
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlyme–Li salt equimolar molten solvates with iodide/triiodide redox anions†
Keisuke Shigenobu,
Azusa Nakanishi,
Kazuhide Ueno *,
Kaoru Dokko and
Masayoshi Watanabe
*,
Kaoru Dokko and
Masayoshi Watanabe
Department of Chemistry and Biotechnology, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama 240-8501, Japan. E-mail: ueno-kazuhide-rc@ynu.ac.jp; Fax: +81-45-339-3951; Tel: +81-45-339-3951
First published on 23rd July 2019
Abstract
Room-temperature-fused Li salt solvates that exhibit ionic liquid-like behaviour can be formed using particular combinations of multidentate glymes and lithium salts bearing weakly coordinating anions, and are now deemed a subset of ionic liquids, viz. solvate ionic liquids (SILs). Herein, we report redox-active glyme–Li salt molten solvates consisting of tetraethyleneglycol ethylmethyl ether (G4Et) and lithium iodide/triiodide, [Li(G4Et)]I and [Li(G4Et)]I3. The coordination structure of the complex ions and the thermal, transport, and electrochemical properties of these molten Li salt solvates were investigated to diagnose whether they can be categorized as SILs. [Li(G4Et)]+ and I3− were found to remain stable as discrete ions and exist as well-dissociated forms in the liquid state, indicating that [Li(G4Et)]I3 can be classified as a good SIL. This study also clarified that the I− and I3− counter anions exhibit an electrochemical redox reaction in the highly concentrated molten Li salt solvates. The redox-active molten Li solvates were further studied as a highly concentrated catholyte for use in rechargeable semi-liquid lithium batteries. Although the cell constructed using [Li(G4Et)]I3 failed to charge after the initial discharge step, the cell containing [Li(G4Et)]I demonstrates reversible charge–discharge behaviour with a high volumetric energy density of 180 W h L−1 based on the catholyte volume.
1. Introduction
Molten salt solvates and highly concentrated electrolyte solutions with increased salt concentration near their saturation limit are in the limelight as possible replacements for conventional organic electrolyte solutions used in electrochemical devices because they afford improved thermal stability, reduced vapor pressure, and enhanced electrochemical stability.1–3 Moreover, certain concentrated electrolytes have been reported to show improved performance in Li-ion batteries, such as higher rate capability and enhanced stable charge–discharge cycling of the high-voltage cathode and metallic Li anode when compared to conventional organic liquid electrolytes.4–9Glyme-based molten Li salt solvates are of particular interest and have been intensively studied because certain stoichiometric mixtures of glymes and Li salts are found to behave like ionic liquids (ILs).10,11 Molten Li salt solvates with long-lived [Li(glyme)]+ complex cations are deemed a new family of ILs, solvate ionic liquids (SILs).12 Numerous combinations of glymes and metal salts have been examined; however, only limited cases using multidentate glymes and metal salts with bulky and weakly coordinating anions, such as bis(trifluoromethanesulfonyl)amide ([TFSA]−), act as good SILs.13 However, these conventional anions only compensate for the positive charge on the Li ions and may have a less active role in the electrochemical properties even though they account for approximately half of the dense electrolyte material weight. In this respect, it is of great research interest to introduce an electrochemically-active functionality into the counter anions used in SILs. The redox-active, “two-in-one” SILs wherein the [Li(glyme)]+ complex cation is responsible for Li ion transport and the redox-active anion imparts reversible redox activity have a potential to be used as a component of catholyte for semi-liquid rechargeable lithium batteries when Li metal is combined as the anode.
In a recent study, one of the authors employed a metal complex anion, tetrabromoferrate ([FeBr4]−), to demonstrate redox-active glyme–Li salt SILs consisting solely of metal complex ions, in which the [FeBr4]− complex anion undergoes reversible redox reaction (i.e., [FeIIIBr4]− + e− ⇄ [FeIIBr4]2−) at ∼3 V vs. Li/Li+.14 However, the SIL with [FeBr4]− needs to be diluted with another SIL for use as a SIL-based catholyte due to its relatively high melting point, which leads to low energy density (37 W h L−1 based on the catholyte volume) as a consequence of the reduced [FeBr4]− concentration.
The highly reversible redox activity of the I−/I3− couple has long been utilized in a variety of electrochemical devices, including dye-sensitized solar cells,15,16 metal–I2 batteries,17–19 and flow batteries.20–22 In recent studies, the I−/I3− couple has also been employed as a redox mediator to improve the charge–discharge performance of high-energy-density Li–O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23,24 and Li–S25,26 batteries. Extensive studies on IL-based electrolytes used for dye-sensitized solar cells have shown that iodide-based ILs have a relatively low melting point and are prone to exist as super-cooled liquids without crystallization.27–29 In this study, we have prepared novel redox-active glyme–Li salt solvates based on the I−/I3− couple in anticipation of melting point depression and anti-crystallization of the glyme–Li salt solvates. The thermal properties, coordination structure, and transport properties of redox-active glyme–Li salt solvates based on the I−/I3− couple were investigated. Furthermore, the two-electron transfer reaction of the I−/I3− couple (i.e., I3− + 2e− ⇄ 3I−) offers an increase in the theoretical capacity of the redox-active SIL when compared to the SIL-based catholyte prepared with [FeBr4]−. This study also reports the use of the neat redox-active SIL as a highly concentrated catholyte (∼3 mol dm−3) in semi-liquid rechargeable lithium batteries.
