
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew molecular architectures containing low-valent cluster centres with di- and trimetalated 2-vinylpyrazine ligands: synthesis and molecular structures of Ru5(CO)15(μ5-C4H2N2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) CH)(μ-H)2 and Ru8(CO)24(μ7-C4H2N2CHC)(μ-H)3†
CH)(μ-H)2 and Ru8(CO)24(μ7-C4H2N2CHC)(μ-H)3†
Md. Monir Hossaina,
Nahid Aktera,
Shishir Ghosh*a,
Vladimir N. Nesterovb,
Michael G. Richmond b,
Graeme Hogarthc and
Shariff E. Kabir*a
b,
Graeme Hogarthc and
Shariff E. Kabir*a
aDepartment of Chemistry, Jahangirnagar University, Savar, Dhaka 1342, Bangladesh. E-mail: sghosh_006@yahoo.com; skabir_ju@yahoo.com
bDepartment of Chemistry, University of North Texas, Denton, TX 76209, USA
cDepartment of Chemistry, King's College London, Britannia House, 7 Trinity Street, London SE1 1DB, UK
First published on 4th July 2019
Abstract
Reaction of 2-vinylpyrazine with Ru3(CO)12 results in multiple C–H bond activations to afford penta- and octa-ruthenium clusters, Ru5(CO)15(μ5-C4H2N2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH)(μ-H)2 (2) and Ru8(CO)24(μ7-C4H2N2CHC)(μ-H)3 (3), in which a Ru3 sub-unit is linked to Ru2 and Ru5 centres via di- and tri-metalated 2-vinylpyrazine ligands, exhibiting novel coordination modes including the loss of ring aromaticity in 2. The bonding of 2 and the mechanism for the fluxional behaviour of the hydrides have been examined by electronic structure calculations.
CH)(μ-H)2 (2) and Ru8(CO)24(μ7-C4H2N2CHC)(μ-H)3 (3), in which a Ru3 sub-unit is linked to Ru2 and Ru5 centres via di- and tri-metalated 2-vinylpyrazine ligands, exhibiting novel coordination modes including the loss of ring aromaticity in 2. The bonding of 2 and the mechanism for the fluxional behaviour of the hydrides have been examined by electronic structure calculations.
Introduction
2-Vinylpyridine is a versatile ligand known for its ability to stabilize transition metal–carbon bonds in a chelating coordination mode, and it has been widely investigated as a feedstock in the exploration of new catalysts for selective olefinic C–H bond activation.1–12 Mononuclear complexes are normally used, chelation occurring either through π-complexation of the alkene functionality or via activation of the β-C–H alkenyl bond(s) in addition to coordination of nitrogen.4–12 At polynuclear centres, diverse substrate coordination modes are feasible, and the binding of nitrogen and vinyl moieties need not occur at the same metal atom. Thus, the metalated derivatives of 2-vinylpyridine have been shown to bind up to four metal atoms, acting as 4–6 electron donors (A–E, Chart 1).13–19 The chemistry of the closely related 2-vinylpyrazine, which contains an additional nitrogen atom within the heterocyclic ring, has been far less studied.20–22 Usually, reactions of these ligands with unsaturated appendages proceed through C–H activation of the vinyl group with concomitant coordination of the adjacent ring nitrogen. However, 2-vinylpyrazine can also coordinate to trimetallic centres through metalation of its aromatic ring utilizing the second ring nitrogen (F, Chart 1), especially if the dominant C–H bond activation pathway does not furnish a kinetically stable product.22 Herein we detail the reaction of Ru3(CO)12 with 2-vinylpyrazine, the aim of which was to synthesize clusters in which all of the donor atoms of the 2-vinylpyrazine ligand participate in bonding to the cluster core. Two new polyruthenium clusters have been isolated in which the 2-vinylpyrazine shows novel and unprecedented coordination modes by utilizing all the available donor atoms for bonding (G and H, Chart 1).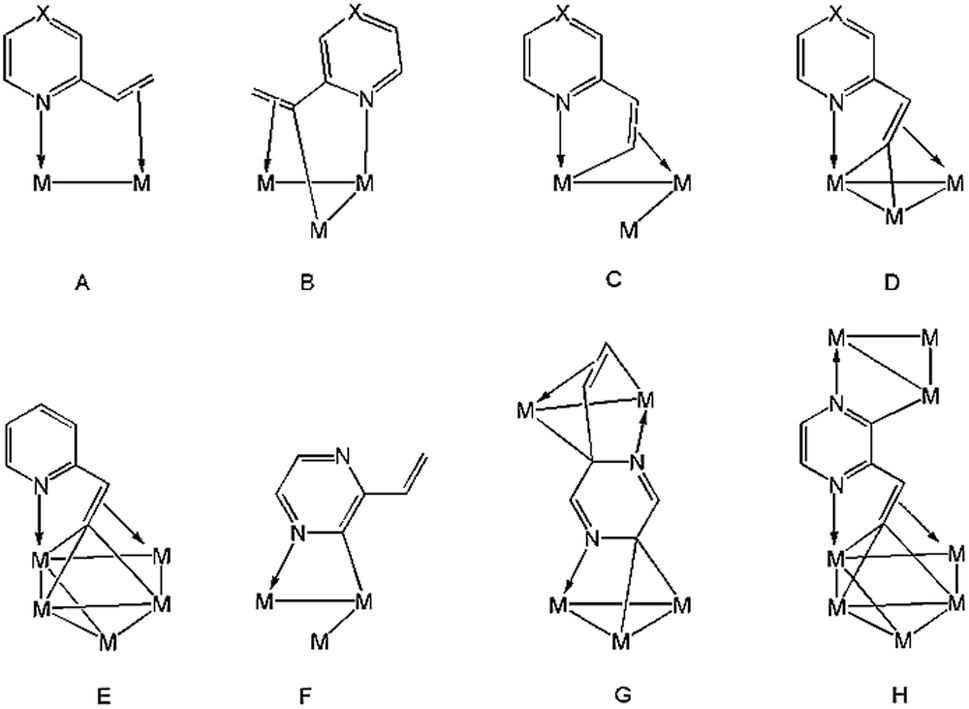 | ||
| Chart 1 Binding modes of 2-vinylpyrazine (X = N) and 2-vinylpyridine (X = CH) at polynuclear cluster centers. | ||
Results and discussion
We first investigated the reaction of 2-vinylpyrazine with Ru3(CO)10(μ-dppm) as it is well-established that bis(diphenylphosphino)methane (dppm) stabilizes the triruthenium framework against fragmentation.19,23 This is indeed the case, and refluxing the two reactants in THF afforded Ru3(CO)6(μ3-C4H3N2CHC)(μ-dppm)(μ-H)2 (1) in 28% yield as the major product resulting from the double C–H activation of the vinyl ligand (Scheme 1). The molecular structure (Fig. 1) shows that as expected the dimetalated 2-vinylpyrazine ligand caps the closed Ru3 triangle, coordinating to Ru(2) and Ru(1) via a ring nitrogen and the vinylic double bond, respectively, and bridging Ru(2)–Ru(3) edge with the β-carbon of the vinyl moiety through double β-C–H bond activation. Spectroscopic data are consistent with the solid-state structure; the 31P{1H} NMR spectrum displays two doublets at δ 44.8 and 32.5 (J 56 Hz), and the 1H NMR spectrum shows two upfield hydride signals at δ −17.15 (dd) and −18.01 (dd) (J 48.5, 2.5 Hz). Formation of 1 is as expected and entirely analogous with the related 2-vinylpyridine chemistry19 with inclusion of the of the second nitrogen centre playing no role in the chemistry.
