DOI:
10.1039/C9RA03911B
(Paper)
RSC Adv., 2019,
9, 20652-20662
Synthesis and biological evaluation of an epidermal growth factor receptor-targeted peptide-conjugated phthalocyanine-based photosensitiser†
Received
24th May 2019
, Accepted 21st June 2019
First published on 2nd July 2019
Abstract
A peptide-conjugated zinc(II) phthalocyanine containing the epidermal growth factor receptor-targeted heptapeptide QRHKPRE has been prepared. The conjugate labelled as ZnPc-QRH* can selectively bind to the cell membrane of HT29 human colorectal adenocarcinoma cells in 10 min followed by internalisation upon prolonged incubation via receptor-mediated endocytosis, leading to localisation in lysosomes eventually. By manipulating the incubation time, the subcellular localisation of the conjugate can be varied and the cell-death pathways induced upon irradiation can also be altered. It has been found that photosensitisation initiated at the cell membrane and in the lysosomes would trigger cell death mainly through necrosis and apoptosis respectively. Intravenous administration of the conjugate into HT29 tumour-bearing nude mice resulted in higher accumulation in the tumour than in most major organs. The selective binding of this conjugate to tumour has also been demonstrated by comparing the results with those of the analogue with a scrambled peptide sequence (EPRQRHK). The overall results indicate that ZnPc-QRH* is a promising EGFR-targeted photosensitiser for photodynamic therapy.
Introduction
Multifunctional theranostic agents for simultaneous imaging and eradication of tumours have received considerable interest.1 Various non-invasive imaging techniques have been used in combination with a range of therapeutic modalities for image-guided therapy. In particular, the combination of fluorescence imaging and photodynamic therapy (PDT) has gained growing attention in recent years.2 PDT is a clinically established treatment modality for a range of superficial and localised cancers, as well as certain non-cancerous conditions, such as wet age-related macular degeneration.3 Its application in photo-eradication of microbials has also been actively explored.4 The treatment involves the action of a photosensitiser, light of an appropriate wavelength and oxygen to induce cytotoxicity via the generation of reactive oxygen species (ROS), such as singlet oxygen. These ROS destroy tumours by multiple mechanisms, including killing malignant cells by apoptosis and/or necrosis, shutting down the tumour vasculature and stimulating the host immune system.5 Compared with the traditional therapeutic methods, such as chemotherapy, radiotherapy and surgery, PDT has several potential advantages. Generally, it is non-invasive and has tolerance of repeated doses, few side effects and high specificity that can be achieved through precise delivery of light. Apart from ROS generation, most of the photosensitisers can also emit fluorescence upon excitation. A balance of these two competitive photochemical and photophysical pathways is important in order to achieve these dual functions. Among the various classes of photosensitisers, such as porphyrins, chlorins, boron dipyrromethenes, indocyanines, aggregation-induced emission dyes and phthalocyanines,6 the last group of functional dyes are particular attractive for fluorescence-guided PDT due to their high ROS generation efficiency and relatively strong fluorescence emission, as well as strong absorption in the red visible region that allows deep tissue penetration, high photostability and ease of chemical and physical modifications.7 To date, a vast number of phthalocyanine-based photosensitisers have been reported, and a few of them have been in clinical use (e.g. Photosens®) or in clinical trial (e.g. Pc 4, CGP55847 and Photocyanine) for PDT of a range of cancers.
One of the drawbacks of phthalocyanines for this application is their poor water solubility and strong aggregation tendency that largely quench their fluorescence emission and ROS generation.7b To circumvent this problem, various nanoplatforms, such as liposomes, polymeric micelles, gold nanoparticles and mesoporous silica nanoparticles have been used to encapsulate or adsorb phthalocyanines for delivery and maintaining their photoactivities.8 Alternatively, various bulky and hydrophilic moieties have been introduced, among which tumour-homing peptides are particularly attractive.9 These biomolecules not only can enhance the water solubility of the hydrophobic macrocycles, but also can deliver the photosensitisers to tumour site selectively via receptor-mediated endocytosis. We have been interested in exploring peptide-conjugated phthalocyanine-based photosensitisers for targeting colonic adenocarcinomas.10 Colorectal cancer (CRC) is one of the most common causes of cancer-related mortality worldwide.11 It typically develops over several years in a linear fashion from adenomatous polyps to carcinomas. Routine colon screening, detection and removal of polyp adenomas at an early stage reduce the incidence of CRC. However, as the disease lacks outward signs or symptoms, most of the cases are detected at a terminal stage. Therefore, early detection and treatment of premalignant lesions are extremely important. Among the various CRC-targeting peptides that have been identified in recent years,12 the heptapeptide QRHKPRE can target the epidermal growth factor receptor (EGFR), which is overexpressed in a variety of human cancer cells, including colonic adenocarcinomas, and can bind specifically to the target cells within a few minutes that can facilitate the bioimaging.12e In fact, EGFR is a validated biomarker for CRC.13 We report herein a novel zinc(II) phthalocyanine conjugated with this peptide sequence, including the synthesis, characterisation, in vitro response against a range of EGFR-positive and negative cell lines, as well as in vivo tumour-localisation property.
Results and discussion
Synthesis and characterisation
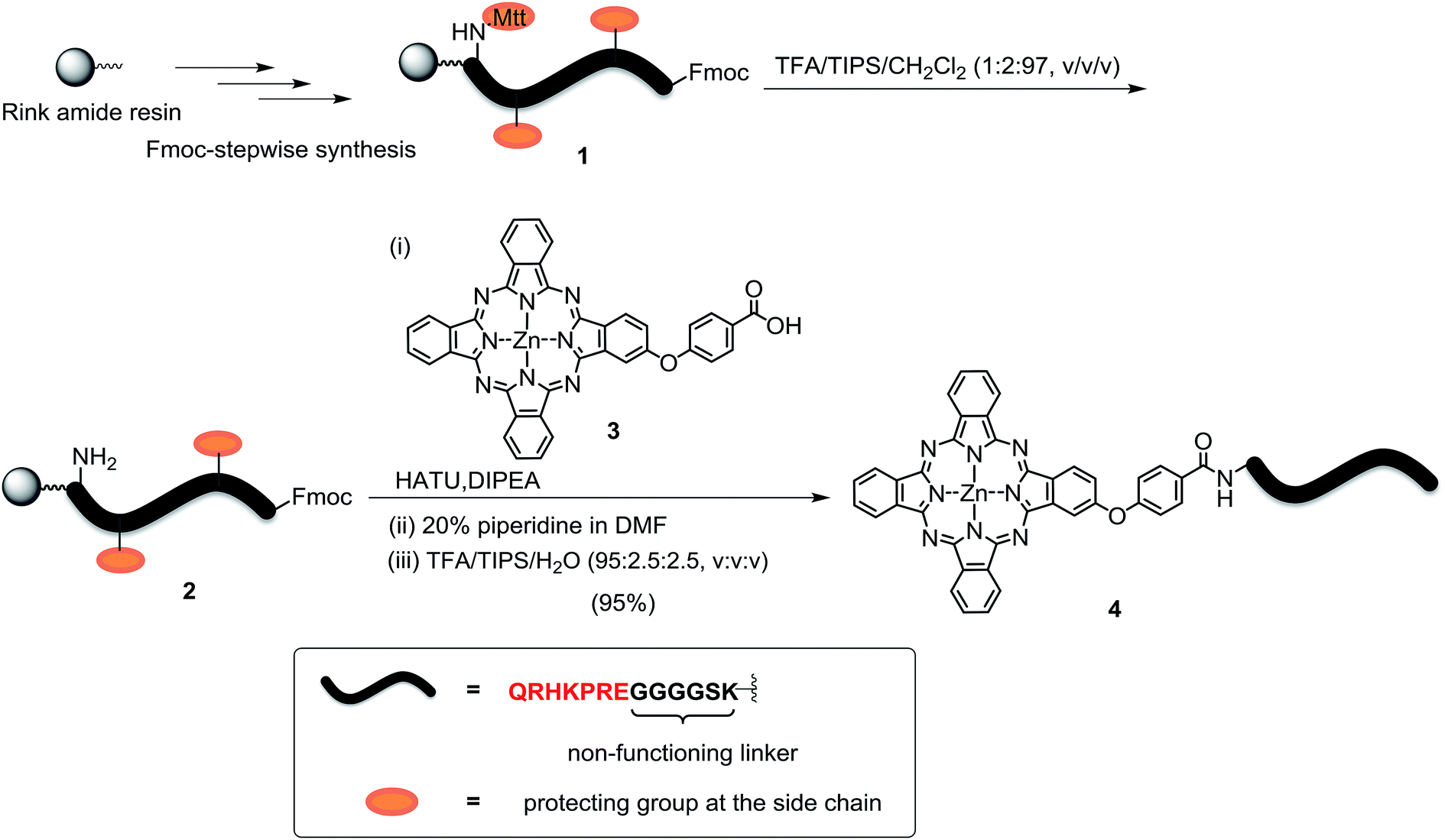
The peptide–phthalocyanine conjugate (compound 4) was prepared according to Scheme 1. The linear peptide QRHKPRE was first linked to the non-functional glycine-serine-lysine GGGGSK spacer by using standard 9-fluorenylmethyloxycarbonyl (Fmoc)-mediated solid-phase synthesis. The lysine at the C-terminal was incorporated as Fmoc-Lys(Mtt)-OH (Mtt = 4-methyltrityl), and the Mtt protecting group in peptide 1 was removed with 1% trifluoroacetic acid (TFA) in CH2Cl2 in the presence of 2% triisopropylsilane (TIPS). The resulting peptide 2 was then coupled with the previously reported carboxy phthalocyanine 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14 on resin overnight in the presence of 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) and N,N-diisopropylethylamine (DIPEA). It was followed by removal of the Fmoc protecting group using 20% piperidine in N,N-dimethylformamide (DMF) and cleavage from the resin with a mixture of TFA/TIPS/H2O (95:2.5:2.5, v/v/v). The target conjugate 4 substituted with the sequence QRHKPREGGGGSK, hereafter labelled as QRH*, was purified with high-performance liquid chromatography (HPLC) and characterised with matrix-assisted laser desorption/ionisation time-of-flight (MALDI-TOF-MS) mass spectrometry (Fig. S1, ESI†). To confirm the targeting effect of the QRHKPRE sequence, an analogue with a scrambled sequence (EPRQRHKGGGGSK, hereafter labelled as EPR*) (conjugate 5, structure not shown) was also prepared for comparison and characterised similarly (Fig. S2, ESI†). With a number of polar and charged amino acid residues, both conjugates possessed good solubility in polar organic solvents and aqueous media.
