
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium catalysed carbonylation of 2-iodoglycals for the synthesis of C-2 carboxylic acids and aldehydes taking formic acid as a carbonyl source†
Ajaz Ahmedab,
Nazar Hussainab,
Monika Bhardwajb,
Anuj Kumar Chhalodiab,
Amit Kumarb and
Debaraj Mukherjee *ab
*ab
aAcademy of Scientific and Innovative Research, India
bNatural Product Chemistry Division, Indian Institute of Integrative Medicine, India. E-mail: dmukherjee@iiim.ac.in
First published on 17th July 2019
Abstract
Pd catalyzed carbonylative reaction of 2-iodo-glycals has been developed taking formic acid as a carbonyl source for the synthesis of 2-carboxylic acids of sugars by the hydroxycarbonylation strategy. The methodology was successfully extended to the synthesis of 2-formyl glycals by using a reductive carbonylation approach. Both ester and ether protected glycals undergo the reaction and furnished sugar acids in good yield which is otherwise not possible by literature methods. The C-2 sugar acids were successfully utilized for the construction of 2-amido glycals, 2-dipeptido-glycal by Ugi reaction and C-1 and C-2 branched glycosyl esters.
Sugar acids constitute a diverse family of carbohydrates1 which play a crucial role in cell–cell recognition, cellular adhesion, and virus–host recognition processes, for protection of cells from pathogen attachment, and in the synthesis of biologically active natural products.2 α,β-Unsaturated sugar acids such as zanamivir and ianinamivir (Fig. 1) are subjects of particular interest because of their application as inhibitors of different glycoproteins such as hemagglutinin (HA) and neuraminidase (NA),3 the major glycoproteins expressed by influenza viruses. While several reports dealing with the synthesis of carboxylic acids at C-6 and C-1 positions of sugars exist,4a,b there is no established procedure for the synthesis of C-2 carboxylic acids. In glycals accessing carboxylic group at C-1 position required t-butyl lithium and carbon dioxide treatment at −78 °C.4c There is only one report available in the literature in which carboxylic group was introduced to the C-2 position of glycals by Furstner et al.4c,d where the C-2 carboxylic acid was derived from the Pinnick oxidation of 2-formyl glycal obtained by classical Vilsmeier–Haack reaction5 and thereafter utilized in the total synthesis of bioactive natural orevactaene (exhibits HIV-1 inhibitory property). This strategy has certain drawbacks like long reaction times with cocktails of oxidants and limited substrate specificity. For example, it works only with ether protected sugars like tri-O-benzyl-D-glycal and fails with other base labile and silyl protecting groups. Further, recovery of 2-formyl glycals after base workup is rather low in our hand. Our experience with glycals6a–f encouraged us to formulate an attractive way to launch carboxylic acid at C-2 position of glycals as shown in Scheme 1 and apply them in the synthesis of C-2 glycoconjugates.
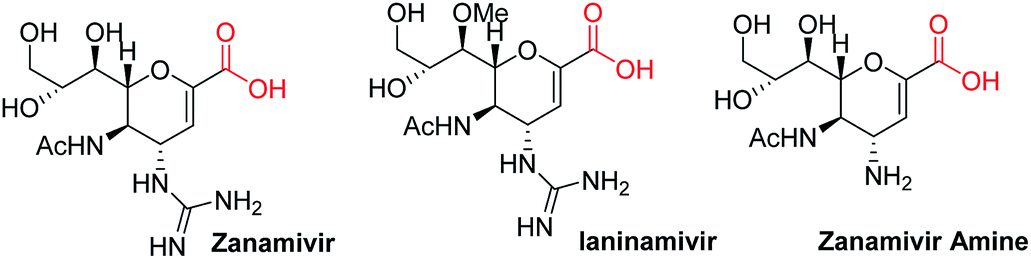 | ||
| Fig. 1 Glycal based acids in drugs. | ||
 | ||
| Scheme 1 Art of launching carboxyl group in sugars. | ||
This is pertinent to mention that carbonylation reactions of C-2 glycals have been successfully carried out by using metal carbonyl for the synthesis of C-2 branched glycoconjugates.7a,b In these reaction stoichiometric amount of costly Mo(CO)6 is required for such transformation that too ends up with some non-carbonylative side products. CO surrogates8,9 such as formic acid, formamide, chloroform and anhydride have been explored in recent times obviating metal carbonyls and CO gas. Among all formic acid is an attractive candidate for insertion of CO in an organic molecule,10 because it liberates one water molecule after releasing one CO molecule thereby making the process environmentally benign. We felt that palladium catalyzed hydroxycarbonylation of stable glycal halides, which are conveniently accessed from glycals in good yield, may prove to be the most effective and environmentally benign method to prepare such molecules. With our continuous interest in synthesis of C-2 branched sugars,11 this time we developed a reagent system for the direct synthesis of C-2 sugar carboxylic acids from 2-iodo glycals using formic acid as carbonyl source. Further, we transformed the synthesized acid for the synthesis of different C-2 branched glycoconjugates.