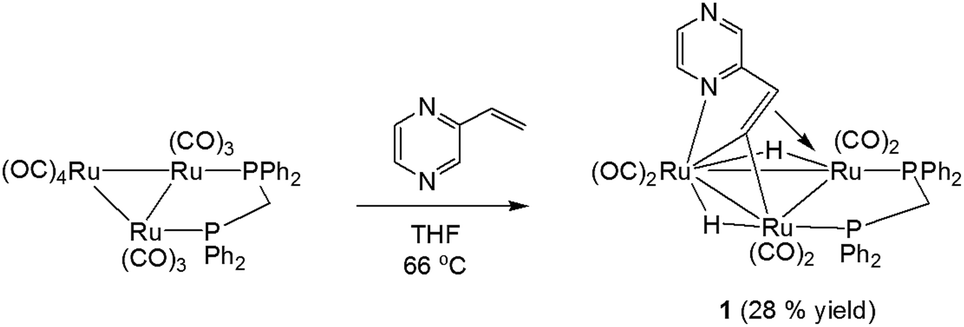 | ||
| Scheme 1 Reaction of Ru3(CO)10(μ-dppm) with 2-vinylpyrazine. | ||
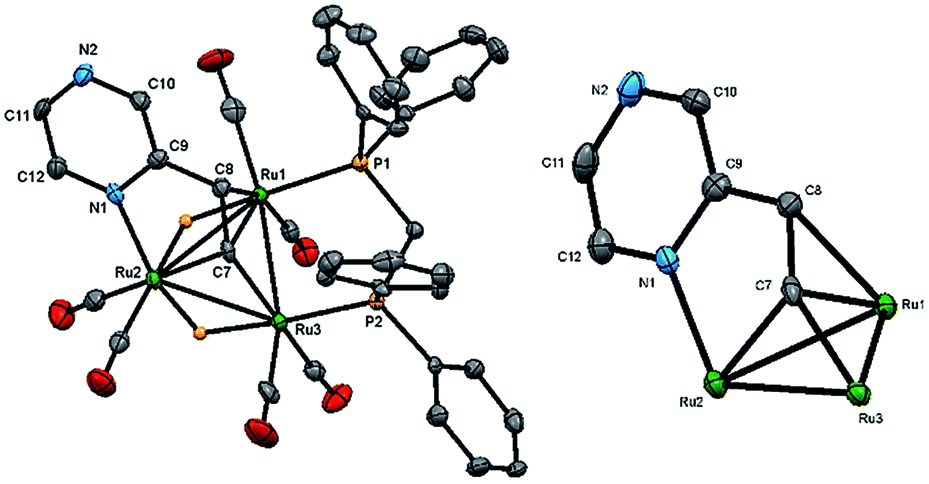 | ||
| Fig. 1 Molecular structure of Ru3(CO)6(μ3-C4H3N2CHC)(μ-dppm)(μ-H)2 (1), showing 50% probability thermal ellipsoids without hydrogen atoms except the bridging hydrides (left) and the cluster core without the dppm, carbonyls and hydrogen atoms (right). Selected bond lengths (Å) and angles (°): Ru(1)–Ru(2) 2.8765(6), Ru(2)–Ru(3) 2.8264(6), Ru(1)–Ru(3) 2.7952(6), Ru(1)–P(1) 2.3273(14), Ru(3)–P(2) 2.2730(14), Ru(2)–N(1) 2.145(4), Ru(1)–C(8) 2.322(5), Ru(1)–C(7) 2.160(5), Ru(2)–C(7) 2.054(5), Ru(3)–C(7) 1.990(5), N(1)–Ru(2)–Ru(1) 83.14(11), N(1)–Ru(2)–Ru(3) 124.02(11), C(7)–Ru(1)–C(8) 36.11(19), C(7)–Ru(1)–Ru(3) 45.12(14), C(7)–Ru(3)–Ru(1) 50.28(15), Ru(1)–C(7)–Ru(3) 84.60(18). | ||
In marked contrast, heating a benzene solution of Ru3(CO)12 and 2-vinylpyrazine afforded penta- and octa-nuclear clusters, Ru5(CO)15(μ5-C4H2N2CHCH)(μ-H)2 (2) and Ru8(CO)24(μ7-C4H2N2CHC)(μ-H)3 (3), in a combined moderate yield of 47%, after workup (Scheme 2). Characterization of both products was made by X-ray crystallography, and the molecular structures are depicted in Fig. 2 and 4.