14 on resin overnight in the presence of 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) and N,N-diisopropylethylamine (DIPEA). It was followed by removal of the Fmoc protecting group using 20% piperidine in N,N-dimethylformamide (DMF) and cleavage from the resin with a mixture of TFA/TIPS/H2O (95:2.5:2.5, v/v/v). The target conjugate 4 substituted with the sequence QRHKPREGGGGSK, hereafter labelled as QRH*, was purified with high-performance liquid chromatography (HPLC) and characterised with matrix-assisted laser desorption/ionisation time-of-flight (MALDI-TOF-MS) mass spectrometry (Fig. S1, ESI†). To confirm the targeting effect of the QRHKPRE sequence, an analogue with a scrambled sequence (EPRQRHKGGGGSK, hereafter labelled as EPR*) (conjugate 5, structure not shown) was also prepared for comparison and characterised similarly (Fig. S2, ESI†). With a number of polar and charged amino acid residues, both conjugates possessed good solubility in polar organic solvents and aqueous media.
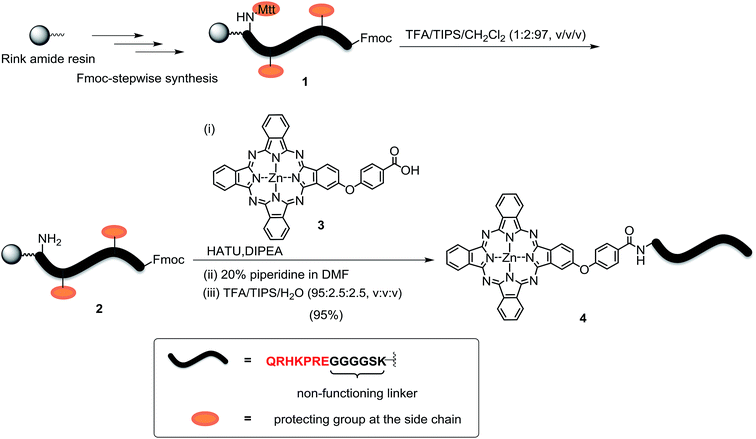 |
| | Scheme 1 Synthesis of conjugate ZnPc-QRH* (4). | |
Spectroscopic and photophysical properties
The electronic absorption and fluorescence spectra of ZnPc-QRH* (4) were recorded in DMF and phosphate buffered saline (PBS) with 0.01% Tween 80 (v/v). As shown in Fig. S3 (ESI†), the conjugate gave an intense and sharp Q band at 672 nm (in DMF) or 680 nm (in PBS), showing that it was soluble and remained non-aggregated in both media as a result of the hydrophilic peptide chain. Upon excitation at 610 nm, the conjugate showed a fluorescence emission at ca. 680 nm. The fluorescence quantum yield (ΦF) in DMF was determined to be 0.24 relative to the unsubstituted zinc(II) phthalocyanine (ZnPc) (ΦF = 0.28).15 To determine the singlet oxygen generation efficiency, 1,3-diphenylisobenzofuran (DPBF) was used as the singlet oxygen scavenger. Conjugate 4 could induce photodegradation of DPBF in both media. Based on the rate of decrease in its absorbance at 417 nm, the singlet oxygen quantum yield (ΦΔ) was determined to be 0.62 in DMF, which was slightly higher than that of ZnPc (ΦΔ = 0.56).16 Both the values of ΦF and ΦΔ of the mono-β-substituted 4 were also higher than those of our previously reported di-α-substituted analogue substituted with another EGFR-targeted peptide GE11 (GYHWYGYTPQNVI) (ΦF = 0.10, ΦΔ = 0.48).17 As expected, the spectral properties and singlet oxygen generation efficiency of the scrambled analogue ZnPc-EPR* (5) were very similar to those of 4. Table 1 summarises the data in DMF for the two conjugates, and Fig. S4 (ESI†) shows that the two conjugates have virtually the same efficiency in generating singlet oxygen for degradation of DPBF.
Table 1 Spectroscopic and photophysical data of conjugates 4 and 5 in DMF
| |
λabs (nm) (logε) |
λema |
ΦFb |
ΦΔc |
| Excited at 610 nm. Relative to ZnPc in DMF (ΦF = 0.28). Relative to ZnPc in DMF (ΦΔ = 0.56). |
| ZnPc-QRH* (4) |
340 (4.49), 607 (4.27), 672 (5.07) |
680 |
0.24 |
0.62 |
| ZnPc-EPR* (5) |
340 (4.47), 607 (4.28), 672 (5.08) |
680 |
0.25 |
0.63 |
Cellular uptake and subcellular localisation
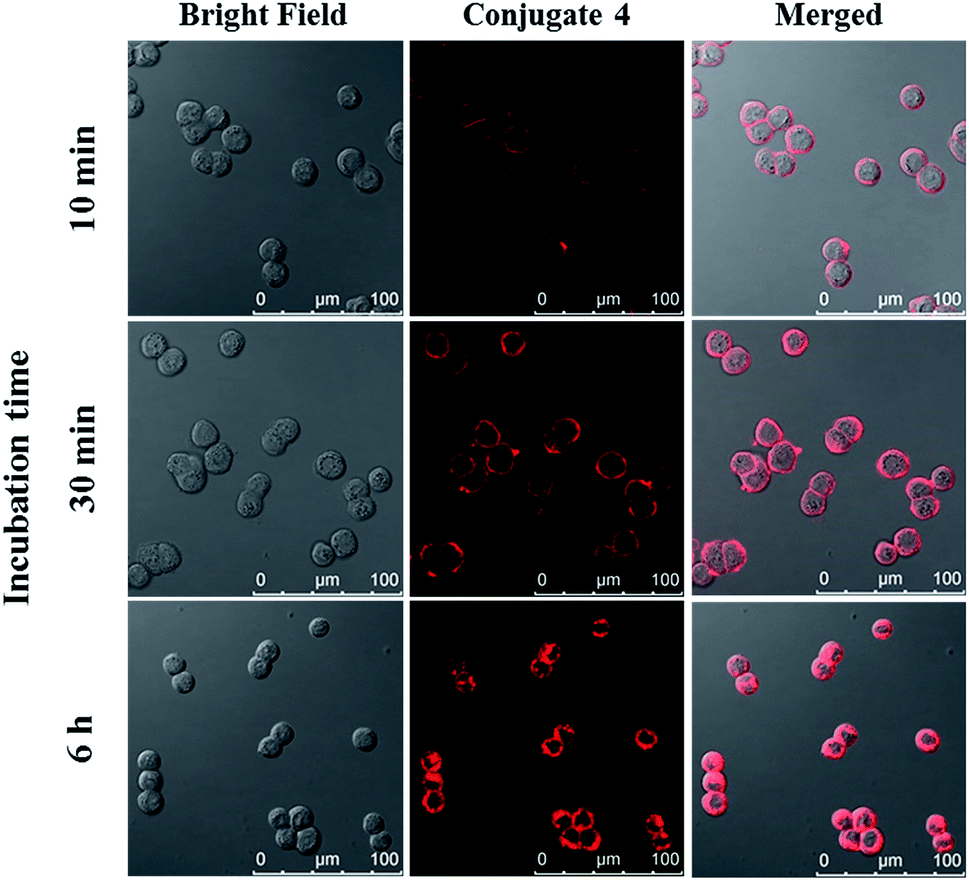
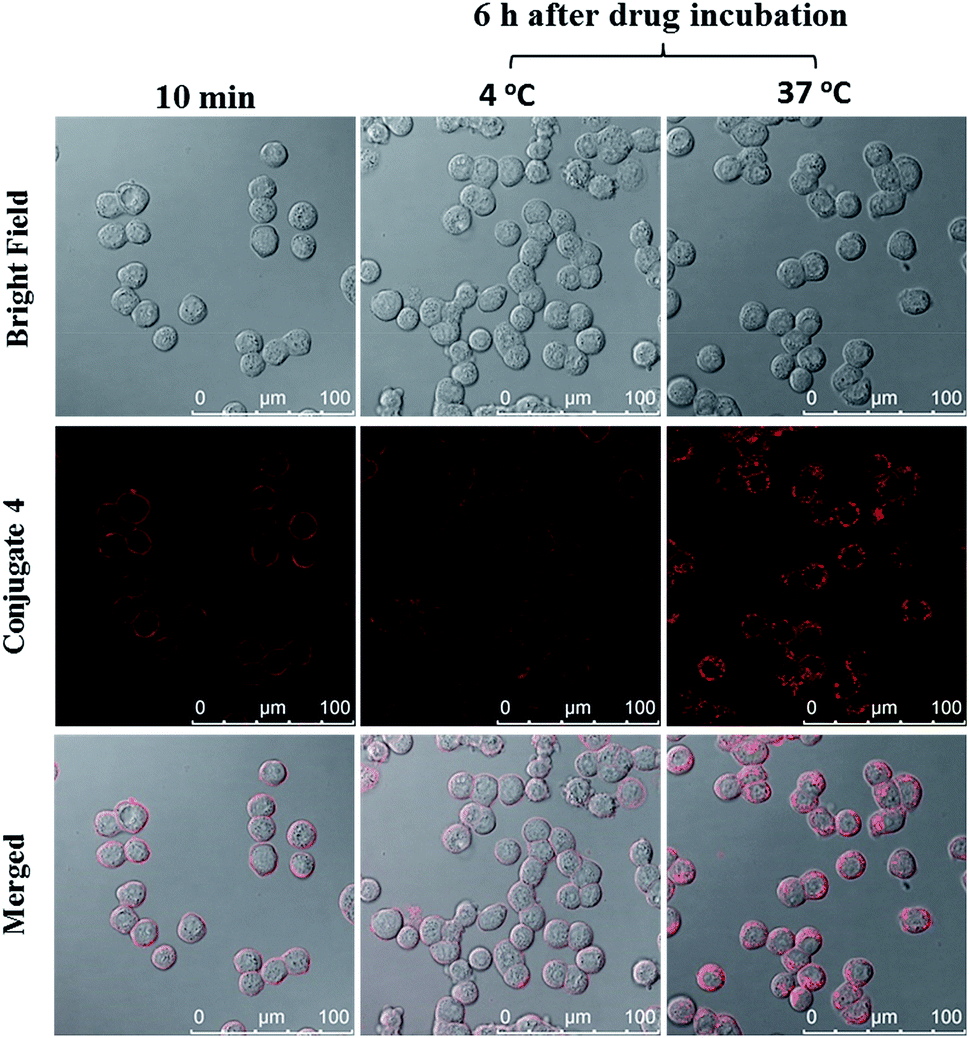
The cellular uptake of ZnPc-QRH* (4) by HT29 human colorectal adenocarcinoma cells was investigated using confocal laser scanning microscopy. Fig. 1 shows the time-dependent uptake process of conjugate 4. It can be seen that fluorescence rings appear in 10 min showing that 4 binds to the cell membrane in a short period of time. It was then gradually internalised into the cells after 30 min and the internalisation became more significant after incubation for 6 h. To confirm whether conjugate 4 was internalised into the cells by endocytosis, the cellular uptake of 4 was monitored at two different temperatures. HT29 cells were first incubated with 4 (2 μM) for 10 min at 37 °C. After washing with PBS for three times, the cells were further incubated in a drug-free medium for a further 6 h at 4 °C or 37 °C. As shown in Fig. 2, after incubation with 4 for 10 min, the conjugate remained on the cell membrane as indicated by its fluorescence. However, after further incubation for 6 h at 37 °C, the drug on the cell membrane was internalised and caused a 3-fold increase in the intracellular fluorescence (Fig. S5, ESI†). The increase in intracellular fluorescence intensity is believed to be caused by the partial quenching of the fluorescence of 4 on the cell membrane which was restored after internalisation. Hence, in the following experiments, the cells were all further incubated in a drug-free medium for a further 6 h after a 10 min drug incubation to avoid unintended fluorescence quenching. By contrast, for the case of post-incubation at 4 °C, the fluorescence images were not significantly changed, showing that internalisation was not significant (Fig. 2). The results suggest that the internalisation of 4 into HT29 cells is mainly through endocytosis.
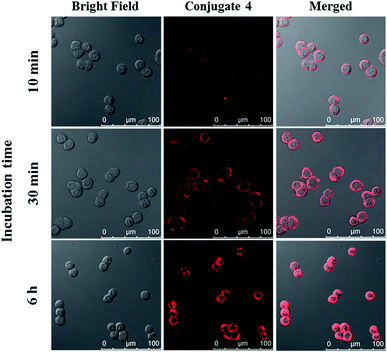 |
| | Fig. 1 Bright field, fluorescence and merged images of HT29 cells after incubation with conjugate 4 (4 μM) for 10 min, 30 min and 6 h respectively. | |
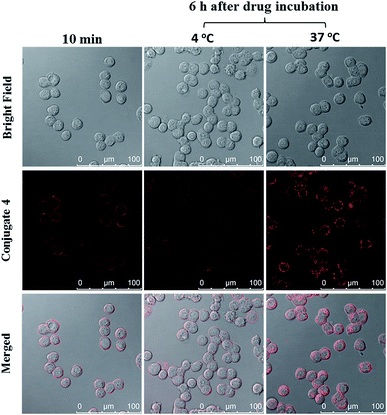 |
| | Fig. 2 Bright-field, fluorescence and merged images of HT29 cells after incubation with conjugate 4 (2 μM) for 10 min, and then the cells were further incubated in a drug-free medium for a further 6 h at 4 °C or 37 °C. | |
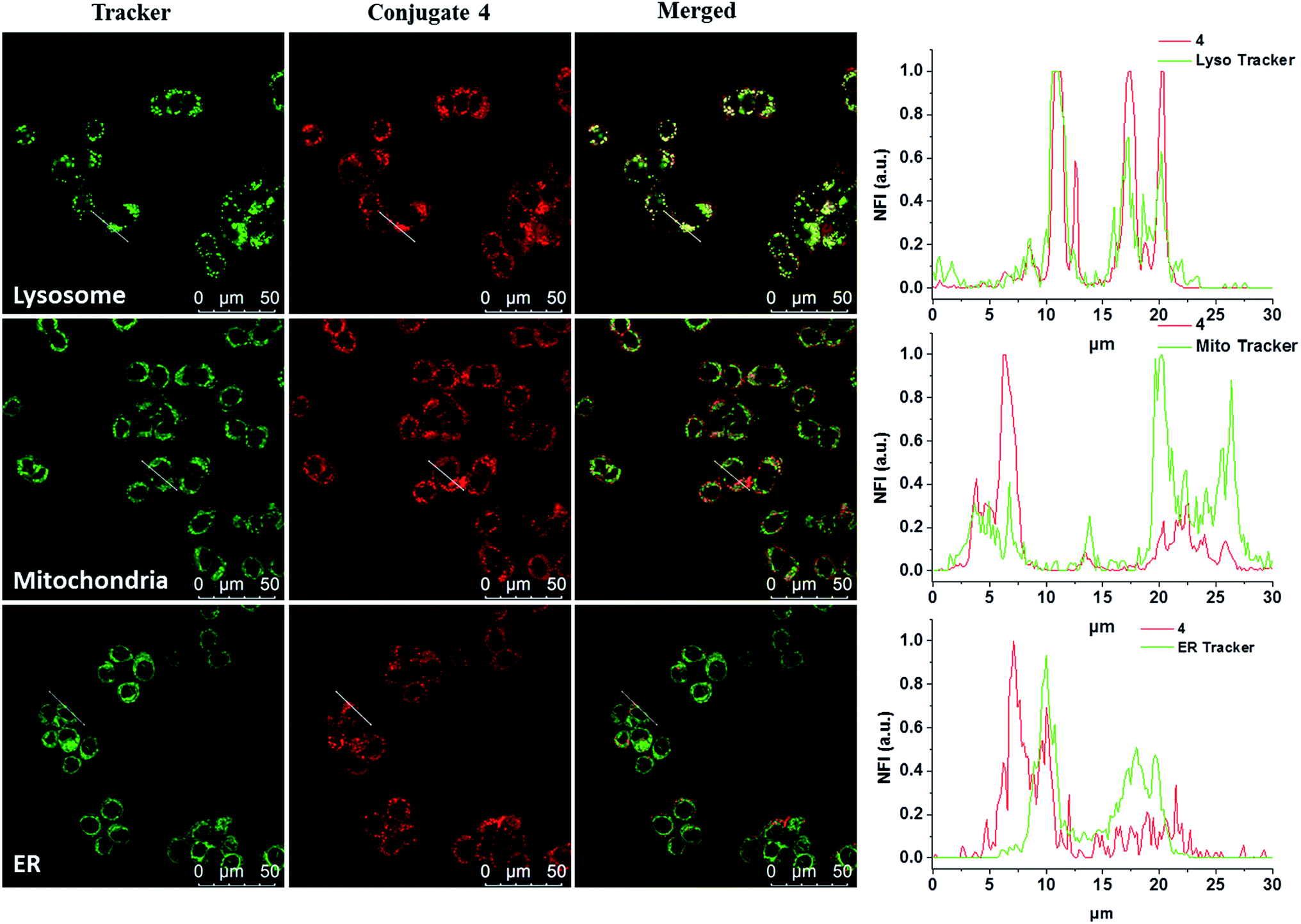
The subcellular localisation of 4 was further examined under the condition of incubation with 4 (2 μM) for 10 min and then in a drug-free medium for a further 6 h at 37 °C. The cells were then stained with LysoTracker Green DND-26, MitoTracker Green FM or ER-Tracker green before being examined with a confocal laser scanning microscope. As shown in Fig. 3, the line profile of the fluorescence intensity of 4 overlaps well with that of LysoTracker Green, but not for the other two trackers, suggesting that the dye is localised mainly in lysosomes, which is the last compartment of the endocytic pathway after internalisation. This result is also consistent with the proposed receptor-mediated endocytosis mechanism.
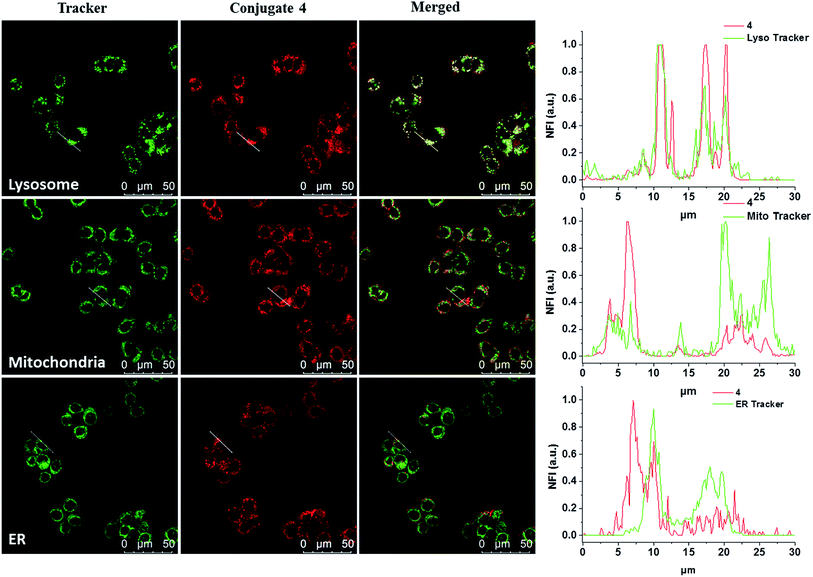 |
| | Fig. 3 Visualisation of the intracellular fluorescence of conjugate 4 and various subcellular trackers, as well as their overlapped images. The cells were first incubated with 4 for 10 min, and then in a drug-free medium for 6 h at 37 °C. They were then further incubated with LysoTracker Green DND-26 (2 μM), MitoTracker Green FM (0.2 μM) or ER-Tracker green (0.2 μM) for 15, 15 and 30 min respectively at 37 °C. The right column shows the fluorescence intensity profiles of 4 and the trackers traced along the white lines in the corresponding images on the left. | |
In vitro specific binding towards HT29 cells
The specific binding of conjugate 4 towards HT29 cells was first validated by competitive inhibition with free QRH* peptide. The cells were pre-incubated with free QRH* at various concentrations (up to 1 mM) for 30 min at 37 °C. They were then washed with PBS and then incubated further with conjugate 4 (2 μM) for 10 min, followed by incubation in a drug-free medium for 6 h. It was found that upon pre-incubation with the free peptide, the intracellular fluorescence intensity decreased as the concentration of the peptide increased (Fig. S6, ESI†). The results indicate that it is the peptide rather than the phthalocyanine unit that binds to the surface of HT29 cells.
