
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePaving the way towards green catalytic materials for green fuels: impact of chemical species on Mo-based catalysts for hydrodeoxygenation†
Diego Valencia *a,
Leonardo Díaz-Garcíaa,
Luis Felipe Ramírez-Verduzcoa,
Amir Qamarb,
Alexander Moewesb and
Jorge Aburtoa
*a,
Leonardo Díaz-Garcíaa,
Luis Felipe Ramírez-Verduzcoa,
Amir Qamarb,
Alexander Moewesb and
Jorge Aburtoa
aDirección de Investigación en Transformación de Hidrocarburos, Instituto Mexicano Del Petróleo, Eje Central Lázaro Cárdenas 152, Col. San Bartolo Atepehuacan, CP 07730, Mexico City, Mexico. E-mail: dvalencia@imp.mx
bDepartment of Physics and Engineering Physics, University of Saskatchewan, 116 Science Place, Saskatoon, SK S7N 5E2, Canada
First published on 11th June 2019
Abstract
A series of Mo-based catalysts were synthesized by tuning the sulfidation temperature to produce mixtures of MoO3 and MoS2 as active phases for the hydrodeoxygenation (HDO) of palmitic acid. Differences in the oxidation states of Mo, and the chemical species present in the catalytic materials were determined by spectroscopic techniques. Palmitic acid was used as a fatty-acid model compound to test the performance of these catalysts. The catalytic performance was related to different chemical species formed within the materials. Sulfidation of these otherwise inactive catalysts significantly increased their performance. The catalytic activity remains optimal between the sulfidation temperatures of 100 °C and 200 °C, whereas the most active catalyst was obtained at 200 °C. The catalytic performance decreased significantly at 400 °C due to a higher proportion of sulfides formed in the materials. Furthermore, the relative proportion of MoO3 to MoS2 is essential to form highly active materials to produce O-free hydrocarbons from biomass feedstock. The transition from MoS2 to MoO3 reveals the importance of Mo–S and Mo–O catalytically active species needed for the HDO process and hence for biomass transformation. We conclude that transitioning from MoS2 to MoO3 catalysts is a step in the right direction to produce green fuels.
1 Introduction
Currently about 82% of the world's energy consumption is based on products derived from fossil fuels. These hydrocarbons containing natural resources are first refined to intermediates, additives, or polymers before they become suitable energy sources for different purposes. However, there are currently several concerns regarding their use.1,2 These are non-renewable energy resources, which take millions of years to form and their extraction has faced continuous challenges due to rapid depletion of known reserves. Furthermore, fossil fuels have been a major contributor to global warming, and we must take steps to investigate new sustainable and more environmental friendly energy sources for the future.3,4Generation of bio-fuels has become a major research and industrial topic during this decade.5,6 Vegetable oils, especially palm oil, are considered good candidates to develop environmental friendly and high quality fuels.7 However, these bio-based oils cannot be used immediately as fuels due to their high oxygen content, which leads to a low heating value, immiscibility with fossil fuels, a tendency for polymerization, thermal instability and high viscosity.8 Catalytic hydrotreating (HDT) is an available technology for the conversion of biomass to liquid biofuels. HDT has been used in past decades to eliminate targeted atoms within hydrocarbon molecules.9 Hydrodeoxygenation (HDO) is the main catalytic process to eliminate oxygen from the organic molecules by hydrogenation and hydrogenolysis reactions.10 This catalytic process takes place at higher H2 pressures and temperatures in the presence of a heterogeneous catalyst, typically MoS2-based active phases supported on metal oxides.11–13
MoS2-based catalysts were developed for the hydrodesulfurization (HDS) process and this metal sulfide is highly active in the elimination of sulfur atom in crude oils. In general, this active phase provides catalytically active sites for hydrogenation and hydrogenolysis reactions, where numerous chemical processes can take place. Its application in biomass conversion has shown a tremendous potential as a catalytic system. Heterogeneous catalysts consisting of MoS2 active species have been tested successfully in the HDO of fatty acids and triacylglycerides. The operational conditions are almost identical to the ones reported for the hydrotreating processes (high temperature and H2 pressure). However, it faced several disadvantages due to the rapid deactivation from coke formation on the catalytic surface or the presence of water that leaches metal species in fluid phase. MoO3 as an active phase is a promising alternative because it does not need any prior sulfidation stage,14,15 but further investigation is still necessary to improve its catalytic performance in HDO reactions. This catalytic active phase has been tested in the HDO of aromatics at low H2 pressure in different oxide supports. It is remarkable that the oxidation states of Mo atoms vary depending on the support used, and these changes might play a crucial role in the elimination of O atoms contained in biomass-derived products.16
The supported MoS2 active phase is prepared from the conversion of MoO3 under sulfidation conditions. Depending on the activation temperature, reaction time, pressure, and the support used, different mixtures of MoO3 and MoS2 are formed within the catalytic materials.17,18 The influence of the sulfidation process on the HDO of aromatics is very important for this type of catalysts.19 The NiMo sulfided catalysts supported on Al2O3 have been tested in the HDO of triacylglycerides and were the best option among different supports to achieve complete deoxygenation mixture of hydrocarbons.20 The use of different supports, metals or combination with zeolites could help to optimize the performance of the newer generation of HDO catalyst.21 It is also desirable to change the MoS2 active phase to an oxidic form with a proper support or zeolite, which is a challenging task, but new generation of biomass-oriented catalysts might help to overcome this obstacle.22–24 Since the oxidation states and oxide/sulfide species play a crucial role in this process, we utilized a catalytic system that allows us to study the importance of the oxidation states of Mo, sulfide or oxide characteristics of these materials, and their impact on the HDO of palmitic acid. In this study, we prepared a series of Mo-based catalysts supported on Al2O3–TiO2 and sulfided at different temperatures to produce mixtures of different Mo catalytically active species. The selection of this support is based on the high activity of the corresponding MoS2-based catalysts that were used in HDS industrial plants in the past. The HDS catalysts supported on Al2O3–TiO2 mixed oxide were proved to be much more active than those supported on pure Al2O3. Our main aim is to understand the importance of oxide vs. sulfide characteristics of these materials, oxidation states of Mo-based catalysts and their effect on the HDO of palmitic acid.
2 Experimental
Preparation of catalysts
First, commercial supports with a composition of γ-Al2O3/TiO2 = 94.3/5.7 wt% were used to prepare oxidic form of the catalysts used as the base material. The catalysts were prepared by co-impregnation of the metal salts dissolved in aqueous solution to the support (γ-Al2O3/TiO2). We used MoO3 (Productos Químicos de Monterrey SA de CV) and nickel(II) carbonate hydroxide tetrahydrate (2NiCO3·3Ni(OH)2·4H2O, Sigma-Aldrich). These metal precursors were added to ammonia solution, 29 wt% (Fermont). This solution was impregnated to the dried support to achieve a homogeneous green pistachio catalyst. The catalyst was then calcined under static air atmosphere at 500 °C for 4 hours. The composition of the final catalyst obtained was 11.9 and 2.9 wt% base oxides (MoO3 and NiO, respectively), while the rest consisted of the support material.The second step involved transformation of the oxide to the sulfide forms. We deliberately chose the sulfidation conditions to produce mixtures of MoO3/MoS2 within the catalytic materials. The catalyst prepared in the first step was sulfided under a mixture of 15 vol% of H2S and H2 at atmospheric pressure. The sulfidation reaction took place with a heating rate of 5 °C min−1 under a continuous flux of 50 cm3 min−1. We prepared four different catalysts by varying the sulfidation temperature. The final temperature was kept constant at 100, 200, 300 and 400 °C, respectively, for 2 hours under the same gas mixture and flow rate as above. The temperature was then lowered to room temperature by simply turning off the oven and letting the samples cool down. Different temperatures were chosen to understand the effect of different Mo species present in the HDO. Each catalyst in this study is identified by its sulfidation temperature.
Characterization of catalysts
The catalysts were characterized by powder X-ray diffraction (XRD), temperature-programmed reduction (TPR), Raman spectroscopy, X-ray photoelectron spectroscopy (XPS), and X-ray absorption (XAS) spectroscopy. XRD patterns were recorded in the 4–80° range on a Siemens 5005 diffractometer, using Cu Kα radiation (λ = 1.5405 Å) and a goniometer speed of 0.02° (2θ) min−1. The TPR experiment was performed using a ZETON Altamira model AMI-200 with a reduction mixture of hydrogen/argon (10 vol% of H2) with a standard thermal conductivity detector (TCD). The samples were studied in a temperature range of 50–850 °C. Micro-Raman measurements were carried out with an excitation wavelength of 532 nm using the objective with magnification 50. A Horiba Jovin Yvo laser instrument was used with incident radiation power of about 9 mW. XPS analysis was performed using a Thermo VG Escalab 250 equipment where Al Kα (1486.6 eV) was used as a source of incident X-rays. The chamber pressure was kept at 3 × 10−8 mbar. The spectra were calibrated using the carbon 1 s excitation at the binding energy of 285 eV. BL 8.0.1 at Advanced light source (ALS)25 was used for O K-edge absorption spectra. Resolving power (E/ΔE) values for the monochromator at this beamline is 7000, which corresponds to an energy resolution value (ΔE) of 0.07 eV at 530 eV. All spectra were collected at room temperature with dwell time of 1 second at each energy step in TFY mode. Energy calibration was performed by measuring O K edge XAS for bismuth germanium oxide (BGO), and comparing its features to the known energy values (532.7 eV and 538.6 eV for XAS).Catalytic activity
The catalysts were tested in the HDO of palmitic acid (PA); Sigma Aldrich >99%; as a model compound of fatty acids. For each test, 0.1 g of the catalyst was used with 40 mL of a solution containing PA (39 mM) in n-dodecane (Sigma Aldrich, anhydrous > 99%). The catalysts were tested in a Parr reactor (100 mL) at 280 °C and 5.2 MPa of H2 pressure for 6 hours. The time evolution of the reaction was observed by withdrawing aliquots each hour and analyzing them on a gas chromatograph. Product identification was analyzed on a Hewlett Packard GC-MS instrument with a FID.3 Results and discussion
Characterization
XRD measurements (Fig. S1†) show the support had a crystalline structure, identified as γ-Al2O3 according to the JCPDS 10-0425. The impregnation of the metal species into the support did not change the crystalline structure of the support, evident from the identical diffraction pattern after the impregnation was done. This parent material, supported with metal species, was then sulfided at different temperatures. No detectable crystalline phase of the catalytically active metals in oxide form was found. Thus, we conclude that the parent material exhibited high dispersion of metal species, which would then be transformed into the sulfide species at higher temperatures.The reduction profile of the oxide supported catalyst is shown in Fig. S2.† The reduction of the Mo species shows hydrogen consumption in a wide temperature range (400–800 °C). The main reduction peak of the catalysts is located at 464 °C, which can be attributed to the partial reduction of Mo in octahedral polymeric species (Mo6+ + 2e− → Mo4+) weakly bound to the Al2O3–TiO2 support. The broad shoulder at higher temperature is associated with the complete reduction of Mo species (Mo4+ + 4e− → Mo0) in polymeric octahedral, tetrahedral and bulk crystalline MoO3.26 The Mo species interacting with the alumina support needs to be reduced and sulfided for the production of MoS2 active species.
Fig. 1 shows the Raman spectra for the catalysts sulfided in this study. MoS2 characteristic peaks, A1g and E2g1 located at 410 and 387 cm−1, respectively,27,28 become stronger as the sulfidation temperature is increased. The in-plane E2g1 mode is due to antisymmetric vibration of two S atoms with respect to the Mo atom. The A1g mode is ascribed to the out-of-plane vibration of the S atoms in opposite directions.29 The catalyst sulfided at 100 °C (S-100) does not show any peaks related to MoS2 species on the surface of the catalyst, which suggests that Mo in the catalyst is mainly in oxide form, and the amount of sulfidation is too low to be detectable. The Raman peaks for the MoS2 first appear for the catalyst sulfided at 200 °C confirming the formation of sulfided species at the catalyst's surface. These Raman features become strongest for the catalyst sulfided at 400 °C (S-400), which is expected due to the more favorable conditions to form these Mo species in this catalyst.
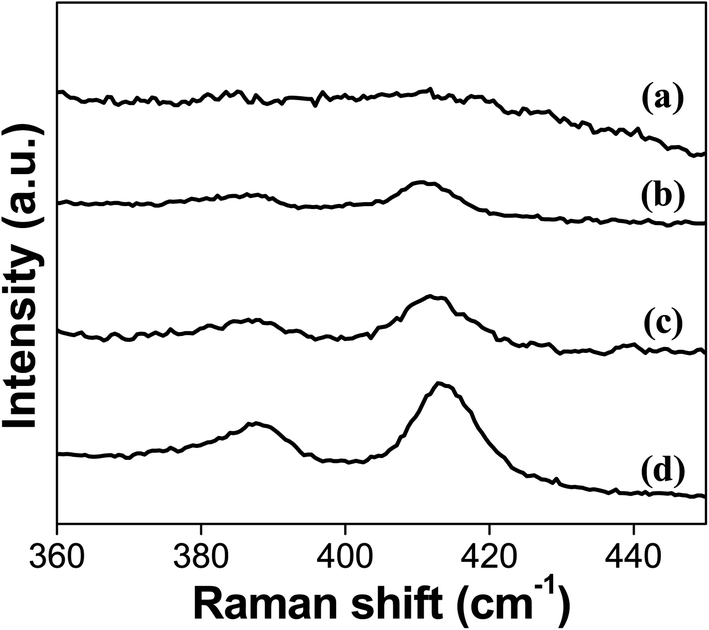 | ||
| Fig. 1 Raman spectra for the catalysts (a) S-100, (b) S-200, (c) S-300 and (d) S-400. | ||
Mo 3d XPS spectra for all the materials sulfided at different temperatures are shown in Fig. 2. The binding energies for the spin–orbit splitting for the Mo 3d5/2 and 3d3/2 are ascribed to Mo6+, Mo5+ and Mo4+. As expected, the relative proportions of Mo species with different oxidation states depend on the sulfidation temperature. For example, the catalyst sulfided at 100 °C (S-100) showed a larger concentration of higher valent molybdenum species (Mo6+), while the Mo4+ was scarce, but nevertheless it was measurable even at this sulfidation temperature (Fig. 2a). A sulfidation temperature of 200 °C resulted in formation of more Mo species with several different oxidation states (Fig. 2b). This spectrum showed several overlapping absorption bands due to contributions from Mo6+, Mo5+ and Mo4+. The catalysts sulfided at 300 and 400 °C also exhibit contributions from all these three different Mo species, but for these two catalysts the predominant contribution comes from Mo4+, which is due to the presence of MoS2 in the catalysts (Fig. 2c and d). Thus, the concentration of Mo species with lower oxidation states increases as the sulfidation temperature was raised.
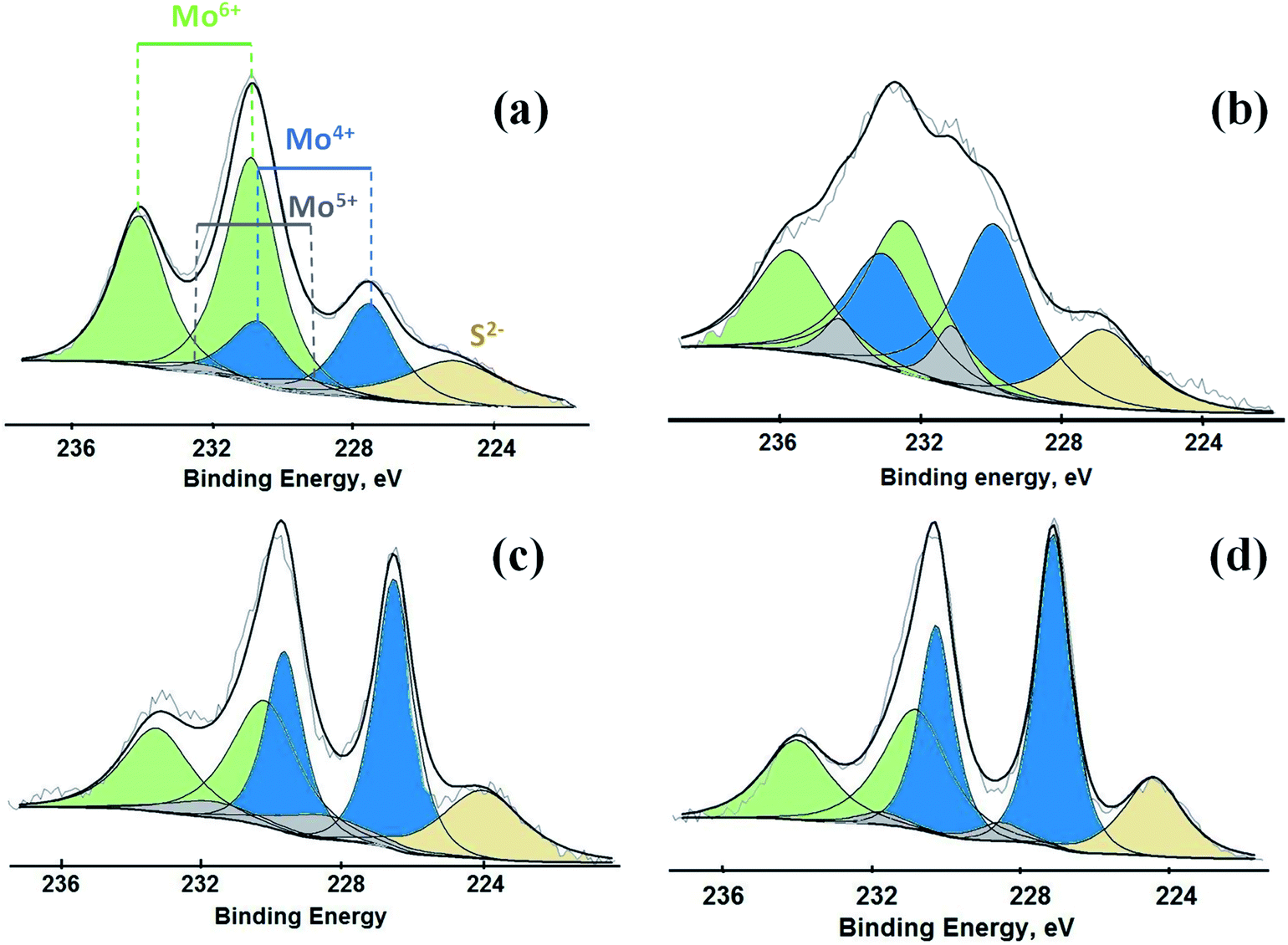 | ||
| Fig. 2 XP spectra for Mo 3d obtained from the catalysts (a) S-100, (b) S-200, (c) S-300 and (d) S-400. | ||
Binding energies and the relative concentrations of different Mo species present in these catalysts are summarized in Table 1. A change in sulfidation temperature causes a shift in the binding energy for the Mo species with same oxidation state, because binding energies are sensitive to not only the oxidation state, but also the local environment around the element probed. For instance, Mo6+ species exhibit a peak at 231.9 eV for S-100 and S-200, while this feature is shifted to 230.8 eV for S-300 and S-400. The catalyst S-100 showed the lowest concentration for Mo4+ (27.3), while highest for Mo6+ (65.9). On the other hand, the catalyst S-400 exhibited the dominant concentration for Mo4+ (55.2), while lowest for Mo6+ (40.3). The relative concentrations of Mo6+ for both S-300 and S-400 are identical. The increment in the Mo4+ species from S-300 to S-400 is related to the reduction of Mo5+ species during the sulfidation process. It is important to mention that the concentration of Mo5+, which is an intermediate species, increased when sulfidation temperature changed from 100 to 200 °C. Mo5+ concentration then decreased when the sulfidation temperature was changed to 400 °C. This is in agreement with the consecutive reduction reactions Mo6+ + e− = Mo5+ + e− = Mo4+ during sulfidation. The largest concentration of Mo5+ was obtained for S-200. We found that the predominant oxidation states in these catalysts were Mo4+ and Mo6+, corresponding to MoS2 and MoO3, respectively. However, the presence of Mo5+ was found in all the catalysts, which might play an important role in the HDO process. In Table 1, we also report calculated ratios of Mo6+/Mo5+ and Mo6+/Mo4+ concentrations to get an estimation of the production of Mo6+ or Mo4+ due to the sulfidation temperature. The Mo6+/Mo5+ ratio is the largest for the catalysts S-100 and S-400 due to the small amount of Mo5+ in these catalysts. Mo6+/Mo4+ ratio reflects the conversion of Mo6+ to Mo4+ during sulfidation. This ratio is almost identical for S-300 and S-400 catalysts. S-400 showed largest difference between Mo6+/Mo5+, and Mo6+/Mo4+ ratios due to an overall decrement in the Mo5+ species. These results are in agreement with the effect of the temperature on the formation of MoS2 under different sulfidation conditions.
| Catalyst | Ta (°C) | Binding energyb (eV) | Relative concentrationc (%) | Ratio of Mo species | |||||
|---|---|---|---|---|---|---|---|---|---|
| Mo4+ | Mo5+ | Mo6+ | Mo4+ | Mo5+ | Mo6+ |
|
|
||
| a Temperature of sulfidation of MoO3 supported catalysts.b The value of binding energy corresponds to Mo 3d5/2.c Concentration of the Mo species was estimated by the integration of the peak area. | |||||||||
| S-100 | 100 | 227.4 | 228.9 | 231.9 | 27.3 | 6.8 | 65.9 | 9.7 | 2.4 |
| S-200 | 200 | 229.6 | 230.9 | 232.4 | 36.8 | 13.2 | 50.0 | 3.8 | 1.4 |
| S-300 | 300 | 227.4 | 228.5 | 230.1 | 49.0 | 10.2 | 40.8 | 4.0 | 0.8 |
| S-400 | 400 | 227.1 | 228.5 | 230.8 | 55.2 | 4.5 | 40.3 | 9.0 | 0.7 |
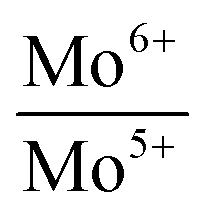
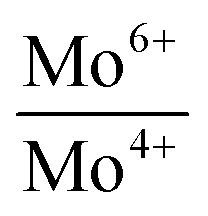
To better understand the incorporation of sulfur-based species into the catalytic materials, S 2p XPS spectra were also collected (Fig. 3). Two different types of sulfur species were detected with binding energies in the range of 160–165 eV and 168–172 eV that belong to S2− and S6+ species, respectively.30–32 These two S species are present due to formation of sulfides and oxysulfides/sulfates in all the catalysts where H2S acted as a source of S. This involves a series of complex chemical reactions dependent on various experimental factors.33,34 Our aim here is not to uncover detailed mechanism for the formation of these S species, but we simply illustrate that different sulfidation conditions result in different proportions of the two sulfur-based species which can have a significant effect on the catalytic performance. The concentration of S6+, which was largest for S-100, decreased by raising the sulfidation temperature due to increasing formation of MoS2. The catalyst S-200 exhibited strong presence of both of these sulfur species, reflected by two convoluted peaks present at binding energies of 158 to 173 eV. The catalyst sulfided at 400 °C exhibited the largest proportion of S2− species identified by their binding energies in the range of 160–164 eV related to the formation of MoS2 in agreement with our Raman results.