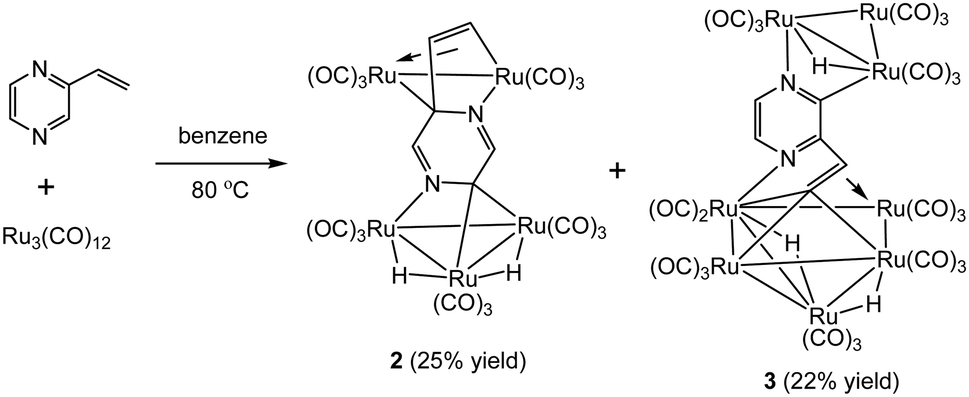 | ||
| Scheme 2 Reaction of Ru3(CO)12 with 2-vinylpyrazine. | ||
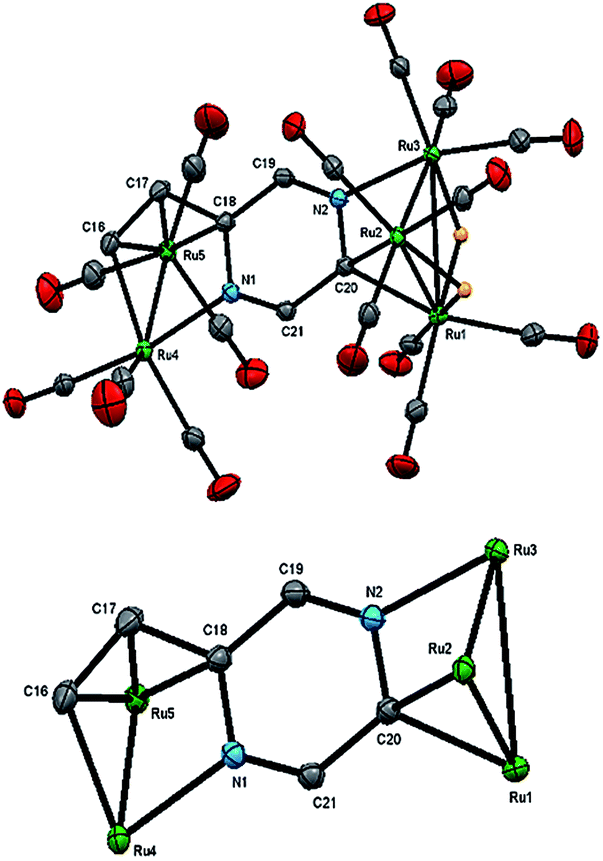 | ||
| Fig. 2 Molecular structure of Ru5(CO)15(μ5-C4H2N2CHCH)(μ-H)2 (2, top), showing 50% probability thermal ellipsoids without hydrogen atoms except the bridging hydrides and the cluster core without carbonyls and hydrogen atoms (bottom). Selected bond lengths (Å) and angles (°): Ru(1)–Ru(2) 2.7905(2), Ru(2)–Ru(3) 2.7742(2), Ru(1)–Ru(3) 2.9263(2), Ru(4)–Ru(5) 2.7409(2), Ru(4)–N(1) 2.1138(13), Ru(3)–N(2) 2.0850(13), Ru(1)–C(20) 2.1549(15), Ru(2)–C(20) 2.1349(15), Ru(5)–C(16) 2.2373(16), Ru(5)–C(17) 2.1858(16), Ru(5)–C(18) 2.2183(15), C(18)–N(1) 1.4425(19), C(18)–C(19) 1.448(2), C(19)–N(2) 1.284(2), C(20)–N(2) 1.4454(19), C(20)–C(21) 1.453(2), C(21)–N(1) 1.2911(19), C(16)–C(17) 1.393(2), C(17)–C(18) 1.454(2), N(2)–Ru(3)–Ru(1) 70.07(3), N(1)–Ru(4)–Ru(5) 72.10(3), Ru(1)–C(20)–Ru(2) 81.15(5). | ||
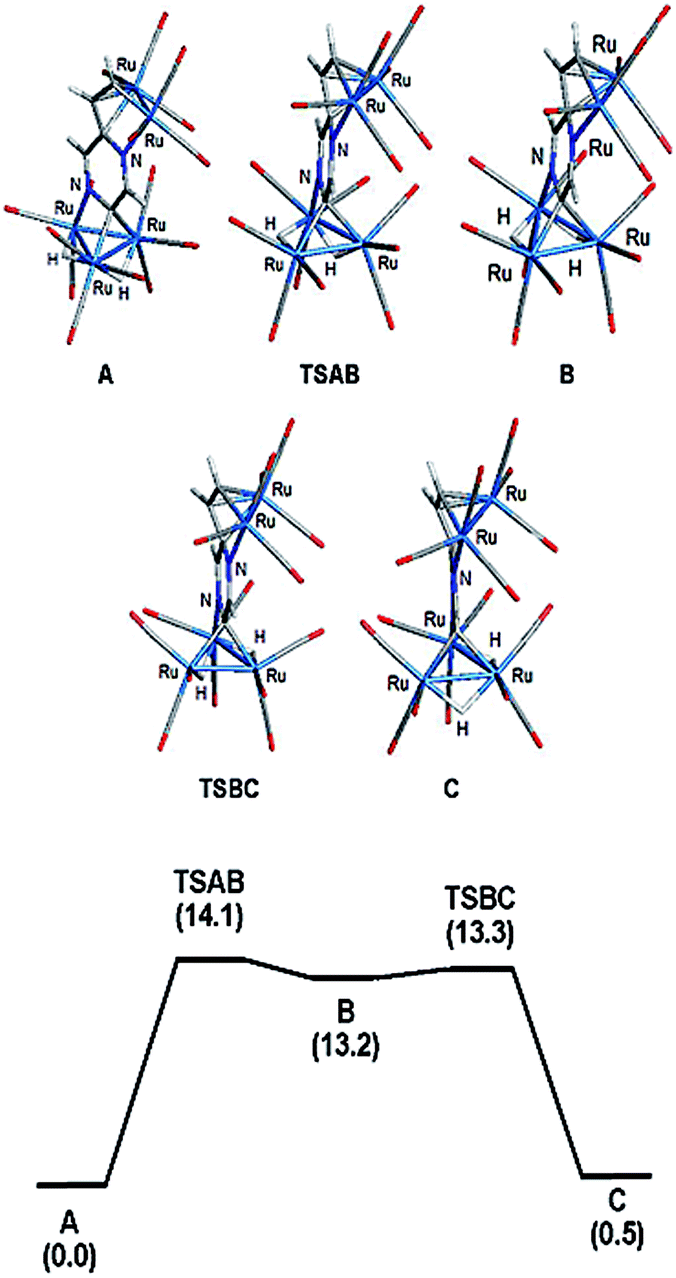 | ||
| Fig. 3 Optimized structures for the isomeric hydrides based on cluster 2 (top) and potential energy profile for hydride scrambling between species A and C (bottom). Energy values (ΔG) are in kcal mol−1 with respect to A. | ||
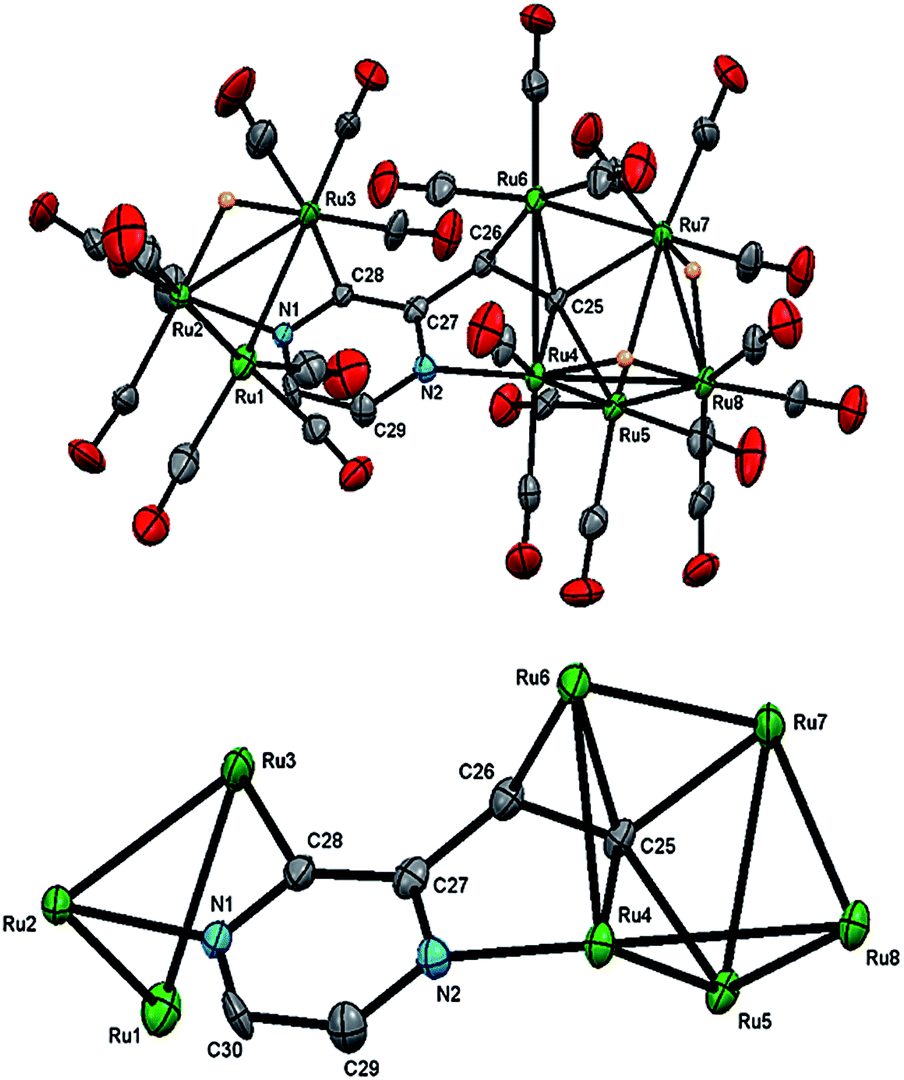 | ||
| Fig. 4 Molecular structure of Ru8(CO)24(μ7-C4H2N2CHC)(μ-H)3 (3, top), showing (a) 50% probability thermal ellipsoids without hydrogen atoms except the bridging hydrides and the cluster core without carbonyls and hydrogen atoms (bottom). Selected bond lengths (Å) and angles (°): Ru(1)–Ru(2) 2.8493(7), Ru(2)–Ru(3) 2.8585(7), Ru(1)–Ru(3) 2.8723(8), Ru(4)–Ru(5) 2.8635(7), Ru(4)–Ru(6) 2.8269(7), Ru(4)–Ru(8) 2.9769(7), Ru(5)–Ru(7) 2.8536(7), Ru(5)–Ru(8) 2.7752(7), Ru(6)–Ru(7) 2.8319(7), Ru(7)–Ru(8) 3.0470(7), Ru(2)–N(1) 2.107(4), Ru(4)–N(2) 2.102(4), Ru(3)–C(28) 2.097(5), Ru(4)–C(25) 2.114(5), Ru(5)–C(25) 2.109(5), Ru(6)–C(25) 2.107(5), Ru(7)–C(25) 2.148(5), Ru(6)–C(26) 2.201(5), C(28)–N(1) 1.336(7), C(28)–C(27) 1.429(7), C(27)–N(2) 1.351(7), C(29)–N(2) 1.349(7), C(29)–C(30) 1.368(8), C(30)–N(1) 1.340(7), C(25)–C(26) 1.447(7), C(26)–C(27) 1.450(8), Ru(5)–Ru(4)–Ru(6) 92.857(19), Ru(6)–Ru(4)–Ru(8) 91.39(2), Ru(4)–Ru(5)–Ru(7) 81.786(18), Ru(4)–Ru(6)–Ru(7) 82.815(18), Ru(5)–Ru(7)–Ru(6) 92.961(19), Ru(6)–Ru(7)–Ru(8) 89.86(2), Ru(4)–Ru(8)–Ru(7) 76.815(18). | ||
