
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDifferential protein expression of a streptomycin-resistant Streptomyces albulus mutant in high yield production of ε-poly-L-lysine: a proteomics study†
Yongjuan Liu,
Xusheng Chen *,
Long Pan and
Zhonggui Mao*
*,
Long Pan and
Zhonggui Mao*
Key Laboratory of Industrial Biotechnology, Ministry of Education, School of Biotechnology, Jiangnan University, 1800 Lihu Road, Wuxi 214122, China. E-mail: chenxs@jiangnan.edu.cn; maozg@jiangnan.edu.cn; Fax: +86-510-85918279; Tel: +86-510-85918279
First published on 2nd August 2019
Abstract
ε-Poly-L-lysine (ε-PL), produced by Streptomyces albulus, is an excellent antimicrobial agent which has been extensively used in the field of food and medicine. In our previous study, we have improved ε-PL production by S. albulus M-Z18 through iterative introduction of streptomycin resistance. To decipher the overproduction mechanism of high-yielding mutant S. albulus SS-62, we conducted a comparative proteomics analysis between S. albulus SS-62 and its parent strain S. albulus M-Z18. Approximately 11.5% of the predicted S. albulus proteome was detected and 401 known or putative regulatory proteins showed statistically differential expression levels. Expression levels of proteins involved in ε-PL precursor metabolism and energy metabolism, and proteins in the pathways related to transcriptional regulation and translation were up-regulated. It was indicated that mutant SS-62 could not only strengthen the ε-PL precursor metabolism and energy metabolism but also tune the pathways related to transcriptional regulation and translation, suggesting a better intracellular metabolic environment for the synthesis of ε-PL in mutant SS-62. To confirm these bioinformatics analyses, qRT-PCR was employed to investigate the transcriptional levels of pls, frr and hrdD and their transcription levels were found to have increased more than 4-fold. Further, overexpression of pls and frr resulted in an increase in ε-PL titer and the yield of ε-PL per unit cell. This report not only represents the first comprehensive study on comparative proteomics in S. albulus, but it would also guide strain engineering to further improve ε-PL production.
Introduction
ε-Poly-L-lysine (ε-PL) is a cationic polypeptide made of 25–35 L-lysine residues linked together via amide bonds between the ε-amino group and the α-carboxylic acid group. It is mainly produced by bacteria belonging to the Streptomycetaceae family as a secondary metabolite.1 ε-PL and its hydrochloride form have been used as natural food preservatives in Japan, South Korea, United States, China and other countries because of their excellent antimicrobial properties. In addition, they are applied in numerous other areas as drug carriers, nanoparticles, gene carriers, liposomes, interferon inducers, lipase inhibitors, hydrogels and coating materials.2 Currently, ε-PL is being industrially produced by aerobic fermentation of Streptomyces albulus. However, high production costs remain a major hurdle to widespread use of this natural antimicrobial agent and highly functional material.Native ε-PL producer S. albulus isolated from soil, like other organisms, has an extremely low capability to biosynthesize the target metabolite. Thus, improving the ability of S. albulus to synthesize ε-PL is a necessary and effective way to reduce its production cost. Different strategies are being developed to improve microbial production of ε-PL. These include ultraviolet and nitroguanidine,3 atmospheric and room temperature plasma,4,5 and genome shuffling.6,7 Despite these strategies achieving production enhancement of ε-PL, the mechanisms remain unclear and require further investigation. In addition, limitation in the knowledge of genetic information, metabolic pathways and regulatory mechanism restrains significant improvement of ε-PL production through genetic and metabolic engineering of S. albulus.8–10 It is well known that one of the effective ways to reveal the metabolic pathway and regulation mechanism is to analyze the high-yield mutants. However, the mutants obtained by physical or chemical mutagenesis, or genome shuffling are not ideal models since the complexity of their mechanisms, making the changes unknown and variable.
