1.88, ddd (13.0, 5.0, 4.6) | 120.0 | 5.27, m |
| 1.40, ddd (13.6, 9.1, 4.6) | 1.13, ddd (13.0, 13.0, 11.1) | |||||
| 11 | 170.9 | 170.9 | 170.4 | |||
| 1′ | 196.4 | 9.38, s | 196.5 | 9.40, s | 197.1 | 9.33, s |
| 2′ | 145.1 | 145.3 | 145.6 | |||
| 3′ | 158.1 | 6.93, d (2.8) | 157.0 | 6.80, d (2.0) | 24.8 | 2.21 |
| 4′ | 36.1 | 2.30 | 40.0 | 2.30 | 32.0 | 1.30 |
| 5′ | 28.7 | 1.57, ddq (14.0, 14.0, 7.4) | 28.8 | 1.59, ddq (14.0, 14.0, 7.4) | 23.8 | 1.30 |
| 1.48, ddq (14.0, 14.0, 7.4) | 1.51, ddq (14.0, 14.0, 7.4) | |||||
| 6′ | 11.8 | 1.05, t (7.4) | 11.5 | 1.03, t (7.4) | 14.2 | 0.91, t (7.0) |
| 1′′ | 100.3 | 4.72, d (7.8) | 100.3 | 4.67, d (7.9) | 100.2 | 4.69, d (7.9) |
| 2′′ | 74.8 | 3.21, dd (9.0, 7.8) | 74.7 | 3.21, dd (9.2, 7.9) | 74.7 | 3.20, dd (9.2, 7.9) |
| 3′′ | 78.0 | 3.39, t (8.7) | 78.0 | 3.38, dd (9.2, 8.7) | 78.0 | 3.37, dd (9.2, 8.9) |
| 4′′ | 71.6 | 3.29 | 71.6 | 3.29 | 71.6 | 3.28, dd (9.7, 8.9) |
| 5′′ | 78.4 | 3.30 | 78.3 | 3.30 | 78.4 | 3.31 |
| 6′′ | 62.8 | 3.90, dd (11.9, 2.1) | 62.7 | 3.86, dd (11.9, 1.9) | 62.7 | 3.89, dd (11.9, 2.2) |
| 3.67, dd (11.9, 5.9) | 3.65, dd (11.9, 5.4) | 3.66, dd (11.8, 5.8) | ||||
The NOE correlations of H-1/H-6, H-1/H-8, and H-1/H-7 revealed that H-1/H-6/H-8 and H-5/H-9 in 3 were of the opposite configurations, suggesting that the relative configurations in 3 were consistent with the common seco-iridoids. Thus, it could be concluded that the absolute configurations in 3 were identical with secologanic acid and defined as 1S/5S/9R. Furthermore, the correlation of H-1′/H-7 observed in the NOESY spectrum revealed that the geometry of the double bond (C-7/C-2′) was E-configuration. In conclusion, 3 was identified as lonimacranalde C.
A plausible biosynthetic pathway, involving an aldol condensation, an oxidative reaction and a key Diels–Alder reaction, was proposed for lonimacranaldes A–C (1, 2 and 3) (Scheme 1). It's obvious that compound 3 was originated from 4 and 5 by an aldol condensation, which occurs frequently in 7-CHO of secologanic acid or secologanin to provide diverse unusual structures.17,19–21 Then, the oxidation of 3 led to the intermediates (6a and 6b), which were never isolated, but might to generate 1 and 2 through a [4 + 2] cycloaddition. The cycloaddition has endo preference and occurs with remote stereocontrol syn to the substituent at the stereogenic center, which can explain the epimerization of 1 and 2 in a certain extent.22,23 This plausible biogenetic pathway of 1–3 has not been validated, but it provides a reference for their biosynthesis and total synthesis.
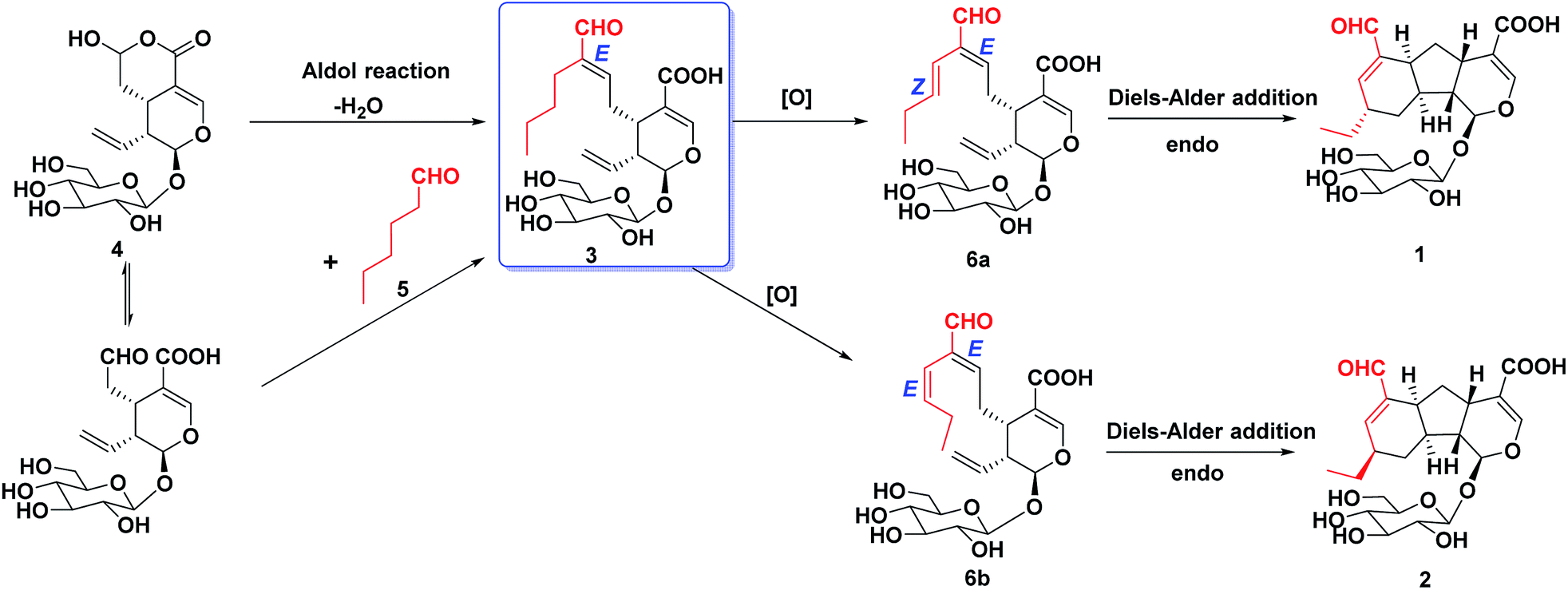 | ||
| Scheme 1 Plausible biosynthetic pathway of 1, 2 and 3. | ||
Anti-inflammatory activities of lonimacranaldes A–C (1, 2 and 3) were evaluated in LPS-stimulated RAW 264.7 cells, including NO (nitric oxide), IL-6 (interleukin 6) and TNF-α (tumor necrosis factor) inhibitory activities. As a result, compound 3 exhibited inhibitory effect on IL-6 with an IC50 value of 6.33 μM (Fig. 6). Unfortunately, none of them displayed TNF-α and NO inhibitory activity at 100 μM.
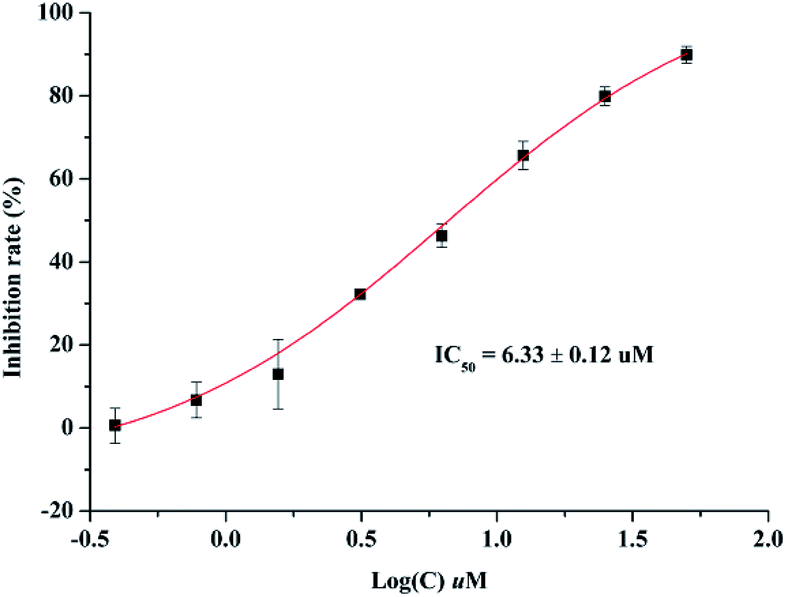 | ||
| Fig. 6 Effect of compound 3 on IL-6 production in LPS-stimulated RAW 264.7 cells. | ||
In order to confirm that compounds 1–3 are indeed natural products, the crude methanol extract of dried flower buds of L. macranthoides was analyzed by UPLC-Q-TOF-MS (see the ESI†). The ion peaks in accord with those of 1–3 were detected, which confirmed the natural occurrence of these compounds.
Conclusions
In conclusion, we have isolated three novel hybrid iridoids, lonimacranaldes A–C (1–3) from the flower buds of Lonicera macranthoides. Their unique architectures and remarkable anti-inflammatory activities of compound 3 should receive considerable attention from synthetic chemists and pharmacologists.Experimental
General experimental procedures
1H and 13C NMR spectra were measured on a Bruker AV 600 (Bruker Co. Ltd., Bremen, German) at 600 and 150 Hz with solvent signals (CD3OD, δH 3.10/δC 49.0) as internal reference. IR spectra were taken on a JASCO FT/IRBCA plus spectrometer (JASCO International Co. Ltd., Tokyo, Japan) with KBr. UV spectra were acquired using a JASCO V-550 UV/Vis spectrometer (JASCO International Co. Ltd., Hachioji, Tokyo, Japan). Optical rotation was measured on JASCO P-1020 digital polarimeter (JASCO International Co. Ltd., Hachioji, Tokyo, Japan). ECD spectra were recorded in methanol using a JASCO J-810 spectrophotometer (Jasco Internal Co. Ltd, Tokyo, Japan). HRESIMS spectra were measured on a Waters Synapt G2 mass spectrometer (Waters, Manchester, U. K.) with a RP-18 column (1.7 m, ϕ 3.0 × 150 mm; BEH). HPLC analyses were performed on a Waters 2695 separations module (Waters, Manchester, U.K.) equipped with a 2998 photodiode array (PDA) detector and an Alltech 3300 evaporative light scattering detector (ELSD; Alltech Inc., Deerfield, Illinois, U.S.A.) using a Phenomenex Gemini C18 column (5 μm, ϕ 4.6 × 250 mm; FLM Inc., Guangzhou, China). The semi-preparative HPLC analyses were performed on a Waters 1515 isocratic HPLC pump (Waters, Manchester, U.K.) coupled with a 2489 UV/Vis detector (Waters, Manchester, U.K.) and a Phenomenex Gemini C18 column (5 μm, ϕ 10 × 250 mm; FLM Inc., Guangzhou, China). The methanol of HPLC grade was purchased from BCR International Co. Ltd. (Shanghai, China), while the acetonitrile was purchased from Merck (Darmstadt, Germany). Diaion HP-20 (Mitsubishi Chemical Co., Tokyo, Japan), silica gel (200–300 mesh, Qingdao Marine Chemical Ltd., Shandong, China), ODS-silica gel (12 nm, S-50 μm, YMC Ltd., Tokyo, Japan) and Sephadex LH-20 (Amersham Pharmacia Biotech, Sweden) were used for chromatography column (CC). TLC was performed on pre-coated silica gel plate (SGF254, 0.2 mm, Yantai Chemical Industry Research Institute, Shandong, China).Plant material
Dried flower buds of L. macranthoides were collected in Shandong Province and purchased from Kunyuan Pharmaceutical Co., Ltd. in Nov, 2014, and identified by Prof. G. X. Zhou of Jinan University. A sample (no. LM201411) is deposited in Jinan University, Guangzhou, China.Extraction and isolation