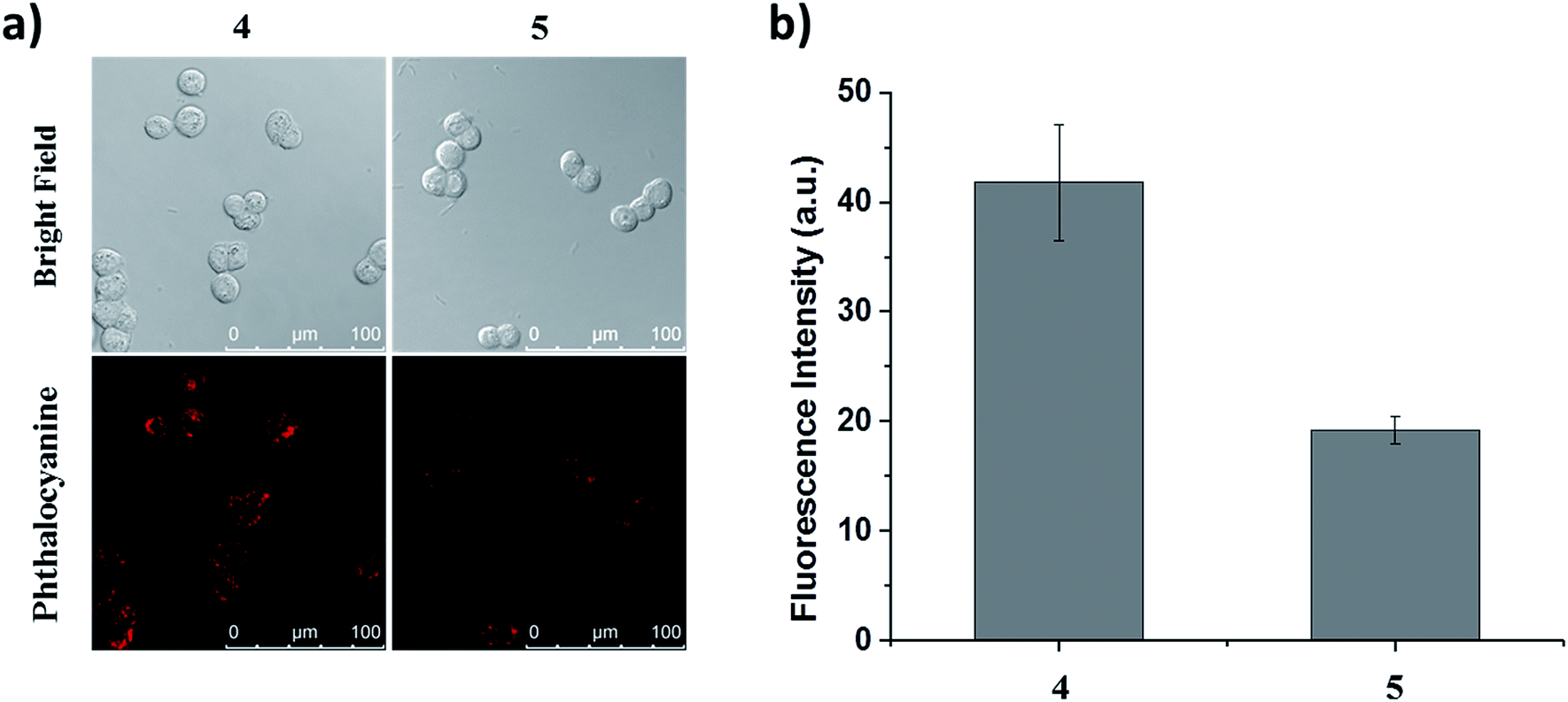
To determine whether the binding of this peptide to HT29 cells was sequence specific, we selected a scrambled sequence (EPR*) for comparison. HT29 cells were incubated with ZnPc-QRH* (4) or ZnPc-EPR* (5) for 10 min, followed by incubation in a drug-free medium for 6 h. As shown in Fig. 4, the intracellular fluorescence intensity for 4 was about 2-fold of that for 5. Although the difference is not very substantial, it is worth noting that the scrambled peptide also contains the same amino acids, which may still form similar secondary structure that can bind to the receptor to some extent. It has also been reported earlier that for the QRH*-conjugated Cy5.5, its intracellular fluorescence intensity is also only about 2-fold of that for the scrambled control against HT29 and SW480 human colon adenocarcinoma cells, which have high and medium EGFR expression respectively.12e The difference in uptake as shown in Fig. 4 shows a high binding affinity of QRH* towards HT29 cells.
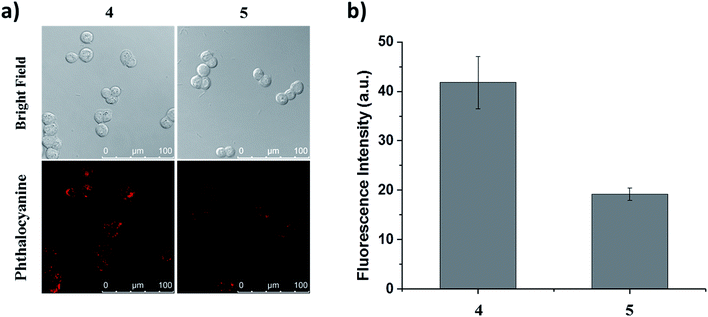 |
| | Fig. 4 (a) Bright-field and fluorescence images of HT29 cells after incubation with conjugate 4 or 5 (2 μM) for 10 min, followed by incubation in a drug-free medium for 6 h. (b) The corresponding quantified fluorescence intensities. Data are expressed as the mean ± standard error of the mean (SEM) of three independent experiments. | |
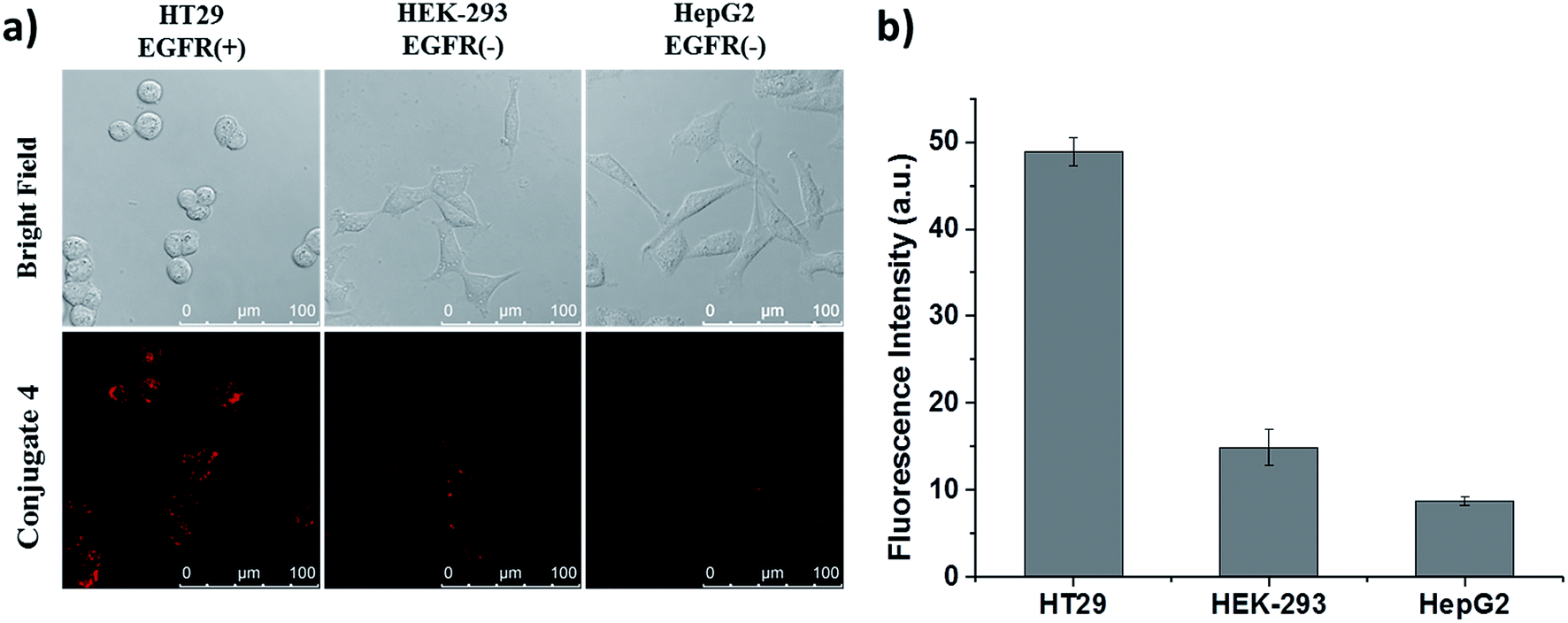
Apart from these studies, we also investigated whether QRH* shows different affinity towards cancer cells with different expression levels of EGFR. Hence, we compared the cellular uptake of conjugate 4 towards HT29 cells, HEK-293 human embryonic kidney cells and HepG2 human hepatocellular carcinoma cells. The latter two were reported to express a low level of EGFR.18 These three kinds of cells were first incubated with 4 for 10 min, followed by incubation in a drug-free medium for 6 h. As shown in Fig. 5, the intracellular fluorescence intensity of 4 in HT29 cells was significantly higher than those in HEK-293 and HepG2 cells by about 4- and 6-fold respectively. These results provide further evidence for the EGFR-targeting property of conjugate 4.
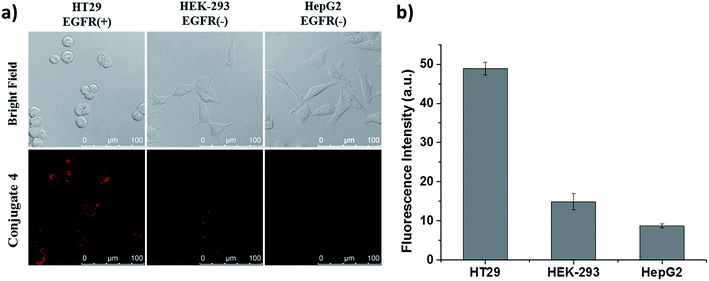 |
| | Fig. 5 (a) Bright-field and fluorescence images of HT29, HEK-293 and HepG2 cells after incubation with conjugate 4 (2 μM) for 10 min, followed by incubation in a drug-free medium for 6 h. (b) The corresponding quantified fluorescence intensities. Data are expressed as the mean ± SEM of three independent experiments. | |
In vitro photodynamic activities
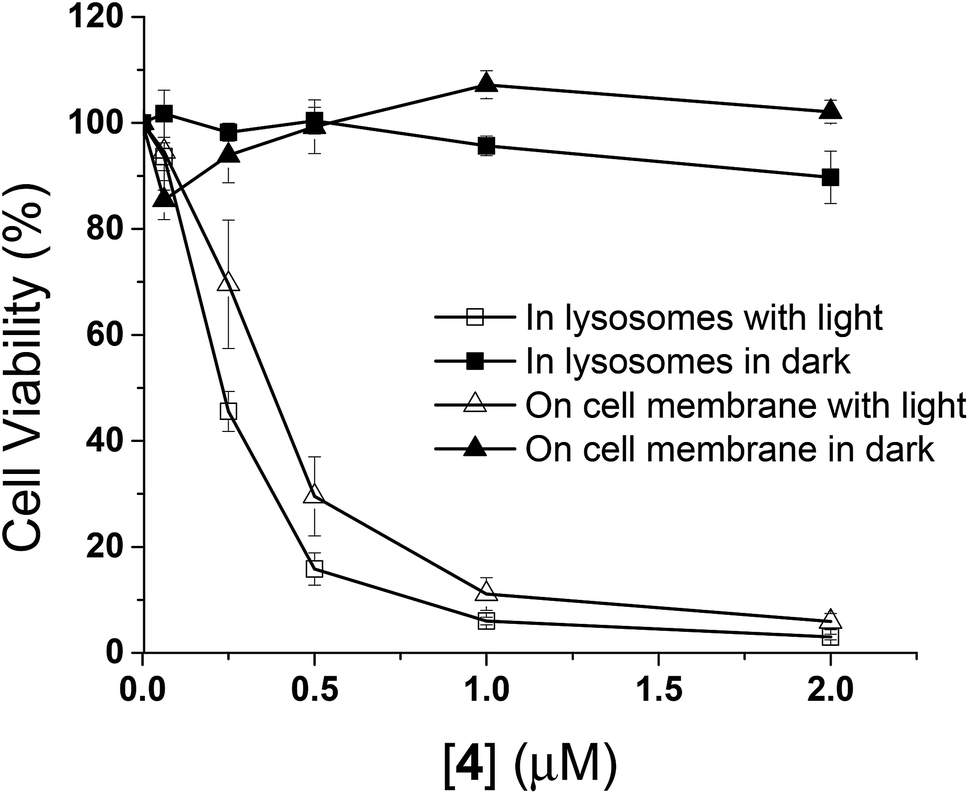
The in vitro photodynamic activities of conjugate 4 were then studied against HT29 cells. As mentioned above, when HT29 cells were incubated with 4 (2 μM) for 10 min, the drug was mainly confined on the cell surface, and internalisation occurred leading to localisation mainly in lysosomes after incubation in a drug-free medium for 6 h. In order to examine if the photocytotoxicity is different when the PDT action is initiated at the cell membrane and in the lysosomes, the cell viabilities were measured by MTT assay [MTT = 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] under these two conditions, both in the absence and presence of light irradiation. As shown in Fig. 6, while conjugate 4 did not show significant dark toxicity towards HT29 cells, it exhibited high cytotoxicity upon illumination for both conditions. The IC50 values defined as the dye concentrations required to kill 50% of the cells (0.37 μM for membrane-based PDT and 0.23 μM for lysosome-based PDT) were found to be similar despite the fluorescence intensity on cell membrane after incubation for 10 min was weaker than that inside the cells after further incubation for 6 h (Fig. S5, ESI†). It has been reported that PDT initiated at the cell membrane usually causes cell death through necrosis, while apoptosis is a dominant cell-death pathway when the photosensitisers are fully internalised.19 The efficiency in cell killing could be different for the two cell-death pathways. In addition, the quenching efficiency for fluorescence emission is usually higher than that for singlet oxygen.20 Hence, the photosensitisation at the cell membrane could be as effective as the process inside the cells even though the quenching of fluorescence seems to be higher in the former compartment. These two factors might be able to account for the discrepancy.
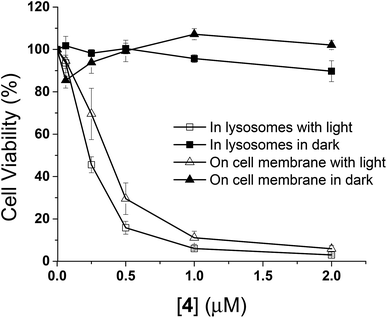 |
| | Fig. 6 Comparison of the cytotoxic effects of conjugate 4 on the cell membrane (triangles) and in lysosomes (squares) of HT29 cells in the absence (closed symbols) and presence (open symbols) of light (λ > 610 nm, 18 mW cm−2, 22 J cm−2). Data are expressed as the mean ± SEM of three independent experiments, each performed in quadruplicate. | |
It is worth mentioning that the photocytotoxicity of 4 is much higher than that of the previously reported peptide–phthalocyanine conjugates for EGFR targeting.21 Among the four conjugates reported, only the two with the cationic charged peptide sequence LARLLT can target EGFR and be internalised into cancer cells, while the other two GE11-conjugated analogues show poor water solubility, which may be due to the presence of three tert-butyl substituents on the phthalocyanine ring. All the four conjugates are non-toxic (IC50 > 100 μM) against HT29 cells, both in the dark and upon irradiation. The photocytotoxicity of 4 also seems to be significantly higher than that of the GE11 conjugate reported by us earlier,17 of which the IC50 value against A431 human epidermoid carcinoma cells was found to be 14 μM. It is believed that the ionic nature of some of the amino acid residues of QRH* plays an important role. It can greatly enhance the water solubility and reduce the aggregation of the phthalocyanine core so that the targeting effect would not be affected. This could be the reason why conjugate 4 could rapidly bind to the EGFR on cell surface within 10 min and induce strong PDT effect.
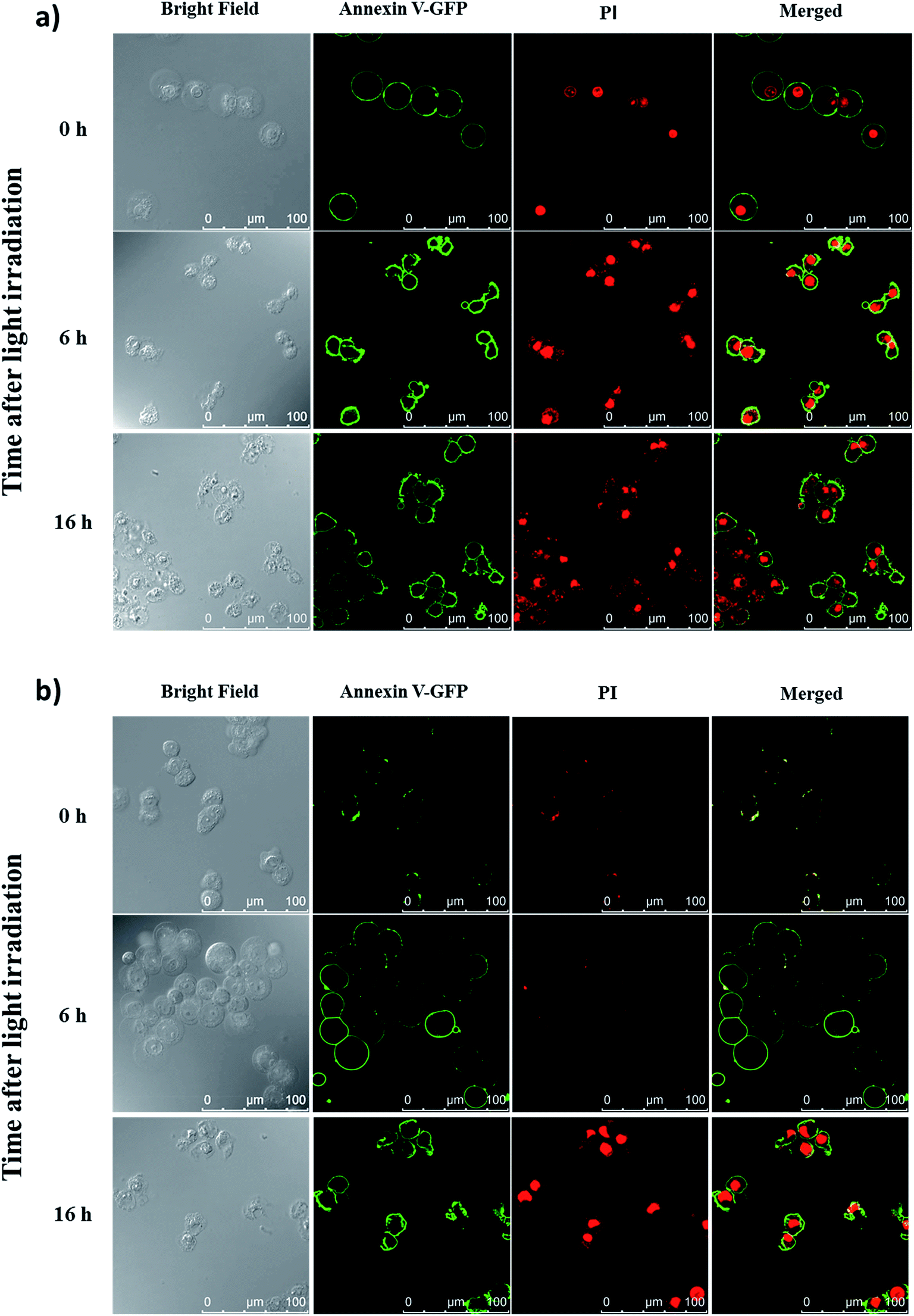