Preliminary experiments were conducted by using 2-iodoglycal 1a, HCOOH as carbonyl source, N,N′-dicyclohexylcarbodiimide, (DCC) as an activator and xantphos as a ligand. When 1a was reacted with 5 mol% Pd(OAc)2, 10 mol% of xantphos, 1 equiv. of DCC, 2 equiv. of formic acid and 2 equiv. of triethyl amine as base in DMF at 90 °C for 16 h the desired product 3a was obtained along with 3a′ in 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 ratio with overall 63% yield (Table 1, entry 1). Perusal of the literature revealed that ligands play a crucial role in such carbonylation reactions. In order to synthesize selectively the desired product 3a, we then decided to screen different ligands starting with the bidentate ligand L2, keeping other parameters as in entry 1. To our delight complete conversion of starting material was observed with 3a as exclusive product in 72% yield (Table 1, entry 2). Encouraged with this result, we next reduced the reaction period to 6 h (Table 1, entry 3) to note that the yield went up to 80%. Further reduction of the time period to 1.5 h still enabled complete conversion along with improved yield (81%) of 3a without any detrimental effect on selectivity (Table 1, entry 4), but reduction in reaction time to 1 h led to decrease in yield (Table 1 entry 5). Other ligands such as L3, L4, L5 and L6 produced mixture of both 3a and 3a′ along with poor overall yields (Table 1 entry 6–9). Next we investigated the role of different Pd catalysts such as Pd(PPh)3, Pd(TFA)2 and PdCl2 (Table 1 entry 10–12) and found that Pd(OAc)2 is the best catalyst for this transformation.
:40 ratio with overall 63% yield (Table 1, entry 1). Perusal of the literature revealed that ligands play a crucial role in such carbonylation reactions. In order to synthesize selectively the desired product 3a, we then decided to screen different ligands starting with the bidentate ligand L2, keeping other parameters as in entry 1. To our delight complete conversion of starting material was observed with 3a as exclusive product in 72% yield (Table 1, entry 2). Encouraged with this result, we next reduced the reaction period to 6 h (Table 1, entry 3) to note that the yield went up to 80%. Further reduction of the time period to 1.5 h still enabled complete conversion along with improved yield (81%) of 3a without any detrimental effect on selectivity (Table 1, entry 4), but reduction in reaction time to 1 h led to decrease in yield (Table 1 entry 5). Other ligands such as L3, L4, L5 and L6 produced mixture of both 3a and 3a′ along with poor overall yields (Table 1 entry 6–9). Next we investigated the role of different Pd catalysts such as Pd(PPh)3, Pd(TFA)2 and PdCl2 (Table 1 entry 10–12) and found that Pd(OAc)2 is the best catalyst for this transformation.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Pd source | Ligand | Time (h) | Conversion (%) | (3a:3a′) |
Yieldb (overall)% |
| a Reaction conditions: 1a (0.18 mmol), 2a (0.36 mmol), Pd(OAc)2 (0.009 mmol), L2 (0.018 mmol), N,N′-dicyclohexylcarbodiimide (DCC) (0.18 mmol), triethylamine (0.36 mmol) at 90 °C for 2 h.b Yield of isolated product. Pd 5 mol% and ligand 10 mol% were used. Ratio of 3a and 3a′ and conversion were determined through 1H NMR. | ||||||
| 1 | Pd(OAc)2 | L1 | 16 | 90 | 60:40 |
63 |
| 2 | Pd(OAc)2 | L2 | 16 | 99 | >99:1 |
72 |
| 3 | Pd(OAc)2 | L2 | 6 | 99 | >99:1 |
80 |
| 4 | Pd(OAc)2 | L2 | 1.5 | 99 | >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1 |
81 |
| 5 | Pd(OAc)2 | L2 | 1 | 90 | >99:1 |
73 |
| 6 | Pd(OAc)2 | L3 | 16 | 40 | 45:55 |
23 |
| 7 | Pd(OAc)2 | L4 | 16 | 30 | 30:70 |
15 |
| 8 | Pd(OAc)2 | L5 | 16 | 10 | — | Traces |
| 9 | Pd(OAc)2 | L6 | 16 | 10 | — | Traces |
| 10 | Pd(PPh)3 | L2 | 2 | 75 | >99:1 |
21 |
| 11 | Pd(TFA)2 | L2 | 2 | 35 | >99:1 |
27 |
| 12 | PdCl2 | L2 | 2 | 23 | >99:1 |
11 |

Utilising the optimised reaction condition (Table 1, entry 4), we then checked the substrate scope (Table 2) using different 2-iodo-glycals. Di-O-benzyl-2-iodo-L-rhamnal 1b was tested to get a better yield of the product 3b, (85%). The galactal substrate also furnished the desired 2-carboxyl galactal 3c in good yield (76%). In order to broaden the substrate scope the reactivity of glycals protected with different protecting groups was next investigated. Gratifyingly, 2, 3-acetonide protected 2-iodo-D-galactal 1d survived under the reaction condition and yielded the product 3d in good yield (75%). Tri-O-ethyl-2-iodoglucal 1e also reacted well and formed the respective acid derivative 3e in (76%) yield. Next we utilized different glycals having silicon based protection or ester protection. Tri-O-acetyl-2-iodoglucal 1f and di-O-acetyl-2-iodoxylal 1g were also well tolerated under the reaction condition and gave the desired products 3f–3g in reasonable yields (73–75%). 2-Bromo-glucal was next tested under the optimized reaction condition and the desired compound 3a was obtained in good yield although it takes 3 h to complete the reaction.
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Time (h) | Yieldb (%) |
| a Reaction conditions: 1 (1 equiv.), 2a (2 equiv.), Pd(OAc)2 (5 mol%), L2 (10 mol%), DCC (1 equiv.), triethylamine (2 equiv.) at 90 °C for 1.5 to 2.5 h in 3 mL of DMF.b Yield of isolated product. | ||||
| 1 | 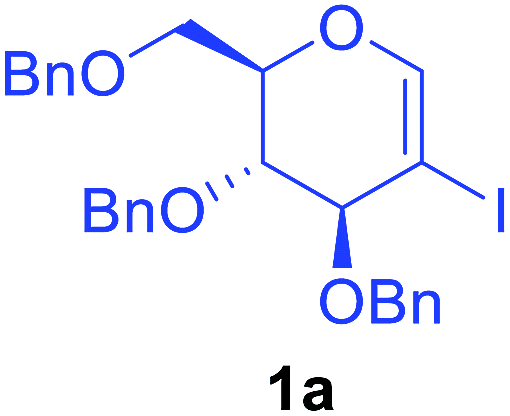 |
 |
1.5 | 81 |
| 2 |  |
 |
1.5 | 85 |
| 3 | 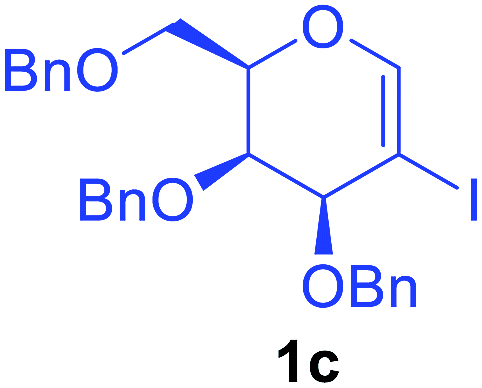 |
 |
1.5 | 76 |
| 4 |  |
 |
1.5 | 75 |
| 5 |  |
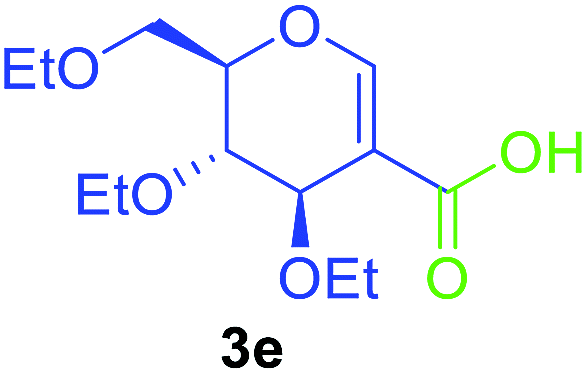 |
2 | 76 |
| 6 |  |
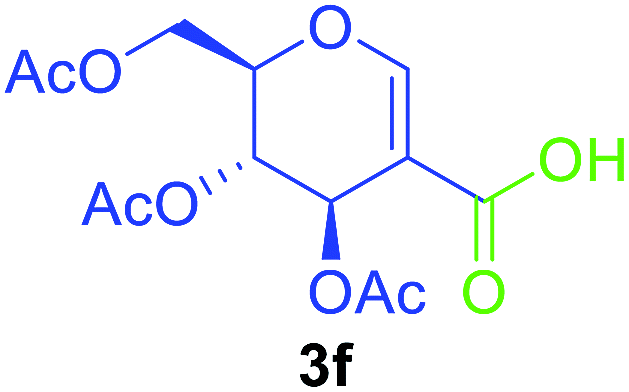 |
2.5 | 73 |
| 7 | 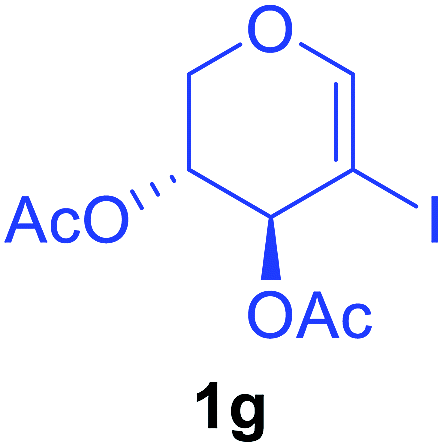 |
 |
2.5 | 75 |

TBS protected sugars acids are used in the total synthesis of various bioactive natural products by activating the anomeric carbon which is otherwise not possible with other protecting groups. In the literature in order to get such types of acids multiple steps are required along with poor overall yield. By utilizing carbonylation strategy under Pd catalysis we were able to synthesis the TBS protected acids in just 2 h when substrate 1h was reacted with HCOOH under Pd catalysis to generate product 3h in good yield.
After successful execution of our strategy for hydroxycarbonylation of 2-iodoglycals, we became interested to apply the same carbonylative approach for the synthesis of 2-formyl glycals. In the literature formylation at C-2 of glycals has been carried out either by Vilsmeier–Haack or XtalFluor-E catalyzed reactions (Scheme 3).5,12 We utilized the reductive carbonylation approach employing triethylsilyl hydride (1.2 equiv.) as a hydride source keeping other reagents same as for acid synthesis. To our delight, the required 2-formyl glycals were obtained in good to excellent yields (Scheme 3, 3aa–3ae).
 | ||
| Scheme 2 Synthesis of silyl protected C-2 carboxylic acid. | ||
 | ||
| Scheme 3 Synthesis of 2-formyl glycals. | ||
To test the utility of sugar acids in the synthesis of C-2 glycoconjugates, sugar acid 3a was successfully utilized for the synthesis of C-2 linked dipeptides via Ugi reaction (Scheme 4, 5a). The simple amide 5b was also synthesised from 3a using thionyl chloride and ammonium hydroxide; the product is otherwise difficult to synthesize via the existing methods. Where substituted amides are synthesized. The acid chloride of 3a could be successfully coupled with 8-aminoquinoline, leading to the amide 5c in good yield. In order to test the reactivity of the acid 3a, we used it as a glycosyl acceptor in glycosylation reaction by treating with a suitable glycosyl donor like 6, when the pseudodisaccharide 7 was isolated in good yield (63%) with a mixture of anomers. On the other hand, treatment with aryne precursor 8 resulted in the formation of compound 9 in excellent yield (84%) via coupling of aryne with the sugar acid. When sugar acid 3a was treated with thiophenol thioester 11 was isolated in excellent yield (Scheme 5).
 | ||
| Scheme 4 Synthesis of amides using 3a. Reaction conditions: (a) 3a (1.5 equiv.), aniline (1 equiv.), anisaldehyde (1 equiv.), cyclohexyl isocyanate (1 equiv.) in ethanol at rt for 48 h; (b) 3a (1.0 equiv.), SOCl2 (1.5 equiv.), NH4OH (37%, 2 mL) in THF for 2 h; (c) 3a (1.0 equiv.), PCl5 (1.2 equiv.), pyridine (6 equiv.), 8-aminoquinoline (1.2 equiv.) in DCM for 5 h. | ||
 | ||
| Scheme 5 Utilization of 3a for establishing ester linkages. | ||
A plausible reaction mechanism has been proposed for hydroxycarbonylation of 2-iodoglycals (Scheme 6). Initially Pd(0) is generated in situ in the presence of ligand Ln. The catalytic cycle then starts with oxidative addition of Pd(0) to 2-iodoglycal A which produces the pallado complex B.
 | ||
| Scheme 6 Plausible mechanism of hydroxycarbonylation reaction. | ||
Coordination and insertion of carbon monoxide generated in situ by the combination of DCC and formic acid leads to the formation of acyl Pd(II) complex C. Formic acid attack on complex C via transmetallation affords the intermediate D with release of HI. Pd(0) could be regenerated for the next catalytic cycle after reductive elimination from complex D with formation of anhydride E. Decomposition of the anhydride with release of one molecule of CO generates the desired sugar acid F.
In conclusion we have developed an efficient and mild Pd catalysed synthetic strategy for hydroxycarbonylation of 2-iodoglycals using the cheap reagent formic acid as CO source and 1 equiv. of DCC as an activator. The methodology was successfully extended to various glycals with different protecting groups like acetonide, ether, ester and silicon based ones. 2-Formyl glycals were also synthesised by using reductive carbonylation approach. The synthesised sugar acid could be used in the synthesis of glycoconjugates, pseudodisaccharides, for aryl ester and thioester.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. A. and N. H. thank CSIR and UGC New Delhi for SRF/JRF. IIIM Publication No. IIIM/2298/2019.Notes and references
- (a) A. Varki, Glycobiology, 1993, 3, 97 CrossRef CAS PubMed; (b) R. Schauer, Glycoconjugate J., 2000, 17, 485 CrossRef CAS; (c) T. Angata and A. Varki, Chem. Rev., 2002, 102, 439 CrossRef CAS.
- (a) A. Varki, Glycobiology, 1993, 3, 97 CrossRef CAS; (b) P. M. Rudd, T. Elliott, P. Cresswell, I. A. Wilson and R. A. Dwek, Science, 2001, 291, 2370 CrossRef CAS; (c) S. Borman, Chem. Eng. News, 2007, 85, 19 CrossRef; (d) P. H. Seeberger and D. B. Werz, Nature, 2007, 446, 1046 CrossRef CAS; (e) D. P. Galonic and D. Y. Gin, Nature, 2007, 446, 1000 CrossRef CAS; (f) B. Ernst and J. L. Magnani, Nat. Rev., 2009, 8, 661 CAS; (g) M. C. Galan, D. Benito-Alifonso and G. M. Watt, Org. Biomol. Chem., 2011, 9, 3598 RSC; (h) T. M. Gloster and D. J. Vocadlo, Nat. Chem. Biol., 2012, 8, 683 CrossRef CAS PubMed; (i) S. J. N. Devi, R. Schneerson, W. Egan, W. F. Vann, J. B. Robbins and J. Shiloach, Infect. Immun., 1991, 59, 732 CAS; (j) H. J. Jennings, R. Roy and F. J. Michon, Immunol, 1985, 134, 2651 CAS.
- (a) N. K. Sauter, J. E. Hanson, G. D. Glick, J. H. Brown, R. L. Crowther, S. J. Park, J. J. Skehel and D. C. Wiley, Biochemistry, 1992, 31, 9609 CrossRef CAS; (b) M. Mammen, G. Dahmann and G. J. Whitesides, J. Med. Chem., 1995, 38, 4179 CrossRef CAS; (c) P. M. Colman, in The influenza Viruses: Influenza Virus Neuraminidase, Enzyme and Antigen, ed. R. M. Krug, Plenum Press, New York, 1989, pp. 175–218 Search PubMed; (d) P. M. Colman, Protein Sci., 1994, 3, 1687 CrossRef CAS PubMed.
- (a) H. Lijun, T. Nardos and H. Xuefei, Chem.–Eur. J., 2006, 12, 5246 CrossRef; (b) S. Combemale, J. N. A. Evoung, S. Houaidji, R. Bibi and V. B. Montero, Molecules, 2014, 19, 1120 CrossRef; (c) P. Lesimple, J. M. Beau, G. Jaurand and P. Sinaji, Tetrahedron Lett., 1986, 27, 6201 CrossRef CAS; (d) J. Preindl, K. Jouvin, D. Laurich, G. Seidel and A. Furstner, Chem.–Eur. J., 2016, 22, 237–247 CrossRef CAS.
- N. G. Ramesh and K. K. Balasubramanian, Tetrahedron Lett., 1991, 32, 3875 CrossRef CAS.
- (a) A. K. Kusunuru, K. Chatanya, M. B. Tatina, V. B. Prasad and D. Mukherjee, Org. Lett., 2015, 17, 3742 CrossRef CAS; (b) M. B. Tatina, A. K. Kusunuru and D. Mukherjee, Org. Lett., 2015, 17, 4624 CrossRef CAS; (c) M. R. Lambu, S. K. Yousuf, D. Mukherjee and S. C. Taneja, Org. Biomol. Chem., 2012, 10, 9090 RSC; (d) M. B. Tatina, S. K. Yousuf and D. Mukherjee, Org. Biomol. Chem., 2012, 10, 5357 RSC; (e) S. K. Yousuf, S. C. Taneja and D. Mukherjee, Org. Lett., 2011, 13, 576 CrossRef CAS; (f) N. Hussain, M. B. Tatina, F. Rasool and D. Mukherjee, Org. Biomol. Chem., 2016, 14, 9989 RSC.
- (a) A. Bordessa, A. Ferry and N. L. Germain, J. Org. Chem., 2016, 81, 12459–12465 CrossRef CAS; (b) M. P. Darbem, K. S. Kanno, I. M. de Oliveira, C. Henrique, A. Esteves, D. C. Pimentac and H. A. Stefani, New J. Chem., 2019, 43, 696–699 RSC.
- (a) Y. L. Zhao, P. L. Jin and A. Lei, J. Am. Chem. Soc., 2008, 130, 9429 CrossRef CAS; (b) H. Zhang, R. Shi, P. Gan, C. Liu, A. Ding, Q. Wang and A. Lei, Angew., Chem. Int. Ed. Engl., 2012, 51, 5204 (Angew. Chem., 2012, 124, 5294) CrossRef CAS; (c) L. R. Odell, F. Russo and M. Larhed, Synlett, 2012, 23, 685 CrossRef CAS; (d) N. F. K. Kaiser, A. Hallberg and M. Larhed, J. Comb. Chem., 2002, 4, 109 CrossRef CAS; (e) J. Wannberg and M. Larhed, J. Org. Chem., 2003, 68, 5750 CrossRef CAS.
- (a) P. Kannaboina, G. Raina, A. Kumar and P. Das, Chem. Commun., 2017, 53, 9446 RSC; (b) P. Hermange, A. T. Lindhardt, R. H. Taaning, K. Bjerglund, D. Lupp and T. Skrydstrup, J. Am. Chem. Soc., 2011, 133, 6061 CrossRef CAS; (c) S. D. Friis, R. H. Taaning, A. T. Lindhardt and T. Skrydstrup, J. Am. Chem. Soc., 2011, 133, 18114 CrossRef CAS; (d) X. Wu, Y. Zhao and H. Ge, J. Am. Chem. Soc., 2015, 137, 4924 CrossRef CAS PubMed; (e) C. Brancour, T. Fukuyama, Y. Mukai, T. Skrydstrup and I. Ryu, Org. Lett., 2013, 15, 2794 CrossRef CAS.
- (a) J. Peng, F. Wu, C. Li, X. Qi and X. Wu, Eur. J. Org. Chem., 2017, 1434 CrossRef CAS; (b) F. Peng, J. Wu, B. Peng, Q. Xinxin and F. W. Xiao, J. Org. Chem., 2017, 82, 9710 CrossRef.
- (a) N. Hussain, M. B. Tatina and D. Mukherjee, Org. Biomol. Chem., 2018, 16, 2666 RSC; (b) N. Hussain, K. Jana, B. Ganguly and D. Mukherjee, Org. Lett., 2018, 20, 1572 CrossRef CAS.
- R. Majdouline, V. Frédéric, M. Julien and F. P. Jean, J. Org. Chem., 2018, 83, 8731 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03626a |
| This journal is © The Royal Society of Chemistry 2019 |