Ribosome engineering, is the introduction of mutations conferring antibiotic resistance to increase metabolite production in bacteria, especially for secondary metabolite production in Streptomyces.11 The mutants resistant to antibiotics frequently possess a point mutation or a deletion mutation within a ribosomal component, including ribosomal protein, rRNA, or translation factor.11 These mutations would change the structure of ribosomes and eventually lead to protein synthesis changes.12,13 In our previous study, we improved ε-PL production of S. albulus M-Z18 by iterative introduction of streptomycin resistance.14 Despite this, current understanding of this improvement remains limited to biochemistry and gene mutation levels, and it is not clear at the molecular level. However, investigation at the molecular level is crucial for engineering S. albulus strains with improved ε-PL production.
In this work, we used comparative proteomics to characterize the differences between parental strain S. albulus M-Z18 and high-yield mutant S. albulus SS-62 by isobaric tag for relative and absolute quantitation (iTRAQ). Based on identification of potential elements involved in controlling ε-PL production in S. albulus, it is possible to further improve the ε-PL production of S. albulus by direct genetic manipulation.
Materials and methods
Strains and recombinant plasmids construction
The strains, plasmids, and primers (restriction sites are italicized) used in this study are listed in Table 1. S. albulus M-Z18 was isolated from the soil as described by Nishikawa and Ogawa,15 preserved in our lab, and were subjected to ultraviolet and nitrosoguanidine mutagenesis as described by Hiraki et al.3 Escherichia coli DH5α was used as the host for gene cloning and subcloning. E. coli strain ET12567 containing pUZ8002 was used as the donor in conjugal transfer of DNA from E. coli to Streptomyces. All recombinant DNA techniques were performed as described by Sambrook et al.16 pIB139 was used as E. coli/Streptomyces shuttle vector. Three open reading frames 561-bp frr, 3960-bp pls and 1002-bp hrdD were amplified from S. albulus M-Z18 genome DNA by using primers in Table 1, respectively, PCR amplification was started at 95 °C for 3 min, followed by 35 cycles of denaturation at 95 °C for 30 s, annealing at 68 °C for 30 s, and elongation at 72 °C for 30 s. The obtained 561-bp frr gene encoded a 186 aa long protein, showing the highest similarity to Frr of S. coelicolor A3(2) (89%, identical amino acids). The PCR fragments were then digested with NdeI and EcoRI, and ligated into vector pIB139 that had been digested by the same enzymes. The genes frr, pls and hrdD were inserted into pIB139 to create plasmids pIB139-frr, pIB139-pls and pIB139-hrdD (ESI Files 1 and 2†). The plasmids pIB139-frr, pIB139-pls and pIB139-hrdD were introduced into E. coli strain ET12567/pUZ8002 and then transferred into S. albulus M-Z18 by intergeneric conjugation to generate recombinant strains.| Strains or plasmids or primers | Description | Source or reference |
|---|---|---|
| a Primers P1 and P2 were used for amplification of frr gene, P3 and P4 were used for amplification of pls gene and P5 and P6 were used for amplification of hrdD gene. | ||
| Strains | ||
| E. coli DH5α | General cloning host | This lab |
| E. coli ET12567 (pUZ8002) | Cmr, Kmr, donor strain for conjugation | Prof. Wang |
| S. albulus M-Z18 | Wild strain, ε-PL producer | This lab |
| S. albulus SS-62 | Mutant strain, ε-PL producer | This work |
| M-Z18-frr | The M-Z18 strain harboring gene frr | This work |
| M-Z18-pls | The M-Z18 strain harboring gene pls | This work |
| M-Z18-hrdD | The M-Z18 strain harboring gene hrdD | This work |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Plasmids | ||
| pIB139 | Derivative of integrative plasmid pSET152, harboring a PermE* promoter, Aprr, OriTRK2, ΦC31 int/attP | Prof. Wang |
| pIB139-frr | frr gene under the control of promoter PermE* in plasmid pIB139 | This work |
| pIB139 pls | pls gene under the control of promoter PermE* in plasmid pIB139 | This work |
| pIB139-hrdD | hrdD gene under the control of promoter PermE* in plasmid pIB139 | This work |
|
||
| Primersa | ||
| P1 | 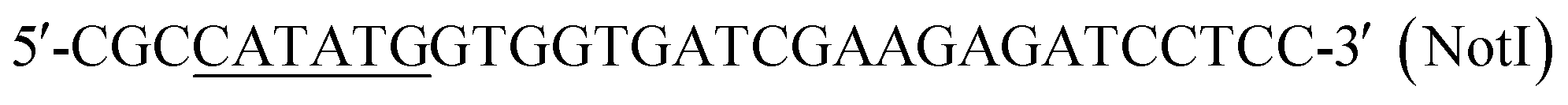 |
This work |
| P2 |  |
This work |
| P3 | 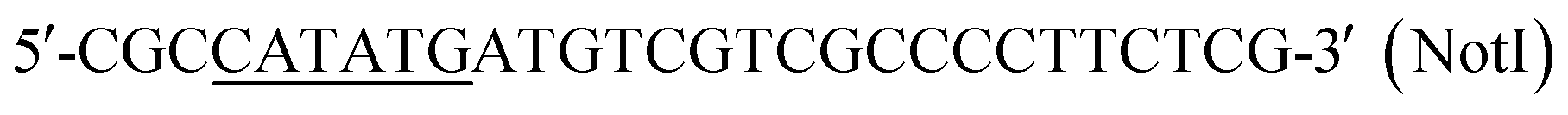 |
This work |
| P4 | 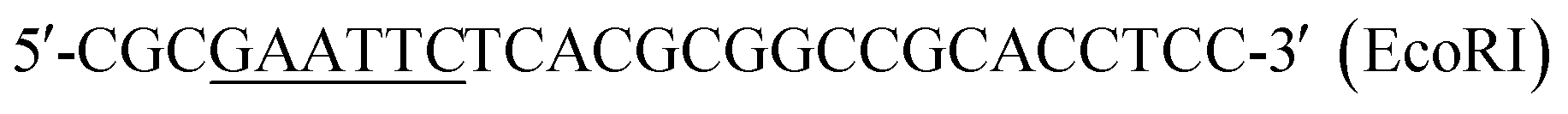 |
This work |
| P5 | 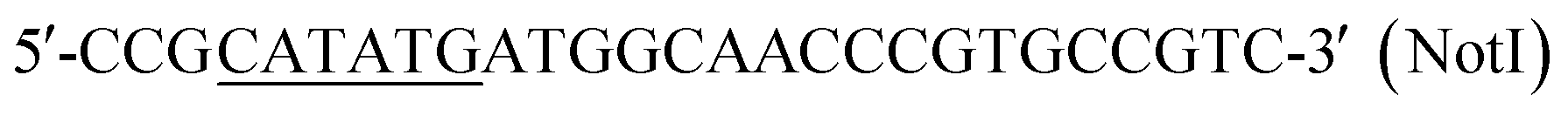 |
This work |
| P6 | 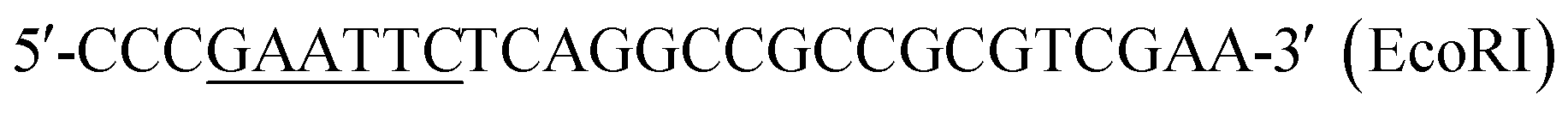 |
This work |
Culture and fermentation media composition
E. coli strains were cultured in liquid or on solid Luria–Bertani (LB) medium at 37 °C. Apramycin (50 μg mL−1), chloramphenicol (25 μg mL−1), ampicillin (100 μg mL−1), or kanamycin (50 μg mL−1) were added when necessary to maintain the plasmids. S. albulus M-Z18 and mutants were maintained on BTN slant medium which contained 10 g L−1 glucose, 1 g L−1 yeast extract, 2 g L−1 peptone, 20 g L−1 agar, pH 7.5. The seed medium (M3G) consists of (g L−1): glucose 50, yeast extract 5, (NH4)2SO4 10, K2HPO4 0.8, KH2PO4 1.36, MgSO4·7H2O 0.5, ZnSO4 0.04, and FeSO4 0.03, pH 6.8. The producing medium consists of (g L−1): glycerol 60, (NH4)2SO4 5, yeast extract 8, K2HPO4 2, MgSO4·7H2O 0.5, ZnSO4 0.04, and FeSO4 0.03, pH 7.5. All media was sterilized at 115 °C for 15 min.Fermentation in a 250 mL Erlenmeyer flask
Strains were incubated on BTN slant medium for 8–10 days at 30 °C until spores mature. Three loops of spores were inoculated into a 250 mL Erlenmeyer flask containing 40 mL of seed medium and incubated at 30 °C for 24 h at 200 rpm. Then 8.0% seed medium was transferred into a 250 mL Erlenmeyer flask containing 40 mL of producing medium. Fermentation was carried out at 30 °C for 72 h on a rotary shaker (HYL-C, Qiang Le Laboratory Equipment Co., Taicang, China) at 200 rpm.Batch fermentation in a 1 l multi-bioreactor system
The batch fermentations were performed in a 1 L glass bioreactor system (T & J Bio-Engineering Co. Ltd., Shanghai, China) consisting of four identical stirred tank reactors with a working volume of 0.6 L. The broth was agitated at 300 rpm with a mechanical stirrer at 30 °C with a 1 vvm aeration rate. The initial pH was adjusted to 6.8 by adding ammonia water (12.5%, v/v), and then the broth was inoculated with 50 mL of 24 h old seed culture. During the fermentation, pH and DO were also monitored online using pH and DO electrodes, respectively (K8S-120 and InPro6800-12-120, Mettler Toledo, Zurich, Switzerland). However, the agitation speed was varied from 300 to 1500 rpm, and aeration rate was manually increased stepwise with steps of 0.5 vvm and a range of 1.0–3.0 vvm.Morphological characterization of strain by scanning electron microscopy