To reveal the cell-death pathways under the two incubation conditions, annexin V-green fluorescent protein (GFP) and propidium iodide (PI) costaining was performed and monitored using confocal laser scanning microscopy. HT29 cells were first incubated with conjugate 4 for 10 min under which the compound was mainly confined on the cell surface. After washing and refilling with a drug-free medium, the cells were irradiated with red light for 20 min (λ > 610 nm, 18 mW cm−2) immediately or after further incubation for 6 h under which the dye had been internalised and was mainly localised in lysosomes. The cells were then costained with annexin V-GFP and PI after 0, 6 and 16 h, and then visualised using confocal laser scanning microscopy. As shown in Fig. 7a, for the cells being incubated with 4 for 10 min, cell necrosis was observed with most of the cells being stained with annexin V-GFP and PI right after light irradiation. For the cells with 6 h post-incubation before light irradiation, they could only be stained with annexin V-GFP at 6 h after light irradiation. After prolonging the incubation time to 16 h after light irradiation, the cells were able to be stained with both annexin V-GFP and PI, indicating a process of early to late apoptosis (Fig. 7b).22 These results demonstrated that when the PDT effect of 4 occurred on cell membrane, it induced cell necrosis, while the cells were killed mainly through apoptosis after the drug was internalised into the cells, which further supported the above hypothesis and were consistent with those observed earlier.19
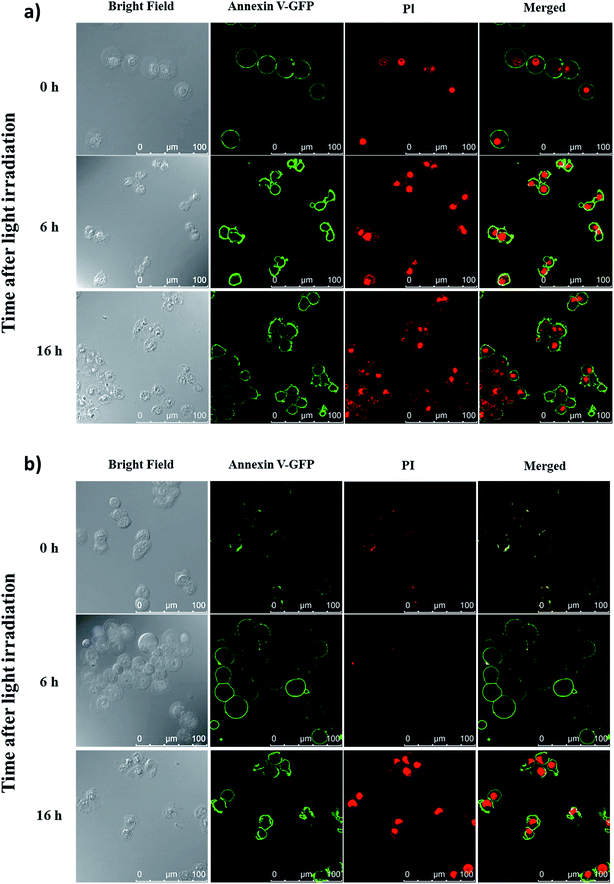 |
| | Fig. 7 Confocal microscopic images of HT29 cells after incubation with conjugate 4 for 10 min and light irradiation (λ > 610 nm, 18 mW cm−2, 20 min) (a) right after drug incubation and (b) 6 h after drug incubation. The cells were then costained with annexin V-GFP and PI after 0, 6 and 16 h. The right-most column shows the merged images of annexin V-GFP (green) and PI (red). | |
In vivo biodistribution
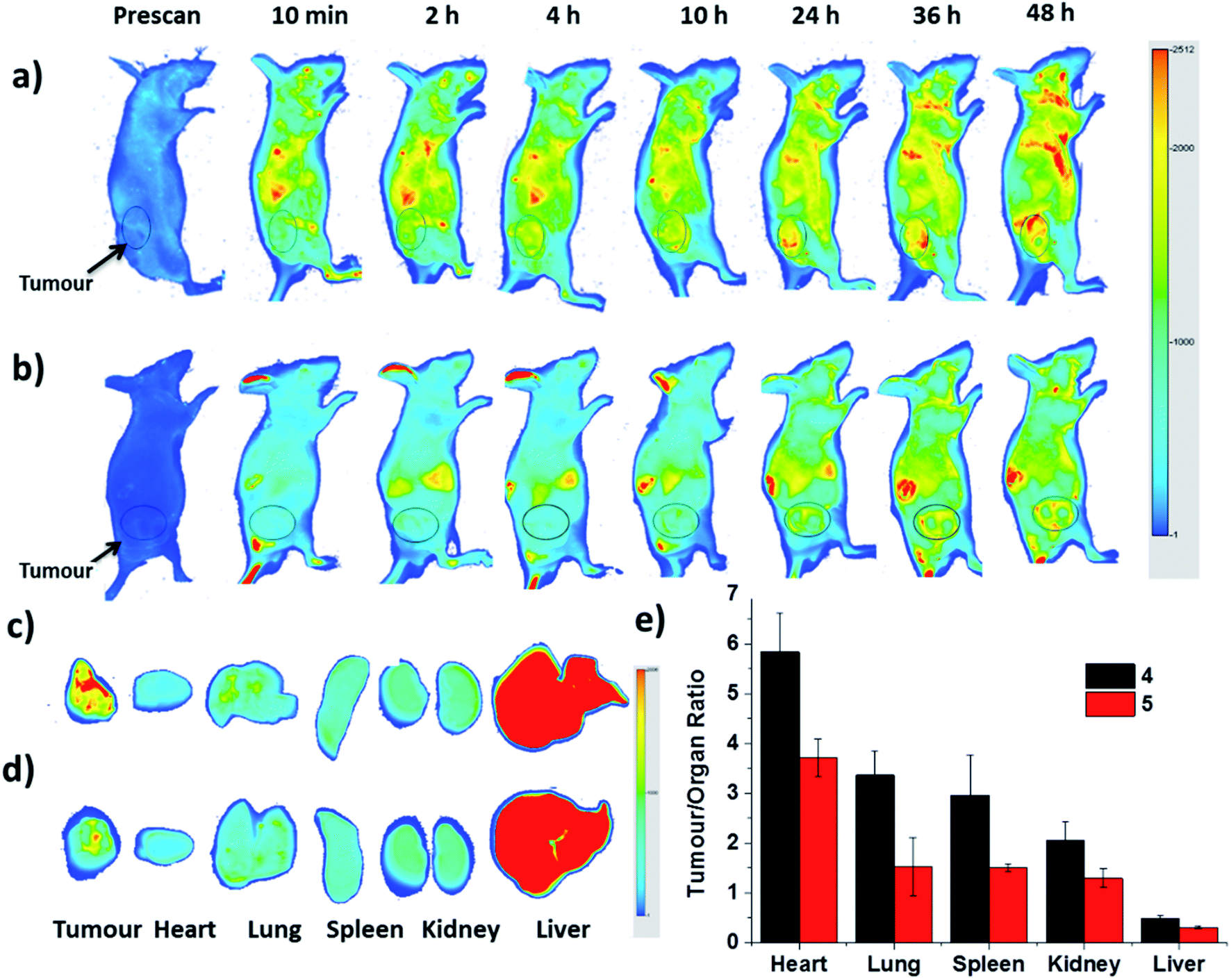
Finally, the biodistribution of ZnPc-QRH* (4) and ZnPc-EPR* (5) in HT29 tumour-bearing nude mice was monitored using an infrared imaging system over a period of 48 h. It can be seen in Fig. 8a that fluorescence was observed as early as 2 h after intravenous injection of 4, and the intensity in the tumour area was increased along with time. In contrast, for the mice being treated with 5, the fluorescence intensity around the tumour remained weak throughout the period (Fig. 8b). To quantify the fluorescence intensities in the tumour and some major organs, the mice were sacrificed after 48 h. The tumour and organs, including heart, lung, spleen, kidney and liver were then harvested, and their relative fluorescence intensities (per unit area) were measured and are reported in tumour/organ ratio (Fig. 8c–e). As shown in Fig. 8e, the accumulation of 4 in tumour was significantly higher than that in the organs examined (with a tumour/organ ratio ranging from 2 to 6) except the liver (with a tumour/organ ratio < 1). For conjugate 5 with a scrambled sequence, the tumour selectivity was consistently lower for all the organs, indicating that the QRH*-conjugated phthalocyanine 4 does have a higher affinity to HT29 tumour compared with the EPR*-conjugated counterpart 5.
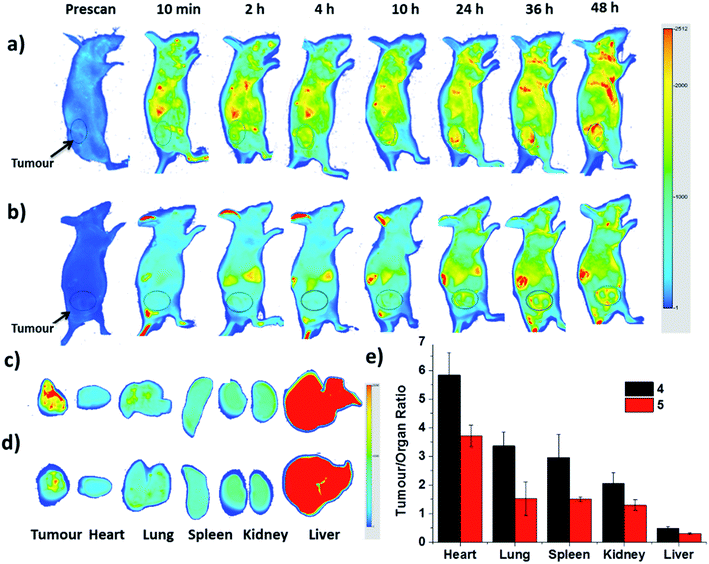 |
| | Fig. 8 In vivo near-infrared fluorescence images of HT29 tumour-bearing nude mice after intravenous injection with (a) 4 and (b) 5 over a period of 48 h. (c) and (d) show the corresponding ex vivo images of the tumour and other major organs harvested at 48 h post-injection. (e) plots the tumour-to-organ ratio for different organs for 4 and 5 at 48 h post-injection based on their relative fluorescence intensities (per unit area). Data are expressed as the mean ± standard deviation of four mice in each group. | |
Conclusions
A zinc(II) phthalocyanine conjugated with a highly hydrophilic QRH* peptide has been synthesised and characterised. The EGFR-targeting effect of this conjugate towards HT29 cells has been validated using a series of in vitro experiments. It has been found that the peptide can promote binding to the cell membrane within 10 min followed by internalisation after prolonged incubation through receptor-mediated endocytosis, leading to localisation in lysosomes eventually. The cellular uptake of this conjugate towards HT29 cells is significantly higher than that for HEK-293 and HepG2 cells used as the EGFR-negative controls. The conjugate also shows high photocytotoxicity towards HT29 cells with an IC50 value of ca. 0.2 μM with the cell-death pathway depending on its localisation where the PDT process is initiated. Finally, the in vivo imaging analysis has also demonstrated the high affinity of this conjugate to HT29 tumour compared with most major organs. The overall results suggest that QRH*, despite having a short peptide sequence, is a very specific ligand for EGFR and the conjugation of this ligand with a simple phthalocyanine-based photosensitiser can make the resulting conjugate highly promising for targeted PDT.
Experimental
General
All reactions were performed under an atmosphere of nitrogen. DMF and CH2Cl2 were dried using an INERT solvent purification system. Chromatographic purification was performed on silica gel (Macherey-Nagel, 230–400 mesh) with the indicated eluents. Size-exclusion chromatography was carried out on Bio-Beads S-X1 beads (200–400 mesh) with the indicated eluents. All other solvents and reagents were of reagent grade and used as received. Phthalocyanine 3 was prepared as described previously.14
MALDI mass spectra were recorded on a Bruker Autoflex Speed MALDI-TOF mass spectrometer. For accurate mass measurements, the lowest m/z value of the isotopic envelope was reported and compared with the theoretical value. Electronic absorption and steady-state fluorescence spectra were taken on a Shimadzu UV-1800 UV-Vis spectrophotometer and a Horiba FluoroMax spectrofluorometer respectively. HPLC experiments were performed on an Apollo-C18 column (5 μm, 4.6 μm × 250 mm) or on an XBridge-C18 column (5 μm, 10 μm × 250 mm) by using a Waters 1525 binary HPLC pump with a Waters 2998 photodiode array detector.
Preparation of 2
This peptide resin was synthesised manually by using a modified Fmoc solid-phase peptide synthesis (SPPS) protocol with the commercially available N-α-Fmoc-protected amino acids. The rink amide resin was used as the solid support, and HATU was used as the carboxyl group activating agent. The swelled resin was treated twice with piperidine (20%) in DMF for 20 min before being coupled with the first amino acid Fmoc-Lys(Mtt)-OH. An excess of the Fmoc-protected amino acids (4 equiv.), DIPEA (8 equiv.) and HATU (4 equiv.) were then used for each coupling. After the final coupling, the peptide resin was washed with DMF and CH2Cl2, and then the Mtt side chain protecting group was selectively removed using TFA (1%) and TIPS (2%) in CH2Cl2 with continuous shaking at room temperature for 60 min. The deprotection was repeated once to ensure successful removal of the Mtt protecting group. To check the purity of the peptide, a portion of the peptide resin was treated with a solution (0.5 mL) containing a mixture of TFA (95%), TIPS (2.5%) and H2O (2.5%) for 2 h to cleave the peptide from the resin and remove the protecting groups. The resin was removed by filtration and the filtrate was precipitated by the addition of diethyl ether. After centrifugation, the supernatant was removed. The yellow solid obtained was redissolved in DMF (0.2 mL) and then precipitated again by the addition of diethyl ether. This purification procedure was repeated twice and the product was dried in vacuo before HPLC analysis.
Preparation of 4
A mixture of 3 (10 mg, 14.0 μmol), DIPEA (20 μL, 112.0 μmol) and HATU (22 mg, 56 μmol) in DMF (4 mL) was added to the peptide resin 2. The mixture was shaken overnight at room temperature. The Fmoc protecting group at the N-terminal was then removed from the resin with piperidine (20%) in DMF, and then the dark-green resin was washed with DMF and CH2Cl2. The resin was then treated with a solution (2 mL) containing TFA (95%), TIPS (2.5%) and H2O (2.5%) for 2 h to remove all the protecting groups on the peptide sequence and cleave the peptide from the resin. The resin was removed by filtration and the filtrate was precipitated by the addition of diethyl ether. After centrifugation, the supernatant was removed. The green solid obtained was redissolved in DMF (0.5 mL), and then precipitated by the addition of diethyl ether. This purification procedure was repeated three times and the product was lyophilised before HPLC purification. The purity of the conjugate 4 was found to be >95% by HPLC analysis (Fig. S1a, ESI†). MS (MALDI-TOF): an isotopic cluster with the lowest m/z value calculated for C95H114N32O20Zn [M + H]+ 2087.825; found: 2087.828 (Fig. S1b, ESI†).
Preparation of 5
This conjugate with a scrambled sequence EPR* was prepared in the same way as 4 described above. The purity of this conjugate was found to be >95% by HPLC analysis (Fig. S2a, ESI†). MS (MALDI-TOF): an isotopic cluster with the lowest m/z value calculated for C95H114N32O20Zn [M + H]+ 2087.825; found: 2087.822 (Fig. S2b, ESI†).
Determination of fluorescence quantum yields (ΦF)
The values of ΦF were determined by the equation ΦF(s) = (Fs/Fref)(Aref/As)(ns2/nref2)Φf(ref),23 where subscript s refers to the sample solutions and ref stands for the reference. F, A and n are the measured fluorescence (area under the emission peak), the absorbance at the excitation position (610 nm) and the refractive index of the solvent respectively. ZnPc in DMF was used as the reference [Φf(ref) = 0.28].15
Determination of singlet oxygen quantum yields (ΦΔ)
The values of ΦΔ were calculated by using ZnPc in DMF as reference (ΦΔ = 0.56)16 and DPBF as the singlet oxygen scavenger. A solution of DPBF (30 μM) and the sample (2 μM) in DMF was irradiated with red light from a 100 W halogen lamp after passing through a water tank for cooling and a colour filter with cut-on wavelength at 610 nm (Newport). The absorption maximum of DPBF at 417 nm was monitored along with time. The ΦΔ values were calculated according to the equation ΦΔ(s) = ΦΔ(ref)(WsIref)/(WrefIs), where ΦΔ(ref) is the ΦΔ of ZnPc in DMF, Ws and Wref are the DPBF photobleaching rates in the presence of the sample and ZnPc respectively, and Is and Iref are the rates of light absorption by the sample and ZnPc respectively.
Cell lines and culture conditions
HT29 human colorectal adenocarcinoma cells (ATCC, no. HTB-38) were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium (Invitrogen, no. 23400-021) supplemented with fetal bovine serum (10%; ThermoFisher Scientific, cat. no. 10270-106) and penicillin–streptomycin solution (100 units per mL and 100 mg mL−1 respectively). HepG2 human hepatocellular carcinoma cells (ATCC, no. HB-8065) and HEK-293 human embryonic kidney cells (ATCC CRL-1573) were maintained in Dulbecco's modified Eagle medium (DMEM) (ThermoFisher Scientific, cat. no. 12100-046) supplemented with fetal bovine serum (10%) and penicillin–streptomycin solution (100 units per mL and 100 μg mL−1 respectively). All the cells were grown at 37 °C in a humidified 5% CO2 atmosphere.
Confocal fluorescence microscopic studies
Approximately 2 × 105 HT29 cells in RPMI medium (2 mL) or HepG2 or HEK-293 cells in DMEM medium (2 mL) were seeded on a confocal dish and incubated overnight at 37 °C with 5% CO2. After removal of the medium, the cells were incubated with corresponding drugs. The medium was then removed and the cells were rinsed with PBS before being viewed with a Leica TCS SP8 high speed confocal microscope equipped with a solid-state 638 nm laser. The ZnPc was excited at 638 nm and monitored at 650–750 nm. The images were digitised and analysed using a Leica Application Suite X software.
Study of subcellular localisation
HT29 cells on glass-bottom dishes of 35 mm diameter (MatTek Corporation, P35G-0-14-C) were incubated in the culture medium with 4 (2 μM) for 10 min at 37 °C. After being washed twice with PBS, the cells were refed with 2 mL of the culture medium and incubated for 6 h, and then stained with 0.2 μM ER-Tracker green (Thermo Fisher Scientific Inc., E34251), 0.2 μM MitoTracker Green FM (Thermo Fisher Scientific Inc., M7514) or 2 μM LysoTracker Green DND-26 (Thermo Fisher Scientific Inc., L7526) in Hank's Balanced Salt Solution (HBSS) at 37 °C for 15, 15 and 30 min respectively. After being washed twice with PBS, the cells were refed with HBSS and observed using a Leica TCS SP8 high speed confocal microscope equipped with a 488 nm argon laser and a 633 nm laser. All the trackers were excited at 488 nm, and their fluorescence was monitored at 500–570 nm, while conjugate 4 was excited at 638 nm and its fluorescence was monitored at 650–760 nm.
Study of photocytotoxicity
Approximately 1 × 104 HT29 cells per well in the culture medium were inoculated in 96-multiwell plates and incubated overnight at 37 °C in a humidified 5% CO2 atmosphere. The conjugate 4 was first dissolved in dimethylsulfoxide (DMSO), and this stock solution was then diluted to respective concentrations with the culture medium. The cells were incubated with 100 μL of these drug solutions for 10 min at 37 °C under 5% CO2. The cells were then rinsed again with PBS, refed with 100 μL of the culture medium and incubated for 0 or 6 h before being illuminated at ambient temperature. The light source consisted of a 300 W halogen lamp, a water tank for cooling and a coloured glass filter (Newport) cut-on at λ = 610 nm. The fluence rate (λ > 610 nm) was 18 mW cm−2. Illumination of 20 min led to a total fluence of 22 J cm−2. Cell viability was determined by means of the colourimetric MTT assay.24 After illumination, the cells were incubated at 37 °C under 5% CO2 for 16 h. A MTT (Sigma) solution in PBS (3 mg mL−1, 50 μL) was added to each well followed by incubation for 4 h under the same environment. DMSO (70 μL) was then added to each well. Solutions in all wells were mixed until homogenous. Absorbance at 490 nm was measured using a plate reader (Tecan Spark 10M Microplate Reader). The average absorbance of the blank wells, which did not contain the cells, was subtracted from the readings of the other wells. The cell viability was then determined by the equation: % viability = [(∑Ai/Acontrol × 100)]/n, where Ai is the absorbance of the ith datum (i = 1, 2…n), Acontrol is the average absorbance of the control wells, in which the conjugate was absent and n (=4) is the number of data points. The percentages of cell viabilities in the cytotoxicity experiments were used as the quantitative expression of the effect of the drugs.
Annexin V-GFP/PI costaining assay
HT-29 cells on glass-bottom dishes of 35 mm diameter (MatTek Corporation, P35G-0-14-C) were incubated in RPMI medium with 4 (0.5 μM) for 10 min at 37 °C. After being washed twice with PBS, the cells were refed with 2 mL of RPMI medium with or without further incubation for 6 h, followed by light irradiation (18 mW cm−2, 20 min). After that, the cells were further incubated at 37 °C under 5% CO2 for 0, 6 or 16 h and then strained with Annexin V-GFP and PI in binding buffer for 15 min before visualisation with confocal fluorescence microscopy. The probes were excited at 488 nm, and the fluorescence was monitored at 500–530 nm (Annexin V-GFP) and 570–680 nm (PI).
In vivo imaging analysis
Female Balb/c nude mice (20–25 g) were obtained from the Laboratory Animal Services Centre at The Chinese University of Hong Kong. All experiments involving live animals were performed in strict accordance with the animal experimentation guidelines and were approved by the Animal Experimentation Ethics Committee of the University (ref. no. 14-044-GRF). Licence to conduct animal experiments was obtained from the Department of Health, Government of the Hong Kong Special Administrative Region. The mice were kept under pathogen-free conditions with free access to food and water. HT29 cells (about 1 × 107 cells in 200 μL HBSS) were inoculated subcutaneously on the back of the mice. Once the tumours had grown to a size of 60–100 mm3, 40 nmol of conjugates 4 and 5 were first dissolved in 400 μL distilled water containing 1% DMSO respectively. These solutions (200 μL, equivalent to 20 nmol of 4 or 5) were injected into the tumour-bearing mice through tail intravenous injection. In vivo fluorescence imaging was captured before and after the injection (at different time points up to 48 h) with an Odyssey infrared imaging system (excitation wavelength = 680 nm, emission wavelength ≥ 700 nm). The images were digitised and analysed by use of the Odyssey imaging system software (9201-500). After in vivo imaging studies, the animals were euthanised at 48 post-injection. Tumours and other major organs were harvested and their fluorescence intensities were measured. Four mice were used for each conjugate.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by a grant from the Research Grants Council of the Hong Kong Special Administrative Region (Ref. No. 14303814).
References
-
(a) K. P. Loh, D. Ho, G. N. C. Chiu, D. T. Leong, G. Pastorin and E. K. H. Chow, Adv. Mater., 2018, 30, 1802368 CrossRef PubMed;
(b) B. del Rosal, B. Jia and D. Jaque, Adv. Funct. Mater., 2018, 28, 1803733 CrossRef;
(c) J. Li and K. Pu, Chem. Soc. Rev., 2019, 48, 38–71 RSC;
(d) C. N. Ko, G. Li, C. H. Leung and D. L. Ma, Coord. Chem. Rev., 2019, 381, 79–103 CrossRef CAS.
-
(a) F. Hu, S. Xu and B. Liu, Adv. Mater., 2018, 30, 1801350 CrossRef PubMed;
(b) S. Jung and X. Chen, Adv. Healthcare Mater., 2018, 7, 1800252 CrossRef PubMed;
(c) S. He, J. Song, J. Qu and Z. Cheng, Chem. Soc. Rev., 2018, 47, 4258–4278 RSC.
-
(a) D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed;
(b) S. B. Brown, E. A. Brown and I. Walker, Lancet Oncol., 2004, 5, 497–508 CrossRef CAS PubMed;
(c) D. van Straten, V. Mashayekhi, H. S. de Bruijn, S. Oliveira and D. J. Robinson, Cancers, 2017, 9, 19 CrossRef PubMed;
(d) X. Li, N. Kwon, T. Guo, Z. Liu and J. Yoon, Angew. Chem., Int. Ed., 2018, 57, 11522–11531 CrossRef CAS PubMed;
(e) S. Monro, K. L. Colón, H. Yin, J. Roque III, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed.
-
(a) C. Mao, Y. Xiang, X. Liu, Y. Zheng, K. W. K. Yeung, Z. Cui, X. Yang, Z. Li, Y. Liang, S. Zhu and S. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 17902–17914 CrossRef CAS PubMed;
(b) J.-f. Lin, J. Li, A. Gopal, T. Munshi, Y.-w. Chu, J.-x. Wang, T.-t. Liu, B. Shi, X. Chen and L. Yan, Chem. Commun., 2019, 55, 2656–2659 RSC.
- A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535–545 CrossRef CAS PubMed.
- M. Lan, S. Zhao, W. Liu, C.-S. Lee, W. Zhang and P. Wang, Adv. Healthcare Mater., 2019, 1900132 CrossRef PubMed.
-
(a) D. K. P. Ng, Future Med. Chem., 2014, 6, 1991–1993 CrossRef CAS PubMed;
(b) R. C. H. Wong, P.-C. Lo and D. K. P. Ng, Coord. Chem. Rev., 2019, 379, 30–46 CrossRef CAS;
(c) X. Li, B.-D. Zheng, X.-H. Peng, S.-Z. Li, J.-W. Ying, Y. Zhao, J.-D. Huang and J. Yoon, Coord. Chem. Rev., 2019, 379, 147–160 CrossRef CAS.
-
(a) R. C. H. Wong, S. Y. S. Chow, S. Zhao, W.-P. Fong, D. K. P. Ng and P.-C. Lo, ACS Appl. Mater. Interfaces, 2017, 9, 23487–23496 CrossRef CAS PubMed;
(b) Y. Sun, H. Hu, N. Zhao, T. Xia, B. Yu, C. Shen and F.-J. Xu, Biomaterials, 2017, 117, 77–91 CrossRef CAS PubMed;
(c) D. Gao and P.-C. Lo, J. Controlled Release, 2018, 282, 46–61 CrossRef CAS PubMed;
(d) J. Ma, D. Chen, Y. Li, Y. Chen, Q. Liu, X. Zhou, K. Qian, Z. Li, H. Ruan, Z. Hou and X. Zhu, J. Controlled Release, 2018, 284, 1–14 CrossRef CAS PubMed;
(e) C. Jiang, R. Wang, H. Ou, A. Li, Y. An, S. Guo and L. Shi, Chem. Commun., 2018, 54, 3985–3988 RSC;
(f) J. Wang, X. Zhuo, X. Xiao, R. Mao, Y. Wang, J. Wang and J. Liu, Nanoscale, 2019, 11, 3386–3395 RSC.
-
(a) C. Zhang, W. Wu, R. Q. Li, W. X. Qiu, Z. N. Zhuang, S. X. Cheng and X. Z. Zhang, Adv. Funct. Mater., 2018, 28, 1804492 CrossRef;
(b) V. Almeida-Marrero, E. van de Winckel, E. Anaya-Plaza, T. Torres and A. de la Escosura, Chem. Soc. Rev., 2018, 47, 7369–7400 RSC.
- M.-R. Ke, S.-L. Yeung, W.-P. Fong, D. K. P. Ng and P.-C. Lo, Chem.–Eur. J., 2012, 18, 4225–4233 CrossRef CAS PubMed.
- S. Kopetz, Nat. Rev. Gastroenterol. Hepatol., 2019, 16, 79–80 CrossRef CAS PubMed.
-
(a) Z. Li, R. Zhao, X. Wu, Y. Sun, M. Yao, J. Li, Y. Xu and J. Gu, FASEB J., 2005, 19, 1978–1985 CrossRef CAS PubMed;
(b) P. L. Hsiung, J. Hardy, S. Friedland, R. Soetikno, C. B. Du, A. P. Wu, P. Sahbaie, J. M. Crawford, A. W. Lowe and T. D. Wang, Nat. Med., 2008, 14, 454–458 CrossRef CAS PubMed;
(c) S. Song, D. Liu, J. Peng, H. Deng, Y. Guo, L. X. Xu, A. D. Miller and Y. Xu, FASEB J., 2009, 23, 1396–1404 CrossRef CAS PubMed;
(d) J. Burggraaf, I. M. C. Kamerling, P. B. Gordon, L. Schrier, M. L. de Kam, A. J. Kales, R. Bendiksen, B. Indrevoll, R. M. Bjerke, S. A. Moestue, S. Yazdanfar, A. M. J. Langers, M. Swaerd-Nordmo, G. Torheim, M. V. Warren, H. Morreau, P. W. Voorneveld, T. Buckle, F. W. B. van Leeuwen, L. I. Ødegårdstuen, G. T. Dalsgaard, A. Healey and J. C. H. Hardwick, Nat. Med., 2015, 21, 955–961 CrossRef CAS PubMed;
(e) J. Zhou, B. P. Joshi, X. Duan, A. Pant, Z. Qiu, R. Kuick, S. R. Owens and T. D. Wang, Clin. Transl. Gastroenterol., 2015, 6, e101 CrossRef CAS PubMed.
- A. R. Sepulveda, S. R. Hamilton, C. J. Allegra, W. Grody, A. M. Cushman-Vokoun, W. K. Funkhouser, S. E. Kopetz, C. Lieu, N. M. Lindor, B. D. Minsky, F. A. Monzon, D. J. Sargent, V. M. Singh, J. Willis, J. Clark, C. Colasacco, R. B. Rumble, R. Temple-Smolkin, C. B. Ventura and J. A. Nowak, Am. J. Clin. Pathol., 2017, 147, 221–260 CrossRef CAS PubMed.
- R. C. H. Wong, D. K. P. Ng, W.-P. Fong and P.-C. Lo, Chem.–Eur. J., 2017, 23, 16505–16515 CrossRef CAS PubMed.
- I. Scalise and E. N. Durantini, Bioorg. Med. Chem., 2005, 13, 3037–3045 CrossRef CAS PubMed.
- M. D. Maree, N. Kuznetsova and T. Nyokong, J. Photochem. Photobiol., A, 2001, 140, 117–125 CrossRef CAS.
- L. Yu, Q. Wang, R. C. H. Wong, S. Zhao, D. K. P. Ng and P.-C. Lo, Dyes Pigm., 2019, 163, 197–203 CrossRef CAS.
-
(a) F. Zhang, S. Wang, L. Yin, Y. Yang, Y. Guan, W. Wang, H. Xu and N. Tao, Anal. Chem., 2015, 87, 9960–9965 CrossRef CAS PubMed;
(b) Q. Zhou, Z. Li, J. Zhou, B. P. Joshi, G. Li, X. Duan, R. Kuick, S. R. Owens and T. D. Wang, Photoacoustics, 2016, 4, 43–54 CrossRef PubMed.
- P. Mroz, A. Yaroslavsky, G. B. Kharkwal and M. R. Hamblin, Cancers, 2011, 3, 2516–2539 CrossRef CAS PubMed.
- J. F. Lovell, J. Chen, M. T. Jarvi, W. G. Cao, A. D. Allen, Y. Liu, T. T. Tidwell, B. C. Wilson and G. Zheng, J. Phys. Chem. B, 2009, 113, 3203–3211 CrossRef CAS PubMed.
- B. G. Ongarora, K. R. Fontenot, X. Hu, I. Sehgal, S. D. Satyanarayana-Jois and M. G. H. Vicente, J. Med. Chem., 2012, 55, 3725–3738 CrossRef CAS PubMed.
- G. Majno and I. Joris, Am. J. Pathol., 1995, 146, 3–15 CAS.
- D. F. Eaton, Pure Appl. Chem., 1988, 60, 1107–1114 CAS.
- H. Tada, O. Shiho, K. Kuroshima, M. Koyama and K. Tsukamoto, J. Immunol. Methods, 1986, 93, 157–165 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03911b |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a