The dried flower buds of L. macranthoides (35 kg) were extracted twice with 70% ethanol (280 L, for 2 h each time) under reflux. The ethanol was evaporated under vacuum, affording a crude extract (8.5 kg), which was suspended in water and then subjected to an HP-20 macroporous resin CC (ϕ 20 × 85 cm) eluted with EtOH–H2O (0:100, 30:70, 50:50, 5:95, v/v) to offer four fractions (LM-1–LM-4). LM-3 (700 g) was chromatographed over a silica gel CC (ϕ 110 × 420 cm) by using a CHCl3–CH3OH–H2O gradient (95:5:0 to 60:40:8, v/v/v), yielding sixteen fractions (3A-3P). Fraction 3E (17.7 g) was separated by ODS (ϕ 3.3 × 33 cm) eluted with CH3OH–H2O (30:70 to 100:0, v/v) to offer eleven subfractions E1–E11. Fractions E6, was separated on a silica gel CC (CHCl3–CH3OH–H2O, 80:20:2, v/v/v), Sephadex LH-20 CC (CH3OH–H2O, 50:50, v/v), followed semipreparative HPLC (CH3CN–H2O–HCOOH, 25:75:0.1, v/v/v) respectively, to yield 1 (tR: 15.3 min, 7.7 mg) and 2 (tR: 17.0 min, 4.7 mg), while fractions E10 was purified by semipreparative HPLC (CH3CN–H2O–HCOOH, 30:70:0.1, v/v/v) to furnish 3 (tR: 22.3 min, 2.0 mg).
ε): 204 (3.9), 232 (3.9), IR (KBr) νmax: 3366, 2880, 2357, 1700, 1414, 1070 cm−1; 1H and 13C NMR data see Table 1.ε): 206 (4.1), 230 (3.9); IR (KBr) νmax: 3389, 2891, 2345, 1678, 1621, 1274, 1070 cm−1; 1H and 13C NMR data see Table 1.ε): 204 (3.9), 234 (4.2); IR (KBr) νmax: 3400, 2926, 2357, 1675, 1627, 1272, 1070 cm−1; 1H and 13C NMR data see Table 1.X-ray crystallographic analysis
017 unique reflections were collected until θmax = 74.16°, in which 4804 reflections were observed [F2 > 4σ(F2)]. The structure was solved by direct methods using the SHELXS-97 program, and refined by the program SHELXL-97 and full-matrix least-squares calculations. In the structure refinements, non-hydrogen atoms were placed on the geometrically ideal positions by the “ride on” method. Hydrogen atoms bonded to oxygen were located by the structure factors with isotropic temperature factors. The final refinement gave R = 0.0360(4804), Rw = 0.0899(4915), S = 1.042, and flack = −0.01(8). Crystallographic data for structure 1 has been deposited at the Cambridge Crystallographic Data Centre (CCDC 1877498).Anti-inflammatory activity assays
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (program no. 81630097, 81602984 and 81803347). The authors are grateful to State Key Laboratory of New-Tech for Chinese Medicine Pharmaceutical Process, Jiangsu Kanion Pharmaceutical Co. Ltd. for their assistance of activity test.Notes and references
- M. Wink, Phytochemistry, 2003, 64, 3–19 CrossRef CAS.
- W. Gu, X. J. Hao, H. X. Liu, Y. H. Wang and C. L. Long, Phytochem. Lett., 2013, 6, 681–685 CrossRef CAS.
- K. Hase, Q. Xiong and S. Kadota, Stud. Nat. Prod. Chem., 2001, 25, 459–482 CAS.
- M. X. Li, X. F. Shang, R. X. Zhang, Z. P. Jia, P. C. Fan, Q. Ying and L. L. Wei, Fitoterapia, 2010, 81, 167–172 CrossRef CAS.