23,24 and Li–S25,26 batteries. Extensive studies on IL-based electrolytes used for dye-sensitized solar cells have shown that iodide-based ILs have a relatively low melting point and are prone to exist as super-cooled liquids without crystallization.27–29 In this study, we have prepared novel redox-active glyme–Li salt solvates based on the I−/I3− couple in anticipation of melting point depression and anti-crystallization of the glyme–Li salt solvates. The thermal properties, coordination structure, and transport properties of redox-active glyme–Li salt solvates based on the I−/I3− couple were investigated. Furthermore, the two-electron transfer reaction of the I−/I3− couple (i.e., I3− + 2e− ⇄ 3I−) offers an increase in the theoretical capacity of the redox-active SIL when compared to the SIL-based catholyte prepared with [FeBr4]−. This study also reports the use of the neat redox-active SIL as a highly concentrated catholyte (∼3 mol dm−3) in semi-liquid rechargeable lithium batteries.
2. Experimental
2.1 Materials
Purified tetraethyleneglycol ethylmethyl ether (G4Et, water content <50 ppm) was kindly supplied by Nippon Nyukazai and lithium bis(trifluoromethanesulfonyl)amide (Li[TFSA], >99.9%, water content <100 ppm) was provided by Solvay Japan. Lithium iodide (LiI, >99.9%, Sigma Aldrich) and iodine (I2, >99.8%, FUJIFILM Wako) were used as received without any further purification. All glyme–Li salt complexes were prepared in an argon-filled glove box ([H2O] <1 ppm; [O2] <1 ppm). G4Et and LiI were mixed in a 1:1 molar ratio at 45 °C to form [Li(G4Et)]I. [Li(G4Et)]I and iodine were mixed in an equimolar ratio and heated at 60 °C to form [Li(G4Et)]I3.
2.2 Measurements
The melting point (Tm) and glass transition temperature (Tg) were determined using differential scanning calorimetry (DSC7020, Hitachi High-Tech Science). All samples were hermetically sealed in aluminium pans in an argon-filled glove box. The sample pans were first pre-heated to 100 °C to erase thermal history. Then they were cooled to −150 °C, and heated to 100 °C at a heating rate of 5 °C min−1 under a nitrogen atmosphere. Both Tm and Tg were calculated from the onset temperatures of the second heating thermograms. The thermal stabilities of the samples were investigated using isothermal thermogravimetric analysis (isothermal TGA) on a TG/DTA STA7200 instrument (Hitachi High-Tech Science) at 100 °C for 140 min under a nitrogen atmosphere.The viscosity and density of [Li(G4Et)]I were measured simultaneously on a Stabinger viscometer (SVM300, Anton Paar). The viscosity and density of [Li(G4Et)]I3 were measured on a density meter (DA-100, Kyoto Denshi Kogyo) and rheometer (Physica MCR 301, Anton Paar), respectively.
Raman spectra were recorded on a laser Raman spectrometer (NRS-4100, JASCO equipped with a 27 mW, 785 nm laser) at a resolution of ∼4 cm−1 and calibrated using a polypropylene standard. The samples were enclosed in a capillary tube to avoid moisture adsorption during the measurement. The sample temperature was adjusted to 30 °C for G4Et and [Li(G4Et)]I, and 60 °C for [Li(G4Et)]I3 using a Peltier microscope stage (TS62, INSTEC) equipped with a temperature controller (mk1000, INSTEC). For the low wavenumber region (100–300 cm−1), [Li(G4Et)]I3 was diluted to 1 mol dm−3 with acetonitrile in order to reduce fluorescence interference. The UV-vis spectra were recorded on a UV-vis spectrometer (UV-2700, Shimadzu) using a cell with an optical path length of 10 mm.
The ionic conductivity was measured using the complex impedance method in the frequency range of 500 kHz to 1 Hz at an alternating voltage amplitude of 200 mV (VMP2, Biologic). Two platinum black electrode cells, whose cell constant was determined using a 0.01 M KCl aqueous solution at 25 °C prior to the measurements, were soaked in each sample and thermally equilibrated at each temperature for at least 1 h within a thermostat chamber.
Cyclic voltammetry was performed on a potentiostat/galvanostat (ModuLab XM ECS, Solartron Analytical) using a three-electrode cell consisting of a platinum disk micro electrode (ca. 10 μm in diameter) as the working electrode, platinum wire as the counter electrode, and lithium metal soaked in 1 mol dm−3 Li[TFSA] in triglyme (G3) as the reference electrode. The diameter of the microelectrode was calibrated using a standard electrolyte solution (0.1 mol dm−3 n-tetrabutylammonium hexafluorophosphate in acetonitrile solution containing 2 mmol dm−3 ferrocene). The Li/Li+ reference electrode was separated from the sample solution by a liquid junction with a Vycor glass frit.
Pulsed field gradient nuclear magnetic resonance (PFG-NMR) measurements were performed on a ECX400 spectrometer (JEOL) equipped with a 9.4 T (400 MHz) narrow bore superconducting magnet, a pulse gradient probe (JEOL), and current amplifier to clarify the self-diffusion coefficients of each component in the sample mixtures, G4Et (1H, 399.7 MHz) and lithium cation (7Li, 155.3 MHz). The experimental procedure for PFG-NMR was described in detail in our previous report.30
The charge and discharge tests were performed using a two-compartment cell separated by a lithium-ion conductive glass ceramic (OHARA, LICGC, 19 mm in diameter), and the iodide/triiodide-based SIL electrolyte used as the catholyte. Carbon paper (TGP-H-060, TORAY, 13 mm in diameter) coated with Ketjen black (EC-600JD, LION) was used as the current collector for the catholyte and the Li metal disk electrode (13 mm in diameter) was used as the anode. For the lithium metal anode, 1 mol dm−3 Li[TFSA] in G4Et was also used as the anolyte to avoid direct contact between the lithium metal and LICGC. A porous glass filter paper (Advantec, GA55, 17 mm in diameter) was employed to retain the catholyte and anolyte in each compartment. Li|anolyte|LICGC|catholyte cells were assembled in an argon-filled glove box. The galvanostatic charge and discharge tests were performed on an automatic charge and discharge instrument (HJ1001SM8A, Hokuto Denko) at 60 °C, and the cut-off voltages were set at 2.2 V and 3.3 V for the discharge and charge steps, respectively. For the cell constructed using the [Li(G4Et)]I3-based catholyte, the charge and discharge cycles were started from the discharge step.
2.3 Density functional theory (DFT) calculations
DFT calculations were performed using the Gaussian 09 program.31 The geometries of the [Li(G4Et)]+ complex cation were fully optimized at the B3LYP/6-311+G** level of theory. Vibrational analysis of the optimized geometry was also carried out at the same level of theory.3. Results and discussion
3.1 Thermal properties
In a previous work, LiI was found to form a 1:1 crystalline solvate with G3 or tetraglyme (G4), but their melting points were higher than room temperature (Tm = 96 °C and 60 °C for [Li(G3)]I and [Li(G4)]I, respectively).32 We previously reported that introducing asymmetry into each end group of the glyme molecules effectively reduces the Tg and Tm of SILs.33 The entropy of fusion increases upon increasing the chain length of the alkyl terminal group in the asymmetric glyme molecules since the conformational degrees of freedom upon melting increases.33,34 Thus, we expected that a low melting 1:1 solvate of LiI may be formed using asymmetric G4Et. The Tm and Tg values of the studied samples are summarised in Table 1. [Li(G4Et)]I was prone to remain as a supercooled liquid in the sample vial over several days, which allowed us to perform further measurements in the liquid state at room temperature, however, [Li(G4Et)]I3 readily crystalized at room temperature although Tm was lower than that of [Li(G4Et)]I. As expected, the Tm of [Li(G4Et)]I was lower than that of [Li(G3)]I and [Li(G4)]I, but still higher than room temperature. Tm of [Li(G4Et)]I3 is lower than that of [Li(G4Et)]I, and the reduction of Tm upon the addition of solid I2 to [Li(G4Et)]I suggests that the cation–anion interaction is weakened via the formation of the charge-delocalized I3−. Indeed, it is known that the addition of a Lewis acid (e.g., AlCl3) to imidazolium halide salts significantly reduces Tm and Tg, and gives room temperature ionic liquids consisting of the imidazolium cation and weakly coordinating Lewis acid–base adduct anions, such as [AlCl4]−.35,36 Likewise, room temperature ionic liquids with polyiodide [Im] (1≤ m ≤ 8) can be formed upon the addition of I2 to imidazolium iodide salts.37,38
| Sample | Tg [°C] | Tm [°C] |
|---|---|---|
| a n.d. = not detected. | ||
| G4Et | n.d. | −29 |
| [Li(G4Et)]I | −56 | 47 |
| [Li(G4Et)]I3 | n.d. | 38 |
The thermal stability of the solvates was studied using isothermal TGA at 100 °C, as shown in Fig. 1. [Li(G4Et)]I shows discernible weight loss, indicating the gradual evaporation of G4Et at 100 °C. On the contrary, the weight loss was negligible for [Li(G4Et)]I3 and its thermal stability was almost the same as that observed for a prototypical SIL, [Li(G4Et)][TFSA], under the same conditions.33 The higher thermal stability of [Li(G4Et)]I3 was attributed to the enhanced interaction between the Li ions and G4Et. The formation of charge-delocalized I3− results in the reduced interaction with Li ions, which in turn enhances the stability of the [Li(G4Et)]+ complex cation.
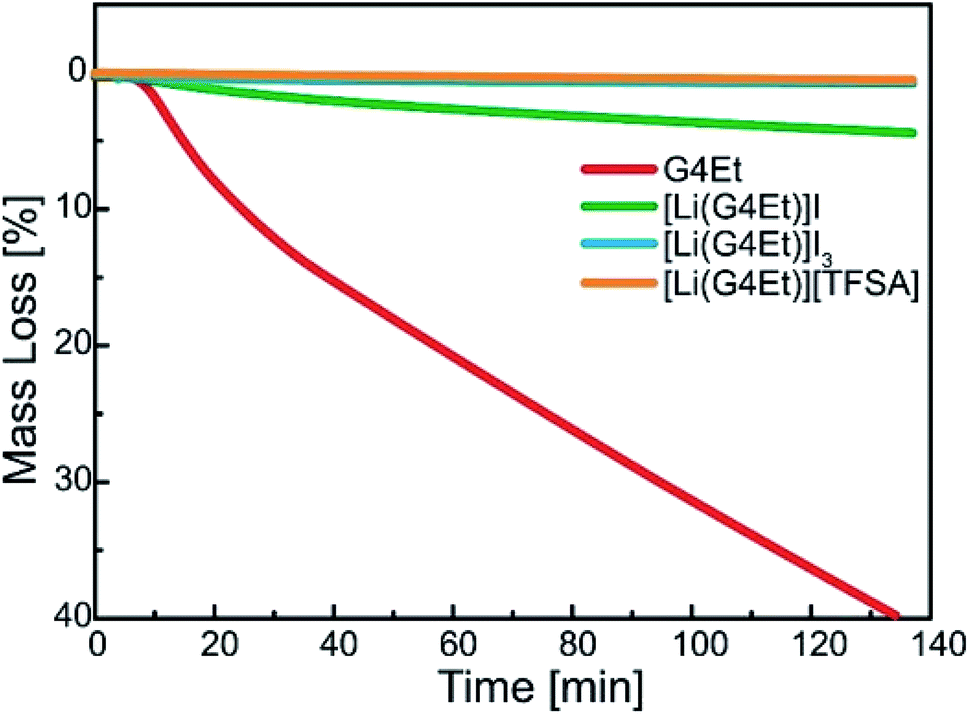 | ||
| Fig. 1 Isothermal thermogravimetric data obtained for G4Et, [Li(G4Et)]I, [Li(G4Et)]I3, and [Li(G4Et)][TFSA] at 100 °C. | ||
3.2 Coordination structure
The Li ion coordination structure has been studied for a series of glyme–Li salt solvates in crystalline and molten states, and a variety of glyme conformations, such as helical polymeric [Lin(glyme)n]n+, dimeric [Li2(glyme)2]2+, and crown-ether-like monomeric [Li(glyme)]+, and has been found to be dependent on the chain length of the glyme molecules and the counter anion of the Li salt used.39–41 In the molten state of G3 and G4-based 1:1 solvates with Li salts, the glyme molecules were found to adopt a crown-ether like conformation to form monomeric [Li(glyme)]+.42 Fig. 2a shows the Raman spectra of the liquid samples, G4Et, [Li(G4Et)]I, and [Li(G4Et)]I3, in the wavenumber range 800–900 cm−1, where the bands can be ascribed to the mixed mode of CH2 rocking and C–O–C stretching vibrations of the glyme molecules.43,44 Except for the neat G4Et, a strong band emerges at ∼870 cm−1 along with weaker bands in the range of 800–850 cm−1. The strong band observed at ∼870 cm−1 can be assigned to the symmetric ring breathing mode of the [Li(G4Et)]+ complex cation with a crown ether-like conformation, similar to the previously reported [Li(G3)]+ and [Li(G4)]+ complex cations.43 A comparison between the experimental and calculated Raman bands confirms the presence of a crown ether-like [Li(G4Et)]+ cation (Fig. 2b); the calculated Raman bands of the optimized [Li(G4Et)]+ complex cation can explain the experimental Raman bands observed for [Li(G4Et)]I and [Li(G4Et)]I3 although the calculated bands appear at a slightly higher wavenumber when compared to the corresponding experimental bands.
 | ||
| Fig. 2 (a) The experimental Raman spectra recorded for G4Et, [Li(G4Et)]I, and [Li(G4Et)]I3 at 60 °C and (b) the calculated Raman spectrum of the optimized [Li(G4Et)]+ complex cation. | ||
The formation of I3− anions was also studied using Raman and UV-vis spectroscopy. Fig. 3a illustrates the Raman spectrum of 1 mol dm−3 [Li(G4Et)]I3 in acetonitrile in the wavenumber range of 125–180 cm−1. The bands observed at 125, 150, and 180 cm−1 can be assigned to the asymmetric triiodide stretching vibration, Fermi resonance, and I–I stretching of the ion pair (namely, [Li(G4Et)]+ I3− in this work), respectively, as previously reported.45 In Fig. 3b, the UV-vis spectra of [Li(G4Et)]I and [Li(G4Et)]I3 diluted with acetonitrile also show characteristic splitting bands from 200 nm to 250 nm for I−, and 290 nm to 360 nm for I3−, respectively. Consequently, the above results confirm that the stable complex ions, [Li(G4Et)]+ and I3−, were successfully formed in the liquid state of [Li(G4Et)]I3.
 | ||
| Fig. 3 (a) The Raman spectrum recorded for 1 mol dm−3 [Li(G4Et)]I3 in acetonitrile. (b) The UV-vis spectra recorded for 1 mmol dm−3 [Li(G4Et)]I and [Li(G4Et)]I3 in acetonitrile. | ||
3.3 Transport and electrochemical properties
The viscosity and ionic conductivity of [Li(G4Et)]I and [Li(G4Et)]I3 are listed in Table 2 along with the density and Li salt concentration at 60 °C. The viscosity of [Li(G4Et)]I3 is one order of magnitude lower than that of [Li(G4Et)]I. This suggests that I3− has a much weaker cation–anion interaction with the complex cation due to charge delocalization. The lower viscosity is responsible for the higher conductivity of [Li(G4Et)]I3, though the salt concentration associated with the number of charge carriers is lower with an equimolar addition of I2.| d [g cm−3] | c [mol dm−3] | η [mPa s] | σ [mS cm−1] | |
|---|---|---|---|---|
| [Li(G4Et)]I | 1.33 | 3.58 | 233 | 1.7 |
| [Li(G4Et)]I3 | 1.86 | 2.98 | 23 | 8.6 |
The redox response of the I−/I3− couple was studied in the molten Li salt solvates and the cyclic voltammograms obtained for the Pt microelectrode in [Li(G4Et)]I and [Li(G4Et)]I3 at 60 °C are shown in Fig. 4. In either case, the I−/I3− redox couple shows a two-electron charge transfer reaction in the molten salt solvates. A typical sigmoidal curve with a limiting current of 0.24 μA was observed at a half wave potential (E1/2) of 3.10 V vs. Li/Li+ for [Li(G4Et)]I, corresponding to the oxidation reaction, 3I− → I3− + 2e− (Fig. 4a). For [Li(G4Et)]I3, the reverse reduction reaction of the I−/I3− couple, I3− + 2e− → 3I−, was detected with a limiting current of 0.32 μA at a more negative E1/2 value of 2.75 V vs. Li/Li+ (Fig. 4b). A relatively large difference of the E1/2 values indicates rather slow kinetics of these redox reactions. In a previous study by Bentley et al., the formal potential of the I−/I3− couple, E° (I−/I3−), was found to change depending on solvent polarity: more positive E° (I−/I3−) value was observed in high polarity and Lewis acidic media in which highly Lewis basic I− is more stabilized.46 The oxidation of I− is more energetically difficult in [Li(G4Et)]I, since the concentration of strong Lewis acidic Li ions (i.e., Li salt concentration, c) is higher for [Li(G4Et)]I. Another possible reason for the more negative value of E1/2 in [Li(G4Et)]I3 is rather slow kinetics of the redox reaction. It is known that I2 and I3− are adsorbed onto Pt electrode surface, giving rise to the large cathodic overpotential.47 The adsorption of I2 and I3− is more likely to occur in [Li(G4Et)]I3, and thereby the E1/2 value may become more negative by a kinetic reason. The subsequent redox response around a potential more positive than 3 V vs. Li/Li+ in [Li(G4Et)]I3 may be assigned to the reaction of triiodide/iodine, I3− → 3/2 I2 + e− (Fig. 4b). In our preliminary study, we found the subsequent redox reaction of I3−/I2 at higher potential in a solvate ionic liquid [Li(G4)][TFSA] containing 5 mmol dm−3 [Li(G4)]I as shown in Fig. S1.†
 | ||
| Fig. 4 The cyclic voltammogram obtained for (a) [Li(G4Et)]I and (b) the reduction of [Li(G4Et)]I3 on the Pt microelectrode at a scan rate of 1 mV s−1 at 60 °C. | ||
3.4 Stability of the complex cations
In our previous studies, low-melting glyme–Li salt complexes were categorized into two electrolyte groups, SILs and concentrated solutions with the stability of the [Li(glyme)]+ complex cations being key to discriminating between the two liquid states.48 The presence of robust complex cations was found to be associated with IL-like properties of the molten salt solvates. The stability of the [Li(G4Et)]+ complex cation was assessed using the ratio of the self-diffusion coefficients of the glyme molecules and Li ions (DG/DLi), as shown in Fig. 5. The diffusivity data for G4Et and the Li ions were obtained using PFG-NMR measurements and the self-diffusion coefficients, DG and DLi, listed in Table S1.† Similar to our previous study,48 DG/DLi was plotted as a function of the molar conductivity ratio (so-called, ionicity of ILs), Λ/ΛNE. Here, Λ is the experimental molar conductivity, whereas ΛNE was calculated from the self-diffusion coefficients of the Li ions and anions, assuming the absence of any correlated motions of the ions on the basis of the Nernst–Einstein equation, ΛNE = F2(DLi + Danion)/RT, where F is the Faraday constant, R is the gas constant, and T is the absolute temperature. Therefore, Λ/ΛNE represents the proportion of ions that contribute to actual ionic conduction from all the diffusing ionic components and reflects the correlation of ionic motions affected by ion–ion interactions.49–51 Because the self-diffusion coefficient of I− and I3− is not measurable using PFG-NMR, the chemical diffusion coefficients determined from the limiting current values (|Ilim| in Fig. 4) were used to calculate the ΛNE values for [Li(G4Et)]I and [Li(G4Et)]I3 (see ESI, Table S1†). In Fig. 5, the data reported for the G3 and G4-based molten Li salt solvates with different anionic structures ([Li(G3)]X and [Li(G4)]X) are also displayed for comparison.33,48 If DG/DLi is unity, the glyme molecules and Li ions are considered to diffuse together in the form of long-lived [Li(glyme)]+ complex cations. Thus, such a molten Li salt solvate can be classified as an SIL. On the other hand, molten Li salt solvates with DG/DLi > 1 are not considered to be as an SIL, but a concentrated solution since the occurrence of a fast ligand exchange between the unstable [Li(glyme)]+ complex cations and counter anions, and the presence of non-coordinating free glyme molecules are suggested. For [Li(G4Et)]I, the relatively low Λ/ΛNE value (0.27) corresponds to the strong attractive interaction and correlated motion (or ionic association) of Li+ and I−. A DG/DLi value slightly higher than 1 suggests that the [Li(G4Et)]+ complex cations are not long-lived and non-coordinating G4Et molecules are present to some extent. Thus, [Li(G4Et)]I is classified as a concentrated solution based on the above classification approach. By contrast, for [Li(G4Et)]I3, both the Λ/ΛNE value and DG/DLi are close to unity. This is indicative of more uncorrelated ionic motions resulting from the weaker interactions observed between Li+ and I3−, and the formation of stable [Li(G4Et)]+ complex cations in molten [Li(G4Et)]I3. The extraordinary high Λ/ΛNE value exceeding 1 may imply Grotthuss-like charge transfer, which involves the exchange of a covalent bond between the I3− anions, as discussed for ionic liquid electrolytes containing I3−.52–54 Consequently, these results and the high thermal stability shown in Fig. 1 support that [Li(G4Et)]I3 may be classified as a SIL. | ||
| Fig. 5 The correlation between DG/DLi and ionicity. Data for [Li(G4)]X and [Li(G4Et)][TFSA] were obtained from ref. 48 and 33 respectively. | ||
3.5 Charge–discharge test
Redox-active SILs containing the I−/I3− couple were studied as the liquid cathode material in 3 V-class semi-liquid rechargeable lithium metal batteries. Recently, stable charge–discharge of lithium polysulfide semi-liquid batteries has been achieved without an ion-selective membrane; the polysulfide shuttle effect that deteriorates the cell performance is eliminated by a passivation layer formed on the lithium metal anode.55 For a commercialized lithium iodine (Li–I2) primary battery, direct contact of I2 with lithium metal instantaneously forms LiI on the lithium electrode as a Li ion conducting passivation layer. Thus, the further reduction reaction of I2 causing self-discharge can be prevented.56 We envisaged that the redox shuttle of I3− in the semi-liquid battery may also be mitigated by the LiI-based passivation layer formed initially on the lithium metal anode. However, the cell voltage of a membrane-free Li|[Li(G4Et)]I3|KB-coated carbon paper cell gradually decreased before discharging the cell, indicating that the stable LiI-based passivation layer was not formed in the catholyte and I3− was continuously reduced to I− on the lithium anode surface (Fig. S2†). Hence, Li ion conductive glass ceramic (LICGC) was employed in order to physically block the I3− redox shuttle.Fig. 6a illustrates the initial discharge curve of the Li|[Li(G4Et)][TFSA]|LICGC|[Li(G4Et)]I3 cell at a low constant current density of 100 μA cm−2 at 60 °C. The cell delivers a low initial capacity of 17 mA h g−1 based on the mass of [Li(G4Et)]I3, which is only 20% of its theoretical capacity (85.9 mA h g−1), and no capacity was obtained in the subsequent charge cycle, even though the redox shuttle effect was no longer a problem. Given the total electrochemical reaction during discharge, [Li(G4Et)]I3 + 2Li → [Li(G4Et)]I + 2LiI (or a more concentrated complex 3[Li(G4Et)1/3]I), the high-melting point products, LiI (Tm = 459 °C) or [Li(G4Et)1/3]I (Tm = 181 °C, see Fig. S3†), may be saturated and solidified/precipitated in the highly concentrated SIL catholyte since the G4Et solvent cannot be transferred between the anolyte and catholyte separated by the LICGC. This will cause a significant increase in the resistance of the catholyte, leading to a high overvoltage in the cell and low utilization of the catholyte. Unlike the cell constructed using [Li(G4Et)]I3, a similar two-compartment cell with neat [Li(G4Et)]I as the initial catholyte enables repeated charge–discharge cycles with an average cell voltage of 2.9 V because the catholyte phase is diluted with the concomitantly generated G4Et during the initial charge step; 3[Li(G4Et)]I → [Li(G4Et)]I3 + 2G4Et + 2Li for the total charge reaction (Fig. 6b). The initial discharge capacity reaches 47.2 mA h g−1, which is 98% of the theoretical capacity of [Li(G4Et)]I (48.3 mA h g−1), although the capacity gradually decreased upon charge–discharge cycling. The capacity fading was attributed to the further oxidation of I3− to form I2 that occurs at a higher potential during charging with a charging cut-off voltage of 3.3 V (i.e., less reversible I3−/I2 redox and precipitation of I2) as suggested by a coulombic efficiency of ∼98%. The volumetric energy density during the initial cycle was calculated to be ∼180 W h L−1 based on the catholyte volume. Due to the high concentration of the redox component in the catholyte, this energy density is approximately five times higher than that of the previously reported semi-liquid lithium battery using the glyme-based SIL catholyte with [FeBr4]−.14 To further study the effects of the salt concentration and charging cut-off voltage on the charge–discharge performance, such as catholyte utilization and cycle life, we also examined the cell with a diluted catholyte, 1 mol dm−3 LiI3 in G4Et with a lower charging cut-off voltage of 3.2 V. As shown in Fig. S4a,† 32.6 mA h g−1 was achieved during the initial cycle with an average cell voltage of 3.0 V. The cell shows relatively stable charge–discharge behaviour and the coulombic efficiency was maintained at 100% over 20 cycles although the volumetric energy density decreased to 110 W h L−1 (Fig. S4b†). As a consequence, suppressing the solidification/precipitation of the reduction products in the catholyte under the optimized cut-off voltage conditions was found to be effective towards improving the cycle ability, coulombic efficiency, and energy density of the semi-liquid Li-polyiodide batteries. To achieve this using thermally stable SIL-based catholytes, the solvent structure and polyhalide composition of the redox active SIL needs to be further optimized. It has been reported that a non-stoichiometric composition of I− and I2 can suppress crystallization in imidazolium polyiodide ILs.38 Increasing one terminal alkyl chain length in the glyme molecules and the use of heteropolyhalides may also hamper the crystallization process, and thereby widen the liquid temperature range of redox-active SILs. Such attempts will allow for long-life operation of semi-liquid lithium metal batteries with high thermal stability and high energy density.
 | ||
| Fig. 6 The charge–discharge curves obtained for the (a) Li|[Li(G4Et)][TFSA]|LICGC|[Li(G4Et)]I3 and (b) Li|[Li(G4Et)][TFSA]|LICGC|[Li(G4Et)]I cells at a constant current density of 100 μA cm−2 at 60 °C. | ||
4. Conclusions
Redox-active glyme–Li salt molten solvates, [Li(G4Et)]I and [Li(G4Et)]I3, were studied in terms of their coordination structures and thermal, transport, and electrochemical properties. [Li(G4Et)]I was excluded from the category of SILs because of its non-negligible volatility at 100 °C, low Λ/ΛNE, and DG/DLi value exceeding 1, wherein rigid ion-pairs and non-coordinating volatile G4Et were suggested to be present to some extent. In contrast, [Li(G4Et)]I3 was thermally stable similar to the SIL, [Li(G4Et)][TFSA]. The crown-ether like [Li(G4Et)]+ and I3− were found to be stable in the liquid state of [Li(G4Et)]I3. The high Λ/ΛNE value close to unity implied weak interaction between uncorrelated ionic motion of [Li(G4Et)]+ and I3−. All these data support that [Li(G4Et)]I3 can be regarded as a good SIL. [Li(G4Et)]I and [Li(G4Et)]I3 underwent two electron oxidation and reduction reactions in the molten Li salt solvates, respectively, and the redox properties were exploited as a catholyte in semi-liquid rechargeable lithium batteries. Whereas the cell constructed using [Li(G4Et)]I3 did not operate as a rechargeable battery due to solidification/precipitation of reduction products of the neat catholyte during discharge, the cell containing [Li(G4Et)]I exhibited relatively stable charge–discharge behaviour with an initial volumetric energy density of 180 W h L−1. However, the volumetric energy density of this cell decreased to 110 W h L−1 after 20 cycles. The present SIL-based catholytes still have room for improvement in their liquid temperature range and cycle stability. However, this study may provide guidelines for applying such concentrated catholytes for semi-liquid lithium batteries with flow battery-like scalability toward large-scale electric energy storage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported in part by the JSPS KAKENHI (Grant No. 16H06053) from the Japan Society for the Promotion of Science (JSPS), and the Advanced Low Carbon Technology Research and Development Program (ALCA) of the Japan Science and Technology Agency (JST).References
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- Y. Yamada, J. H. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CAS.
- D. W. McOwen, D. M. Seo, O. Borodin, J. Vatamanu, P. D. Boyle and W. A. Henderson, Energy Environ. Sci., 2014, 7, 416–426 RSC.
- R. Petibon, C. P. Aiken, L. Ma, D. Xiong and J. R. Dahn, Electrochim. Acta, 2015, 154, 287–293 CrossRef CAS.
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J.-G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS PubMed.
- L. Suo, W. Xue, M. Gobet, S. G. Greenbaum, C. Wang, Y. Chen, W. Yang, Y. Li and J. Li, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1156–1161 CrossRef CAS PubMed.
- J. Wang, Y. Yamada, K. Sodeyama, C. H. Chiang, Y. Tateyama and A. Yamada, Nat. Commun., 2016, 7, 12032 CrossRef CAS PubMed.
- Y. Yamada, M. Yaegashi, T. Abe and A. Yamada, Chem. Commun., 2013, 49, 11194–11196 RSC.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 13121–13129 CrossRef CAS PubMed.
- M. Watanabe, K. Dokko, K. Ueno and M. L. Thomas, Bull. Chem. Soc. Jpn., 2018, 91, 1660–1682 CrossRef CAS.
- C. Austen Angell, Y. Ansari and Z. Zhao, Faraday Discuss., 2012, 154, 9–27 RSC.
- T. Mandai, K. Yoshida, K. Ueno, K. Dokko and M. Watanabe, Phys. Chem. Chem. Phys., 2014, 16, 8761–8772 RSC.
- Y. Kemmizaki, H. Tsutsumi and K. Ueno, Electrochemistry, 2018, 86, 46–51 CrossRef CAS.
- H. J. Snaith and L. Schmidt-Mende, Adv. Mater., 2007, 19, 3187–3200 CrossRef CAS.
- J. Wu, Z. Lan, J. Lin, M. Huang, Y. Huang, L. Fan and G. Luo, Chem. Rev., 2015, 115, 2136–2173 CrossRef CAS PubMed.
- Y. Zhao, L. Wang and H. R. Byon, Nat. Commun., 2013, 4, 1896 CrossRef PubMed.
- H. Tian, T. Gao, X. Li, X. Wang, C. Luo, X. Fan, C. Yang, L. Suo, Z. Ma, W. Han and C. Wang, Nat. Commun., 2017, 8, 14083 CrossRef CAS PubMed.
- H. Tian, S. Zhang, Z. Meng, W. He and W.-Q. Han, ACS Energy Lett., 2017, 2, 1170–1176 CrossRef.
- M. Yu, W. D. McCulloch, D. R. Beauchamp, Z. Huang, X. Ren and Y. Wu, J. Am. Chem. Soc., 2015, 137, 8332–8335 CrossRef CAS PubMed.
- G.-M. Weng, Z. Li, G. Cong, Y. Zhou and Y.-C. Lu, Energy Environ. Sci., 2017, 10, 735–741 RSC.
- Z. Li, G. Weng, Q. Zou, G. Cong and Y.-C. Lu, Nano Energy, 2016, 30, 283–292 CrossRef CAS.
- H.-D. Lim, H. Song, J. Kim, H. Gwon, Y. Bae, K.-Y. Park, J. Hong, H. Kim, T. Kim, Y. H. Kim, X. Lepró, R. Ovalle-Robles, R. H. Baughman and K. Kang, Angew. Chem., Int. Ed., 2014, 53, 3926–3931 CrossRef CAS PubMed.
- W.-J. Kwak, D. Hirshberg, D. Sharon, H.-J. Shin, M. Afri, J.-B. Park, A. Garsuch, F. F. Chesneau, A. A. Frimer, D. Aurbach and Y.-K. Sun, J. Mater. Chem. A, 2015, 3, 8855–8864 RSC.
- S. Meini, R. Elazari, A. Rosenman, A. Garsuch and D. Aurbach, J. Phys. Chem. Lett., 2014, 5, 915–918 CrossRef CAS PubMed.
- F. Wu, J. T. Lee, N. Nitta, H. Kim, O. Borodin and G. Yushin, Adv. Mater., 2015, 27, 101–108 CrossRef CAS PubMed.
- V. K. Thorsmølle, D. Topgaard, J. C. Brauer, S. M. Zakeeruddin, B. Lindman, M. Grätzel and J.-E. Moser, Adv. Mater., 2012, 24, 781–784 CrossRef PubMed.
- Y. U. Paulechka and A. V. Blokhin, J. Chem. Thermodyn., 2014, 79, 94–99 CrossRef CAS.
- C. Krause, J. R. Sangoro, C. Iacob and F. Kremer, J. Phys. Chem. B, 2010, 114, 382–386 CrossRef CAS PubMed.
- Y. Ugata, M. L. Thomas, T. Mandai, K. Ueno, K. Dokko and M. Watanabe, Phys. Chem. Chem. Phys., 2019, 21, 9759–9768 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- W. A. Henderson, J. Phys. Chem. B, 2006, 110, 13177–13183 CrossRef CAS PubMed.
- T. Tamura, K. Yoshida, T. Hachida, M. Tsuchiya, M. Nakamura, Y. Kazue, N. Tachikawa, K. Dokko and M. Watanabe, Chem. Lett., 2010, 39, 753–755 CrossRef CAS.
- K. Hashimoto, S. Suzuki, M. L. Thomas, T. Mandai, S. Tsuzuki, K. Dokko and M. Watanabe, Phys. Chem. Chem. Phys., 2018, 20, 7998–8007 RSC.
- J. Estager, J. D. Holbrey and M. Swadźba-Kwaśny, Chem. Soc. Rev., 2014, 43, 847–886 RSC.
- C. A. Angell, N. Byrne and J.-P. Belieres, Acc. Chem. Res., 2007, 40, 1228–1236 CrossRef CAS PubMed.
- V. K. Thorsmølle, J. C. Brauer, S. M. Zakeeruddin, M. Grätzel and J.-E. Moser, J. Phys. Chem. C, 2012, 116, 7989–7992 CrossRef.
- H. Abe, H. Kishimura, M. Takaku, M. Watanabe and N. Hamaya, Faraday Discuss., 2018, 206, 49–60 RSC.
- W. A. Henderson, N. R. Brooks, W. W. Brennessel and V. G. Young, Chem. Mater., 2003, 15, 4679–4684 CrossRef CAS.
- W. A. Henderson, N. R. Brooks and V. G. Young, Chem. Mater., 2003, 15, 4685–4690 CrossRef CAS.
- W. A. Henderson, F. McKenna, M. A. Khan, N. R. Brooks, V. G. Young and R. Frech, Chem. Mater., 2005, 17, 2284–2289 CrossRef CAS.
- K. Ueno, R. Tatara, S. Tsuzuki, S. Saito, H. Doi, K. Yoshida, T. Mandai, M. Matsugami, Y. Umebayashi, K. Dokko and M. Watanabe, Phys. Chem. Chem. Phys., 2015, 17, 8248–8257 RSC.
- D. Brouillette, D. E. Irish, N. J. Taylor, G. Perron, M. Odziemkowski and J. E. Desnoyers, Phys. Chem. Chem. Phys., 2002, 4, 6063–6071 RSC.
- J. Grondin, J.-C. Lassegues, M. Chami, L. Servant, D. Talaga and W. A. Henderson, Phys. Chem. Chem. Phys., 2004, 6, 4260–4267 RSC.
- P. H. Svensson and L. Kloo, Chem. Rev., 2003, 103, 1649–1684 CrossRef CAS PubMed.
- C. L. Bentley, A. M. Bond, A. F. Hollenkamp, P. J. Mahon and J. Zhang, J. Phys. Chem. C, 2015, 119, 22392–22403 CrossRef CAS.
- K. J. Hanson and C. W. Tobias, J. Electrochem. Soc., 1987, 134, 2204–2210 CrossRef CAS.
- K. Ueno, K. Yoshida, M. Tsuchiya, N. Tachikawa, K. Dokko and M. Watanabe, J. Phys. Chem. B, 2012, 116, 11323–11331 CrossRef CAS PubMed.
- H. Tokuda, S. Tsuzuki, M. A. B. H. Susan, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2006, 110, 19593–19600 CrossRef CAS PubMed.
- K. R. Harris, J. Phys. Chem. B, 2010, 114, 9572–9577 CrossRef CAS PubMed.
- H. K. Kashyap, H. V. R. Annapureddy, F. O. Raineri and C. J. Margulis, J. Phys. Chem. B, 2011, 115, 13212–13221 CrossRef CAS PubMed.
- V. K. Thorsmølle, G. Rothenberger, D. Topgaard, J. C. Brauer, D.-B. Kuang, S. M. Zakeeruddin, B. Lindman, M. Grätzel and J.-E. Moser, ChemPhysChem, 2011, 12, 145–149 CrossRef PubMed.
- R. Kawano and M. Watanabe, Chem. Commun., 2005, 2107–2109 RSC.
- J. G. McDaniel and A. Yethiraj, J. Phys. Chem. B, 2018, 122, 250–257 CrossRef CAS PubMed.
- Y. Yang, G. Zheng and Y. Cui, Energy Environ. Sci., 2013, 6, 1552–1558 RSC.
- C. Holmes, ECS Trans., 2007, 6, 1–7 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Cyclic voltammogram in [Li(G4)][TFSA] containing 5 mM [Li(G4)]I, numerical data of diffusion coefficients, molar conductivity, and Λ/ΛNE, time-dependent cell voltage of a membrane-free Li|[Li(G4Et)]I3|KB-coated carbon paper cell, DSC curve for [Li(G4Et)1/3]I. Charge–discharge performance of Li|1 mol dm−3 Li[TFSA] in G4Et|LICGC|1 mol dm−3 LiI3 in G4Et cell at 60 °C. See DOI: 10.1039/c9ra03580j |
| This journal is © The Royal Society of Chemistry 2019 |