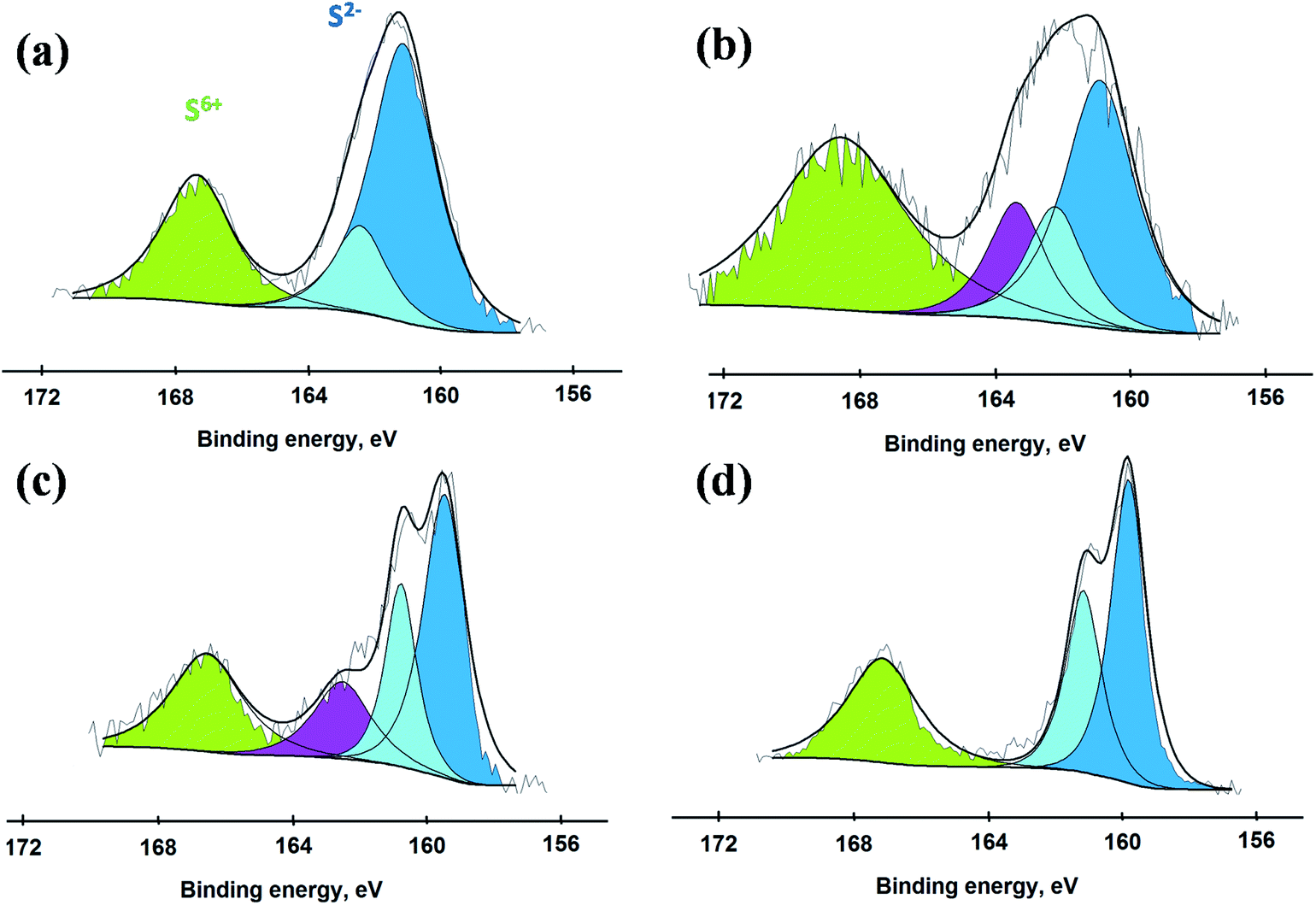 | ||
| Fig. 3 XP spectra for S 2p obtained from the catalysts (a) S-100, (b) S-200, (c) S-300 and (d) S-400. | ||
Synchrotron-based measurements were carried out to get more information about the atomic species in the catalysts under study. These measurements allowed us to selectively probe the local electronic environment of oxygen sites in the catalytic materials. Oxygen K-edge X-ray absorption spectra (XAS) were obtained in the total fluorescence yield (TFY) mode for the materials under study. XAS spectra collected for the two reference materials, MoO2 and MoO3 pure compounds, look identical (Fig. S3†) where the main absorption energy is located around 531 eV. The positions of the pre-edge and edge spectral regions does not show any significant differences between Mo6+ and Mo4+. To study the effect of reduction on the electronic structure of O, we reduced some of the catalysts under H2 atmosphere without performing any sulfidation process. O K-edge spectra for these materials are shown in Fig. S4.† Two main common aspects were found between the pure MoO2/MoO3 and the ones supported on Al2O3–TiO2. Reduction of these catalysts shows no effect on the electronic structure of O atoms. The spectral features and the intensity of the absorption bands are very similar for pure MoO2/MoO3 and for the samples reduced. However, we found important differences in the O K-edge XAS for the catalysts which were sulfided in the range of 100–400 °C (Fig. 4). The catalyst sulfided at 100 °C showed the highest intensity for band located at 531.1 eV near pre-edge, but it decreased for S-200 catalyst. This feature, due to MoO2 and MoO3, is present for both the pure Mo-oxide samples and for samples on Al2O3–TiO2 support. The decrement in intensity of this feature indicates that Mo–O species are transformed into sulfides (Mo–S) during sulfidation. Importantly, the O K-edge displayed an energy shift in this peak when the sulfidation temperature changed from 200 to 300 °C. The intensity of this feature remained almost the same, but it belongs to a different species as evident from the shifted position of this band at 531.5 eV. For S-400, this peak remains at 531.5 eV, but its intensity decreases compared to S-300. This implies increasing sulfidation temperature not only reduces the concentration of oxygen based species, but also causes formation of species with different local environment around the O atoms. S-100 and S-200 have one type of O sites, while S-300 and S-400 have a different type of O sites.
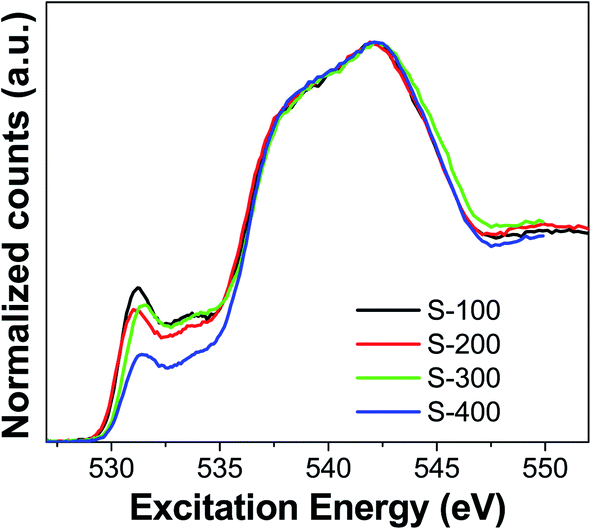 | ||
| Fig. 4 O K-edge X-ray absorption spectra (TFY mode) for the catalysts sulfided at different temperatures. | ||
All the different experimental techniques discussed above indicate one common theme that a significant structural change occurs around 200 °C to 300 °C of sulfidation temperature. Raman spectra indicated the formation of sulfur species in the catalyst's surface starts about 200 °C of sulfidation temperature, in agreement with the S 2p XPS results. The O K-edge XAS measurements also show that Mo–O compounds present in the catalytic materials changed in the range of 200–300 °C.
Hydrodeoxygenation of palmitic acid
We used palmitic acid (PA) as a model compound of fatty acids contained in vegetable oils or animal fats to test the performance of these catalysts. The conversion of PA is shown in Fig. 5. The catalyst without prior sulfidation, in its oxide form, showed very little activity. All the catalysts sulfided were active under these HDO conditions. However, the catalysts sulfided at lower temperatures were more active. The catalysts sulfided between 100–300 °C reached a conversion of 100% after 6 h of HDO reaction. The catalyst sulfided at 200 °C (S-200) showed the highest conversion of PA after 1 h of reaction. The sulfidation temperature of these catalysts played a major role in their performance. We discovered differences in the oxidation states of Mo, and presence of Mo–S and Mo–O compounds in the catalytic materials discussed above. We also found changes in the sulfur species due to different reaction temperatures, where a mixture of sulfates/sulfides was formed. The sulfidation process depends on two different variable, temperature and the type of gas present. H2 in the mixture of the atmosphere can reduce MoO3 to lower oxidation states. On the other hand, the presence of H2S can also produce MoS2 species that are active in the HDS petroleum-based process, but not necessarily in the HDO reactions. We also detected several intermediate species from sulfur and oxygen that are also present in the catalytic materials.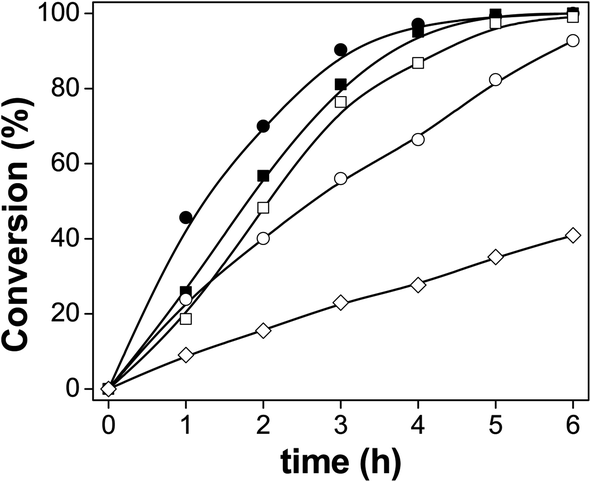 | ||
| Fig. 5 Conversion of palmitic acid over the catalysts: (■) S-100, (●) S-200, (□) S-300, (○) S-400 and (◊) non-sulfide reference. | ||
In order to clarify the questions about the effects of the sulfidation conditions on the catalytic performance, we tested some catalysts without any source of sulfur present during their thermal treatment. PA was converted up to only 41% over the oxide catalyst without any thermal or chemical pretreatment, which is too low for a good candidate for the HDO of biomass feedstocks. We tested the catalysts characterized by XAS (Fig. S4†) reduced under H2 atmosphere without any sulfur source, to elucidate the effect of Mo–S compounds and their impact on the catalytic performance. We chose two temperatures, 200 and 400 °C, for the reduction under H2 atmosphere with the same conditions as the sulfided counterparts, but using only H2 gas in the reaction chamber. These catalysts can convert PA about 38% and 48%, respectively. The conversion of PA over the catalyst reduced at 200 °C is very similar to the one obtained with the oxide catalyst. The catalytic activity slightly increased at higher reducing temperature, but was still lower compared with the sulfided counterparts. We found very similar Mo–O species for these catalytic materials by XAS measurements which explains their similar behavior in HDO. Therefore, we stress that the presence of Mo–O and Mo–S in the catalysts is crucial for their performance to more effectively convert PA.
We enlist the kinetic parameters in Table 2 for the catalysts under study. The highest conversion of PA was achieved over the S-200 catalyst, which reached 50% after only 1.2 h, while for S-400 it took 2.7 hours. The kinetic parameters of the catalysts follow a similar trend as the conversion. The pseudo-rate constants for the S-100 and S-200 have the highest values of 11.13 and 11.73 h−1 gcat−1, respectively. Pseudo-rate constants normalized per mmol of Mo atoms within the catalytic materials are also higher for the lower temperature sulfidation materials. S-100, S-200, and S-300 catalysts have almost one order of magnitude higher pseudo-rates compared to the non-sulfided oxide supported catalyst (Ox). The normalized constants are given per mmol of Mo atoms, which shows only a tiny amount is needed to convert the PA over these catalysts, especially S-200 (14.10 h−1 mmolMo−1). The fact that these higher conversions and pseudo-rates are achieved at relatively low sulfidation temperatures is an important step toward transitioning from sulfide-based catalysts (originally developed for petroleum industry) to MoO3 based catalysts for biomass feedstock. We have shown that it is not necessary to have only MoS2 present in these materials to achieve high conversion of PA, mixture of Mo–O and Mo–S active species work equally well to convert PA. The change of MoS2 for MoO3 as active species in these catalysts is a greener alternative since no H2S is needed for sulfidation. We consider that tuning properly the chemical or redox properties can provide high active materials based on MoO3 for biomass conversion in the future. On the other hand, MoO3 can be handled and stored easier than MoS2.
| Catalyst | t50a (h) | Conversionb (%) | kc (h−1 gcat−1) | kd (h−1 mmolMo−1) | roe, 102 (mol L−1 h−1 gcat−1) |
|---|---|---|---|---|---|
| a Time to reach 50% conversion of PA.b Conversion of HDO for 6 h of reaction.c Pseudo first-order rate constant per gram of catalyst.d Pseudo first-order rate constant per mmol of Mo in the catalyst.e Initial rate of reaction. | |||||
| S-100 | 1.8 | 100 | 11.13 | 13.14 | 8.22 |
| S-200 | 1.2 | 100 | 11.76 | 14.10 | 8.70 |
| S-300 | 2.1 | 100 | 7.95 | 9.53 | 7.76 |
| S-400 | 2.7 | 93 | 4.06 | 4.87 | 5.53 |
| Ox | nd | 41 | 0.83 | 0.99 | 1.97 |
The HDO network of this PA consists of consecutive hydrogenation reactions of the carboxylic acid moiety to the aldehyde, then to its alcohol version, and finally to n-hexadecane.35 Parallel reactions take place such as decarboxylation or decarbonylation. The reaction network of the PA obtained over these catalysts is shown in Fig. 6. The hydrogenation reactions in the HDO of fatty acids over MoS2-based catalysts are favored due to the easy detection of the products. In this case, we found a very small amount of the decarbonylation or decarboxylation product (n-pentadecane). We also found an unexpected product from condensation of PA with its alcohol product, which is the ester product formed from these catalysts. This product has been scarcely observed, but recently reported.36–38
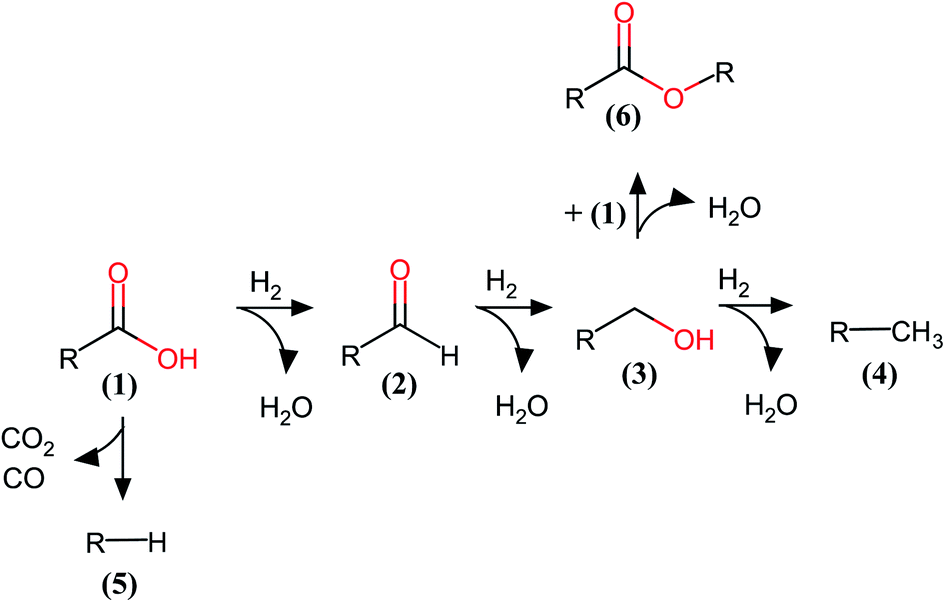 | ||
| Fig. 6 Reaction network for the Mo-based catalysts in the HDO of palmitic acid (R = C15H31). (1) Palmitic acid, (2) hexadecanal, (3) 1-hexadecanol, (4) n-hexadecane, (5) n-pentadecane and (6) cetyl palmitate. | ||
In Fig. 7, we have plotted the distribution of HDO products as a function of conversion of PA. The relative proportions of the products strongly depend on the sulfidation temperature of the catalyst. Table 3 lists the distribution of products obtained at the same conversion for all the catalysts, where we can easily compare the HDO routes over this series of catalysts. The main products are 1-hexadecanol and n-hexadecane for the materials which were sulfided, while the oxide catalyst produces large amounts of 1-hexadecanol, hexadecanal, and cetyl palmitate. The oxide form of the catalyst is less active in the HDO, and the mixture of products contains large amounts of oxygenated molecules. This specific catalyst is not able to convert PA and the main products are poor in O-free molecule yields. Therefore the effect of sulfidation of these catalysts is positive on the HDO of fatty acids for its catalytic activity and selectivity. The highly active catalyst (S-200) produces larger quantities of n-hexadecane (0.51) and 1-hexadecanol (0.32) at 40% of PA conversion. Importantly, changes in sulfidation temperature change the selectivity because n-pentadecane starts appearing while 1-hexadecanol decreases as temperature is increased. We analyzed the composition of products for 6 h of reaction, revealing that the products contained mostly deoxygenated molecules. It is important to mention that PA showed three main products over the S-200 catalyst at 6 h (100% conversion) of reaction, n-hexadecane (0.87), n-pentadecane (0.05) and 1-hexadecanol (0.08). The distribution of products in the HDO of PA over the S-400 catalyst at 6 h is different, where the presence of O-containing compounds were still present. We found a composition of products of n-hexadecane (0.61), 1-hexadecanol (0.19), n-pentadecane (0.13) and cetyl palmitate (0.06) over this catalyst after 6 h of reaction. The products of the HDO over the catalyst sulfided at 400 °C contained larger amounts of O-containing molecules that are undesirable for the purpose of green fuels. Compared to the products at 40% of conversion, the composition at the end of the HDO test showed that the highly active catalyst is also the most selective (S-200) toward the complete HDO product (n-hexadecane). The production of n-pentadecane due to decarboxylation or decarbonylation in both S-200 and S-400 occurs only at the beginning of the HDO process for these catalysts.
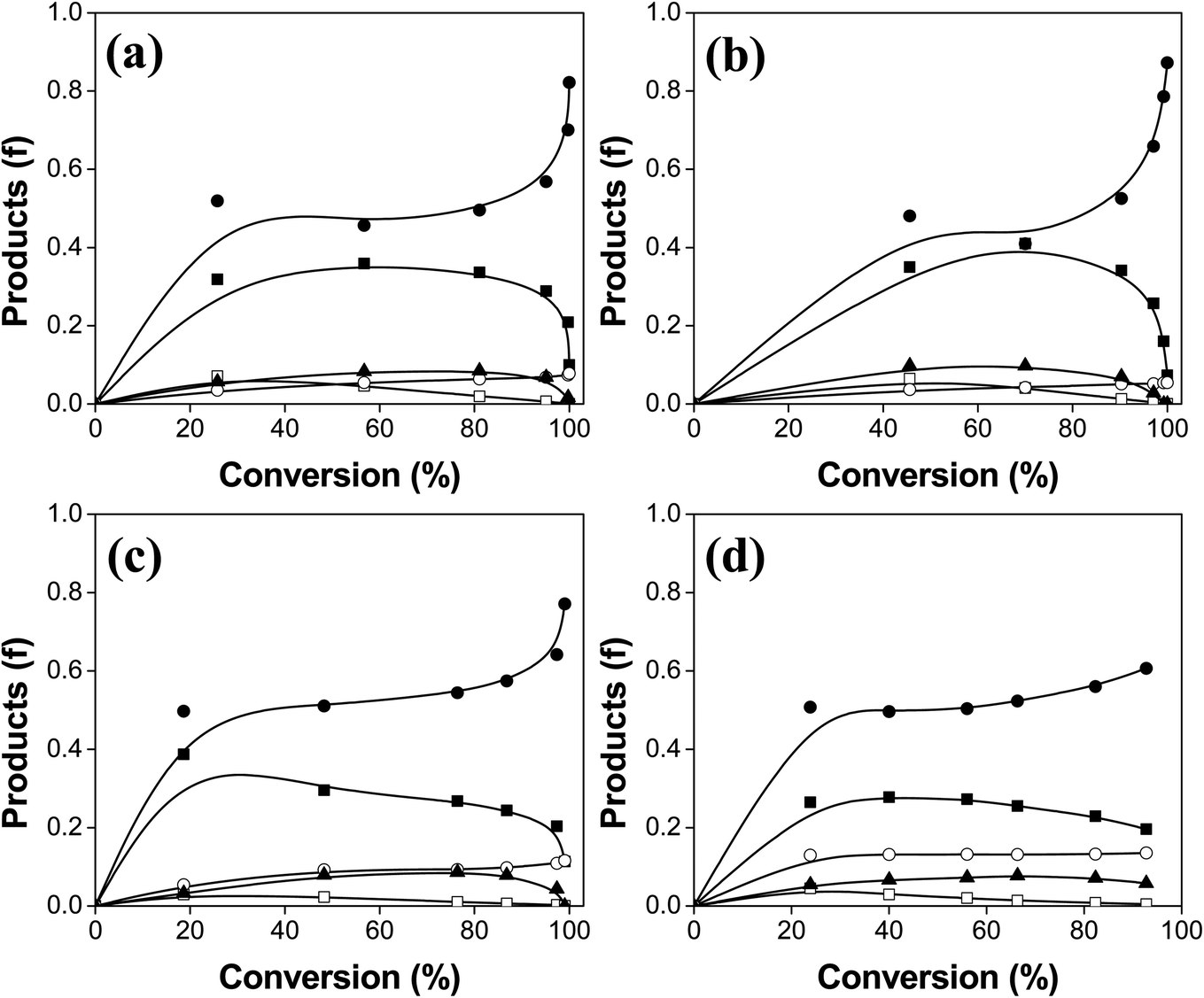 | ||
| Fig. 7 Distribution of HDO products over the catalysts (a) S-100, (b) S-200, (c) S-300 and (d) S-400. The products identified are: (■) 1-hexadecanol, (●) n-hexadecane, (□) hexadecanal, (○) n-pentadecane and (▲) cetyl palmitate. | ||
| Catalyst | 1-Hexadecanol | Hexadecanal | n-Hexadecane | n-Pentadecane | Cetyl palmitate |
|---|---|---|---|---|---|
| S-100 | 0.34 | 0.07 | 0.48 | 0.04 | 0.07 |
| S-200 | 0.32 | 0.05 | 0.51 | 0.05 | 0.07 |
| S-300 | 0.32 | 0.02 | 0.51 | 0.08 | 0.06 |
| S-400 | 0.28 | 0.03 | 0.50 | 0.13 | 0.06 |
| Ox | 0.34 | 0.39 | 0.06 | 0.00 | 0.21 |
The catalytic performance of the Mo-based catalysts in this study showed the importance of intermediate oxide/sulfide active sites being present. The pure oxide form of these catalysts is not very active in the HDO of palmitic acid and more importantly, neither is the selectivity directed toward O-free molecules. The sulfidation of the catalysts significantly improved their performance, with the most active catalyst achieved at sulfidation temperature at 200 °C. The amount of sulfide species increased at higher sulfidation temperatures, but the catalytic performance degraded. We also found a decrement in the Mo–O species with increasing temperature, and a formation of O species in different local environment for S-300 and S-400 than S-100 and S-200 catalysts. Spectroscopic methods provide strong evidence for the participation of both Mo–O and Mo–S species to achieve higher conversion of PA and selectivity towards O-free products, more specifically for the catalysts S-100 and S-200. Therefore, a proper balance of Mo species (6+, 5+ and 4+) with different oxide/sulfide characteristics is of paramount importance to synthesize highly active catalysts for HDO of fatty acids and related compounds. This mixture of species in the catalyst for HDO also results in higher yields of O-free products for these catalysts. This S-200 catalyst was the best in terms of conversion of palmitic acid as well as the production of O-free hydrocarbons. Catalyst S-200 exhibited a relative concentration of 50%, 13.2% and 36.8% for Mo6+, Mo5+ and Mo4+, respectively. This was the best catalytic material in the HDO of PA, and it contained half of the Mo in the high valent species, while the rest was in the low valent species. On the other hand, Mo5+ showed the largest relative concentration in this catalyst compared to all the series of materials, which might indicate that the performance strongly depends on a proper balance of all the Mo species. This study illustrates that the catalytic performance depends on the Mo species, but increasing Mo4+ species is not beneficial for the activity and selectivity of these Mo-based materials. We hypothesize that a catalytic material with Mo6+ (50%) and increasing the Mo5+ species with a relative concentration of Mo4+ between 27–35 would be a highly active catalytic material in the HDO of fatty acids.
We have established differences in the characteristics of the Mo species in the supported catalysts. The oxidation states of Mo atoms played a major role in their catalytic performance in the HDO of fatty acids, depending on the sulfidation temperature. Sulfur is necessary to produce highly active materials by introducing sulfides or sulfate species in the catalysts. We illustrate the catalytic cycles of Mo species for the reduction processes (palmitic acid and derived products in the HDO) and the oxidation reaction (hydrogen) in Fig. 8. It is important to note that lower oxidation states for Mo atoms would be involved in the reduction reactions from the fatty acids and their derivatives, while higher Mo oxidation states would activate hydrogen to be added into the fatty acids and related products. The number of electrons are also important for the complete catalytic cycle. We expect that a new Mo-based catalyst needs to possess a combination of those chemical species to become highly active and selective towards HDO reactions. The Mo-based catalysts do not need to be fully sulfided to be highly active in the HDO process. This finding can play an important role to developing cleaner catalytic materials for the production of clean fuels, and provides a way for improving the catalytic performance of Mo-based heterogeneous catalysts by tuning their oxidation states and chemical species.
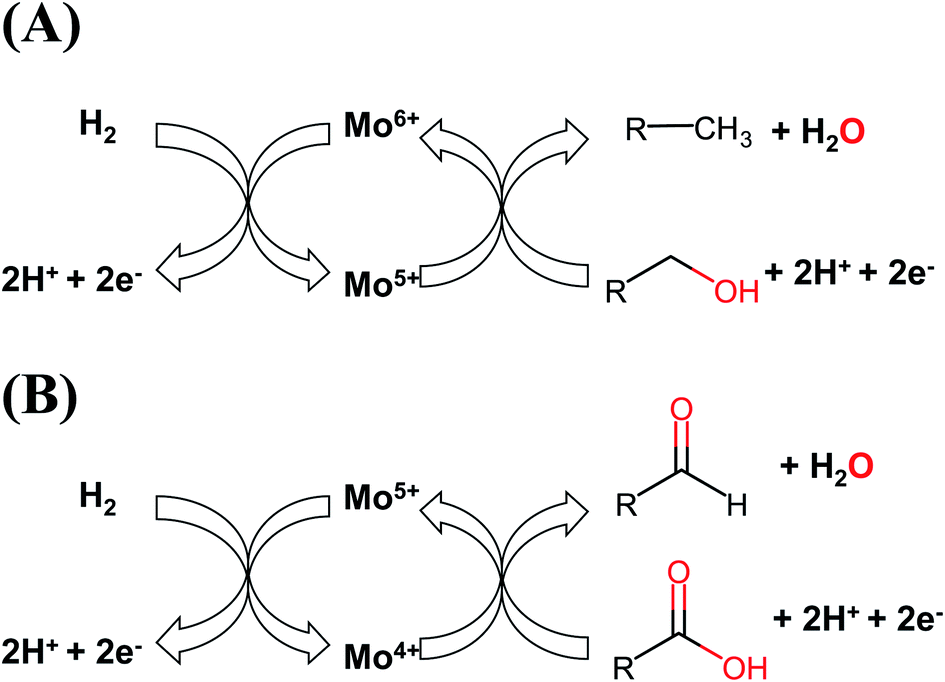 | ||
| Fig. 8 Catalytic cycles for the active Mo species. (A) Activation of H2 and hydrodeoxygenation of fatty alcohol by Mo5+/Mo6+ and (B) activation of H2 and reduction of fatty acid by the Mo4+/Mo5+ active species. | ||
4 Conclusions
We have successfully synthesized a series of Mo-based catalysts with different chemical characteristics by using a range of sulfidation temperatures. Different conditions during the sulfidation process result in formation of different chemical species and performance of the catalysts. The sulfidation of these catalysts, supported on Al2O3–TiO2, significantly improved their performance for the HDO of palmitic acid. However, catalysts with high formation of sulfides resulted in lower catalytic performance. Thus, a proper combination of Mo oxidation states as well as chemical characteristic (such as oxide, sulfide or intermediate) of the materials is needed to produce highly active and selective heterogeneous catalysts for oxygen elimination in fatty acids. The presence of different Mo species is necessary to promote different reactions under HDO conditions. We suggest that a new class of Mo-based catalysts for the production of green fuels should be considered, which incorporate mixed active species ranging from oxides to sulfides to achieve higher yields of O-free compounds.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors wish to thank Project Y.61023 CEMIE-BIO Clúster Bioturbosina (Instituto Mexicano del Petróleo) and Fondos SENER-CONACYT (248090) for financial support. R. I. Conde is gratefully acknowledged for her help with batch reactors. J. Navarrete, D. Zaragoza and P. Morales are also acknowledged for sulfidation of samples, GC-MS, XRD, Raman spectroscopic and XPS measurements, respectively. The synchrotron-based measurements were supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Canada Research Chair program.References
- E. Vine, Energy Efficiency, 2008, 1, 49–63 CrossRef.
- R. Baños, F. Manzano-Agugliaro, F. G. Montoya, C. Gil, A. Alcayde and J. Gómez, Renewable Sustainable Energy Rev., 2011, 15, 1753–1766 CrossRef.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS.
- G. S. Alemán-Nava, V. H. Casiano-Flores, D. L. Cárdenas-Chávez, R. Díaz-Chavez, N. Scarlat, J. Mahlknecht, J.-F. Dallemand and R. Parra, Renewable Sustainable Energy Rev., 2014, 32, 140–153 CrossRef.
- A. Corma, G. W. Huber, L. Sauvanaud and P. ÓConnor, J. Catal., 2007, 247, 307–327 CrossRef CAS.
- L. Wu, T. Moteki, A. A. Gokhale, D. W. Flaherty and F. D. Toste, Chem, 2016, 1, 32–58 CrossRef CAS PubMed.
- P. Tamunaidu and S. Bhatia, Bioresour. Technol., 2007, 98, 3593–3601 CrossRef CAS PubMed.
- M. Mohammad, T. K. Hari, Z. Yaakob, Y. C. Sharma and K. Sopian, Renewable Sustainable Energy Rev., 2013, 22, 121–132 CrossRef CAS.
- P. M. Mortensen, J.-D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS.
- E. Furimsky, Appl. Catal., A, 2000, 199, 147–190 CrossRef CAS.
- M. J. Girgis and B. C. Gates, Ind. Eng. Chem. Res., 1991, 30, 2021–2058 CrossRef CAS.
- T. V. Choudhary and C. B. Phillips, Appl. Catal., A, 2011, 397, 1–12 CrossRef CAS.
- M. Toba, Y. Abe, H. Kuramochi, M. Osako, T. Mochizuki and Y. Yoshimura, Catal. Today, 2011, 164, 533–537 CrossRef CAS.
- T. Prasomsri, M. Shetty, K. Murugappan and Y. Román-Leshkov, Energy Environ. Sci., 2014, 7, 2660–2669 RSC.
- D. Valencia, I. García-Cruz, L. F. Ramírez-Verduzco and J. Aburto, ACS Omega, 2018, 3, 14165–14172 CrossRef CAS.
- M. Shetty, K. Murugappan, T. Prasomsri, W. H. Green and Y. Román-Leshkov, J. Catal., 2015, 331, 86–97 CrossRef CAS.
- N. Chen, S. Gong and E. W. Qian, Appl. Catal., B, 2015, 174–175, 253–263 CrossRef CAS.
- L. van Haandel, G. M. Bremmer, E. J. M. Hensen and Th. Weber, J. Catal., 2016, 342, 27–39 CrossRef CAS.
- M. Ferrari, S. Bosmans, R. Maggi, B. Delmon and P. Grange, Catal. Today, 2001, 65, 257–264 CrossRef CAS.
- J. Horácek, Z. Tišler, V. Rubáš and D. Kubicka, Fuel, 2014, 121, 57–64 CrossRef.
- T. L. Chew and S. Bhatia, Bioresour. Technol., 2009, 100, 2540–2545 CrossRef CAS PubMed.
- M. Shahinuzzaman, Z. Yaakob and Y. Ahmed, Renewable Sustainable Energy Rev., 2017, 77, 1375–1384 CrossRef CAS.
- N. Arun, R. V. Sharma and A. K. Dalai, Renewable Sustainable Energy Rev., 2015, 48, 240–255 CrossRef CAS.
- Y. Shi, Y. Cao, Y. Duan, H. Chen, Y. Chen, M. Yang and Y. Wu, Green Chem., 2016, 18, 4633–4648 RSC.
- J. J. Jia, T. A. Callcott, J. Yurkas, A. W. Ellis, F. J. Himpsel, M. G. Samant, J. Stohr, D. L. Ederer, J. A. Carlisle, E. A. Hudson, L. J. Terminello, D. K. Shuh and R. C. C. Perera, Rev. Sci. Instrum., 1995, 66, 1394–1397 CrossRef CAS.
- R. López Cordero and A. López Agudo, Appl. Catal., A, 2000, 202, 23–35 CrossRef.
- Y. Wang, C. Cong, C. Qiu and T. Yu, Small, 2013, 17, 2857–2861 CrossRef PubMed.
- J. Jadwiszczak, C. ÓCallaghan, Y. Zhou, D. S. Fox, E. Weitz, D. Keane, C. P. Cullen, I. Ó'Reilly, C. Downing, A. Shmeliov, P. Maguire, J. J. Gough, C. McGuinness, M. S. Ferreira, A. L. Bradley, J. J. Boland, G. S. Duesberg, V. Nicolosi and H. Zhang, Sci. Adv., 2018, 4, 5031 CrossRef PubMed.
- P. A. Bertrand, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 5745–5749 CrossRef CAS.
- A. Galtayries, S. Wisniewski and J. Grimblot, J. Electron Spectrosc. Relat. Phenom., 1997, 87, 31–44 CrossRef CAS.
- D. Nilolova, R. Edreva-Kardjieva, G. Gouliev, T. Grozeva and P. Tzvetkov, Appl. Catal., A, 2006, 297, 135–144 CrossRef.
- E. J. Romano and K. H. Schulz, Appl. Surf. Sci., 2005, 246, 262–270 CrossRef CAS.
- A. G. Vandeputte, M.-F. Reyniers and G. B. Marin, J. Phys. Chem. A, 2010, 114, 10531–10549 CrossRef CAS PubMed.
- L. Díaz-García, V. Santes, T. Viveros-García, A. Sánchez-Trujillo, J. Ramírez-Salgado, C. Ornelas and E. Rodríguez-Castellón, Catal. Today, 2017, 282, 230–239 CrossRef.
- D. Valencia, I. García-Cruz, V. H. Uc, L. F. Ramírez-Verduzco, M. A. Amezcua-Allieri and J. Aburto, Biomass Bioenergy, 2018, 112, 37–44 CrossRef CAS.
- E. Kordouli, B. Pawelec, K. Bourikas, C. Kordulis, J. L. G. Fierro and A. Lycourghiotis, Appl. Catal., B, 2018, 229, 139–154 CrossRef CAS.
- G. Zafeiropoulos, N. Nikolopoulos, E. Kordouli, L. Sygellou, K. Bourikas, C. Kordulis and A. Lycourghiotis, Catalysts, 2019, 9, 210 CrossRef.
- E. Kordouli, L. Sygellou, C. Kordulis, K. Bourikas and A. Lycourghiotis, Appl. Catal., B, 2017, 209, 12–22 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03208h |
| This journal is © The Royal Society of Chemistry 2019 |