- Z. D. Nan, M. B. Zhao, K. W. Zeng, S. H. Tian, W. N. Wang, Y. Jiang and P. F. Tu, Chin. J. Nat. Med., 2016, 14, 61–65 Search PubMed.
- C. Wang, P. Xin, Y. Wang, X. Zhou, D. Wei, C. Deng and S. Sun, Fitoterapia, 2018, 124, 152–159 CrossRef CAS.
- C. Wang, X. Zhou, Y. Wang, D. Wei, C. Deng, X. Xu, P. Xin and S. Sun, Molecules, 2017, 22, 2101 CrossRef.
- N. Chaipukdee, K. Kanokmedhakul, S. Kanokmedhakul, R. Lekphrom and S. G. Pyne, Fitoterapia, 2016, 113, 97–101 CrossRef CAS.
- A. E. Ayeb-Zakhama, A. Beyaoui, S. B. Salem, L. Sakka-Rouis, J. Bouajila, H. B. Jannet and F. Harzallah-Skhiri, Ind. Crops Prod., 2015, 76, 653–659 CrossRef.
- R. D. Sonawane, V. B. Deore, S. D. Patil, C. R. Patil, S. J. Surana and R. K. Goyal, Physiol. Behav., 2015, 144, 66–72 CrossRef CAS.
- C. M. Camero, M. P. Germanò, A. Rapisarda, V. D'Angelo, S. Amira, F. Benchikh, A. Braca and M. D. Leo, Rev. Bras. Farmacogn., 2018, 28, 374–377 CrossRef CAS.
- E. L. Ghisalberti, Phytomedicine, 1998, 5, 147–163 CrossRef CAS.
- J. A. Pérez, J. M. Hernández, J. M. Trujillo and H. López, Stud. Nat. Prod. Chem., 2005, 32, 303–363 Search PubMed.
- Y. D. Mei, N. Zhang, W. Y. Zhang, J. S. Tang, H. Zhou, Y. Yu and X. S. Yao, Chin. J. Nat. Med., 2019, 17(4), 27–32 Search PubMed.
- Y. D. Mei, D. B. Pan, Y. N. Jiang, W. Y. Zhang, X. J. Yao, Y. Dai, Y. Yu and X. S. Yao, Fitoterapia, 2019, 134, 297–304 CrossRef CAS.
- N. Ikeda, M. Ishihara, T. Tsuneya, M. Kawakita, M. Yoshihara, Y. Suzuki, R. Komaki and M. Inui, Flavour Fragrance J., 1994, 9, 325–331 CrossRef CAS.
- Y. Yu, W. X. Song, C. G. Zhu, S. Lin, F. Zhao, X. L. Wu, Z. G. Yue, B. Liu, S. J. Wang, S. P. Yuan, Q. Hou and J. G. Shi, J. Nat. Prod., 2011, 74, 2151–2160 CrossRef CAS.
- R. E. Krull and F. R Stermitz, Phytochemistry, 1998, 49, 2413–2415 CrossRef CAS.
- K. Miettinen, L. Dong, N. Navrot, T. Schneider, V. Burlat, J. Pollier, L. Woittiez, S. V. D. Krol, R. Lugan, T. Ilc, R. Verpoorte, K. Oksman-Caldentey, E. Martinoia, H. Bouwmeester, A. Goossens, J. Memelink and D. Werck-Reichhar, Nat. Commun., 2014, 5, 4175 CrossRef CAS.
- W. X. Song, S. Li, S. Wang, Y. Wu, J. C. Zi, M. L. Gan, Y. L. Zhang, M. T. Liu, S. Lin, Y. C. Yang and J. G. Shi, J. Nat. Prod., 2008, 71, 922–925 CrossRef CAS.
- Y. Yu, C. G. Zhu, S. J. Wang, W. X. Song, Y. C. Yang and J. G. Shi, J. Nat. Prod., 2013, 76, 2226–2233 CrossRef CAS.
- R. Iafe and K. N. Houk, Org. Lett., 2006, 8, 3469–3472 CrossRef CAS.
- B. Moreau, M. Ginisty and D. A. Alberico, J. Org. Chem., 2007, 72, 1235–1240 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra of all new compounds. CCDC 1877498. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra04029c |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |