
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLewis acidic FeCl3 promoted 2-aza-Cope rearrangement to afford α-substituted homoallylamines in dimethyl carbonate†
Karthik Gadde ,
Jonas Daelemans,
Bert U. W. Maes and
Kourosch Abbaspour Tehrani*
,
Jonas Daelemans,
Bert U. W. Maes and
Kourosch Abbaspour Tehrani*
Organic Synthesis, Department of Chemistry, University of Antwerp, Groenenborgerlaan 171, 2020 Antwerp, Belgium. E-mail: kourosch.abbaspourtehrani@uantwerpen.be; Fax: +32 32653233; Tel: +32 32653226
First published on 7th June 2019
Abstract
The iron(III)-catalyzed efficient strategy for the synthesis of α-substituted homoallylamines was accomplished via a cationic 2-aza-Cope rearrangement of aldimines, generated in situ by condensation of commercially available aldehydes and easily synthesizable 1,1-diphenylhomoallylamines. This reaction features a broad substrate scope with high yields and is conducted in an eco-friendly solvent, i.e. dimethyl carbonate.
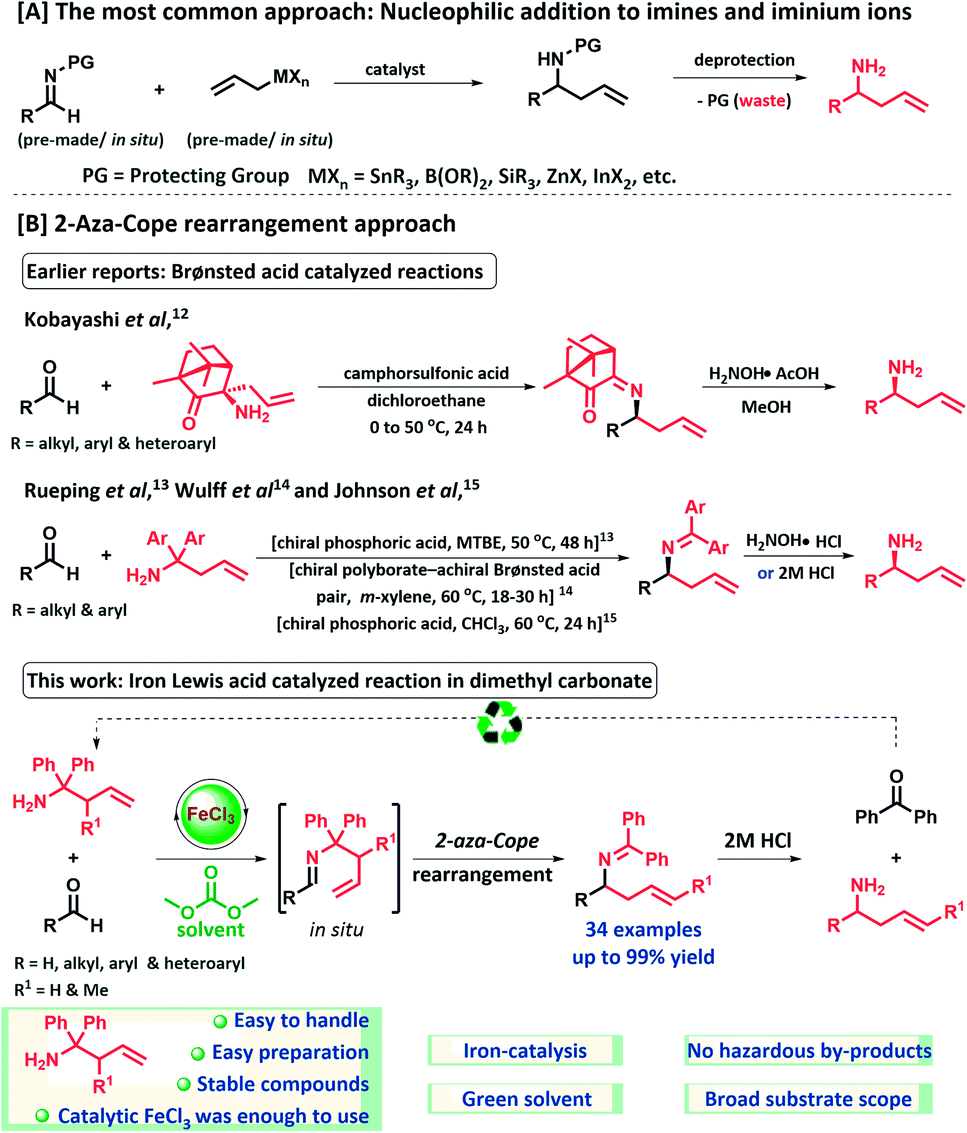
The use of sustainable catalysts and green solvents are the most influential factors to reduce the environmental impact of chemical synthesis. In this context, abundant base-metal catalysis, especially the field of iron catalysis has gained significant attention in organic synthesis.1 Iron has many advantages over other transition metals, such as low cost, low toxicity, its natural abundance, environmentally benign nature and often straightforward protocols for conversion to its corresponding salts and complexes. During the last three decades, there has been a growing interest in using green solvents in chemical processes. In recent solvent selection guidelines, dimethyl carbonate (DMC) has been classified as a “recommended” solvent.2 The green merits of dimethyl carbonate are fast biodegradability, low toxicity, mild odor, low evaporation rate, low density and good environmental compatibility.3 The latter can be explained by its low photochemical ozone creation potential (POCP) in comparison to common volatile organic compounds (VOCs) (2.5; e.g. ethylene = 100) and its recent production methods using CO2 as a raw material.3f–g
Homoallylamines are valuable intermediates and important structural motifs in organic chemistry for the synthesis of heterocycles, natural products and pharmaceutical compounds.4 The most common approach for α-substituted homoallylamine synthesis is the direct nucleophilic addition of allylic organometal or metalloid derivatives to imines (Scheme 1A).5 However, the addition of allyl Grignard reagents to imines are limited and mainly restricted to non-enolizable imines.6 Aldimines which contain α-hydrogens generally fail to give acceptable yields of secondary amines due to the poor electrophilicity of the imine carbon and competing α-deprotonation.6a–e In this regard, less basic reagents such as allyl stannanes, allyl silanes, allyl boronates and allyl boranes have been used. Despite significant utility of these reported methods, many of these procedures still show drawbacks, including generation of stoichiometric amounts of metal-containing (toxic) waste. Furthermore, often harsh deprotection conditions are required to obtain N-unprotected homoallylamines, also generating extra waste, which in most of the cases cannot be recovered and reused for reagent synthesis.5–7
 | ||
| Scheme 1 Synthesis of α-substituted homoallylamines. | ||
On the other hand, the [3,3]-sigmatropic rearrangement approach for homoallylamine synthesis is much less developed. In 1950, Horowitz and Geissman first reported a 2-aza-Cope rearrangement.8 This reaction has rarely been utilized in organic synthesis due to the inherent problem of the reversibility of the process.9 In pioneering studies, Overman and co-workers devised an aza-Cope–Mannich reaction sequence to overcome this problem and have applied this development successfully in numerous alkaloid syntheses.10 Recently, an aza-Cope rearrangement strategy has been utilized in fluorescent probes for imaging formaldehyde in biological systems.11 In literature, very few reports of 2-aza-Cope rearrangements are known for the synthesis of homoallylamines from aldehydes (Scheme 1B).12–15 In 2006, Kobayashi and co-workers developed a Brønsted acid catalyzed method for the synthesis of chiral homoallylamines from aldehyde and chiral camphorquinone derived homoallylamine.12 Unfortunately, highly hazardous dichloroethane was employed as solvent. In 2008, Rueping and Antonchick reported the first chiral phosphoric acid catalyzed aza-Cope rearrangement for the synthesis of chiral homoallylamines.13 Although the α-substituted homoallylamines were isolated in moderate to good yield (up to 87%), in good to high enantiomeric excesses (up to 94% ee) at 50 °C in methyl tert-butyl ether, this method limited to aromatic aldehydes and requires longer reaction times (48 h), which is a drawback. In 2011, Wulff et al., developed chiral polyborate – achiral Brønsted acid pair catalyzed reaction at 60 °C in m-xylene to improve the enantioselection with broader substrate scope for homoallylamines.14 Most recently, Johnson and co-workers reported an enantioconvergent method for the synthesis of chiral β-amino amides from racemic β-formyl amides and diphenylhomoallylamines at 60 °C in chloroform.15 To the best of our knowledge, no examples of Lewis acid catalyzed 2-aza-Cope rearrangement for the synthesis of homoallyamines from aldehydes in green solvent have been reported. Inspired by above catalytic methodologies and as a continuation of our efforts in imine activation reactions,16 herein we report an efficient and eco-friendly synthesis of homoallylamines via an iron(III)-catalyzed 2-aza-Cope rearrangement of in situ generated aldimines from readily accessible 1,1-diphenylhomoallylamines and aldehyde feedstocks. In this developed protocol, non-toxic and inexpensive iron(III) chloride has been used as a catalyst and the reaction has been carried out in dimethyl carbonate.
We commenced the optimization studies using benzaldehyde (1a) and 1,1-diphenylhomoallylamine (2a) as the model substrates. In the light of the recent advances in the field, the choice of sterically hindered 1,1-diphenylhomoallylamine was crucial to drive the reaction from the aldimine intermediate to the ketimine product.13–15 Our preliminary experiments with screening of various Lewis acids revealed that, the aza-Cope rearrangement on intermediate 3a can be achieved by using 10 mol% of Fe(OTf)3 (see the ESI Section-2† for details). Further, we continued our study from benzaldehyde (1a, 1.0 equiv.) and 1,1-diphenylhomoallylamine (2a, 1.0 equiv.) using iron salts as catalysts in standardly used dichloromethane in a sealed vial at 50 °C (Table 1). Screening of a number of iron salts revealed that FeCl3 was the best catalyst in terms of higher yield and relative cost (entries 1–5). Interestingly from solvent screening studies, dimethyl carbonate was found to be the best alternative for dichloromethane and the most of the solvents were compatible solvents for the reaction except polar solvents such as water, DMSO, DMF and MeOH (entries 5–20). Further, elevating the reaction temperature increased the reaction rate with complete conversion (entries 18–20). The FeCl3 catalyst furnished a complete and clean conversion within 6 h at 90 °C in dimethyl carbonate (99%, entry 20) and no traces of aldimine intermediate 3a were found in the reaction mixture.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (10 mol%) | Solvent | Temp (°C) | Time (h) | Yieldb [%] | |
| 3a | 4a | |||||
| a Reaction conditions: 1a (0.25 mmol, 1.0 equiv.), 2a (0.25 mmol, 1.0 equiv.), catalyst (10 mol%), 4 Å MS (100 mg) and solvent (0.5 mL).b Yields were determined by 1H NMR analysis with 1,3,5-trimethoxybenzene as internal standard.c Without MS.d 5 mol% of FeCl3 was used.e 100 mol% of BF3·Et2O was used. DMC = dimethyl carbonate. MS = molecular sieves. | ||||||
| 1 | Fe(OTf)3 | CH2Cl2 | 50 | 18 | 17 | 78 |
| 2 | FeCl3·6H2O | CH2Cl2 | 50 | 18 | 22 | 76 |
| 3 | FeCl2·4H2O | CH2Cl2 | 50 | 18 | 54 | 29 |
| 4 | FeCl2 | CH2Cl2 | 50 | 18 | 38 | 44 |
| 5 | FeCl3 | CH2Cl2 | 50 | 18 | 13 | 85 |
| 6 | FeCl3 | ClCH2CH2Cl | 85 | 18 | 0 | 98 |
| 7c | FeCl3 | H2O | 50 | 18 | 65 | 0 |
| 8 | FeCl3 | DMSO | 50 | 18 | 58 | 0 |
| 9 | FeCl3 | DMF | 50 | 18 | 47 | 0 |
| 10 | FeCl3 | MeCN | 82 | 18 | 0 | 94 |
| 11 | FeCl3 | MeOH | 65 | 18 | 84 | 14 |
| 12 | FeCl3 | 1,4-Dioxane | 101 | 18 | 0 | 94 |
| 13 | FeCl3 | EtOAc | 78 | 18 | 0 | 89 |
| 14 | FeCl3 | 2-MeTHF | 80 | 18 | 0 | 94 |
| 15 | FeCl3 | n-BuOAc | 126 | 18 | 0 | 94 |
| 16 | FeCl3 | Toluene | 110 | 18 | 0 | 91 |
| 17c | FeCl3 | Neat | 50 | 18 | 48 | 0 |
| 18 | FeCl3 | DMC | 25 | 18 | 9 | 71 |
| 19 | FeCl3 | DMC | 50 | 18 | 3 | 90 |
| 20 | FeCl3 | DMC | 90 | 6 | 0 | 99 |
| 21c | FeCl3 | DMC | 90 | 18 | 1 | 59 |
| 22d | FeCl3 | DMC | 90 | 18 | 64 | 24 |
| 23 | No catalyst | DMC | 90 | 18 | 91 | 6 |
| 24 | Fe(OTf)3 | DMC | 90 | 18 | 0 | 91 |
| 25 | InCl3 | DMC | 90 | 18 | 0 | 94 |
| 26 | AlCl3 | DMC | 90 | 18 | 70 | 11 |
| 27e | BF3·Et2O | DMC | 90 | 18 | 0 | 81 |
The formation of the product 4a was reduced when the reaction was carried out in the absence of molecular sieves (entry 21). Further, upon decreasing the amount of FeCl3 to 5 mol%, the yield of the product 4a was reduced and unsurprisingly, 3a was observed as major product without catalyst (entries 22–23). Under the optimized condition, similar yields were achieved with Fe(OTf)3 and InCl3 (entries 24–25), while other Lewis acids such as AlCl3 and BF3·Et2O afforded the product in lower yield (entries 26–27). Based on our Lewis acid catalyst screening (see the ESI Section-2† for details), FeCl3, InCl3, BiCl3, Fe(OTf)3, In(OTf)3, Sc(OTf)3 and Yb(OTf)3 were found to be effective catalysts and furnished the expected rearranged product in above 75% yield, while FeCl2, CuCl2, AlCl3, Ni(OTf)2 and other Lewis acids did not lead to good yields. These results seems to be in good agreement with literature17 data on classification of Lewis acids with respect to their activities in aldimine reactions. The low-toxic and inexpensive FeCl3 catalyst was selected for further investigation of the scope of the reaction.
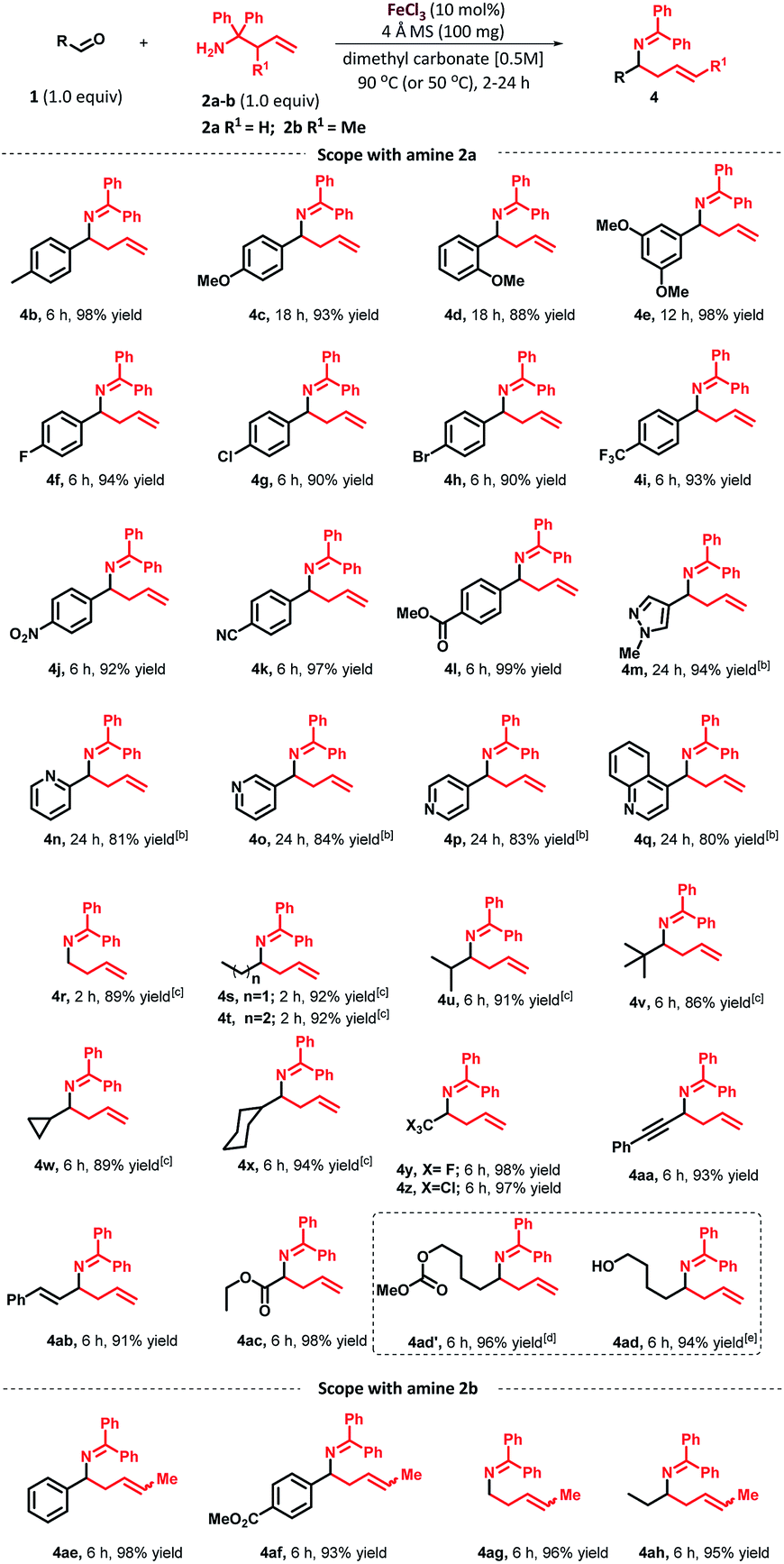
As depicted in Table 2, the scope of aldehydes was first investigated with 1,1-diphenyl homoallylamine (2a). To our delight, the scope of aldehydes was broad, providing high yields and showcasing the generality of this method. Aryl aldehydes bearing ortho-, meta- and para-substituents at the arene ring were all well tolerated under the optimized reaction conditions. As tabulated, p-tolualdehyde furnished the desired product 4b in 98% yield. In the case where the aryl ring contains an electron-donating methoxy group at the para-position (4c) and ortho-position (4d), it took 18 h to drive the reaction to completion. In general, we observed that electron rich aromatic aldehydes require longer reaction time than electron-poor aromatic aldehydes. The reaction of 3,5-dimethoxybenzaldehyde furnished the desired product 4e in 98% yield. Similarly, benzaldehyde with flouro-, chloro-, bromo- or trifluoromethyl substituents at the para-position afforded the desired products 4f–i in excellent yields (94%, 90%, 90% and 93% respectively). In addition, substituted benzaldehydes containing a variety of functional groups such as nitro (4j), nitrile (4k) and ester (4l) were compatible with the reaction conditions. Interestingly, heteroaromatic aldehydes containing a pyrazole, pyridine and quinoline ring were also successful and provided the desired products 4m–q in good yields, although a higher catalyst loading (25 mol%) was required. The use of a higher catalyst loading and slow progress of the reaction can be explained by the strong coordination between the nitrogen from the heterocyclic aldehyde with the metal catalyst, rendering the metal catalyst unavailable for participation in the catalytic cycle. As expected, the reaction of paraformaldehyde afforded 4r in 89% yield. Furthermore, linear, branched, and cyclic aliphatic aldehydes such as propionaldehyde, butyraldehyde, isobutyraldehyde, pivalaldehyde, cyclopropane- and cyclohexanecarboxaldehyde furnished the corresponding homoallylamines 4s–x, at 50 °C in 92%, 92%, 91%, 86%, 89% and 94% yields, respectively. Interestingly, transformation with trifluoroacetaldehyde hydrate and chloral hydrate provided the desired products 4y and 4z in excellent yields. In addition, it is worth noting that a variety of functional groups such as phenylpropargyl (4aa), trans-cinnamyl (4ab) and ester (4ac) were all well tolerated in the transformation, which would offer the potential for increasing molecular complexity via further functionalization. In the case of aldehyde substrate containing a hydroxy group, under the optimized reaction conditions, the methoxycarbonylated product 4ad′ was obtained in 96% yield instead of the expected product 4ad.18 To overcome this particular substrate issue, switching solvent system to propylene carbonate afforded the desired product 4ad in 94% yield. Next, we examined the scope of aldehydes with 2-methyl-1,1-diphenylhomoallylamine (2b). Under the optimized conditions, these reactions furnished E/Z mixtures of the corresponding homoallylamines 4ae–ah in good to excellent yields. The most of the examples in Table 2 did not require column chromatography purification.
| a Reaction conditions: 1 (0.25 mmol, 1 equiv.), 2a or 2b (0.25 mmol, 1 equiv.), FeCl3 (10 mol%), 4 Å molecular sieves (100 mg) in dimethyl carbonate (0.5 mL, 0.5 M), 90 °C, isolated yield.b 25 mol% of FeCl3 was used.c Reactions were carried out at 50 °C.d 96% yield of methoxycarbonylation of alcohol product was obtained.e In place of dimethyl carbonate, propylene carbonate was used as solvent at 90 °C. |
|---|
 |
To further demonstrate the potential of this method, a gram-scale reaction was performed. The reaction of benzaldehyde (1a, 5 mmol) and 1,1-diphenyl homoallylamine (2a, 5 mmol) under standard reaction conditions furnished the desired product 4a in 94% yield (Scheme 2)
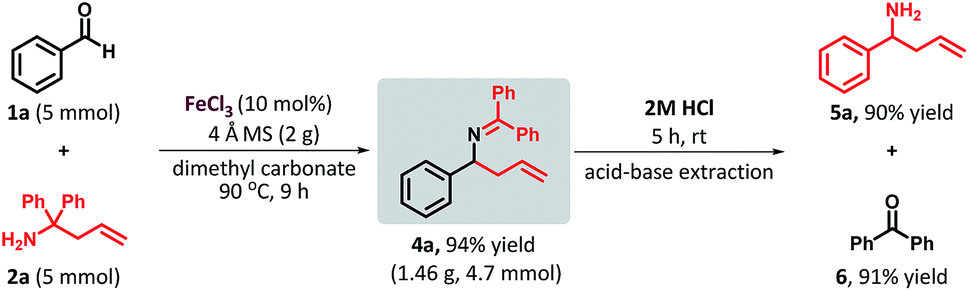 | ||
| Scheme 2 Scale-up reaction, hydrolysis of the ketimine and recovery of benzophenone. | ||
Based on our observations (see the ESI Section-3† for details) and in light of recent reports,13–15,16a the following reaction mechanism is proposed (Scheme 3)
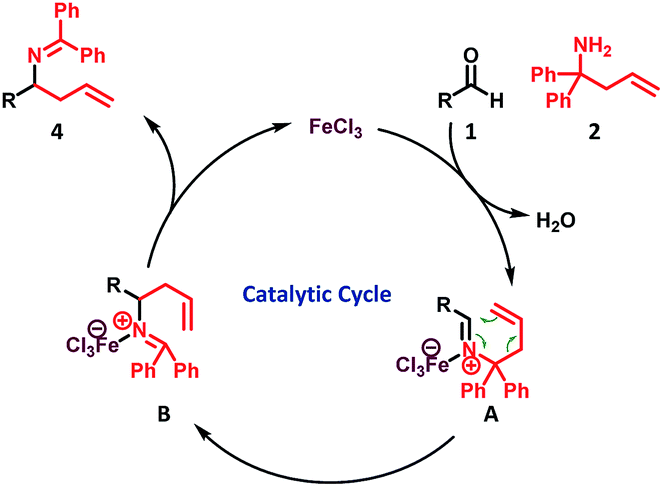 | ||
| Scheme 3 Plausible catalytic cycle. | ||
In summary, this study demonstrate the first example of iron catalyzed 2-aza-Cope rearrangement for the synthesis of a wide variety of α-substituted homoallylamines from readily accessible starting materials in a green solvent, producing water as the sole by-product. By this protocol, the synthesis of α-alkyl-, alkenyl-, aryl- and heteroaryl homoallylamines are achieved with high yields. Notably, N-unprotected (free NH2) α-substituted homoallyamines can be easily generated by mild acidic treatment of the rearranged benzophenone imines.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the University of Antwerp (BOF) and the Hercules Foundation. The authors thank Prof. Filip Lemière (University of Antwerp) for HRMS measurements.Notes and references
- For selected reviews on iron catalysis, see: (a) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217 CrossRef CAS PubMed; (b) B. Plietker, in Iron Catalysis in Organic Chemistry, ed. B. Plietker, WILEY-VCH Verlag, Weinheim, 2008 CrossRef; (c) S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317 CrossRef CAS PubMed; (d) A. Correa, O. García Mancheño and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108 RSC; (e) B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500 CrossRef CAS PubMed; (f) W. M. Czaplik, M. Mayer, J. Cvengroš and A. Jacobi von Wangelin, ChemSusChem, 2009, 2, 396 CrossRef CAS PubMed; (g) R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282 RSC; (h) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (i) K. Gopalaiah, Chem. Rev., 2013, 113, 3248 CrossRef CAS PubMed; (j) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170 CrossRef CAS PubMed; (k) A. Fürstner, ACS Cent. Sci., 2016, 2, 778 CrossRef PubMed; (l) A. Guérinot and J. Cossy, Top. Curr. Chem., 2016, 374, 49 CrossRef PubMed; (m) R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086 CrossRef CAS PubMed.
- (a) D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288 RSC; (b) H. E. Gottlieb, G. Graczyk-Millbrandt, G. G. A. Inglis, A. Nudelman, D. Perez, Y. Qian, L. E. Shuster, H. F. Sneddon and R. J. Upton, Green Chem., 2016, 18, 3867 RSC; (c) C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879 RSC; (d) N. R. Babij, E. O. McCusker, G. T. Whiteker, B. Canturk, N. Choy, L. C. Creemer, C. V. De Amicis, N. M. Hewlett, P. L. Johnson, J. A. Knobelsdorf, F. Li, B. A. Lorsbach, B. M. Nugent, S. J. Ryan, M. R. Smith and Q. Yang, Org. Process Res. Dev., 2016, 20, 661 CrossRef CAS.
- (a) P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706 CrossRef CAS PubMed; (b) B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554 CrossRef PubMed; (c) F. Aricò and P. Tundo, Russ. Chem. Rev., 2010, 79, 479 CrossRef; (d) S.-H. Pyo, J. H. Park, T.-S. Chang and R. Hatti-Kaula, Curr. Opin. Green Sustain. Chem., 2017, 5, 61 CrossRef; (e) G. Fiorani, A. Perosa and M. Selva, Green Chem., 2018, 20, 288 RSC; (f) F. Rivetti, Green Chemistry: Challenging Perspectives, ed. P. T. Anastas and P. Tundo, Oxford University Press, 2000, p. 201 Search PubMed; (g) M. E. Jenkin and G. D. Hayman, Atmos. Environ., 1999, 33, 1275 CrossRef CAS.
- Representative examples, see: (a) C. O. Puentes and V. Kouznetsov, J. Heterocycl. Chem., 2002, 39, 595 CrossRef CAS; (b) T. Saloranta and R. Leino, Tetrahedron Lett., 2011, 52, 4619 CrossRef CAS; (c) I. Bosque, J. C. González-Gómez, A. Guijarro, F. Foubelo and M. Yus, J. Org. Chem., 2012, 77, 10340 CrossRef CAS PubMed; (d) S. Zhao, G. Sirasani, S. Vaddypally, M. J. Zdilla and R. B. Andrade, Angew. Chem., Int. Ed., 2013, 52, 8309 CrossRef CAS PubMed; (e) A. Feula, S. S. Dhillon, R. Byravan, M. Sangha, R. Ebanks, M. A. H. Salih, N. Spencer, L. Male, I. Magyary, W. P. Deng, F. Müller and J. S. Fossey, Org. Biomol. Chem., 2013, 11, 5083 RSC; (f) B. Su, H. Zhang, M. Deng and Q. Wang, Org. Biomol. Chem., 2014, 12, 3616 RSC; (g) S. Munagala, G. Sirasani, P. Kokkonda, M. Phadke, N. Krynetskaia, P. Lu, F. J. Sharom, S. Chaudhury, M. D. M. Abdulhameed, G. Tawa, A. Wallqvist, R. Martinez, W. Childers, M. Abou-Gharbia, E. Krynetskiy and R. B. Andrade, Bioorg. Med. Chem., 2014, 22, 1148 CrossRef CAS PubMed; (h) P.-F. Chiang, W.-S. Li, J.-H. Jian, T.-S. Kuo, P.-Y. Wu and H.-L. Wu, Org. Lett., 2018, 20, 158 CrossRef CAS PubMed; (i) T. Druzhenko, Y. Skalenko, M. Samoilenko, A. Denisenko, S. Zozulya, P. O. Borysko, M. I. Sokolenko, A. Tarasov and P. K. Mykhailiuk, J. Org. Chem., 2018, 83, 1394 CrossRef CAS PubMed; (j) T. Guo, B.-H. Yuan and W.-J. Liu, Org. Biomol. Chem., 2018, 16, 57 RSC.
- For selected reviews, see: (a) Y. Yamamoto and N. Asao, Chem. Rev., 1993, 93, 2207 CrossRef CAS; (b) D. Enders and U. Reinhold, Tetrahedron: Asymmetry, 1997, 8, 1895 CrossRef CAS; (c) R. Bloch, Chem. Rev., 1998, 98, 1407 CrossRef CAS PubMed; (d) S. Kobayashi and H. Ishitani, Chem. Rev., 1999, 99, 1069 CrossRef CAS PubMed; (e) G. Alvaro and D. Savoia, Synlett, 2002, 2002, 651 CrossRef; (f) S. Kobayashi, Y. Mori, J. S. Fossey and M. M. Salter, Chem. Rev., 2011, 111, 2626 CrossRef CAS PubMed; (g) M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2011, 111, 7774 CrossRef CAS.
- (a) H. Thies and H. Schonenberger, Chem. Ber., 1956, 89, 1918 CrossRef CAS; (b) H. Gilman and J. Eisch, J. Am. Chem. Soc., 1957, 79, 2150 CrossRef CAS; (c) G. Stork and S. R. Dowd, J. Am. Chem. Soc., 1963, 85, 2178 CrossRef CAS; (d) R. W. Layer, Chem. Rev., 1963, 63, 489 CrossRef CAS; (e) A. R. Katritzky, Q. Hong and Z. Yang, J. Org. Chem., 1994, 59, 7947 CrossRef CAS; (f) A. Desmarchelier, P. Ortiz and S. R. Harutyunyan, Chem. Commun., 2015, 51, 703 RSC.
- P. G. M. Wuts, Greene's Protective Groups in Organic Synthesis, John Wiley & Sons, Hoboken, 5th edn, 2014 Search PubMed.
- R. M. Horowitz and T. A. Geissman, J. Am. Chem. Soc., 1950, 72, 1518 CrossRef CAS.
- (a) D. J. Hart and Y. M. Tsai, Tetrahedron Lett., 1981, 22, 1567 CrossRef CAS; (b) P. M. M. Nossin, J. A. M. Hamersma and W. N. Speckamp, Tetrahedron Lett., 1982, 23, 3807 CrossRef CAS; (c) H. Ent, H. de Koning and W. N. Speckamp, Tetrahedron Lett., 1983, 24, 2109 CrossRef CAS; (d) H. Ent, H. de Koning and W. N. Speckamp, Tetrahedron Lett., 1985, 26, 5105 CrossRef CAS; (e) E. Vedejs, S. C. Fields, R. Hayashi, S. R. Hitchcock, D. R. Powell and M. R. Schrimpf, J. Am. Chem. Soc., 1999, 121, 2460 CrossRef CAS; (f) P. Müller, J.-L. Toujas and G. Bernardinelli, Helv. Chim. Acta, 2000, 83, 1525 CrossRef; (g) Z. D. Aron and L. E. Overman, Org. Lett., 2005, 7, 913 CrossRef CAS PubMed; (h) P. Merino, T. Tejero and V. Mannucci, Tetrahedron Lett., 2007, 48, 3385 CrossRef CAS; (i) E. Marca, I. Delso, T. Tejero, J. T. Vazquez, R. L. Dorta and P. Merino, Tetrahedron Lett., 2009, 50, 7152 CrossRef CAS; (j) M. P. McCormack, T. Shalumova, J. M. Tanski and S. P. Waters, Org. Lett., 2010, 12, 3906 CrossRef CAS PubMed; (k) I. Delso, T. Tejero, A. Goti and P. Merino, J. Org. Chem., 2011, 76, 4139 CrossRef CAS PubMed; (l) I. Delso, A. Melicchio, A. Isasi, T. Tejero and P. Merino, Eur. J. Org. Chem., 2013, 2013, 5721 CrossRef CAS; (m) M. P. McCormack and S. P. Waters, J. Org. Chem., 2013, 78, 1176 CrossRef CAS PubMed; (n) S. Toulot, Q. Bonnin, V. Comte, L. Adriaenssens, P. Richard and P. Le Gendre, Eur. J. Org. Chem., 2013, 2013, 736 CrossRef CAS; (o) I. Bosque, F. Foubelo and J. C. Gonzalez-Gomez, Org. Biomol. Chem., 2013, 11, 7507 RSC; (p) J. Liu, C. G. Cao, H. B. Sun, X. Zhang and D. Niu, J. Am. Chem. Soc., 2016, 138, 13103 CrossRef CAS PubMed.
- (a) L. E. Overman and M.-A. Kakimoto, J. Am. Chem. Soc., 1979, 101, 1310 CrossRef CAS; (b) L. E. Overman and L. T. Mendelson, J. Am. Chem. Soc., 1981, 103, 5579 CrossRef CAS; (c) L. E. Overman, L. T. Mendelson and L. A. Flippin, Tetrahedron Lett., 1982, 23, 2733 CrossRef CAS; (d) L. E. Overman, M. Kakimoto, M. E. Okazaki and G. P. Meier, J. Am. Chem. Soc., 1983, 105, 6622 CrossRef CAS; (e) M. Brüggemann, A. I. McDonald, L. E. Overman, M. D. Rosen, L. Schwink and J. P. Scott, J. Am. Chem. Soc., 2003, 125, 15284 CrossRef PubMed; (f) W. G. Earley, J. E. Jacobsen, A. Madin, G. P. Meier, C. J. O'Donnell, T. Oh, D. W. Old, L. E. Overman and M. J. Sharp, J. Am. Chem. Soc., 2005, 127, 18046 CrossRef CAS PubMed; (g) C. L. Martin, L. E. Overman and J. M. Rohde, J. Am. Chem. Soc., 2008, 130, 7568 CrossRef CAS PubMed; (h) T. B. Dunn, J. M. Ellis, C. C. Kofink, J. R. Manning and L. E. Overman, Org. Lett., 2009, 11, 5658 CrossRef CAS PubMed; (i) C. L. Martin, L. E. Overman and J. M. Rohde, J. Am. Chem. Soc., 2010, 132, 4894 CrossRef CAS PubMed.
- (a) T. F. Brewer and C. J. Chang, J. Am. Chem. Soc., 2015, 137, 10886 CrossRef CAS PubMed; (b) A. Roth, H. Li, C. Anorma and J. Chan, J. Am. Chem. Soc., 2015, 137, 10890 CrossRef CAS PubMed.
- M. Sugiura, C. Mori and S. Kobayashi, J. Am. Chem. Soc., 2006, 128, 11038 CrossRef CAS PubMed.
- M. Rueping and A. P. Antonchick, Angew. Chem., Int. Ed., 2008, 47, 10090 CrossRef CAS PubMed.
- (a) H. Ren and W. D. Wulff, J. Am. Chem. Soc., 2011, 133, 5656 CrossRef CAS PubMed; (b) H. Ren and W. D. Wulff, Org. Lett., 2013, 15, 242 CrossRef CAS PubMed.
- C. G. Goodman and J. S. Johnson, J. Am. Chem. Soc., 2015, 137, 14574 CrossRef CAS PubMed.
- (a) S. Stas, K. Abbaspour Tehrani and G. Laus, Tetrahedron, 2008, 64, 3457 CrossRef CAS; (b) C. C. Malakar, B. U. W. Maes and K. Abbaspour Tehrani, Adv. Synth. Catal., 2012, 354, 3461 CrossRef CAS; (c) C. C. Malakar, S. Stas, W. Herrebout and K. Abbaspour Tehrani, Chem.–Eur. J., 2013, 19, 14263 CrossRef CAS PubMed; (d) W. E. Van Beek, J. Van Stappen, P. Franck and K. Abbaspour Tehrani, Org. Lett., 2016, 18, 4782 CrossRef CAS PubMed.
- S. Kobayashi, T. Busujima and S. Nagayama, Chem.–Eur. J., 2000, 6, 3491 CrossRef CAS.
- S. Jin, A. J. Hunt, J. H. Clark and C. R. McElroy, Green Chem., 2016, 18, 5839 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03277k |
| This journal is © The Royal Society of Chemistry 2019 |