The morphological characteristics of aerial hyphae and spores of the S. albulus M-Z18 and mutant SS-62 were observed after 2 and 7 days of incubation at 30 °C by using scanning electron microscopy (SEM). Cut agar blocks were washed twice in phosphate-buffered saline solution and then immersed in 5% glutaraldehyde. After that, the samples were immobilized with 1% osmium acid (0.1 M PBS, pH 7.2) and then washed with 0.1 M PBS. After an ethanol gradient dehydration, samples were taken at the critical point dried, and pasted on the sample table of the ion sputtering instrument (SCD 005, BAL-TEC, California, America) was coated and scanned under scanning electron microscope (Quanta 200, FEI, Hillsboro, America).GUS assay
Plasmid PIB139-gusA* harboring the gusA gene was a gift from Dr Ma from China Jiliang University (Hangzhou, Zhejiang). The plasmid PIB139-gusA* was introduced into E. coli strain ET12567/pUZ8002 and then transferred into S. albulus M-Z18 and mutant strain SS-62 by intergeneric conjugation to generate S. albulus M-Z18-GUS and SS-62-GUS, respectively. Mycelia were harvested by centrifugation, washed once with distilled water, and resuspended in lysis buffer (50 mM phosphate buffer [pH 7.0], 5 mM dithiothreitol [DTT], 0.1% Triton X-100, 1 mg mL−1 lysozyme). Lysis was performed at 37 °C for 15 min. Lysates were centrifuged at 4000 × g for 10 min. Then, 0.5 mL of lysate was mixed with 0.5 mL of dilution buffer [50 mM phosphate buffer (pH 7.0), 5 mM DTT, 0.1% Triton X-100] supplemented with 5 μL 0.2 M p-nitrophenyl-β-d-glucuronide. The optical density at 415 nm was measured after 20 min of incubation at 37 °C. As a reference, a 1:1 mixture of lysate and dilution buffer was used.17
Sample preparation, iTRAQ labeling and LC-MS/MS analysis
Ten mL of cultures grown for 30 h were harvested by centrifugation at 10000 × g, 4 °C for 15 min to remove spent media, following which the pellets were washed three times with 50 mm PBS (pH 7.2) for protein extractions. SDT buffer was added to the sample, and transferred to 2 mL tubes with amount quartz sand (another 1/4 inch ceramic bead MP 6540-424 for tissue samples). The lysate was homogenized by MP homogenizer (24 × 2, 6.0 M S−1, 60 s, twice). The homogenate was sonicated and boiled for 15 min. The boiled homogenate was centrifuged at 14000 × g for 40 min and the resulting supernatant was filtered with 0.22 μm filters. The protein content in the filtrate was quantified using the BCA Protein Assay Kit (Bio-Rad, USA). 200 μg of proteins for each sample were incorporated into 30 μL SDT buffer (4% SDS, 100 mM DTT, 150 mM Tris–HCl pH 8.0). The detergent, DTT and other low-molecular-weight components were removed using UA buffer (8 M urea, 150 mM Tris–HCl pH 8.0) by repeated ultrafiltration (Microcon units, 10 kD). Then 100 μL iodoacetamide (100 mM IAA in UA buffer) was added to block reduced cysteine residues and the samples were incubated for 30 min in darkness. The filters were washed with 100 μL UA buffer three times and then 100 μL dissolution buffer (DS buffer) twice. Finally, the protein suspensions were digested with 4 μg trypsin (Promega) in 40 μL DS buffer overnight at 37 °C, and the resulting peptides were collected as a filtrate. The peptides of each sample were desalted on C18 Cartridges (Empore™ SPE Cartridges C18 (standard density), bed ID 7 mm, volume 3 mL, Sigma), concentrated by vacuum centrifugation and reconstituted in 40 μL of 0.1% (v/v) formic acid. The peptide content was estimated by UV light spectral density at 280 nm using an extinctions coefficient of 1.1 of 0.1% (g l−1) solution that was calculated on the basis of the frequency of tryptophan and tyrosine in vertebrate proteins. 100 μg peptide mixture of each sample was labeled using iTRAQ reagent according to the manufacturer's instructions (Applied Biosystems).
iTRAQ labeled peptides were fractionated by SCX chromatography using the AKTA Purifier system (GE Healthcare). The dried peptide mixture was reconstituted and acidified with buffer A (10 mM KH2PO4 in 25% of ACN, pH 3.0) and loaded onto a PolySULFOETHYL 4.6 × 100 mm column (5 μm, 200 Å, PolyLC Inc, Maryland, U.S.A.). The peptides were eluted at a flow rate of 1 mL min−1 with a gradient of 0–8% buffer B (500 mM KCl, 10 mM KH2PO4 in 25% of ACN, pH 3.0) for 22 min, 8–52% buffer B during 22–47 min, 52–100% buffer B during 47–50 min, 100% buffer B during 50–58 min, and buffer B was reset to 0% after 58 min. The elution was monitored by absorbance at 214 nm, and fractions were collected every 1 min. The collected fractions were desalted on C18 Cartridges (Empore™ SPE Cartridges C18 (standard density), bed ID 7 mm, volume 3 mL, Sigma) and concentrated by vacuum centrifugation. Each fraction was injected for nanoLC-MS/MS analysis. The peptide mixture was loaded onto a reverse phase trap column (Thermo Scientific Acclaim PepMap100, 100 μm × 2 cm, nanoViper C18) connected to the C18-reversed phase analytical column (Thermo Scientific Easy Column, 10 cm long, 75 μm inner diameter, 3 μm resin) in buffer A (0.1% formic acid) and separated with a linear gradient of buffer B (84% acetonitrile and 0.1% formic acid) at a flow rate of 300 nL min−1 controlled by IntelliFlow technology.
LC-MS/MS analysis was performed on a Q Exactive mass spectrometer (Thermo Scientific) that was coupled to Easy nLC (Thermo Fisher Scientific). The mass spectrometer was operated in positive ion mode. MS data was acquired using a data-dependent top 10 method dynamically choosing the most abundant precursor ions from the survey scan (300–1800 m/z) for HCD fragmentation. Automatic gain control (AGC) target was set to 3 × 106, and maximum inject time to 10 ms. Dynamic exclusion duration was 40.0 s. Survey scans were acquired at a resolution of 70000 at m/z 200 and resolution for HCD spectra was set to 17500 at m/z 200, and isolation width was 2 m/z. Normalized collision energy was 30 eV and the underfill ratio, which specifies the minimum percentage of the target value likely to be reached at maximum fill time, was defined as 0.1%.
Gene expression analysis by quantitative real-time PCR
Total RNAs of S. albulus M-Z18 and SS-62 at 30 h (early stationary phase) during fermentation were extracted and purified by RNA extraction kit (MiniBEST, TaKaRa Biotechnology Company, Dalian, China). RNAs were reverse transcribed using a PrimeScript RT Reagent Kit (TaKaRa Biotechnology Company, Dalian, China) according to the manufacturer's instructions. Quantitative RT-PCR was carried out by mixing SYBR Premix Ex Taq (10 μL), forward primer (0.4 μM, 0.8 μL), reverse primer (0.4 μM, 0.8 μL), ROX (0.4 μL), and cDNA (100 ng, 2 μL); a final volume of 20 μL was prepared by adding ddH2O. The mixture was amplified using a LightCycler®48 II (Applied Biosystems, USA) under the following parameters: 10 min at 94 °C, followed by 40 cycles of 94 °C for 15 s, 60 °C for 1 min. All of these operations were repeated three times and quantitative RT-PCR results were subjected to 2−ΔΔCt method18 for relative quantification with 16S rDNA as endogenous control gene. Primers used for quantitative RT-PCR are listed in Table 2 and designed by Primer 5.0.| Gene | Gene product | DNA sequencea (5′–3′) |
|---|---|---|
| a All primers were designed according to the sequence of the S. albulus M-Z18. | ||
| pls | ε-PL synthase | F: TCTTCACGCTCCTGACCACT |
| R: CCGAGGCTGGCGTAGAG | ||
| hrdD | RNA polymerase sigma factor | F: GCCAAGGAGGTGGAGCTGTCCC |
| R: CGGGCGACGGCGACGACC | ||
| frr | Ribosome recycling factor | F: CAACATCATCCGTGTGAC |
| R: CTTGTTCTTGGCGACCTT | ||
| relA | ppGpp | F: GCATGAACACCATCAAGT |
| R: CGATCTCGTCGTACATCT | ||
| 16S rDNA | Control gene | F: CAGGCTAGAGTTCGGTAG |
| R: CGGTGTTCCTCCTGATAT | ||
Data analysis
MS/MS spectra were searched using MASCOT engine (Matrix Science, London, UK; version 2.2) embedded into Proteome Discoverer 1.4. Protein identification was performed according to the database of Universal Protein (UniProt). The differential proteins of fold change >1.2 or <0.83 and p < 0.05 was finalized by the use of biological replicate method. Each protein function was identified by the Gene Ontology (GO) terms and classified by the GO enrichment analysis approach (http://www.geneontology.org/). The online reference system Kyoto Encyclopedia of Genes and Genomes (KEGG) was used for the systematic interpretation of differentially expressed proteins (http://geneontology.org/).Analytical methods
The samples were obtained from the Erlenmeyer flasks and bioreactor for analysis. The broth was centrifuged (4500 × g, 10 min), and the precipitate was collected, washed twice with distilled water, and then dried at 105 °C to a constant weight to determine dry cell weight (DCW) of the culture. The concentration of glucose was determined using a biosensor (SBA-40C, Biology Institute of Shandong Academy of Sciences, Shandong, China). The supernatant was used to determine the ε-PL concentration using the methyl orange precipitation method according to Itzhaki.19 In brief, an equal volume of sample with 0.06–0.12 g L−1 ε-PL diluted with 0.07 mM phosphate buffer (pH 6.90) and 0.7 mM methyl orange solution were mixed together, which were reacted at 30 °C with shaking for 30 min. The ε-PL concentration can be estimated from the absorbance at 465 nm of the methyl orange remaining in the supernatant solution through standard curve calculation.Statistical analysis
All described experiments were performed in independent biological triplicates, unless stated otherwise. