
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall molecule PROTACs: an emerging technology for targeted therapy in drug discovery
Haixiang Peia,
Yangrui Peng a,
Qiuhua Zhao*b and
Yihua Chen*a
a,
Qiuhua Zhao*b and
Yihua Chen*a
aShanghai Key Laboratory of Regulatory Biology, The Institute of Biomedical Sciences, School of Life Sciences, East China Normal University, Shanghai 200241, China. E-mail: yhchen@bio.ecnu.edu.cn
bSchool of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200241, China. E-mail: qhzhao@chem.ecnu.edu.cn
First published on 30th May 2019
Abstract
Curing malignant carcinomas is a grand ambition in the development of human health. Over the past decades, targeted therapies have become one of the most successful ways of achieving this. Of these approaches, small molecule inhibitors and monoclonal antibodies are two major methods, however several barriers to their development and clinical use still exist. The use of proteolysis-targeting chimeras (PROTACs) is a new technology through utilizing a intracellular ubiquitin-proteasome system to induce targeted protein degradation, is receiving much attention in the field of targeted therapies. Hetero-bifunctional PROTACs have the potential to eliminate the “undruggable” proteome that comprises about 85% of human proteins, which indicates their great prospects in therapeutic fields. However, there are some hurdles preventing current PROTACs moving from bench to clinic, such as delivery and bioavailability. This review provides an overview of the development of PROTAC technology and will briefly summarize the future possible directions of this approach.
1. Introduction
Since Hippocrates proposed the concept of carcinomas, complicated and changeable malignant tumors have always been great threats to human health. Monoclonal antibodies and small molecule inhibitors have shown much success in fighting cancer over the past decades. The primary advantages of monoclonal antibodies derive from their high selectivity, high binding affinity and prolonged pharmacokinetic profile for blocking extracellular protein–protein interactions. However, the large size of antibodies restricts them from crossing cell membranes. Small molecule inhibitors have high cell permeability relative to monoclonal antibodies, which makes it possible to target intracellular proteins. Besides, their oral bioavailability and specific pharmacokinetic properties lead to small molecule inhibitors taking the role of being the main targeted therapy towards intracellular proteins.However, there are still several deficiencies of small molecule inhibitors that limit their utility against all target proteins. Firstly, small molecule inhibitors have a militating effect through occupying active sites. A majority of human proteins, such as transcription factors, non-enzymatic proteins and scaffolding proteins, lack active sites or have an active site scarcely suitable for occupying and are thus dubbed “undruggable” targets.1 Secondly, high drug dosages are necessary as treatment progresses for therapeutic efficiency, which may always cause off-target effects.2 Thirdly, continuous exposure to small molecule inhibitors always induces the mutation of target proteins, which will cause drug-resistance.3 In addition, the compensatory feedback activation of downstream signaling may happen when inhibiting only one specific target.4,5 Moreover, the long-term inhibition of target proteins may lead to compensatory protein overexpression and intracellular protein accumulation, which may lead to incomplete inhibition, and thus an increase in drug dosages.6 All these limitations seriously obstruct the discovery and durable clinical effectiveness of small molecule inhibitors.
A novel concept called induced protein degradation has the potential to overcome most of the limitations of small molecule inhibitors. This strategy proposes that a small molecule just needs a brief interaction with its target protein, leading to the loss of function of the target. Compared to the “occupancy-driven” mode of small molecule inhibitors, this “event-driven” pharmacology leads the target protein to attain loss-of-function via a transient binding event, which is based on employing the cellular ubiquitin-proteasome system (UPS) to induce target protein ubiquitination for destructive purposes7. After target-protein destruction, this small-molecule drug can survive and carry on another cycle of target-protein degradation (Fig. 1). This sub-stoichiometric activity is catalytic in nature, avoiding maintaining a high level of drug dosage.8 As the target protein must be resynthesized, the overexpression and accumulation of the target protein can be averted. Theoretically, this approach has the potential to target the “undruggable” proteome that limits traditional drugs, as the warhead needs only slight binding affinity to recruit the protein of interest (POI) rather than high inhibition activity.9 HSP inhibitors and hydrophobic tagging strategies were the first generation of drugs targeting protein degradation, however poor druggability and pharmacological defects impeded the clinical application of these methods.7
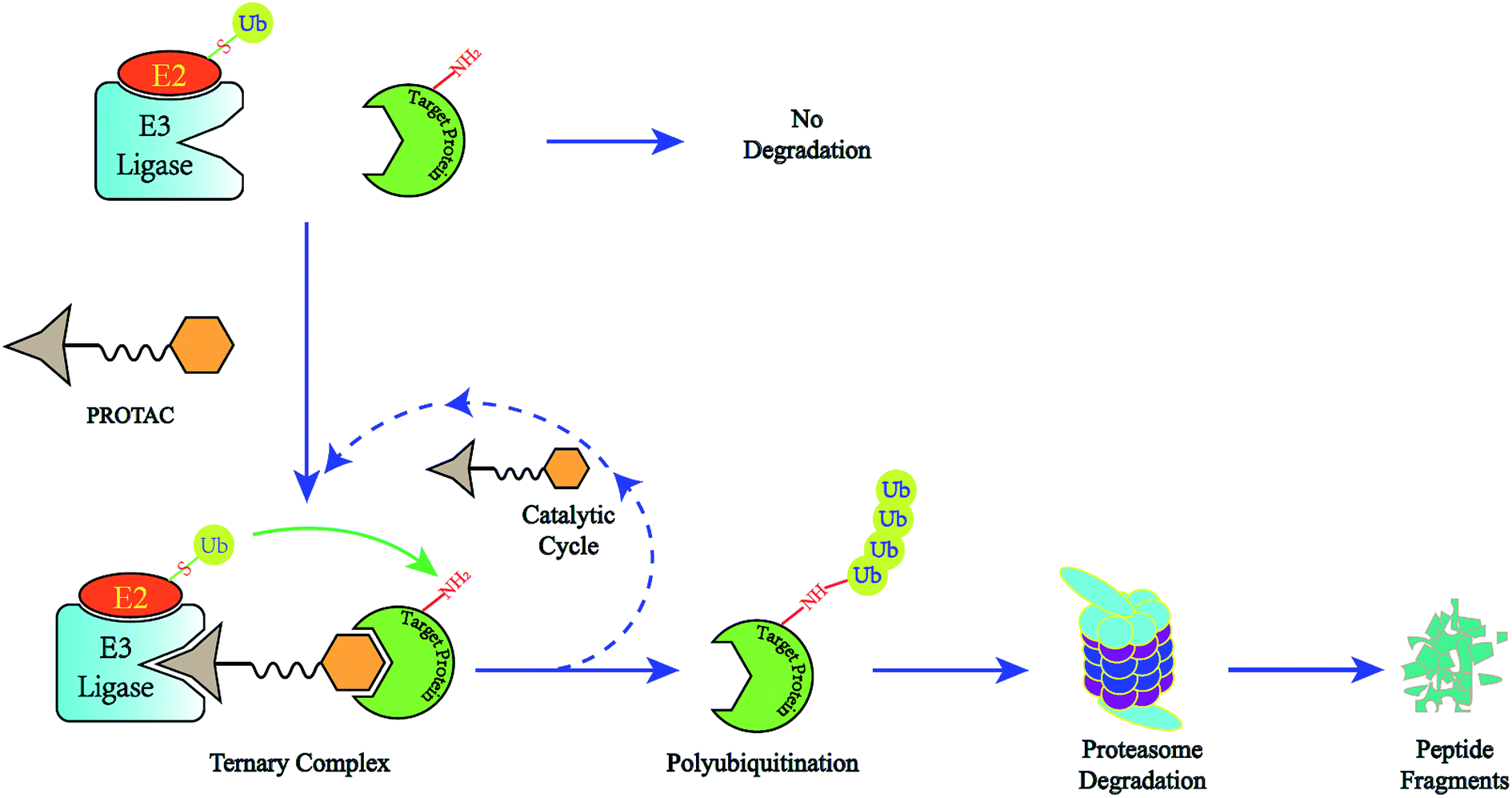 | ||
| Fig. 1 The degradation cycle of an E3 ligase using PROTACs. The blue oval represents the protein of interest (POI) and the orange oval represents the E3 ligase for recruiting. A PROTAC molecule comprises a warhead to target the POI, an E3 ligand to recruit the E3 ligase and a linker to connect them. When the PROTAC molecule draws the POI and E3 ligase close, E3 will employ an E2 ubiquitin-conjugating enzyme to transfer ubiquitin to the surface of the targeted protein. Then the proteasome will recognize the polyubiquitination signal and degrade the POI. At the same time, the PROTAC molecule will separate from the ternary complex and participate in another degradation cycle. | ||
The use of proteolysis-targeting chimeras (PROTACs) is a new area of induced protein degradation, which employs hetero-bifunctional molecules that induce a ligand to bind with the target protein, another ligand to recruit an E3 ubiquitin ligase, and a linker to concatenate the two ligands. Once the ternary complex (target-PROTAC-E3) is formed, the recruited E3 will employ an E2 ubiquitin-conjugating enzyme to transfer ubiquitin to the surface of the targeted protein. A polyubiquitination signal will be recognized by the proteasome to promote the degradation of the targeted proteins. To date, PROTAC technology has been used to induce the degradation of various proteins, including kinases, skeleton proteins, nuclear receptors and transcriptional factors, as well as regulatory proteins. In recent years, this technology has drawn much attention from researchers worldwide in different fields. According to data from Web of Science, over 200 papers have been reported from 2001; a growth spurt was observed in the past five years, especially (Fig. 2). Also, PROTAC technology holds great promise for development for clinic use. Several companies, such as Arvinas, C4 Therapeutics, Kymera Therapeutics and Captor Therapeutics, are setting about pushing the development of PROTAC. The pioneer Arvinas leads the way and is making great efforts to push its two PROTAC molecules into clinic trails. According to the latest news, the FDA has cleared an application from Arvinas for phase I trials of the oral PROTAC ARV-110 drug, which targets the androgen receptor in metastatic castration-resistant prostate cancer. These encouraging superiorities have aroused much interest from many big pharmaceutical companies. Arvinas has announced research collaboration and a licensing agreement with Pfizer and obtained over one hundred million dollars of financing to advance PROTAC technology. C4 therapeutics has gained a strategic collaboration with Roche and launched with 73 million dollars of series A financing.
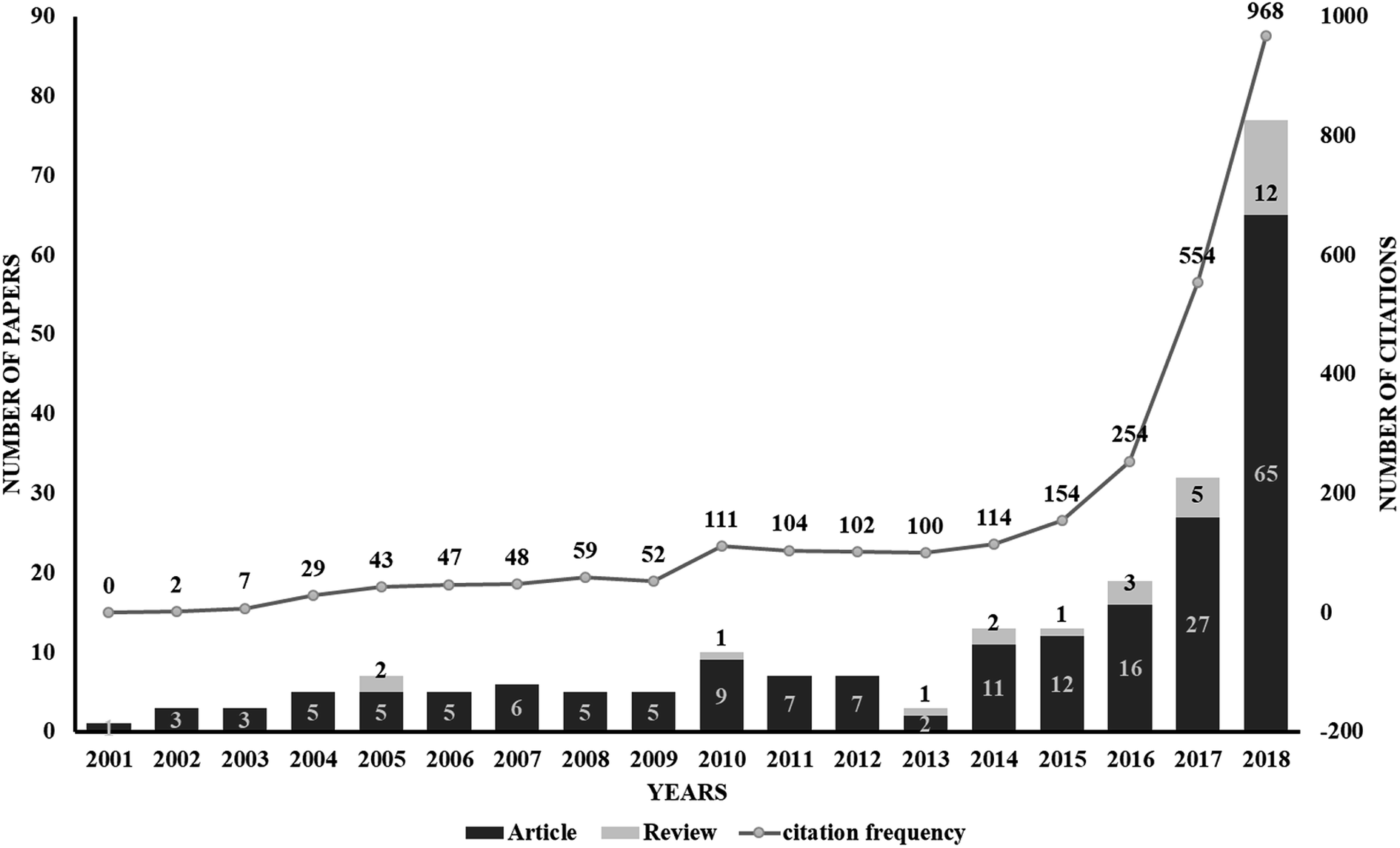 | ||
| Fig. 2 A graph showing the publications associated with PROTAC technology. Data from Web of Science (http://apps.webofknowledge.com). This graph reflects the number of articles and reviews. As can be seen, a drastic increase from 2014 indicates the rapid development of PROTAC technology. | ||
This review will summarize the development of PROTAC technology. We place emphasis on elaborating the advantages of small-molecule PROTACs compared to small molecule inhibitors, together with the associated challenges and potential development directions. In addition, we also analyze deficiencies and forecast the future development of PROTACs.
2. PROTAC technology
2.1 Peptide-based PROTACs
In 2001, Crews and his colleagues put forward the concept of PROTACs for the first time.10 The first-generation PROTACs contain a phosphopeptide that binds to the E3 ligase β-TRCP, and a small-molecule Ovalicin that targets MetAP-2. The lack of cell permeability of the peptide moieties is the major defect of this series of PROTACs, which limits their utility as chemical probes. The first cell-permeable PROTACs, coupling a poly-D-Arg sequence to a peptide derived from HIF-1α that binds to the VHL (von Hippel Lindau) E3 ligase, were developed in 2004.11 These peptide-based PROTACs showed the efficient degradation of targets in cultured cells (e.g., FKBP12F36V, MetAP-2, and androgen and estrogen receptors). Despite many efforts having been made to optimize peptide-based PROTACs, inherent defects such as high molecular weight, low potency, poor cell permeability and peptide bond lability restrict the potential clinical use of peptide-based PROTACs.2.2 Small-molecule PROTACs
Although many shortcomings exist, the first-generation peptide-based PROTACs proved that recruiting the E3 ligase to induce protein degradation is a feasible strategy to generate a new direction for drug development. All-small-molecule PROTACs have been proposed to achieve better cell permeability and rapid target degradation. So far, with the successful development of small-molecule ligands, four E3 ligases (MDM2, cIAP1, CRBN (cereblon) and von Hippel-Lindau (VHL)) have been used as the basis for all-small-molecule PROTACs to selectively degrade target proteins. | ||
| Fig. 3 An MDM2-based PROTAC (AR targeting). | ||
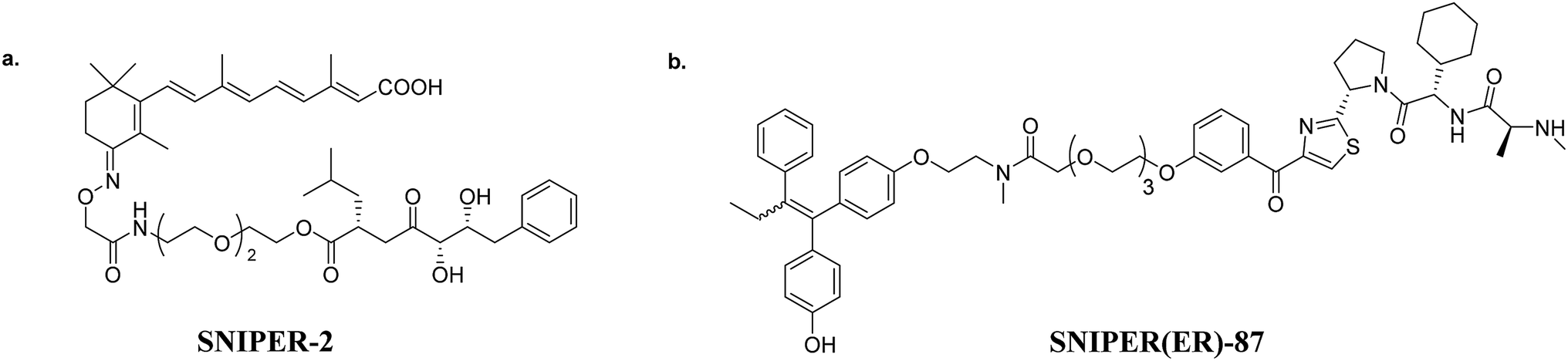 | ||
| Fig. 4 (a) A cIAP-based PROTAC (CRABPII targeting), and (b) XIAP-based PROTAC (ER targeting). | ||
 | ||
| Fig. 5 CRBN-based PROTACs: (a) dBET1 (BRD4-targeting); (b) ARV-825 (BRD4-targeting); and (c) P13I (BTK-targeting). | ||
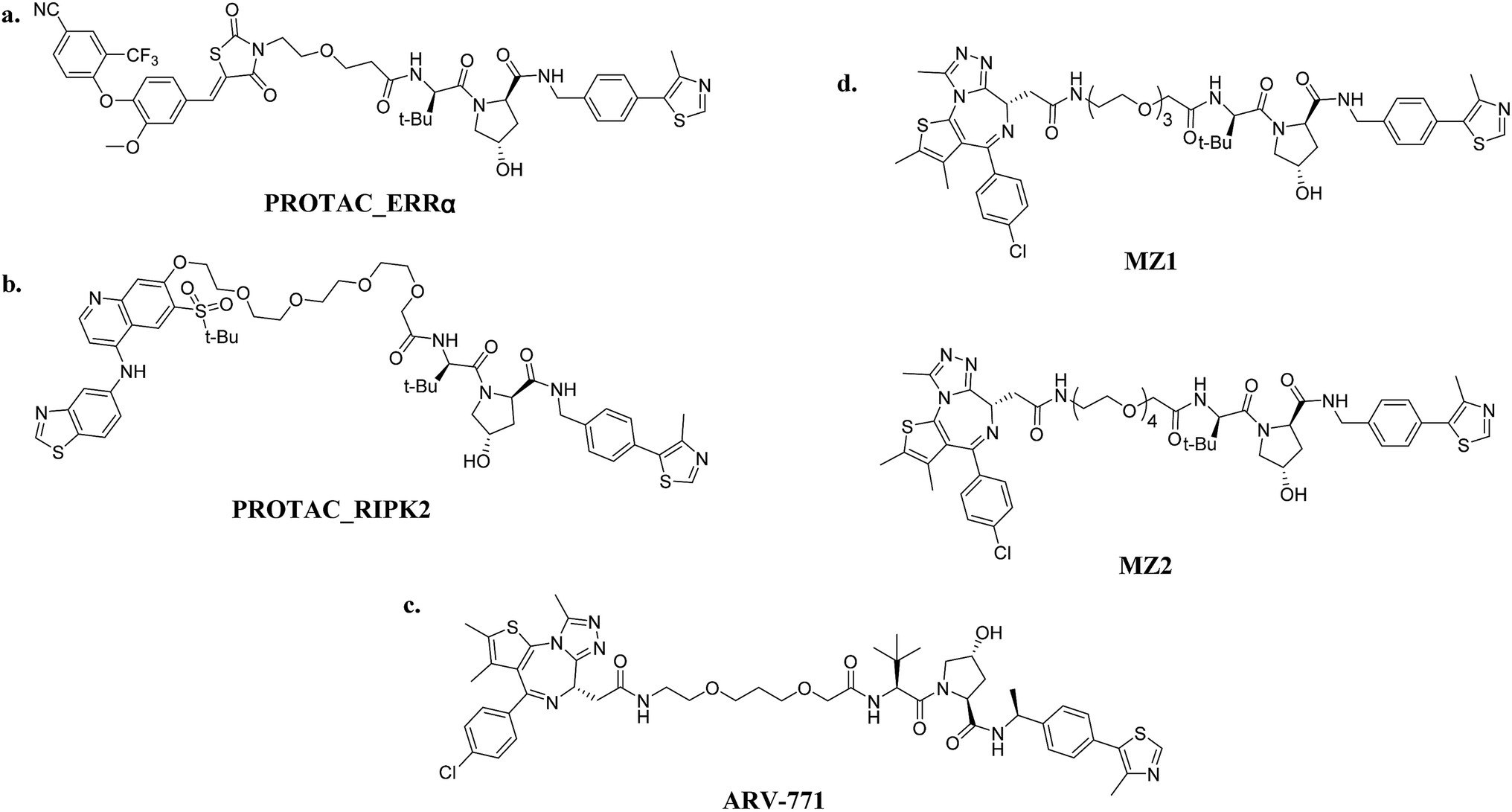 | ||
| Fig. 6 VHL-based PROTACs: (a) PROTAC_ERRα (ERRα-targeting); (b) PROTAC_RIPK2 (RIPK2-targeting); (c) ARV-771 (BRD4-targeting); and (d) MZ1/MZ2 (BRD4-targeting). | ||
Small-molecule VHL-based PROTACs have also been used to degrade BRD4. ARV-771, a picomolar VHL-based pan-BET degrader, which is under clinical study now, exerted efficiency against a CRPC mouse xenograft with activity much better than CRBN-based PROTAC dBET1 and ARV-825, revealing the capacity of PROTAC technology to combat solid tumors42 (Fig. 6c). Another two BRD4-targeting PROTACs MZ1 and MZ2 both induce the degradation of BRD4 within 24 h (ref. 43) (Fig. 6d). Interestingly, these PROTACs induce the selective removal of BRD4 over BRD2 and BRD3, in contrast with dBET1 and ARV-825, indicating that the recruitment of different E3 ligases may lead to different selectivity profiles. Further studies utilizing the tyrosine kinase inhibitors bosutinib and dasatinib that target chimeric BCR-ABL and ABL were carried out to compare VHL- and CRBN-based PROTAC in a head-to-head manner.32 Surprisingly, the selectivities of PROTACs depend on the identity of the inhibitor warhead as well as the recruited E3 ligase, despite target management. Moreover, the results indicated that the CRBN E3 ligase is more indiscriminate in its degradation profile against VHL, maybe owing to the flexible cullin 4A scaffold of the CRBN E3 ligase complex.44,45 In order to know more about the selective degradation of PROTACs, a promiscuous warhead Foretinib, which binds to 133 kinases, was chosen.46 Interestingly, the results indicated that both CRBN- and VHL-based PROTACs dramatically increase the degradation selectivities, in contrast with the parent warhead. Steric clashes that impact the stability of the ternary complex may influence the selectivity of the warhead. These studies inspire us as regards the potential of PROTAC to convert a nonselective or weak-affinity inhibitor into a selective degrader. To date, VHL-based PROTACs targeting DHODH,47 AR,48,49 ER,50 TRIM24,51 Fak,52 FLT-3,53 ALK,54 p38 (ref. 55) and TANK56 have also been reported.
3. Challenges and opportunities
3.1 Expanding the library in use
Though the prospect of PROTAC technology has attracted wide interest, some challenges still exist to be overcome. E3 ubiquitin ligases are a huge family with over 600 members, but only CRBN, VHL and IAP are presently widely used in PROTACs. A vast space remains to be excavated to allow more E3 ligases to be applied to PROTAC technology. Several obstacles lie in the way of developing new E3 ligases for PROTACs. Exploiting some small-molecule ligands with high affinities for one E3 ligase is paramount, since it is very difficult to develop inhibitors based on protein–protein interactions. Fortunately, over the past decades, some E3 ligase inhibitors have been discovered, which have the potential to be exploited as novel E3 ligase ligands for PROTACs.The MDM2-based small-molecule PROTAC was discovered in 2008, but it was not widely used because of its poor activity.12 This research lagged for 10 years until the well-reported study of MDM2 inhibitors. In 2018, C. M. Crews and his group reported A1874, a new MDM2-based PROTAC that comprises the BRD4 ligand JQ1 and an MDM2 antagonist idasanutlin with nanomolar potency.57 As MDM2-based PROTACs have the ability both to degrade target proteins and stabilize p53, A1874 was discovered to perform better in the anti-proliferation of many cancer cell lines with wide-type p53 than the corresponding VHL-based PROTAC. This research emphasizes the untapped potential of MDM2-based PROTACs and may expand the library of E3 ligases that can be used in PROTAC technology.
Keap1 is a member of the cullin3-based E3 ligase group, which directs its substrate Nrf2 towards degradation. Developing novel inhibitors that interrupt Keap1-Nrf2 PPI to enhance Nrf2 for the prevention of cancer is a feasible direction.58–60 Z. Y. Jiang and his group developed a class of small-molecule inhibitors that target Keap1-Nrf2 PPI with nanomolar activities.61–65 Besides, they also synthesized a Keap-dependent peptide PROTAC to knockdown Tau,66 indicating that a new class of small-molecule PROTACs based on the Keap1 E3 ligase can be expected and its development is only just a matter of time.
TRIM24 is a p53-induced E3-ubiquitin ligase that targets endogenous p53 for degradation.67,68 In 2015, J. Bennett and W. S. Palmer reported a series of TRIM24 inhibitors (e.g., IACS-7e and IACS-9571) with nanomolar potencies.69,70 A TRIM24 degrader was synthesized as a chemical probe for acute leukemia.51 Besides, IACS-9571 was reported in a patent to act as an E3 ligand to form a PROTAC for the degradation of EZH1/EZH2.71
Interestingly, very similar to the discovery of the CRBN E3-ubiquitin ligase as the primary target of thalidomide,19,20 anticancer sulfonamides (e.g., indisulam, E7820 and CQS) were reported to induce CAPERα (also known as RBM39) degradation by recruiting the DCAF15 E3 ligase.72,73 Much evidence has proved that DCAF15 is the main target of anticancer sulfonamides. In addition, the E3 ligase DCAF15 belongs to the CUL4A/B family, just like CRBN,74 and they show similar chemical modulation, which suggests that transforming sulfonamides into bifunctional PROTACs is a potential direction through recruiting the DCAF15 E3 ligase.75
3.2 Homo-PROTACs
Except for recruiting E3 ligases to deplete target proteins, the over expression of several E3 ligases is often observed in various cancers; this is always correlated with chemo-resistance and poor prognoses.76 Inducing the degradation of the E3 ligase itself using PROTACs may be an effective strategy, which has thus produced the concept of homo-PROTACs. The first homo-PROTAC, CM11, was constituted of two VHL inhibitors and one linker.77 This symmetric homodimerized compound induced the potent, rapid self-degradation of VHL in different cell lines. Like CM11, another CRBN-based homo-PROTAC was reported, which had the potent effect of antagonism with pomalidomide.783.3 Optimizing PROTAC technology
In addition, various problems remain in existence before PROTAC technology can mature for clinical applications, such as off-target effects, in vivo metabolic stability, cellular permeability and large molecule weight. According to Lipinski's rule of five, many efforts should be made to convert PROTAC molecules to be more “drug-like”. Beyond that, it is hard to synthesize and optimize bifunctional molecules; these are also important obstacles in research and manufacture. On the bright side, many groups have tried to break through these barriers. An advanced technology called CLIPTAC (in-cell click-formed PROTAC) has been proposed, which separates the bifunctional molecule into two pairs, one pair with a tetrazine tagged thalidomide derivative and another pair with a trans-cyclo-octene (TCO)-tagged ligand of the target protein in the cell79 (Fig. 7). In the concept of click chemistry, these two pairs are able to unite as one to form a PROTAC via a rapid reaction in a cell. Both of the pairs have lower molecular weights and better cell penetrability than the original PROTACs. This strategy can be expanded to induce the degradation of any target protein via an in-cell click-formed PROTAC. This click chemistry concept also provides a rapid synthesis method for PROTACs in vitro.80 Another technology named HaloPROTAC has been reported to target the degradation of intracellular GFP (green fluorescent protein)-HaloTag7 fused proteins, which greatly simplifies the optimization procedure of PROTACs in terms of finding appropriate linker lengths and linker attachment points81 (Fig. 8). Further, HaloTag7 was fused with different E3 ligases to degrade GFP, to evaluate the E3 ligase role in PROTAC technology.82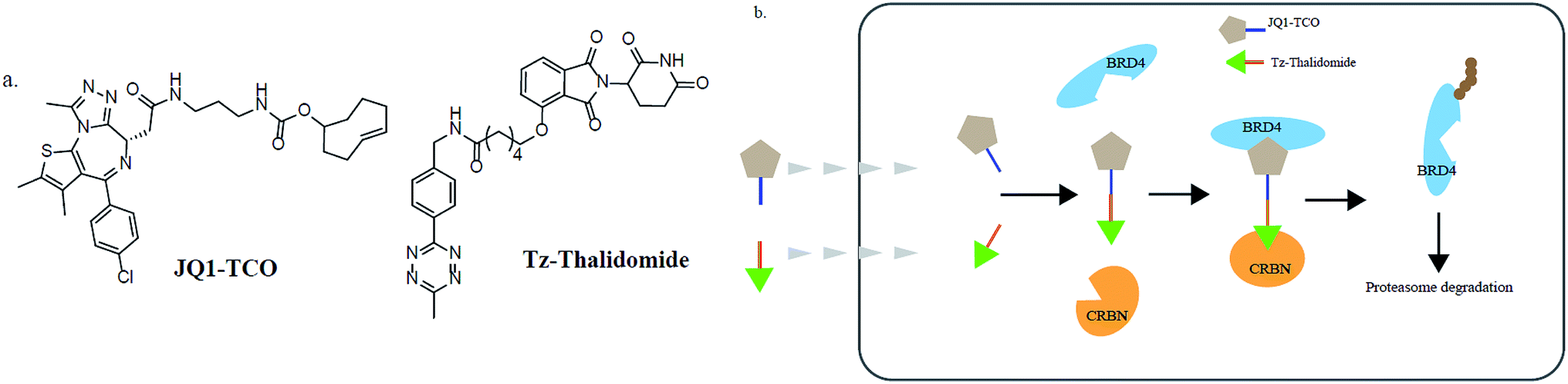 | ||
| Fig. 7 (a) The structures of JQ1-TCO and Tz-thalidomide. (b) The mechanism of CLIPTACs. Two precursor molecules penetrate the cytomembrane in succession and form a CLIPTAC intracellularly, resulting in the proteasome degradation of BRD4. | ||
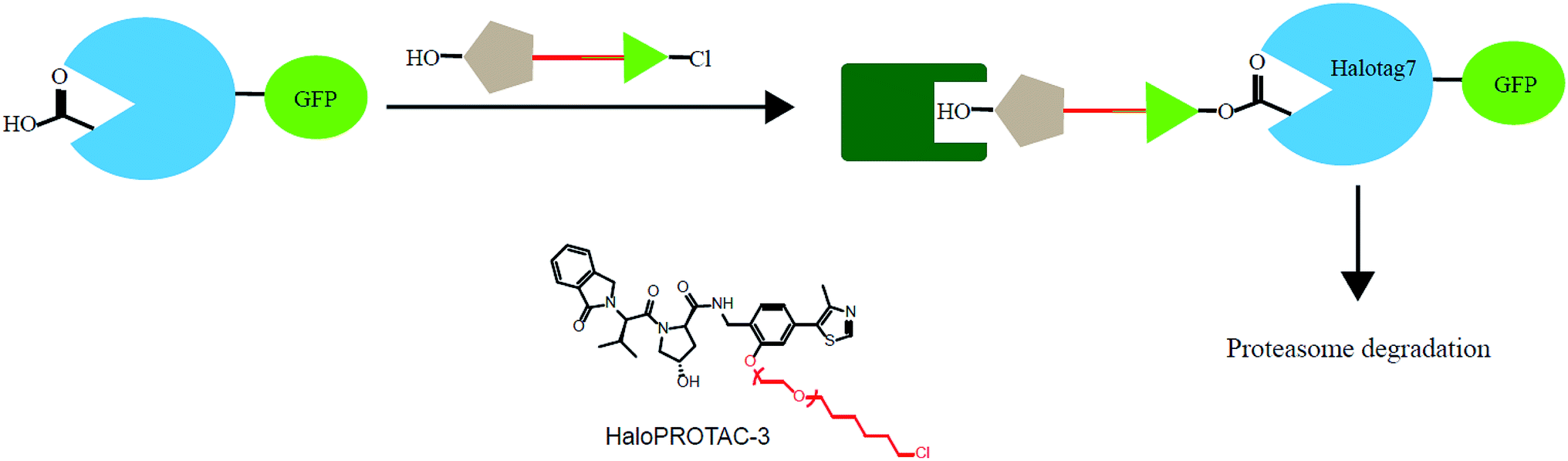 | ||
| Fig. 8 A schematic diagram showing a HaloPROTAC. The HaloPTORAC utilizes the specific recognition by HaloTag7 of hexyl chloride tags and thus induces the degradation of HaloTag7-GFP fused proteins. This strategy simplifies the optimization of PROTACs. | ||
4. Conclusions and future prospects
In summary, PROTAC technology has shown significant improvements in recent decades, mainly from all-peptide to small-molecule compounds, and in terms of the cellular permeabilities, solubilities, stabilities and affinities of PROTACs. PROTAC technology shows many advantages over small-molecule inhibitors, such as inducing the durative and fast depletion of target proteins and downstream signaling cascades, overcoming mutation-caused drug resistance, enhancing target selectivities and modifying potencies, and reducing dosage due to its sub-stoichiometric catalytic nature. Despite small molecules having achieved much success in being applied to classically druggable proteins in recent years, the most promising aspect of this technology is its potential to target “undruggable” proteins. Approximately 85% of the human proteome, such as transcription factors, scaffolding proteins, and non-enzyme proteins, has a lack of enzymatic activity or functional interactions. However, the event-driven nature of PROTACs permits their binding to any site of the target protein, instead of a stationary active site, thus driving these “undruggable” proteins to become pharmaceutically vulnerable.Therefore, utilizing PROTACs to induce protein degradation has the potential of being a promising therapeutic strategy. To date, although most research into PROTACs has specially focused on applications to cancer therapy, the treatment of other diseases may benefit from this burgeoning technology. So far, only one PROTAC compound has been pushed to clinical trials recently. Therefore, the clinical outcome of PROTAC technology remains uncertain. Nevertheless, it is a long journey to advance PROTAC technology from the bench to the clinic. Overall, a large amount of effort is being made to develop PROTAC technology, and we are hoping for a bright future.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by grants from the National Key R&D Program of China (2018YFA0507001), the National Natural Science Foundation of China (81673304), and the Innovation program of the Shanghai Municipal Education Commission (2017-01-07-00-05-E00011).References
- S. Gu, D. Cui, X. Chen, X. Xiong and Y. Zhao, BioEssays, 2018, 40, 1700247 CrossRef PubMed.
- M. Toure and C. M. Crews, Angew. Chem., Int. Ed. Engl., 2016, 55, 1966–1973 CrossRef CAS PubMed.
- S. Dogan, R. Shen, D. C. Ang, M. L. Johnson, S. P. D'Angelo, P. K. Paik, E. B. Brzostowski, G. J. Riely, M. G. Kris, M. F. Zakowski and M. Ladanyi, Clin. Cancer Res., 2012, 18, 6169–6177 CrossRef CAS PubMed.
- E. Rozengurt, H. P. Soares and J. Sinnet-Smith, Mol. Cancer Ther., 2014, 13, 2477–2488 CrossRef CAS PubMed.
- E. Buck, A. Eyzaguirre, M. Rosenfeld-Franklin, S. Thomson, M. Mulvihill, S. Barr, E. Brown, M. O'Connor, Y. Yao, J. Pachter, M. Miglarese, D. Epstein, K. K. Iwata, J. D. Haley, N. W. Gibson and Q.-S. Ji, Cancer Res., 2008, 68, 8322–8332 CrossRef CAS PubMed.
- J. Spiegel, P. M. Cromm, G. Zimmermann, T. N. Grossmann and H. Waldmann, Nat. Chem. Biol., 2014, 10, 613–622 CrossRef CAS PubMed.
- A. C. Lai and C. M. Crews, Nat. Rev. Drug Discovery, 2017, 16, 101–114 CrossRef CAS PubMed.
- J. Salami and C. M. Crews, Science, 2017, 355, 1163–1167 CrossRef CAS PubMed.
- P. M. Cromm and C. M. Crews, Cell Chem. Biol., 2017, 24, 1181–1190 CrossRef CAS PubMed.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS PubMed.
- J. S. Schneekloth, F. N. Fonseca, M. Koldobskiy, A. Mandal, R. Deshaies, K. Sakamoto and C. M. Crews, J. Am. Chem. Soc., 2004, 126, 3748–3754 CrossRef CAS PubMed.
- A. R. Schneekloth, M. Pucheault, H. S. Tae and C. M. Crews, Bioorg. Med. Chem. Lett., 2008, 18, 5904–5908 CrossRef CAS PubMed.
- Y. Itoh, M. Ishikawa, M. Naito and Y. Hashimoto, J. Am. Chem. Soc., 2010, 132, 5820–5826 CrossRef CAS PubMed.
- Y. Itoh, R. Kitaguchi, M. Ishikawa, M. Naito and Y. Hashimoto, Bioorg. Med. Chem., 2011, 19, 6768–6778 CrossRef CAS PubMed.
- K. Okuhira, Y. Demizu, T. Hattori, N. Ohoka, N. Shibata, T. Nishimaki-Mogami, H. Okuda, M. Kurihara and M. Naito, Cancer Sci., 2013, 104, 1492–1498 CrossRef CAS PubMed.
- N. Ohoka, K. Nagai, T. Hattori, K. Okuhira, N. Shibata, N. Cho and M. Naito, Cell Death Dis., 2014, 5, e1513 CrossRef CAS PubMed.
- Y. Demizu, N. Shibata, T. Hattori, N. Ohoka, H. Motoi, T. Misawa, T. Shoda, M. Naito and M. Kurihara, Bioorg. Med. Chem. Lett., 2016, 26, 4865–4869 CrossRef CAS PubMed.
- N. Ohoka, K. Okuhira, M. Ito, K. Nagai, N. Shibata, T. Hattori, O. Ujikawa, K. Shimokawa, O. Sano, R. Koyama, H. Fujita, M. Teratani, H. Matsumoto, Y. Imaeda, H. Nara, N. Cho and M. Naito, J. Biol. Chem., 2017, 292, 4556–4570 CrossRef CAS PubMed.
- J. Kronke, N. D. Udeshi, A. Narla, P. Grauman, S. N. Hurst, M. McConkey, T. Svinkina, D. Heckl, E. Comer, X. Y. Li, C. Ciarlo, E. Hartman, N. Munshi, M. Schenone, S. L. Schreiber, S. A. Carr and B. L. Ebert, Science, 2014, 343, 301–305 CrossRef PubMed.
- T. Ito, H. Ando, T. Suzuki, T. Ogura, K. Hotta, Y. Imamura, Y. Yamaguchi and H. Handa, Science, 2010, 327, 1345–1350 CrossRef CAS PubMed.
- G. E. Winter, D. L. Buckley, J. Paulk, J. M. Roberts, A. Souza, S. Dhe-Paganon and J. E. Bradner, Science, 2015, 348, 1376–1381 CrossRef CAS PubMed.
- J. Lu, Y. Qian, M. Altieri, H. Dong, J. Wang, K. Raina, J. Hines, J. D. Winkler, A. P. Crew, K. Coleman and C. M. Crews, Chem. Biol., 2015, 22, 755–763 CrossRef CAS PubMed.
- B. Zhou, J. T. Hu, F. M. Xu, Z. Chen, L. C. Bai, E. Fernandez-Salas, M. Lin, L. Liu, C. Y. Yang, Y. J. Zhao, D. McEachern, S. Przybranowski, B. Wen, D. X. Sun and S. M. Wang, J. Med. Chem., 2018, 61, 462–481 CrossRef CAS PubMed.
- C. Qin, Y. Hu, B. Zhou, E. Fernandez-Salas, C.-Y. Yang, L. Liu, D. McEachern, S. Przybranowski, M. Wang, J. Stuckey, J. Meagher, L. Bai, Z. Chen, M. Lin, J. Yang, D. N. Ziazadeh, F. Xu, J. Hu, W. Xiang, L. Huang, S. Li, B. Wen, D. Sun and S. Wang, J. Med. Chem., 2018, 61, 6685–6704 CrossRef CAS PubMed.
- Y. Sun, X. Zhao, N. Ding, H. Gao, Y. Wu, Y. Yang, M. Zhao, J. Hwang, Y. Song, W. Liu and Y. Rao, Cell Res., 2018, 28, 779–781 CrossRef CAS PubMed.
- A. D. Buhimschi, H. A. Armstrong, M. Toure, S. Jaime-Figueroa, T. L. Chen, A. M. Lehman, J. A. Woyach, A. J. Johnson, J. C. Byrd and C. M. Crews, Biochemistry, 2018, 57, 3564–3575 CrossRef CAS PubMed.
- C. M. Robb, J. I. Contreras, S. Kour, M. A. Taylor, M. Abid, Y. A. Sonawane, M. Zahid, D. J. Murry, A. Natarajan and S. Rana, Chem. Commun., 2017, 53, 7577–7580 RSC.
- C. M. Olson, B. Jiang, M. A. Erb, Y. Liang, Z. M. Doctor, Z. Zhang, T. Zhang, N. Kwiatkowski, M. Boukhali, J. L. Green, W. Haas, T. Nomanbhoy, E. S. Fischer, R. A. Young, J. E. Bradner, G. E. Winter and N. S. Gray, Nat. Chem. Biol., 2018, 14, 163–170 CrossRef CAS PubMed.
- A. Zorba, C. Nguyen, Y. R. Xu, J. Starr, K. Borzilleri, J. Smith, H. Y. Zhu, K. A. Farley, W. D. Ding, J. Schiemer, X. D. Feng, J. S. Chang, D. P. Uccello, J. A. Young, C. N. Garcia-Irrizary, L. Czabaniuk, B. Schuff, R. Oliver, J. Montgomery, M. M. Hayward, J. Coe, J. S. Chen, M. Niosi, S. Luthra, J. C. Shah, A. El-Kattan, X. Y. Qiu, G. M. West, M. C. Noe, V. Shanmugasundaram, A. M. Gilbert, M. F. Brown and M. F. Calabrese, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E7285–E7292 CrossRef PubMed.
- K. Yang, Y. Song, H. Xie, H. Wu, Y.-T. Wu, E. D. Leisten and W. Tang, Bioorg. Med. Chem. Lett., 2018, 28, 2493–2497 CrossRef CAS PubMed.
- C. Zhang, X.-R. Han, X. Yang, B. Jiang, J. Liu, Y. Xiong and J. Jin, Eur. J. Med. Chem., 2018, 151, 304–314 CrossRef CAS PubMed.
- A. C. Lai, M. Toure, D. Hellerschmied, J. Salami, S. Jaime-Figueroa, E. Ko, J. Hines and C. M. Crews, Angew. Chem., Int. Ed., 2016, 55, 807–810 CrossRef CAS PubMed.
- M. Schiedel, D. Herp, S. Hammelmann, S. Swyter, A. Lehotzky, D. Robaa, J. Olah, J. Ovadi, W. Sippl and M. Jung, J. Med. Chem., 2018, 61, 482–491 CrossRef CAS PubMed.
- W. Li, C. Gao, L. Zhao, Z. Yuan, Y. Chen and Y. Jiang, Eur. J. Med. Chem., 2018, 151, 237–247 CrossRef CAS PubMed.
- Y. Zou, D. Ma and Y. Wang, Cell Biochem. Funct., 2019, 37, 21–30 CrossRef CAS PubMed.
- D. L. Buckley, I. Van Molle, P. C. Gareiss, H. S. Tae, J. Michel, D. J. Noblin, W. L. Jorgensen, A. Ciulli and C. M. Crews, J. Am. Chem. Soc., 2012, 134, 4465–4468 CrossRef CAS PubMed.
- C. Galdeano, M. S. Gadd, P. Soares, S. Scaffidi, I. Van Molle, I. Birced, S. Hewitt, D. M. Dias and A. Ciulli, J. Med. Chem., 2014, 57, 8657–8663 CrossRef CAS PubMed.
- P. Soares, M. S. Gadd, J. Frost, C. Galdeano, L. Ellis, O. Epemolu, S. Rocha, K. D. Read and A. Ciulli, J. Med. Chem., 2018, 61, 599–618 CrossRef CAS PubMed.
- X. Lucas, I. Van Molle and A. Ciulli, J. Med. Chem., 2018, 61, 7387–7393 CrossRef CAS PubMed.
- D. P. Bondeson, A. Mares, I. E. Smith, E. Ko, S. Campos, A. H. Miah, K. E. Mulholland, N. Routly, D. L. Buckley, J. L. Gustafson, N. Zinn, P. Grandi, S. Shimamura, G. Bergamini, M. Faelth-Savitski, M. Bantscheff, C. Cox, D. A. Gordon, R. R. Willard, J. J. Flanagan, L. N. Casillas, B. J. Votta, W. den Besten, K. Famm, L. Kruidenier, P. S. Carter, J. D. Harling, I. Churcher and C. M. Crews, Nat. Chem. Biol., 2015, 11, 611–617 CrossRef CAS PubMed.
- R. J. Deshaies, Nat. Chem. Biol., 2015, 11, 634–635 CrossRef CAS PubMed.
- K. Raina, J. Lu, Y. Qian, M. Altieri, D. Gordon, A. M. Rossi, J. Wang, X. Chen, H. Dong, K. Siu, J. D. Winkler, A. P. Crew, C. M. Crews and K. G. Coleman, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7124–7129 CrossRef CAS PubMed.
- M. Zengerle, K. H. Chan and A. Ciulli, ACS Chem. Biol., 2015, 10, 1770–1777 CrossRef CAS PubMed.
- E. S. Fischer, A. Scrima, K. Boehm, S. Matsumoto, G. M. Lingaraju, M. Faty, T. Yasuda, S. Cavadini, M. Wakasugi, F. Hanaoka, S. Iwai, H. Gut, K. Sugasawa and N. H. Thomae, Cell, 2011, 147, 1024–1039 CrossRef CAS PubMed.
- J. Liu and R. Nussinov, J. Biol. Chem., 2011, 286, 40934–40942 CrossRef CAS PubMed.
- D. P. Bondeson, B. E. Smith, G. M. Burslem, A. D. Buhimschi, J. Hines, S. Jaime-Figueroa, J. Wang, B. D. Hamman, A. Ishchenko and C. M. Crews, Cell Chem. Biol., 2018, 25, 78–87.e5 CrossRef CAS PubMed.
- J. T. Madak, C. R. Cuthbertson, W. Chen, H. D. Showalter and N. Neamati, Chem. - Eur. J., 2017, 23, 13875–13878 CrossRef CAS PubMed.
- T. K. Neklesa, M. Jin, A. P. Crew, A. K. Rossi, R. R. Willard, H. Dong, K. Siu, J. Wang, D. A. Gordon, X. Chen, C. Ferraro, C. M. Crews, K. Coleman and J. D. Winkler, J. Clin. Oncol., 2016, 34(suppl. 2), 267 Search PubMed.
- X. Han, C. Wang, C. Qin, W. Xiang, E. Fernandez-Salas, C.-Y. Yang, M. Wang, L. Zhao, T. Xu, K. Chinnaswamy, J. Delproposto, J. Stuckey and S. Wang, J. Med. Chem., 2019, 62, 941–964 CrossRef CAS PubMed.
- J. Hu, B. Hu, M. Wang, F. Xu, B. Miao, C.-Y. Yang, M. Wang, Z. Liu, D. F. Hayes, K. Chinnaswamy, J. Delproposto, J. Stuckey and S. Wang, J. Med. Chem., 2019, 1420–1442 CrossRef CAS PubMed.
- L. N. Gechijian, D. L. Buckley, M. A. Lawlor, J. M. Reyes, J. Paulk, C. J. Ott, G. E. Winter, M. A. Erb, T. G. Scott, M. Xu, H. S. Seo, S. Dhe-Paganon, N. P. Kwiatkowski, J. A. Perry, J. Qi, N. S. Gray and J. E. Bradner, Nat. Chem. Biol., 2018, 14, 405–412 CrossRef CAS PubMed.
- P. M. Cromm, K. T. G. Samarasinghe, J. Hines and C. M. Crews, J. Am. Chem. Soc., 2018, 140, 17019–17026 CrossRef CAS PubMed.
- G. M. Burslem, J. Song, X. Chen, J. Hines and C. M. Crews, J. Am. Chem. Soc., 2018, 140, 16428–16432 CrossRef CAS PubMed.
- C. H. Kang, D. H. Lee, C. O. Lee, J. D. Ha, C. H. Park and J. Y. Hwang, Biochem. Biophys. Res. Commun., 2018, 505, 542–547 CrossRef CAS PubMed.
- B. E. Smith, S. L. Wang, S. Jaime-Figueroa, A. Harbin, J. Wang, B. D. Hamman and C. M. Crews, Nat. Commun., 2019, 10, 131 CrossRef PubMed.
- A. P. Crew, K. Raina, H. Q. Dong, Y. M. Qian, J. Wang, D. Vigil, Y. V. Serebrenik, B. D. Hamman, A. Morgan, C. Ferraro, K. Siu, T. K. Neklesa, J. D. Winkler, K. G. Coleman and C. M. Crews, J. Med. Chem., 2018, 61, 583–598 CrossRef CAS PubMed.
- J. Hines, S. Lartigue, H. Dong, Y. Qian and C. M. Crews, Cancer Res., 2019, 79, 251–262 CrossRef CAS PubMed.
- T. Suzuki, H. Motohashi and M. Yamamoto, Trends Pharmacol. Sci., 2013, 34, 340–346 CrossRef CAS PubMed.
- M. C. Jaramillo and D. D. Zhang, Genes Dev., 2013, 27, 2179–2191 CrossRef CAS PubMed.
- M. C. Lu, J. A. Ji, Z. Y. Jiang and Q. D. You, Med. Res. Rev., 2016, 36, 924–963 CrossRef CAS PubMed.
- Z. Y. Jiang, M. C. Lu, L. L. Xu, T. T. Yang, M. Y. Xi, X. L. Xu, X. K. Guo, X. J. Zhang, Q. D. You and H. P. Sun, J. Med. Chem., 2014, 57, 2736–2745 CrossRef CAS PubMed.
- Z. Y. Jiang, M. C. Lu and Q. D. You, J. Med. Chem., 2016, 59, 10837–10858 CrossRef CAS PubMed.
- M. Lu, H. S. Zhou, Q. D. You and Z. Jiang, J. Med. Chem., 2016, 59, 7305–7310 CrossRef CAS PubMed.
- M. C. Lu, S. J. Tan, J. A. Ji, Z. Y. Chen, Z. W. Yuan, Q. D. You and Z. Y. Jiang, ACS Med. Chem. Lett., 2016, 7, 835–840 CrossRef CAS PubMed.
- H.-P. Sun, Z.-Y. Jiang, M.-Y. Zhang, M.-C. Lu, T.-T. Yang, Y. Pan, H.-Z. Huang, X.-J. Zhang and Q.-d. You, Med. Chem. Commun., 2014, 5, 93–98 RSC.
- M. Lu, T. Liu, Q. Jiao, J. Ji, M. Tao, Y. Liu, Q. You and Z. Jiang, Eur. J. Med. Chem., 2018, 146, 251–259 CrossRef CAS PubMed.
- K. Allton, A. K. Jain, H. M. Herz, W. W. Tsai, S. Y. Jung, J. Qin, A. Bergmann, R. L. Johnson and M. C. Barton, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 11612–11616 CrossRef CAS PubMed.
- A. K. Jain, K. Allton, A. D. Duncan and M. C. Barton, Mol. Cell. Biol., 2014, 34, 2695–2709 CrossRef PubMed.
- J. Bennett, O. Fedorov, C. Tallant, O. Monteiro, J. Meier, V. Gamble, P. Savitsky, G. A. Nunez-Alonso, B. Haendler, C. Rogers, P. E. Brennan, S. Muller and S. Knapp, J. Med. Chem., 2016, 59, 1642–1647 CrossRef CAS PubMed.
- W. S. Palmer, G. Poncet-Montange, G. Liu, A. Petrocchi, N. Reyna, G. Subramanian, J. Theroff, A. Yau, M. Kost-Alimova, J. P. Bardenhagen, E. Leo, H. E. Shepard, T. N. Tieu, X. Shi, Y. Zhan, S. Zhao, M. C. Barton, G. Draetta, C. Toniatti, P. Jones, M. Geck Do and J. N. Andersen, J. Med. Chem., 2016, 59, 1440–1454 CrossRef CAS PubMed.
- J. Jin, WO2018081530A1, 2018.
- T. Han, M. Goralski, N. Gaskill, E. Capota, J. Kim, T. C. Ting, Y. Xie, N. S. Williams and D. Nijhawan, Science, 2017, 356, eaal3755 CrossRef PubMed.
- T. Uehara, Y. Minoshima, K. Sagane, N. H. Sugi, K. O. Mitsuhashi, N. Yamamoto, H. Kamiyama, K. Takahashi, Y. Kotake, M. Uesugi, A. Yokoi, A. Inoue, T. Yoshida, M. Mabuchi, A. Tanaka and T. Owa, Nat. Chem. Biol., 2017, 13, 675–680 CrossRef CAS PubMed.
- J. Cheng, J. Guo, B. J. North, K. Tao, P. Zhou and W. Wei, Biochim. Biophys. Acta, Rev. Cancer, 2018, 1871, 138–159 CrossRef PubMed.
- G. E. Winter, Nat. Chem. Biol., 2017, 13, 575–576 CrossRef CAS PubMed.
- Y. Sun, Neoplasia, 2006, 8, 645–654 CrossRef CAS PubMed.
- C. Maniaci, S. J. Hughes, A. Testa, W. Chen, D. J. Lamont, S. Rocha, D. R. Alessi, R. Romeo and A. Ciulli, Nat. Commun., 2017, 8, 830 CrossRef PubMed.
- C. Steinebach, S. Lindner, N. D. Udeshi, D. C. Mani, H. Kehm, S. Kopff, S. A. Carr, M. Gutschow and J. Kronke, ACS Chem. Biol., 2018, 13, 2771–2782 CrossRef CAS PubMed.
- H. Lebraud, D. J. Wright, C. N. Johnson and T. D. Heightman, ACS Cent. Sci., 2016, 2, 927–934 CrossRef CAS PubMed.
- R. P. Wurz, K. Dellamaggiore, H. Dou, N. Javier, M. C. Lo, J. D. McCarter, D. Mohl, C. Sastri, J. R. Lipford and V. J. Cee, J. Med. Chem., 2018, 61, 453–461 CrossRef CAS PubMed.
- D. L. Buckley, K. Raina, N. Darricarrere, J. Hines, J. L. Gustafson, I. E. Smith, A. H. Miah, J. D. Harling and C. M. Crews, ACS Chem. Biol., 2015, 10, 1831–1837 CrossRef CAS PubMed.
- P. Ottis, M. Toure, P. M. Cromm, E. Ko, J. L. Gustafson and C. M. Crews, ACS Chem. Biol., 2017, 12, 2570–2578 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |