
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDecorated single-enantiomer phosphoramide-based silica/magnetic nanocomposites for direct enantioseparation†
Fatemeh Karimi Ahmadabad a,
Mehrdad Pourayoubi*a and
Hadi Bakhshib
a,
Mehrdad Pourayoubi*a and
Hadi Bakhshib
aDepartment of Chemistry, Faculty of Science, Ferdowsi University of Mashhad, Mashhad, Iran. E-mail: pourayoubi@um.ac.ir
bMacromolecular Chemistry II, University of Bayreuth, Universitätsstraße 30, 95440 Bayreuth, Germany
First published on 29th August 2019
Abstract
The nano-composites Fe3O4@SiO2@(–O3Si[(CH2)3NH])P(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)(NH-R(+)CH(CH3)(C6H5))2 (Fe3O4@SiO2@PTA(+)) and Fe3O4@SiO2@(–O3Si[(CH2)3NH])P(O)(NH-S(−)CH(CH3)(C6H5))2 (Fe3O4@SiO2@PTA(−)) were prepared and used for the chiral separation of five racemic mixtures (PTA = phosphoric triamide). The separation results show chiral recognition ability of these materials with respect to racemates belonging to different families of compounds (amine, acid, and amino-acid), which show their feasibility to be potential adsorbents in chiral separation. The nano-composites were characterized by FTIR, TEM, SEM, EDX, XRD, and VSM. The VSM curves of nano-composites indicate their superparamagnetic property, which is stable after their use in the separation process. Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@PTA(+) and Fe3O4@SiO2@PTA(−) are regularly spherical with uniform shape and the average sizes of 17–20, 18–23, 36–47 and 43–52 nm, respectively.
O)(NH-R(+)CH(CH3)(C6H5))2 (Fe3O4@SiO2@PTA(+)) and Fe3O4@SiO2@(–O3Si[(CH2)3NH])P(O)(NH-S(−)CH(CH3)(C6H5))2 (Fe3O4@SiO2@PTA(−)) were prepared and used for the chiral separation of five racemic mixtures (PTA = phosphoric triamide). The separation results show chiral recognition ability of these materials with respect to racemates belonging to different families of compounds (amine, acid, and amino-acid), which show their feasibility to be potential adsorbents in chiral separation. The nano-composites were characterized by FTIR, TEM, SEM, EDX, XRD, and VSM. The VSM curves of nano-composites indicate their superparamagnetic property, which is stable after their use in the separation process. Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@PTA(+) and Fe3O4@SiO2@PTA(−) are regularly spherical with uniform shape and the average sizes of 17–20, 18–23, 36–47 and 43–52 nm, respectively.
Introduction
Chirality is a major concern in modern pharmaceuticals,1,2 pesticides,3 flavorings,4 food additives,5 non-linear optical materials5,6 and asymmetric catalysis.7–10 Chiral recognition/resolution is of great interest and ever-increasing importance, thus large efforts have been made towards techniques suitable for this aim such as chromatography,11–14 enzyme resolution,15 membrane separation,16–18 and chemical recognition.19 In this regard, the applications of chiral materials/chiral selectors in the enantioseparations based on the formation of different diastereomeric adducts, were studied,20–24 and the interactions such as hydrogen bonding, hydrophobic/hydrophilic, π–π, dipole–dipole, and ionic were reported in the adduct formation.25–28Since their discovery, silica-based materials have emerged as a source of immense potential for applications like for example in selective adsorption, separation, catalysis, and preparation of novel functional materials.29–36 Chemically modifiable surfaces and superparamagnetic property of magnetic modified silica nanoparticles have attracted much attention.31,37–41 The usefulness of magnetic adsorbents is due to their easily and rapidly separation under an external magnetic field, and in such purposes Fe2O3 nanorods, Fe3O4 and Fe–Pt alloy nanoparticles, and Fe3O4–polymer composites were studied.41–49 Recently, with regard to chiral discrimination, some magnetic nano-structured materials decorated with appropriate chiral molecules have been inspected.50–56 Typical examples are Fe3O4@SiO2@PNCD and Fe3O4@SiO2@CBDMPC, where the chiral selectors PNCD and CBDMPC are substituted β-cyclodextrin and cellulose, respectively, and their most important functional groups are OH and ether oxygen. Direct separation of enantiomers via such decorated magnetic nanomaterials would show great advantages compared with traditional methods of chiral separation.29,57 One example is the application of R(+)-α-methylbenzylamine-modified magnetic chiral sorbent in enantioseparation of mandelic acid.58
From our previous studies on phosphorus–oxygen, phosphorus–nitrogen and phosphorus–sulfur compounds,59–64 the PO group was found as the best hydrogen-bond acceptor with respect to the other possible acceptor groups which usually exist in the molecules, typically CO, SO, N–O, PS, ester and ether oxygen atoms, nitrogen, and π-system, examined with X-ray diffraction study, statistical analysis based on the data deposited in the Cambridge Structural Database (CSD)65,66 and quantum chemical calculations in some cases. The structures investigated include both achiral and chiral phosphorus compounds.63,67–70
With this background in mind, in the current article, we synthesized chiral phosphoramide enantiomers including Si–OCH3 segment in the achiral moiety, which is sensitive to hydrolysis condensation. This characteristic helps to immobilize silicon-based chiral phosphoric triamide (PTA) on Fe3O4@SiO2, to produce composite materials which dually possess the chiral and superparamagnetic properties. The target materials include the PO functional group which allows acting as a good selector, due to relatively strong interactions with OH and NH units. The full characterizations of prepared composites and their behavior as adsorbents in direct enantioseparation have been investigated.
Experimental
Materials
Phosphoryl chloride (POCl3, 99%), (R)-(+)-α-methylbenzylamine (RMBA, 98%), (S)-(−)-α-methylbenzylamine (SMBA, 98%) and (±)-α-methylbenzylamine (MBA, 98%) were bought from Sigma-Aldrich. Methanol, ethanol, tetrahydrofuran (THF), sodium chloride (NaCl), ferric chloride hexahydrate (FeCl3·6H2O), ferrous chloride tetrahydrate (FeCl2·4H2O), ammonium hydroxide (25% (w/w)), glycerol, tetraethyl orthosilicate (TEOS), 3-aminopropyltrimethoxysilane (APTMS), DL-tartaric acid (99%), DL-phenylalanine (99%), DL-valine (99%) and triethylamine (TEA, 99%) were purchased from Merck.Instruments
Fourier transformed infrared (FTIR) spectra were obtained by a Buck scientific spectrometer (model EQUINOX 55) using KBr disks. The size and morphology of nanoparticles were monitored using a transmission electron microscope (TEM) from Leo (model 920 AB) with accelerating voltage of 35 kV and resolution of 1 nm. A Tescan instrument (model Mira) was also used for scanning electron microscopy (SEM) with an accelerating voltage of 35 kV and a resolution of 4 nm. Energy-dispersive X-ray spectroscopy (EDX) was performed by an Oxford Instrument (model INCA). XRD patterns were recorded by a Bruker instrument (model D8 Advance) using Ni-filtered Cu Kα radiation. 31P, 1H- and 13C-nuclear magnetic resonance (NMR) spectra were recorded on a Bruker instrument (model Avance 300) using DMSO-d6/THF and CDCl3/CH3OH/H2O as solvents. Circular dichroism (CD) measurements were carried out on an Aviv circular dichroism spectrometer (model 215) using solutions in THF (5 g L−1). Magnetic susceptibility measurements were carried out using a vibrating sample magnetometer (VSM) (BHV-55, Riken, Japan) in the magnetic field range from −8000 to 8000 Oe at room temperature.Synthesis of single-enantiomer phosphoric triamides
The procedures for the synthesis of P(O)(NH-R-(+)CH(CH3)C6H5)2(NH(CH2)3Si(OCH3)3), PTA(+), and P(O)(NH-S-(−)CH(CH3)C6H5)2(NH(CH2)3Si(OCH3)3), PTA(−), are similar and the only difference is related to the enantiomeric form of amine used. Therefore, the procedure is typically described for PTA(+) as follows: a solution of RMBA (4.6 mmol) and TEA (4.6 mmol) in dry THF (15 mL) was added dropwise to a stirring solution of POCl3 (2.3 mmol) in dry THF (10 mL) at 0 °C. After stirring for 5 h, the obtained white solid (triethylammonium chloride salt) was filtered off and a solution of APTMS (2.3 mmol) and TEA (2.3 mmol) in dry THF (10 mL) was added dropwise to the filtrate at 0 °C. After stirring for 4 h, the generated triethylammonium chloride salt was filtered off. Since the Si(OCH3)3 moiety of PTA was moisture sensitive, a few amounts of THF solution were directly used for the NMR measurements, after adding two droplets of DMSO-d6 to the NMR tube for adjustment requirement. Finally, the THF solvent was removed under vacuum and the prepared phosphoric triamide was used for the synthesis of Fe3O4@SiO2@PTA. [α]RTD (in dry THF, 5 g L−1) = +34.1°. Selected peaks in FTIR (cm−1): 3396, 3248 (N–H), 3028 (C–Haromatic), 2949 (C–H), 2844, 1190 (PO), 1082 (Si–O). 31P{1H}-NMR (THF/DMSO-d6, ppm): 13.35. 13C{1H}-NMR (THF/DMSO-d6, ppm): 6.42 (s, C2), 21.63 (s, C6+11), 23.76 (s, C6+11), 25.09 (d, C3, 3JP–C = 6.5 Hz), 46.22 (s, C4), 50.10–50.90 (C1, C5+12), (126.26, 126.35, 126.62, 128.10, 128.53) (C8–10,14–16), 146.44 (d, C7+13), 147.70 (d, C7+13, 3JP–C = 7.2 Hz). 1H-NMR (THF/DMSO-d6, ppm): 0.56 (m, 2H, CH2), 1.38 (m, 6H, CH3), 1.57 (m, 2H, CH2), 2.02 (m, 2H, CH2), 3.40 (t, 1H, NH), 3.48 (m, 9H, CH3), 3.88 (t, 1H, NH), 3.98–4.22 (m, 2H, CH), 4.28 (b, 1H, NH), 7.15–7.50 (10H, C–Haromatic).
Preparation of chiral phosphoramide-modified hybrid magnetic/silica nanoparticles, Fe3O4@SiO2@PTA(+) and Fe3O4@SiO2@PTA(−)
Fe3O4, as nanoparticles, was prepared according to a previously reported method72 as follows: to a solution of FeCl3·6H2O (8.6 mmol) and FeCl2·4H2O (4.3 mmol) in H2O (40 mL) at 85 °C, NH3 (3 mL, 25%) was added (under bubbling of N2 gas). After 20 min, the magnetic nanoparticles were separated and washed with an aqueous solution of NaCl (0.02 M, 50 mL) and then twice with deionized water (2 × 50 mL), followed by drying under vacuum at 60 °C for 5 h.Fe3O4@SiO2 was prepared according to a published sol–gel method.72 The synthesis procedure is as follows: Fe3O4 (1 g) was dispersed in deionized water (100 mL) under ultrasonication for 10 min. After that, an aqueous solution of TEOS (10% v/v, 100 mL) and then glycerol (50 mL) were added to this mixture. The pH was adjusted to 4.5 by using glacial acetic acid and the reaction mixture was stirred continuously for 2 h at 90 °C. The product was separated with a magnet and then washed four times with ethanol. Finally, the obtained Fe3O4@SiO2 was dried under vacuum at 60 °C for 6 h.
At the end step, chiral phosphoric triamide was deposited on the Fe3O4@SiO2 nano-composite. The procedures are similar for deposition of PTA(+) or PTA(−) and typically is given for PTA(+). For this aim, Fe3O4@SiO2 (2 g) was ultrasonicated in ethanol (50 mL) for 10 min, then PTA(+) (0.5 g) in 2 mL ethanol was added under nitrogen atmosphere and the mixture refluxed for 12 h. After, the residue (Fe3O4@SiO2@PTA(+)) was magnetically separated and washed with methanol/H2O. The condensation of Si–OCH3 in phosphoric triamide and Si–OH in Fe3O4@SiO2 leads to deposition through the formation of Si–O–Si linkage.
Methods
Specific rotations of PTA(+) and PTA(−), [α], were measured with an automatic digital polarimeter (WZZ-2B, sodium lamp), using solutions in THF (5 g L−1), according to the following equation:| [α] = α/(L × c) | (1) |
The averages of particle sizes for nano-adsorbents were calculated by using the Scherrer equation, as follows:71
D = Kλ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ cosθ
| (2) |
Fe3O4@SiO2@PTA(+) and Fe3O4@SiO2@PTA(−) were examined for chiral separation of five racemic mixtures, i.e., (±)-α-methylbenzylamine, DL-tartaric acid, DL-alanine, DL-phenylalanine, and DL-valine. For this aim, 1 g adsorbent was mixed with 25 mL solution of each racemic mixture (5 g L−1) in H2O/CH3OH (80/20, v/v). The mixture was continuously stirred for 1 h. After that, the adsorbent was collected with a magnet and the optical purity of supernatant was determined through measuring its optical rotatory according to the following equation:52,53
| Optical purity = [α]supernatant/[α]pure enantiomer | (3) |
Results and discussion
Syntheses, spectroscopic features and optical properties of PTA(+) and PTA(−)
The route of synthesis and chemical structure of P(O)(NH-R-(+)CH(CH3)C6H5)2(NH(CH2)3Si(OCH3)3), PTA(+), and P(O)(NH-S-(−)CH(CH3)C6H5)2(NH(CH2)3Si(OCH3)3), PTA(−), are shown in Fig. 1a and b. For the synthesis of these chiral compounds, the prepared reagents P(O)(NH-R-(+)CH(CH3)C6H5)2Cl and P(O)(NH-S-(−)CH(CH3)C6H5)2Cl were treated with APTMS in order to the replacement of Cl atom by (CH3O)3Si(CH2)3NH moiety in the presence of triethylamine as an HCl scavenger. The P–Cl bond in the reagent and Si–OCH3 bond in PTA are moisture sensitive; hence, dry THF was used as the reaction medium. The usefulness of this solvent is also related to the high solubility of chiral reagent and chiral product and the low solubility of the triethylammonium chloride salt, which allows to a nearly clean reaction and easy purification.
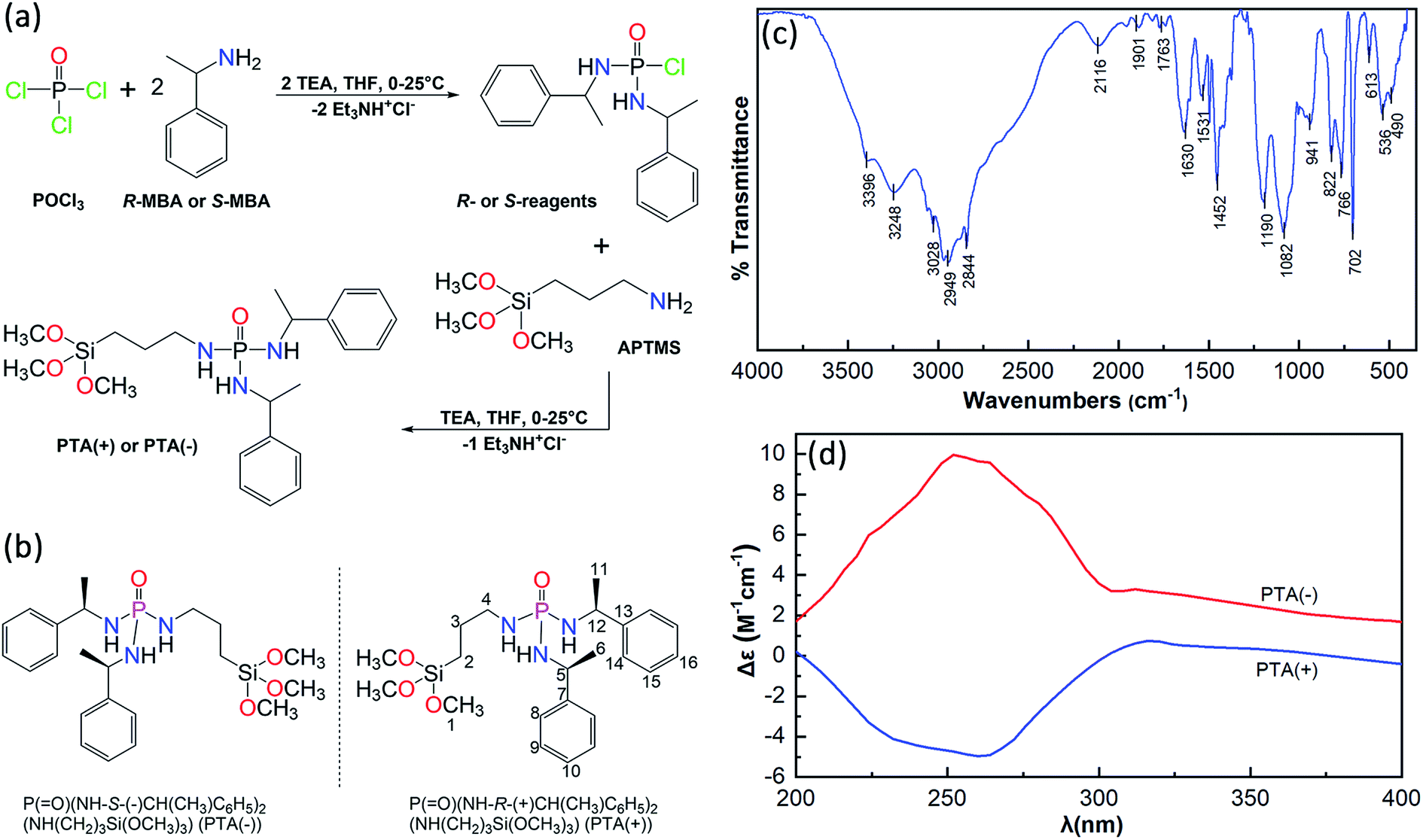 | ||
| Fig. 1 Synthesis route (a), chemical structure (b), FTIR spectrum (c) and CD spectra (d). | ||
It should be noted that the removing of triethylammonium chloride salt (or other organic salt depending on HCl scavenger used) can be done by H2O for the phosphoramides non-sensitive to moisture.68,73 For the water-sensitive compounds, a solvent with different solubilities of phosphoramide and organic salt is used. For examples, the syntheses of moisture-sensitive phosphoramides rac-XP(O)(OC6H5)(Cl) [X = (NHC6H4-p-CH3) and (NH-cyclo-C6H11)] were reported68 in dry chloroform, with low solubilities of p-toluidine hydrochloride and cyclohexylamine hydrochloride salts and high solubilities of phosphoramides. Moreover, the synthesis of P(O)(NH-R-(+)CH(CH3)C6H5)2Cl and P(O)(NH-S-(−)CH(CH3)C6H5)2Cl have been reported.63
The FTIR spectrum of PTA is shown in Fig. 1c, the characteristic peaks of N–H, C–H, PO and Si–O bonds confirm the correctness of the structure. In the 31P{1H} NMR spectrum (Fig. S1†) a singlet is observed at 13.35 ppm in comparison with the value of 3.75 ppm (in DMSO-d6) for [2,6-F2-C6H3C(O)NH][R-(+)(C6H5)CH(CH3)NH]2P(O), which is a closely related phosphoric triamide published in point of view of the chiral amine used.74
The specific rotations of P(O)(NH-R-(+)CH(CH3)C6H5)2(NH(CH2)3Si(OCH3)3) and P(O)(NH-S-(−)CH(CH3)C6H5)2(NH(CH2)3Si(OCH3)3) were obtained as +34.1° and −33.5°, respectively. The circular dichroism (CD) spectra of these two enantiomers are shown in Fig. 1d. The CD spectra exhibit opposing signals, a negative peak with a minimum at 260 nm belonged to PTA(+) and a positive peak with a maximum at 252 nm for PTA(−). On the other hand, the CD patterns are almost mirrored images with respect to one another in the range of 200–400 nm.
In the 13C{1H}-NMR spectrum (Fig. 2a), two sets of signals are observed for the carbon atoms of two NHCH(CH3)C6H5 groups. Typically, two singlets at 21.63 and 23.76 ppm are related to methyl carbon atoms and two doublets at 146.44 and 147.70 ppm correspond to the ipso-carbon atoms. The corresponding peaks for the (CH3O)3SiCH2CH2CH2 moiety are the singlets at 6.42, 46.22 and 50.10 ppm and a doublet at 25.09 ppm. The ortho, meta and para carbon atoms of two phenyl rings appear in the range of 126.26–128.53 ppm. For the assignment of the signals of chiral amine, the NMR spectra of [2,6-F2-C6H3C(O)NH][R-(+)(C6H5)CH(CH3)NH]2P(O)74 were inspected and for the assignment of the signals of silicon-based amine, the NMR spectra of Fe3O4@SiO2@–O2(OCH3)Si[(CH2)3NH2]75 and the newly synthesized ((CH3O)3SiCH2CH2CH2NH)P(O)(OC6H5)2 compound were investigated. The structure, details of the synthesis procedure of ((CH3O)3SiCH2CH2CH2NH)P(O)(OC6H5)2 and NMR data are given in the ESI (Fig. S2–S5†). The 1H-NMR spectrum shows the characteristic peaks for chiral and silicon-based amine fragments, which includes the signals at 0.56, 1.57, 2.02 and 3.48 ppm (multiplets) and at 4.28 ppm (broad) related to three CH2, CH3 groups and one NH for silicon-based amine, and the signals at 1.38, 3.40, 3.88 and 3.98–4.22 ppm, respectively for CH3, two NH units and CH groups of the chiral amine segments. The aromatic protons appeared at 7.15–7.50 ppm (Fig. 2b).
 | ||
| Fig. 2 The 13C-NMR (a) and 1H-NMR (b) spectra of PTA. The peaks marked with * are related to triethylammonium chloride. | ||
Syntheses and characterization of Fe3O4@SiO2@PTA(+) and Fe3O4@SiO2@PTA(−)
The Fe3O4 nanoparticles were prepared by precipitation from iron(II) and (III) ions in a basic solution under nitrogen atmosphere and then coated with a thin silica layer, formed by hydrolysis and condensation of TEOS in an acidic alcohol/water solution via a sol–gel process. Then, the immobilization of the chiral selector was carried out through the formation of Si–O–Si linkage,76 between the silica layer and silicon-based chiral selector. The procedure for the preparation of Fe3O4@SiO2@PTA(+) and Fe3O4@SiO2@PTA(−) is illustrated in Scheme 1. | ||
| Scheme 1 Preparation steps for the fabrication of Fe3O4@SiO2@PTA. | ||
FTIR
Grafting of chiral phosphoric triamide on Fe3O4@SiO2 was verified by FTIR. The FTIR spectra of Fe3O4, Fe3O4@SiO2, and Fe3O4@SiO2@PTA(+) are shown in Fig. 3, respectively. In all spectra, the broad peak in the range of 540–578 cm−1 is attributed to the Fe–O vibration. For silicon-containing nanoparticles, the strong and broad bands at about 1000–1100 cm−1 demonstrate the presence of Si–O–Si and Si–OH stretching vibrations.77,78 In the IR spectrum of Fe3O4@SiO2@PTA(+), the band at 1214 cm−1 is related to the PO stretching vibration of PTA(+) and the peaks in the range 2840–3248 cm−1 are related to C–H and N–H stretches. These peaks confirmed the occurrence of immobilization.
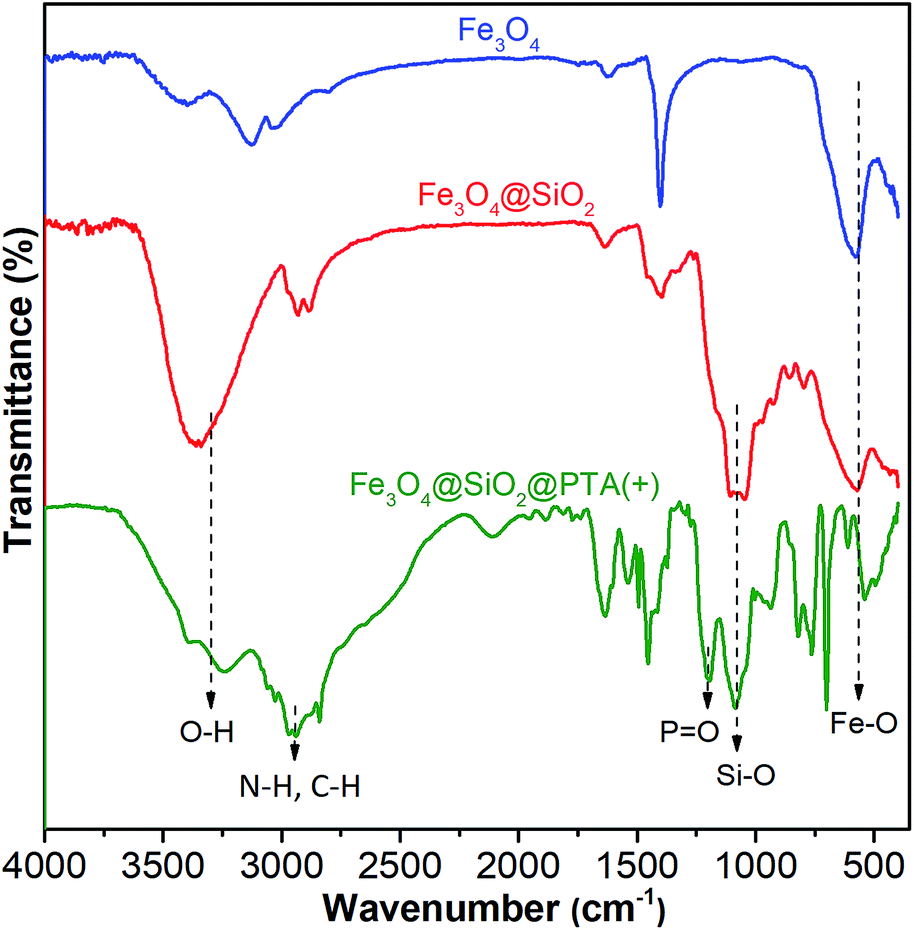 | ||
| Fig. 3 The FTIR spectra of Fe3O4, Fe3O4@SiO2, and Fe3O4@SiO2@PTA(+). | ||
VSM
The magnetic properties of Fe3O4, Fe3O4@SiO2, and Fe3O4@SiO2@PTA were studied by the hysteresis loops at room temperature by using vibrating sample magnetometry (VSM), with the magnetization curves as shown in Fig. 4. The magnetization curves exhibited superparamagnetic properties of Fe3O4 and Fe3O4@SiO2 with the saturation magnetizations of about 80 emu g−1 and 76 emu g−1, respectively. For Fe3O4@SiO2@PTA(+), the saturation magnetizations were measured before and after using in the chiral separation process, giving to be about 47 emu g−1 and 45 emu g−1, respectively, showing the stability of superparamagnetic property during the separation process.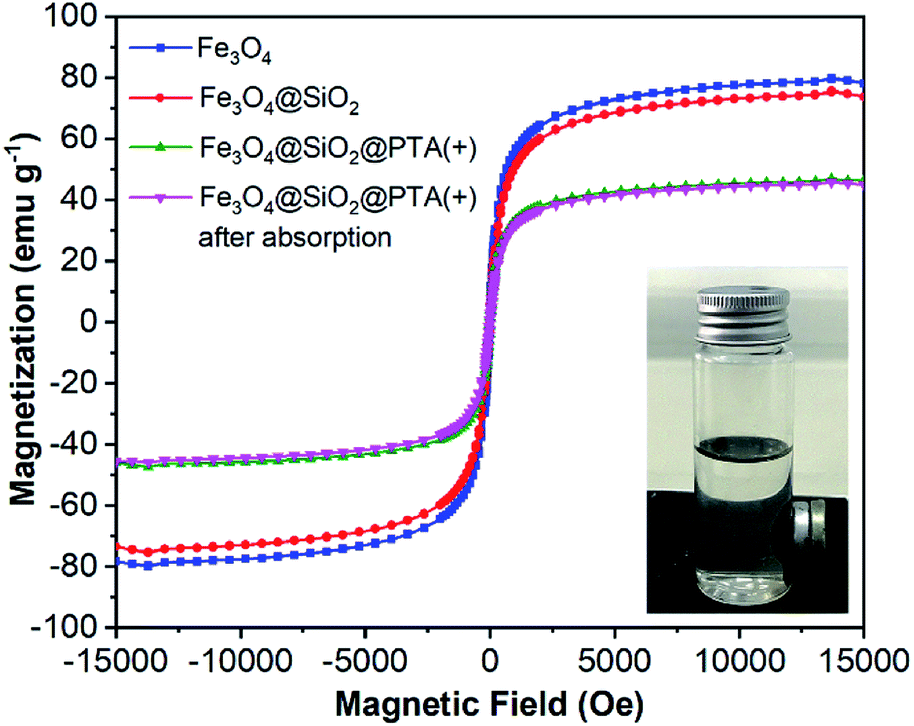 | ||
| Fig. 4 Magnetization curves for Fe3O4, Fe3O4@SiO2, and Fe3O4@SiO2@PTA(+) nanoparticles before and after using as an adsorbent. | ||
XRD
The crystalline structures of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@PTA(+) and Fe3O4@SiO2@PTA(−) were analyzed via XRD (Fig. 5). Characteristic diffraction peaks for Fe3O4 were observed at 30.1° (202), 35.5° (311), 43.1° (400), 53.6° (422), 57° (511), 62.6° (440), 71° (602) and 74° (533), in agreement with the crystalline cubic spinel Fe3O4 structure (JCPDS no. 19-0629).79 The similar characteristic peaks observed for Fe3O4@SiO2, Fe3O4@SiO2@PTA(+) and Fe3O4@SiO2@PTA(−) indicated the stability of the crystalline phase of Fe3O4 in the composite materials. The averages of particle sizes for nano-composites were calculated by using the Scherrer equation, which yielded the values of 17, 18, 36 and 43 nm for Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@PTA(+) and Fe3O4@SiO2@PTA(−), respectively, in good agreement with microscopy results. The expanded XRD spectra and related details have been shown in Fig. S6–S9.†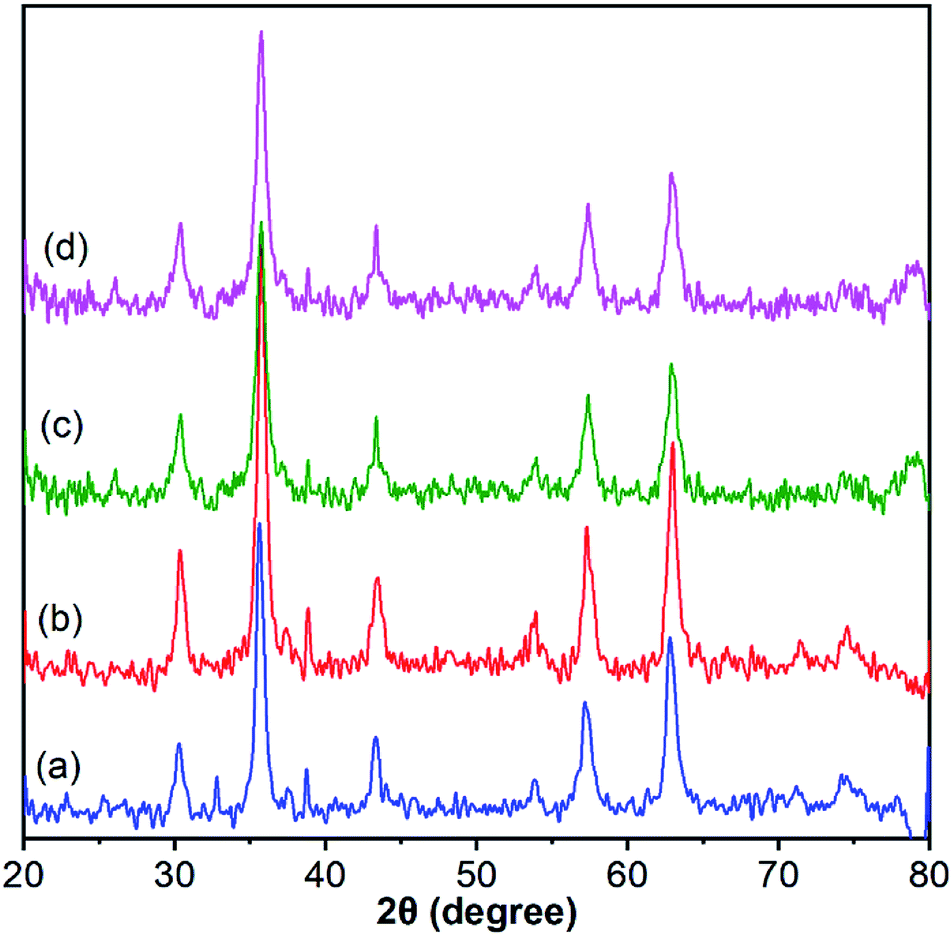 | ||
| Fig. 5 X-Ray powder diffraction patterns for (a) Fe3O4, (b) Fe3O4@SiO2, (c) Fe3O4@SiO2@PTA(+) and (d) Fe3O4@SiO2@PTA(−). The expanded spectra and related details have been shown in Fig. S6–S9.† | ||
TEM, SEM, and EDX
The morphologies of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@PTA(+) and Fe3O4@SiO2@PTA(−) were identified with SEM and TEM, as shown in Fig. 6. The images exhibit a nearly mono-dispersed structure for Fe3O4, with a rough surface because of the numerous reunited nanoparticles. After coating with SiO2, the surface of nanoparticles became smooth, and a SiO2 layer was observed around the Fe3O4 core. Deposition of chiral phosphoric triamide causes an increase in the particles' diameters. The averages of particle sizes are about 20, 23, 47 and 52 nm for Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@PTA(+) and Fe3O4@SiO2@PTA(−), respectively. | ||
| Fig. 6 SEM and TEM images of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@PTA(+) and Fe3O4@SiO2@PTA(−). | ||
EDX spectra of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@PTA(+) and Fe3O4@SiO2@PTA(−) were recorded. In Fe3O4@SiO2@PTA(+) and Fe3O4@SiO2@PTA(−) nanocomposites energetic lines for silicon, iron, carbon, oxygen, and phosphorus have been observed that confirm the immobilization of chiral selectors on the surface of Fe3O4@SiO2 (Fig. 7).
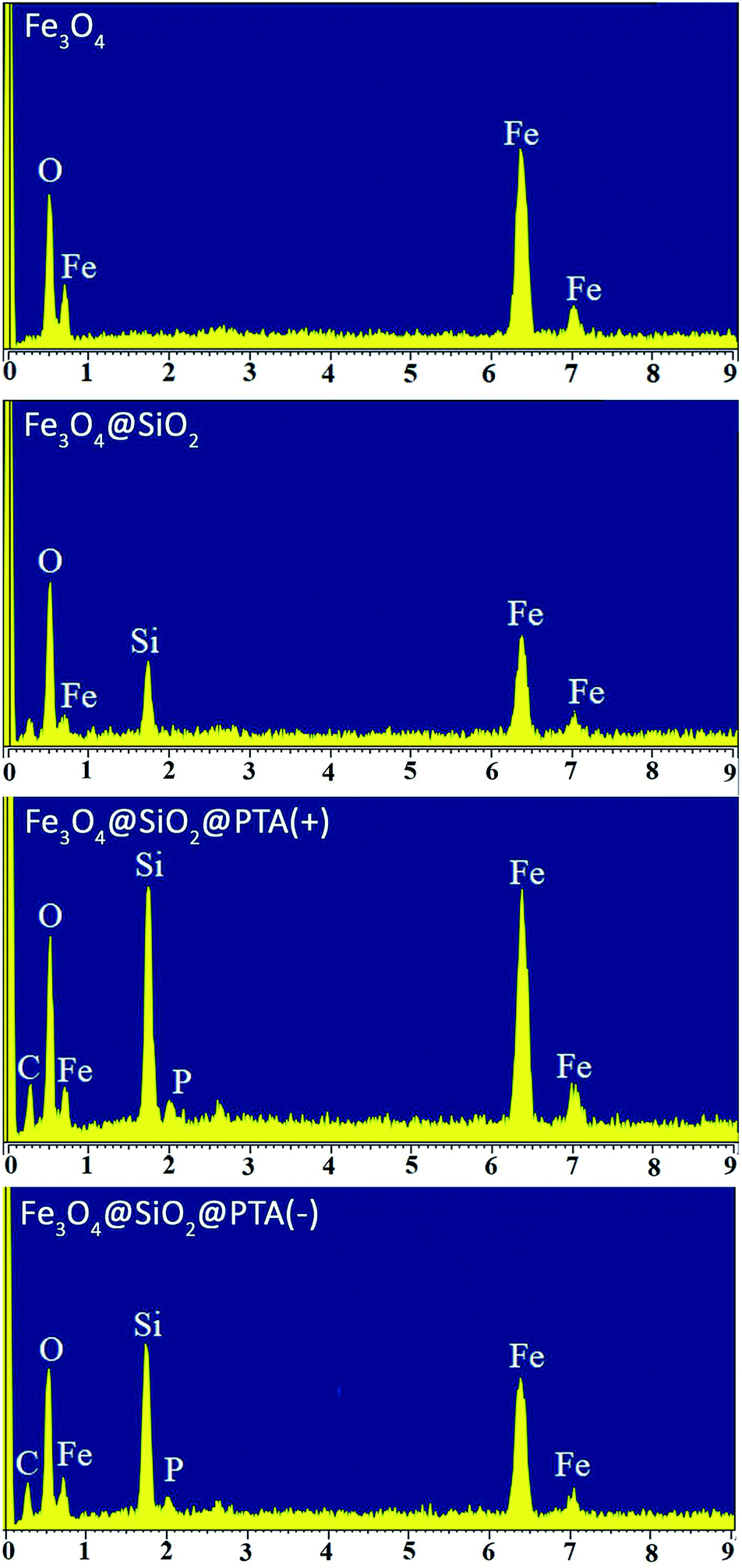 | ||
| Fig. 7 EDX images of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@PTA(+) and Fe3O4@SiO2@PTA(−). | ||
Direct separation of racemic compounds
Enantioseparation was carried out according to details specified in the experimental section for five racemic compounds (±)-α-methylbenzylamine, DL-tartaric acid, DL-alanine, DL-phenylalanine, and DL-valine. Before treatment with adsorbent, all five racemic compounds showed no optical activity because of the equal amounts of optical (+) and (−) enantiomers in the solutions. The results of enantioseparation processes are given in Table 1 and the chemical structures of racemic compounds are shown in Scheme 2. The optical rotations for supernatants showed that Fe3O4@SiO2@PTA(+) adsorbed (−) enantiomers of valine, alanine, tartaric acid and phenylalanine, and (+) enantiomer of α-methylbenzylamine, while Fe3O4@SiO2@PTA(−) separated the other enantiomers (optical purities 18–82%). To study the possibility of cleavage of chiral selector in the presence of racemic mixture, NMR spectroscopy was typically done for the supernatant of the enantioseparation experiment including Fe3O4@SiO2@PTA(+) in the presence of (±)-MBA solution. The 1H-NMR and 13C-NMR spectra merely showed the signals of α-methylbenzyl amine and the 31P-NMR spectrum did not show any signal, which confirmed the stability of adsorbent in the racemic mixture. The 1H-NMR spectrum is shown in Fig. S10.† The recycling of nano-composite adsorbents was also checked after ending the separation process. So, adsorbents were gathered by an external magnet, washed in a mixture of H2O/CH3OH under ultrasonic radiation for 10 min and dried in a vacuum oven for 12 h in 25 °C. Due to the release of the adsorbed enantiomer, the washing solution showed the optical rotatory.| Samples | [α]RTλ (°)/O.P. (%) | |
|---|---|---|
| Fe3O4@SiO2@PTA(+) | Fe3O4@SiO2@PTA(−) | |
| (±)-MBA | −16.8/82 | +14.7/72 |
| DL-Alanine | +8.9/79 | −8.0/71 |
| DL-Valine | +16.4/72 | −14.1/62 |
| DL-Tartaric acid | +2.4/19 | −2.3/18 |
| DL-Phenylalanine | +18.4/58 | −20.1/63 |
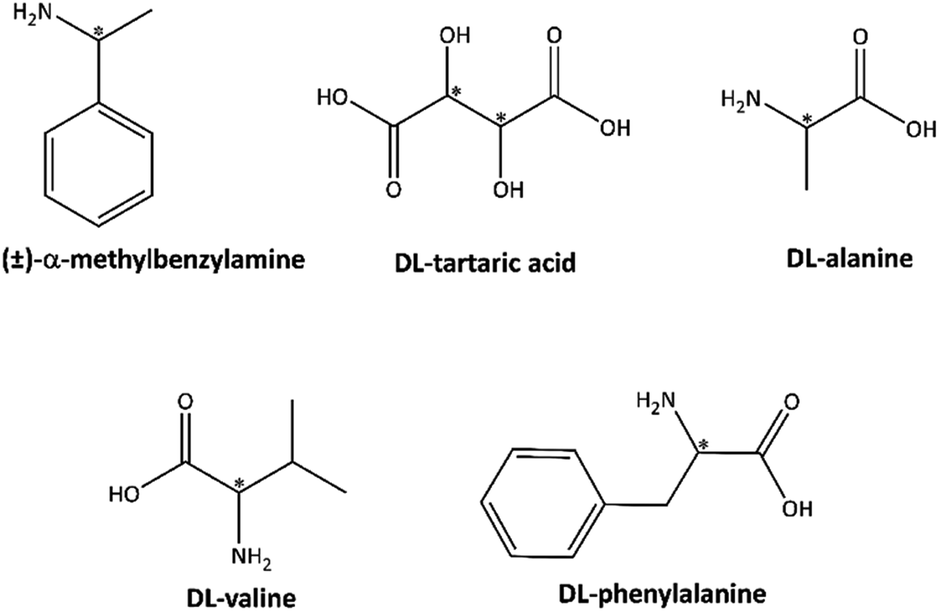 | ||
| Scheme 2 Chemical structures of racemic mixtures. | ||
Typically, the dried (+)-adsorbent was added to 25 mL of (±)-MBA solution (5 g L−1, water/methanol 80/20, v/v) and continuously stirred for 1 h. After that, the specific rotation of solution changed to −10.1° showing that the chiral adsorbent can be recycled with little loss of activity. This slight loss of activity may be due to a few changes in the morphology of nanoparticles in the presence of H2O/CH3OH/(±)-MBA, which is usual for nano-compounds in the presence of chemicals, however, it was not further investigated.53,80
A comparison of chiral adsorbents presented here with other chiral Fe3O4@SiO2-based adsorbents is given in Table 2. The Fe3O4@SiO2@PTA(+) and Fe3O4@SiO2@PTA(−) possess competitive advantages, such as high selectivity and recycling in the direct enantioseparation. Furthermore, we examined the racemic samples belonging to both acid and base families, which were successfully separated by these adsorbents. Preparation of both chiral (+) and (−) adsorbents is another advantage of this work, which helps to facilitate the separation process.
| Chiral adsorbents | Examined racemates | Optical purity (%) | Reference |
|---|---|---|---|
| Cellulose derivative-modified magnetic silica | (±)-2-Phenoxypropionic acid, benzoin methyl ether, promethazine hydrochloride | Above 80 | 57 |
| Teicoplanin-modified magnetic silica microspheres | DL-Tryptophan, DL-phenylalanine, DL-mandelic acid, N-benzoyl-DL-alanine | 8–35 | 53 |
| β-CD modified magnetic silica microspheres | Dansyl DL-valine, dansyl DL-phenylalanine, dansyl DL-leucine | 10–12 | 81 |
| HSA modified magnetic silica microspheres | DL-Tryptophan | 10–38 | 82 and 83 |
| Fe3O4@SiO2@PNCD | DL-Tryptophan | 80 | 28 |
| Fe3O4@SiO2@PTA(+) | (±)-MBA, DL-alanine, DL-valine, DL-tartaric acid, DL-phenylalanine | 18–82 | This work |
| Fe3O4@SiO2@PTA(−) |
Conclusion
Two single enantiomer nano-composites Fe3O4@SiO2@PTA(+) and Fe3O4@SiO2@PTA(−) were prepared and used for direct enantioseparation of some racemic mixtures. The racemates examined belong to both acid and base families. Excellent separation was achieved for examined basic racemic mixtures and comparative results were obtained for acidic ones with regard to previously reported Fe3O4@SiO2-based adsorbents. The nanocomposites were characterized by FTIR, TEM, SEM, EDX, XRD, and VSM. The nanoparticles have a uniform shape with an average diameter of around 40–50 nm. The superparamagnetic property is observed in nano-composites and is also stable after enantioseparation.Conflicts of interest
The authors have no conflicts of interest regarding this work.Acknowledgements
Authors gratefully acknowledge the financial support for this study by Ferdowsi University of Mashhad (Project No. 41927/3).References
- G. N. Sas, 9.17 Industrial Applications of Chiral Chromatography, 2012, vol. 9 Search PubMed.
- G.-Q. Lin, Q.-D. You and J.-F. Cheng, Chiral drugs: chemistry and biological action, John Wiley & Sons, 2011 Search PubMed.
- N. C. P. de Albuquerque, D. B. Carrão, M. D. Habenschus and A. R. M. de Oliveira, J. Pharm. Biomed. Anal., 2018, 147, 89–109 CrossRef PubMed.
- F. He, Y. L. Qian and M. C. Qian, Food Chem., 2018, 239, 622–630 CrossRef CAS.
- K. W. Smith, S. Link and W.-S. Chang, J. Photochem. Photobiol., C, 2017, 32, 40–57 CrossRef CAS.
- D. S. Bradshaw, J. M. Leeder, M. M. Coles and D. L. Andrews, Chem. Phys. Lett., 2015, 626, 106–110 CrossRef CAS.
- K. Itoh, F. Odate, T. Karikomi, K. Obe, T. Miyamori, H. Kamiya, K. Yoza, K. Nagai, H. Fujii and H. Suga, RSC Adv., 2019, 9, 12365–12369 RSC.
- Y. Miyagi, T. Sotani, T. Yajima, N. Sano and F. Sanda, Polym. Chem., 2018, 9, 1772–1779 RSC.
- Q. Zhang and L. Zhao, Tetrahedron Lett., 2018, 59, 310–316 CrossRef CAS.
- N. Yoshinari and T. Konno, Bull. Chem. Soc. Jpn., 2018, 91, 790–812 CrossRef CAS.
- G. D'Orazio, C. Fanali, M. Asensio-Ramos and S. Fanali, TrAC, Trends Anal. Chem., 2017, 96, 151–171 CrossRef.
- Z. Shedania, R. Kakava, A. Volonterio, T. Farkas and B. Chankvetadze, J. Chromatogr. A, 2018, 1557, 62–74 CrossRef CAS PubMed.
- R. S. Hegade, M. De Beer and F. Lynen, J. Chromatogr. A, 2017, 1515, 109–117 CrossRef CAS.
- J. Martens and R. Bhushan, J. Pharm. Biomed. Anal., 1990, 8, 259–269 CrossRef CAS.
- O. Jurček, M. Wimmerová and Z. Wimmer, Coord. Chem. Rev., 2008, 252, 767–781 CrossRef.
- J. J. Keating, S. Bhattacharya and G. Belfort, J. Membr. Sci., 2018, 555, 30–37 CrossRef CAS.
- C. Meng, Y. Sheng, Q. Chen, H. Tan and H. Liu, J. Membr. Sci., 2017, 526, 25–31 CrossRef CAS.
- T. Gumí, C. Minguillón and C. Palet, Polymer, 2005, 46, 12306–12312 CrossRef.
- S. Marchesan, C. D. Easton, K. E. Styan, L. J. Waddington, F. Kushkaki, L. Goodall, K. M. McLean, J. S. Forsythe and P. G. Hartley, Nanoscale, 2014, 6, 5172–5180 RSC.
- L. Wang, K. Yu, B.-B. Zhou, Z.-H. Su, S. Gao, L.-L. Chu and J.-R. Liu, Dalton Trans., 2014, 43, 6070–6078 RSC.
- Y. Dai, S. Wang, J. Zhou, Y. Liu, D. Sun, J. Tang and W. Tang, J. Chromatogr. A, 2012, 1246, 98–102 CrossRef CAS PubMed.
- Z. Shedania, R. Kakava, A. Volonterio, T. Farkas and B. Chankvetadze, J. Chromatogr. A, 2018, 1557, 62–74 CrossRef CAS PubMed.
- R. Gutierrez-Climente, A. Gomez-Caballero, A. Guerreiro, D. Garcia-Mutio, N. Unceta, M. A. Goicolea and R. J. Barrio, J. Chromatogr. A, 2017, 1508, 53–64 CrossRef CAS PubMed.
- G.-H. Zhang, S. Liang, S. Tang, W. Chen and Z.-W. Bai, Anal. Methods, 2019, 11, 1604–1612 RSC.
- R. Wang, Y. Zheng, X. Li, J. Chen, J. Cui, J. Zhang and X. Wan, Polym. Chem., 2016, 7, 3134–3144 RSC.
- B. Kafková, Z. Bosáková, E. Tesařová and P. Coufal, J. Chromatogr. A, 2005, 1088, 82–93 CrossRef PubMed.
- Y. Li, X. Chen and N. Gu, J. Phys. Chem. B, 2008, 112, 16647–16653 CrossRef CAS.
- Y. K. Ye and R. W. Stringham, Chirality, 2006, 18, 519–530 CrossRef CAS PubMed.
- L. Guo, Y. Song, H. Yu, L. Pan and C. Cheng, Appl. Surf. Sci., 2017, 407, 82–92 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard and S. B. McCullen, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- Y. Yu, L. Yu, K. Shih and J. P. Chen, J. Colloid Interface Sci., 2018, 521, 252–260 CrossRef CAS PubMed.
- W. Cai, M. Guo, X. Weng, W. Zhang and Z. Chen, Mater. Sci. Eng., C, 2019, 98, 65–73 CrossRef CAS PubMed.
- M. Shao, F. Ning, J. Zhao, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2012, 134, 1071–1077 CrossRef CAS PubMed.
- T. Zhao, X. Zhu, C.-T. Hung, P. Wang, A. Elzatahry, A. A. Al-Khalaf, W. N. Hozzein, F. Zhang, X. Li and D. Zhao, J. Am. Chem. Soc., 2018, 140, 10009–10015 CrossRef CAS.
- Y. Li, J. J.-Y. Suen, E. Prince, E. M. Larin, A. Klinkova, H. Thérien-Aubin, S. Zhu, B. Yang, A. S. Helmy and O. D. Lavrentovich, Nat. Commun., 2016, 7, 12520 CrossRef CAS.
- Z. Asgharpour, F. Farzaneh and A. Abbasi, RSC Adv., 2016, 6, 95729–95739 RSC.
- C. Maccato, G. Carraro, D. Peddis, G. Varvaro and D. Barreca, Appl. Surf. Sci., 2018, 427, 890–896 CrossRef CAS.
- N. Liang, X. Hou, P. Huang, C. Jiang, L. Chen and L. Zhao, Sci. Rep., 2017, 7, 13844 CrossRef.
- Q. Gao, W. Xie, Y. Wang, D. Wang, Z. Guo, F. Gao, L. Zhao and Q. Cai, RSC Adv., 2018, 8, 4321–4328 RSC.
- H. Bagheri, N. Pajooheshpour, A. Afkhami and H. Khoshsafar, RSC Adv., 2016, 6, 51135–51145 RSC.
- S. Tural, B. Tural, M. Ş. Ece, E. Yetkin and N. Özkan, J. Sep. Sci., 2014, 37, 3370–3376 CrossRef CAS PubMed.
- Q. Wei, P. Song, Z. Yang and Q. Wang, Phys. E, 2018, 103, 156–163 CrossRef CAS.
- S. R. Firoozabadi, M. Bonyadi and A. Lashanizadegan, J. Nat. Gas Sci. Eng., 2018, 59, 374–386 CrossRef.
- Q. Zheng, Z. R. Zhang, J. Du, L. L. Lin, W. X. Xia, J. Zhang, B. R. Bian and J. P. Liu, J. Mater. Sci. Nanotechnol., 2019, 35, 560–567 CrossRef.
- M. Gadhvi, Mater. Sci. Eng., B, 2010, 168, 106–110 CrossRef CAS.
- X. Deng, W. Li, G. Ding, T. Xue and X. Chen, Sep. Purif. Rev., 2019, 48, 14–29 CrossRef CAS.
- W. Wang, B. Tang, B. Ju, Z. Gao, J. Xiu and S. Zhang, J. Mater. Chem. A, 2017, 5, 958–968 RSC.
- X. Liu, Z. Ma, J. Xing and H. Liu, J. Magn. Magn. Mater., 2004, 270, 1–6 CrossRef CAS.
- B. Tural, E. Ertaş and S. Tural, Desalin. Water Treat., 2016, 57, 26153–26164 CrossRef CAS.
- B. Tural, E. Ertaş, B. Enez, S. A. Fincan and S. Tural, J. Environ. Chem. Eng., 2017, 5, 4795–4802 CrossRef CAS.
- S. Tural, M. Ş. Ece and B. Tural, Ecotoxicol. Environ. Saf., 2018, 162, 245–252 CrossRef CAS.
- Y. Wei, A. Tian, Y. Li, X. Wang and B. Cao, J. Mater. Chem., 2012, 22, 8499–8504 RSC.
- J. Wu, P. Su, J. Huang, S. Wang and Y. Yang, J. Colloid Interface Sci., 2013, 399, 107–114 CrossRef CAS PubMed.
- H. J. Choi and M. H. Hyun, Chem. Commun., 2009, 6454–6456 RSC.
- K. Li, Z. Zeng, J. Xiong, L. Yan, H. Guo, S. Liu, Y. Dai and T. Chen, Colloids Surf., A, 2015, 465, 113–123 CrossRef CAS.
- L.-Y. Guo, S.-Y. Zeng, Z. Jagličić, Q.-D. Hu, S.-X. Wang, Z. Wang and D. Sun, Inorg. Chem., 2016, 55, 9006–9011 CrossRef CAS PubMed.
- J. M. Chem, Y. Wei, A. Tian, Y. Li, X. Wang and B. Cao, J. Mater. Chem., 2012, 22, 8499–8504 RSC.
- T. Tarhan, B. Tural, S. Tural and G. Topal, Chirality, 2015, 27, 835–842 CrossRef CAS PubMed.
- E. Torabi Farkhani, M. Pourayoubi, M. Izadyar, P. V. Andreev and E. S. Shchegravina, Acta Crystallogr., Sect. C: Struct. Chem., 2018, 74, 847–855 CrossRef CAS.
- B. Vahdani Alviri, M. Pourayoubi, A. Farhadipour, M. Nečas and V. Bertolasi, Acta Crystallogr., Sect. C: Struct. Chem., 2018, 74, 1610–1621 CrossRef CAS.
- F. Sabbaghi, M. Pourayoubi, M. Nečas and K. Damodaran, Acta Crystallogr., Sect. C: Struct. Chem., 2019, 75, 77–84 CrossRef CAS PubMed.
- S. Akbari, R. S. Khoshnood, F. Karimi Ahmadabad, M. Pourayoubi, M. Dušek and E. S. Shchegravina, RSC Adv., 2019, 9, 9153–9159 RSC.
- F. Karimi Ahmadabad, M. Pourayoubi and H. Bakhshi, J. Appl. Polym. Sci., 2019, 136, 48034 CrossRef.
- B. Vahdani Alviri, M. Pourayoubi, A. Saneei, M. Keikha, A. Van Der Lee, A. Crochet, A. A. Ajees, M. Nečas, K. M. Fromm, K. Damodaran and T. A. Jenny, Tetrahedron, 2018, 74, 28–41 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS.
- F. Sabbaghi, M. Dušek, S. Bayat and M. Nečas, Struct. Chem., 2016, 27, 1831–1844 CrossRef CAS.
- M. Pourayoubi, F. Karimi Ahmadabad, H. Eshtiagh-Hosseini, M. Kučeráková, V. Eigner and M. Dušek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 1181–1185 CrossRef CAS PubMed.
- M. Toghraee, M. Pourayoubi and V. Divjakovic, Polyhedron, 2011, 30, 1680–1690 CrossRef CAS.
- A. Tarahhomi, M. Pourayoubi, J. A. Golen, P. Zargaran, B. Elahi, A. L. Rheingold, M. A. Leyva Ramírez and T. Mancilla Percino, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 260–270 CrossRef CAS PubMed.
- U. Holzwarth and N. Gibson, Nat. Nanotechnol., 2011, 6, 534 CrossRef CAS PubMed.
- X. Liu, Z. Ma, J. Xing and H. Liu, J. Magn. Magn. Mater., 2004, 270, 1–6 CrossRef CAS.
- M. Keikha, M. Pourayoubi, A. Tarahhomi and A. van der Lee, Acta Crystallogr., Sect. C: Struct. Chem., 2016, 72, 251–259 CrossRef CAS PubMed.
- M. Eghbali Toularoud, M. Pourayoubi, M. Dušek, V. Eigner and K. Damodaran, Acta Crystallogr., Sect. C: Struct. Chem., 2018, 74, 608–617 CrossRef CAS PubMed.
- S. Radi, S. Tighadouini, M. Bacquet, S. Degoutin, F. Cazier, M. Zaghrioui and Y. N. Mabkhot, Molecules, 2013, 19, 247–262 CrossRef PubMed.
- H. Naeimi, Z. S. Nazifi and Z. Sadat, J. Nanopart. Res., 2013, 15, 2026 CrossRef PubMed.
- L.-D. Guo, Y.-Y. Song, H.-R. Yu, L.-T. Pan and C.-J. Cheng, Appl. Surf. Sci., 2017, 407, 82–92 CrossRef CAS.
- T. Kieu, H. Ta, M. Trinh, N. V. Long, T. Thanh, M. Nguyen, T. Lien, T. Nguyen, T. L. Thuoc, B. T. Phan, D. Mott, S. Maenosono, H. Tran-van and V. H. Le, Colloids Surf., A, 2016, 504, 376–383 CrossRef.
- T. Yang, C. Shen, Z. Li, H. Zhang, C. Xiao, S. Chen, Z. Xu, D. Shi, J. Li and H. Gao, J. Phys. Chem. B, 2005, 109, 23233–23236 CrossRef CAS PubMed.
- F. Karimi Ahmadabad, M. Pourayoubi and H. Bakhshi, Mater. Chem. Phys., 2017, 199, 79–87 CrossRef.
- J. Wu, P. Su, D. Guo, J. Huang and Y. Yang, New J. Chem., 2014, 38, 3630–3636 RSC.
- F. Edwie, Y. Li and T.-S. Chung, J. Membr. Sci., 2010, 362, 501–508 CrossRef CAS.
- J. Wu, P. Su, Y. Yang, J. Huang, Y. Wang and Y. Yang, J. Mater. Chem. B, 2014, 2, 775–782 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03260f |
| This journal is © The Royal Society of Chemistry 2019 |