The structure of 2 consists of individual Ru3 and Ru2 units linked via a dimetalated μ5-C4H2N2CHCH ligand formed upon the expected activation of a β-C–H alkenyl bond, and metalation of the ring proton directly trans to the vinyl group. The Ru2 unit is coordinated by one of the ring nitrogen atoms and the metalated vinyl moiety of the heterocyclic ligand in such a way that one ruthenium is bonded to the ring nitrogen and the β-carbon of the vinyl moiety, while the other ruthenium makes a σ,π-vinyl type interaction with the vinylic double bond and ipso carbon, C(18). The heterocyclic ligand is coordinated to the Ru3 unit by capping one face of the ruthenium triangle using the second ring nitrogen atom and an adjacent ring carbon; thus importantly, two of the ring carbon atoms are now sp3-hybridized. The change in hybridization is supported by a careful examination of C–C and C–N bond distances, being only two double bonds within the ring that are fully localized between the N(1)–C(21) and N(2)–C(19) bonds. The other two carbon atoms of the ring, C(18) and C(20), can now be considered sp3-hybridized which is in accord with the widening of the acute angle involving these carbons [C(17)–C(18)–Ru(5) 69.52(9) and Ru(1)–C(20)–Ru(2) 81.15(5)°]. The alternating C–C and C–N bond distances in the heterocycle ring confirm the loss in aromaticity upon adoption of the face-capping and edge-bridging of the Ru3 and Ru2 units, respectively. The Ru–C distances involving the σ,π-vinyl type interaction with Ru(5) are quite similar [Ru(5)–C(16) 2.2373(16), Ru(5)–C(17) 2.1858(16) and Ru(5)–C(18) 2.2183(15) Å] which also corroborate this assumption. Each ruthenium atom achieves an 18-electron configuration considering the μ5-C4H2N2CHCH ligand acts as 10-electron donor.
The 1H NMR spectrum of 2 displays two doublets at δ 7.91 and 5.84 (J 6.0 Hz) attributed to the vinyl protons and two singlets at δ 6.49 and 6.05 for the pyrazine ring protons. The hydride region of the spectrum does not show any discernible hydride resonances (except for a slight upward curving of the baseline at ca. δ −15.2) indicating rapid movement of both hydrides about the three ruthenium–ruthenium edges within the Ru3 unit at room temperature. Upon cooling to 273 K, two broad singlets appeared at δ −13.96 and −16.58 and these resonances sharpened further as the temperature was lowered (Fig. S1 and S2†). Based on a Δν value of 1048 Hz for the two hydrides in the slow-exchange region and a coalescence temperature of 298 K, we estimate a ΔG‡ of 12.4 kcal mol−1 for hydride scrambling.24
The bonding in 2 was examined by electronic structure calculations, and the optimized structure for species C depicted in Fig. 3 reproduces the important structural features found in the experimental structure. The two hydrides bridge adjacent Ru–Ru bonds with one of the hydrides sharing a common metallic edge with the benzylidene moiety. The other hydride bridges the Ru–Ru bond that lies on the side of the Ru3 cluster below the external Ru2 appendage. We next investigated the regiochemical preference for hydride disposition about the triangular core due to the presence of an additional Ru–Ru bond that could serve as a site for hydride coordination. Knowledge of other possible isomers would provide an overall stability ordering of the isomeric hydrides not to mention insight into the fluxional pathway exhibited by the hydride ligands.
Two additional hydride isomers corresponding to A and B (Fig. 3) were computed as viable ground-state species with A the most stable of the three isomers. Migration of the hydride that lies below the Ru2 appendage in C to the non-hydride-bridged Ru–Ru bond affords A, which is 0.5 kcal mol−1 more stable than C. Isomer B is the least stable of the three isomers, and it lies 13.2 kcal mol−1 above A. Species B may be viewed as the product of hydride migration from the edge-bridged Ru–Ru bond ligated by the benzylidene moiety to the adjacent non-hydride-bridged Ru–Ru bond. These data, which confirm the regiochemical preference for a hydride to share a common Ru–Ru bond with the edge-bridging benzylidene moiety, are not unlike those recently reported by us in heterocyclic-bridged triosmium clusters.25
The fluxional behavior of the two hydrides in 2 was computationally investigated, and the scrambling of both hydrides about the Ru3 core proceeds through species B.26 This transient intermediate B effectively facilitates the exchange of both hydrides about the benzylidene-bridged Ru–Ru bond and one of the two other Ru–Ru bonds in 2. The potential energy profile for the rearrangement is shown in the bottom portion of Fig. 3, and the process is analogous to a reversible first-order reaction involving an A ⇌ B ⇌ C sequence. The energy difference (ΔG‡) between the two transition states (TSAB and TSBC) is 0.8 kcal mol; TSAB represents the rate-limiting step for the isomerization reaction with a barrier of 14.1 kcal mol−1 that closely matches the experimental value of 12.4 kcal mol−1. Scheme 3 illustrates the hydride exchange between the participating ground-state species starting from A.
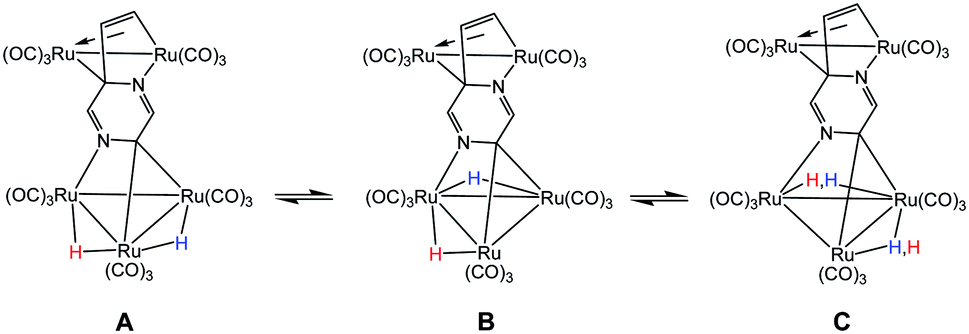 | ||
| Scheme 3 Hydride scrambling between species A and C through intermediate B. | ||
The structure of 3 (Fig. 4) consists of Ru3 and Ru5 sub-units linked by a μ7-C4H2N2CHC ligand. The large sub-unit consists of an edge-bridged butterfly arrangement in which the vinyl pyrazine coordination is similar to that seen in 1; both β-hydrogens of the vinyl moiety have oxidatively added and the vicinal nitrogen atom is also coordinated. Thus the ligand caps a square face of the pentagonal ruthenium core with the Ru–C distances varying between 2.107(5) and 2.148(5) Å, the shortest of which is to Ru(6), also coordinated by the α-carbon [Ru(6)–C(26) 2.201(5) Å]. The ligand is coordinated to the Ru3 sub-unit by via the second nitrogen atom and metalation of an adjacent ring proton. Unlike the metalated ligand found in 2, the aromaticity of the pyrazine ring is fully maintained in 3. The three hydrides were located and refined in the structural analysis; one bridges the Ru(2)–Ru(3) edge of the Ru3 unit, while the other two span across Ru(4)–Ru(8) and Ru(7)–Ru(8) edges of the Ru5 cluster. There are ten formal ruthenium–ruthenium bonds ranging between 2.7752(7) and 3.0470(7) Å and each ruthenium has VEC of 18 with the μ7-C4H2N2CHC ligand acting as a 9-electron donor. The 1H NMR spectrum of 3 clearly indicates the triple C–H bond activation of the 2-vinylpyrazine ligand, displaying a sharp up-field singlet at δ −14.30, and two overlapping singlets at δ −21.20 and −21.21; two further doublets at δ 7.71 and 7.36 (J 4.0 Hz) and a singlet at δ 5.19 being associated with the pyrazine ring. The hydride region also shows another set of resonances (at δ −14.48, −21.30 and −21.31) indicating the presence of another isomer in solution (ca. 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). No isomer equilibration was found when the mixture was examined by EXSY 1H NMR at room temperature (Fig. S3†), and VT 1H NMR examination over the temperature range 298–233 K confirmed an invariant isomeric composition (Fig. S4 and S5†).
:1). No isomer equilibration was found when the mixture was examined by EXSY 1H NMR at room temperature (Fig. S3†), and VT 1H NMR examination over the temperature range 298–233 K confirmed an invariant isomeric composition (Fig. S4 and S5†).
The precise mode of formation of 1–3 remains unknown, but a plausible reaction scheme may be posited (Scheme 4). Initial coordination of the nitrogen vicinal to the vinyl group is followed by the first β-H activation to afford key intermediate A. A second β-H activation then affords 1, the inclusion of the electron-donor diphosphine making further CO loss unfavorable. In the absence of the diphosphine, the putative Ru3(CO)8(μ3-C4H3N2CHC)(μ-H)2 reacts with additional Ru3(CO)12 to afford a pentanuclear derivative akin to that found in the reaction of Ru3(CO)12 with 2-vinylpyridine,19 but with the pyrazine derivative further nitrogen coordination and C–H ring activation furnish 3. Formation of pentanuclear 2 is proposed to result from the interception of intermediate A with Ru3(CO)12 which binds to nitrogen and also undergoes C–H ring activation; cluster 2 results from hydride transfer between the two cluster sub-units which also leads to loss of aromaticity of the ring.
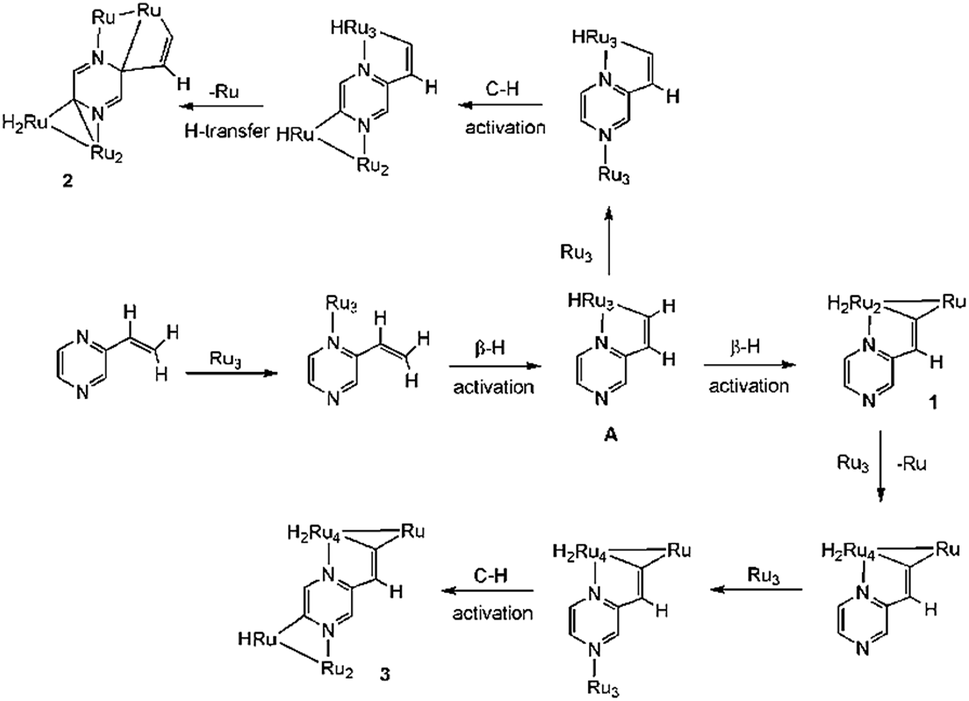 | ||
| Scheme 4 Proposed scheme for the formation of 1–3. | ||
Conclusions
In summary, we have shown that the reaction of 2-vinylpyrazine with Ru3(CO)12 leads to multiple C–H bond activations, affording unusual clusters with novel coordination modes. This reactivity is significantly different from that seen in related triosmium chemistry,20 in which only a single C–H bond activation, akin to the chemistry of 2-vinylpyridine at triosmium centers, is observed.19,20 The reactivity of 2-vinylpyrazine towards Ru3(CO)12 is also quite different from that of 2-vinylpyridine19 due to the presence of the additional nitrogen in the heterocyclic ring that can facilitate further C–H bond activation by coordinating to a metal center. The relatively weak metal–metal bonds in Ru3(CO)12 as compared to Os3(CO)12 and Ru3(CO)10(μ-dppm) also play an important role in facilitating coordination of both nitrogen atoms.Experimental
General and instrumentation details
All reactions were carried out under a nitrogen atmosphere using standard Schlenk techniques unless otherwise stated. Reagent-grade solvents were dried using appropriate drying agents and distilled by standard methods prior to use. Ru3(CO)12 was purchased from Strem Chemicals Inc. and used without further purification. 2-Vinylpyrazine and dppm were purchased from Acros Organics and used as received. Ru3(CO)10(μ-dppm) was prepared according to a literature method.27 Products were separated without any special precautions on TLC plates coated with 0.25 mm silica gel (HF254-type 60, E. Merck, Germany). IR spectra were recorded on a Shimadzu FTIR Prestige 21 spectrophotometer and NMR spectra on a Bruker DPX 400 instrument. All chemical shifts are reported in δ units and are referenced to the residual protons of the deuterated solvents (1H) or to external H3PO4 (31P), whose chemical shift is assigned to δ = 0.0. Elemental analyses were performed by the Microanalytical laboratory of Wazed Miah Science Research Centre at Jahangirnagar University.Reaction of Ru3(CO)10(μ-dppm) with 2-vinylpyrazine
A thf solution (25 mL) of Ru3(CO)10(μ-dppm) (0.10 g, 0.10 mmol) and 2-vinylpyrazine (11 mg, 0.10 mmol) was heated to reflux for 3.5 h. After removal of the solvent under reduced pressure the residue was chromatographed by TLC on silica gel. Elution with hexane/CH2Cl2 (1:1 v/v) gave three bands. The third band afforded [Ru3(CO)6(μ3-C4H3N2CHC)(μ-dppm)(μ-H)2] (1) (30 mg, 28%) as orange crystals after recrystallization from hexane/CH2Cl2 at 4 °C. The content of the other two bands were too small for characterization. Analytical and spectroscopic data for 1: anal. calcd for C37H28N2O6P2Ru3·CH2Cl2: C, 43.60; H, 2.89; N, 2.68. Found: C, 44.01; H, 2.94; N, 2.71%. IR (ν(CO), CH2Cl2): 2024s, 1998sh, 1986s, 1959s, 1942sh cm−1. 1H NMR (CDCl3): δ 8.24 (d, J 3.0 Hz, 1H), 7.99 (m, 2H), 7.88 (d, J 3.0 Hz, 1H), 7.78 (d, J 3.0, 1H), 7.58–7.35 (m, 12H), 7.27 (m, 2H), 7.12 (m, 2H), 6.91 (m, 2H), 4.58 (m, 1H), 4.30 (m, PCH2P, 1H), 3.23 (m, PCH2P, 1H), −17.15 (dd, 2JPH 48.5, 2JHH 2.5 Hz, 1H), −18.01 (dd, 2JPH 48.5, 2JHH 2.5 Hz, 1H). 31P{1H} NMR (CDCl3): δ 44.8 (d, 3JPP 56 Hz, 1P), 32.5 (d, 3JPP 56 Hz, 1P).
Reaction of Ru3(CO)12 with 2-vinylpyrazine
A benzene solution (25 mL) of Ru3(CO)12 (0.10 g, 0.16 mmol) and 2-vinylpyrazine (34 mg, 0.32 mmol) was heated to reflux for 6 h. The solvent was removed by rotary evaporation under vacuum and the residue chromatographed by TLC on silica gel. Elution with cyclohexane/CH2Cl2 (4:1, v/v) developed two major and several very minor bands. The major bands afforded, in order of elution, [Ru5(CO)15(μ5-C4H2N2CHCH)(μ-H)2] (2) (20 mg, 25%) as red crystals after recrystallization from hexane/chlorobenzene and [Ru8(CO)24(μ7-C4H2N2CHC)(μ-H)3] (3) (18 mg, 22%) as orange crystals from hexane/dichloromethane at 4 °C. The minor bands were too small for complete characterization. Analytical and spectroscopic data for 2: anal. calcd for C21H6N2O15Ru5: C, 24.45; H, 0.59; N, 2.72. Found: C, 24.76; H, 0.62; N, 2.75%. IR (ν(CO), CH2Cl2): 2105s, 2080s, 2074vs, 2053vs, 2037vs, 2006s cm−1. 1H NMR (CD2Cl2, 233 K): δ 7.91 (d, J 6.0 Hz, 1H), 6.49 (s, 1H), 6.05 (s, 1H), 5.84 (d, J 6.0 Hz, 1H), −13.96 (s, 1H), −16.58 (s, 1H). Analytical and spectroscopic data for 3: anal. calcd for C30H6N2O24Ru8: C, 22.70; H, 0.38; N, 1.77. Found: C, 22.97; H, 0.41; N, 1.81%. IR (ν(CO), CH2Cl2): 2104s, 2094s, 2069s, 2066s, 2062s, 2044s, 2015s cm−1. 1H NMR (CD2Cl2, 298 K): δ 7.71 (d, J 4.0 Hz, 1H), 7.36 (d, J 4.0 Hz, 1H), 5.19 (s, 1H); hydride region: major isomer: δ −14.30 (s, 1H), −21.20 (s, 1H), −21.21 (s, 1H); minor isomer: δ −14.48 (s, 1H), −21.30 (s, 1H), −21.31 (s, 1H). Major:minor ca. 5:1.
X-ray crystallography
Single crystals suitable for X-ray diffraction analysis were grown by slow diffusion of hexane into a chlorobenzene (for 1) or CH2Cl2 (for 2 and 3) solution containing each product. Suitable crystals were mounted on a Nonius Kappa CCD diffractometer using a Nylon loop and Paratone oil and the diffraction data were collected at 150(1) K using Mo-Kα radiation (λ = 0.71073). Unit cell determination, data reduction, and absorption corrections were carried out using CrysAlisPro.28 The structures were solved with the ShelXS29 structure solution program by direct methods and refined by full-matrix least-squares on the basis of F2 using ShelXL29 within the OLEX2 (ref. 30) graphical user interface. All non-hydrogen atoms were anisotropically refined while the hydrogen atoms (except those directly bonded to metals and were isotropically refined) were included using a riding model. Pertinent crystallographic parameters are given in Table S1.† Two structures were found in the unit of 2 that showed no significant differences, allowing us to limit the bonding discussion to the one structure presented in the main body of the paper.Computational methodology and modeling details
The reported calculations were performed with the hybrid meta exchange–correlation functional M06,31 as implemented by the Gaussian 09 program package.32 The Ru atoms were described by Stuttgart–Dresden effective core potentials (ecp) and an SDD basis set, while a 6-31G(d') basis set was employed for the remaining atoms.The reported geometries represent fully optimized ground states (positive eigenvalues) and transition states (one imaginary eigenvalue) as verified from the analytical Hessian. The computed frequencies were used to make zero-point and thermal corrections to the electronic energies; the reported free energies (ΔG) are quoted in kcal mol−1 relative to the specified standard. The geometry-optimized structures have been drawn with the JIMP2 molecular visualization and manipulation program.33
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the Wazed Miah Science Research Center, Jahangirnagar University for collection of X-ray intensity data and for NMR spectroscopic data. Financial support from The Robert A. Welch Foundation through Grant B-1093 is appreciated, and the use of the computational resources through the High-Performance Computing Services and CASCaM (NSF-supported facility CHE-1531468) at the University of North Texas are acknowledged.Notes and references
- S. Oi, K. Sakai and Y. Inoue, Org. Lett., 2005, 7, 4009–4011 CrossRef CAS PubMed.
- (a) G. Dyker, Angew. Chem., Int. Ed., 1999, 38, 1698–1712 CrossRef; (b) F. Kakiuchi and S. Murai, Acc. Chem. Res., 2002, 35, 826–834 CrossRef CAS PubMed.
- S. Oi, Y. Tanaka and Y. Inoue, Organometallics, 2006, 25, 4773–4778 CrossRef CAS.
- A. G. Wong-Foy, L. M. Henling, M. Day, J. A. Labinger and J. E. Bercaw, J. Mol. Catal. A: Chem., 2002, 189, 3–16 CrossRef CAS.
- J. Navarro, E. Sola, M. Martín, I. T. Dobrinovitch, F. J. Lahoz and L. A. Oro, Organometallics, 2004, 23, 1908–1917 CrossRef CAS.
- H.-F. Klein, S. Camadanli, R. Beck, D. Leukel and U. Flörke, Angew. Chem., Int. Ed., 2005, 44, 975–977 CrossRef CAS PubMed.
- O. V. Ozerov, M. Pink, L. A. Watson and K. G. Caulton, J. Am. Chem. Soc., 2004, 126, 2105–2113 CrossRef CAS PubMed.
- M. L. Buil, M. A. Esteruelas, E. Goni, M. Oliván and E. Oñate, Organometallics, 2006, 25, 3076–3083 CrossRef CAS.
- J. N. Coalter III, W. E. Streib and K. G. Caulton, Inorg. Chem., 2000, 39, 3749–3756 CrossRef.
- P. Barrio, M. A. Esteruelas and E. Oñate, Organometallics, 2004, 23, 3627–3639 CrossRef CAS.
- M. A. Esteruelas, F. J. Fernández-Alvarez, M. Oliván and E. Oñate, J. Am. Chem. Soc., 2006, 128, 4596–4597 CrossRef CAS PubMed.
- B. Eguillor, M. A. Esteruelas, M. Oliván and E. Oñate, Organometallics, 2005, 24, 1428–1438 CrossRef CAS.
- K. Burgess, H. D. Holden, B. F. G. Johnson, J. Lewis, M. B. Hursthouse, N. P. C. Walker, A. J. Deeming, P. J. Manning and R. Peters, J. Chem. Soc., Dalton Trans., 1985, 85–90 RSC.
- W.-Y. Wong and W.-T. Wong, J. Organomet. Chem., 1996, 513, 27–29 CrossRef CAS.
- J. P.-K. Lau and W.-T. Wong, Inorg. Chem. Commun., 2003, 6, 174–177 CrossRef CAS.
- S. S. Chan, W.-Y. Wong and W.-T. Wong, J. Organomet. Chem., 1994, 474, C30–C33 CrossRef CAS.
- W.-Y. Wong, S. Chan and W.-T. Wong, J. Organomet. Chem., 1995, 493, 227–229 CrossRef.
- S. E. Kabir, F. Ahmed, A. Das, M. R. Hassan, D. T. Haworth, S. V. Lindeman, G. M. G. Hossain, T. A. Siddiquee and D. W. Bennett, J. Organomet. Chem., 2008, 693, 1696–1702 CrossRef CAS.
- K. A. Azam, D. W. Bennett, M. R. Hassan, D. T. Haworth, G. Hogarth, S. E. Kabir, S. V. Lindeman, L. Salassa, S. R. Simi and T. A. Siddiquee, Organometallics, 2008, 27, 5163–5166 CrossRef CAS.
- M. R. Al-Mamun, S. Ghosh, S. E. Kabir and G. Hogarth, J. Organomet. Chem., 2017, 849–850, 80–87 CrossRef CAS.
- R. Azpíroz, A. D. Giuseppe, V. Passarelli, J. J. Pérez-Torrente, L. A. Oro and R. Castarlenas, Organometallics, 2018, 37, 1695–1707 CrossRef.
- R. Beck, S. Camadanli, U. Flörke and H.-F. Klein, Eur. J. Inorg. Chem., 2015, 2543–2559 CrossRef CAS.
- S. E. Kabir and G. Hogarth, Coord. Chem. Rev., 2009, 253, 1285–1315 CrossRef CAS.
- Here the free energy of activation has been computed using the Eyring equation: ΔG‡ = RTc[ln(kBTc/hkc)], where the physical constants R, kB, and h have their normal meanings, and Tc and kc represent the coalescence temperature and the rate constant at coalescence. The value for kc has been estimated as a function of Δν according to the equation: kc = (0.5)1/2πΔν..
- S. A. Begum, M. A. H. Chowdhury, S. Ghosh, D. A. Tocher, M. G. Richmond, L. Yang, K. I. Hardcastle, E. Rosenberg and S. E. Kabir, RSC Adv., 2018, 8, 32672–32683 RSC.
- The exchange process involving hydride migration (H) between the non-benzylidene-bridged Ru–Ru bonds in 2 may be eliminated from consideration because both hydrides undergo exchange broadening at comparable rates. This phenomenon is inconsistent with the below process where the hydride (H) associated with the benzylidene-bridged Ru–Ru bond does not undergo site exchange with the other hydride (H)
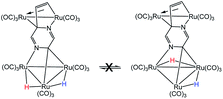 .
. - M. I. Bruce, B. K. Nicholson and M. L. Williams, Inorg. Synth., 1990, 26, 265–280 Search PubMed.
- CrysAlisPro, Oxford Diffraction, Yarnton, England, 2015 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- (a) JIMP2, version 0.091, a free program for the visualization and manipulation of molecules: M. B. Hall and R. F. Fenske, Inorg. Chem., 1972, 11, 768–775 CrossRef CAS; (b) J. Manson, C. E. Webster and M. B. Hall, Texas A&M University, College Station, TX, 2006, http://www.chem.tamu.edu/jimp2/index.html.
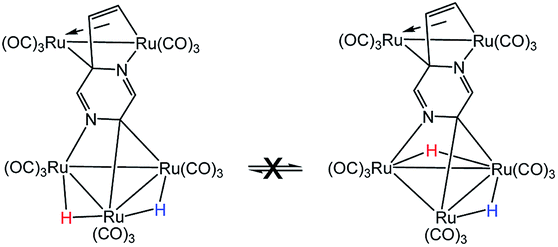
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1881604 (1), 1877756 (2), and 1877757 (3) contain the supplementary crystallographic data for compounds 1–3. Atomic coordinates of all DFT-optimized stationary points and transition states. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra03841h |
| This journal is © The Royal Society of Chemistry 2019 |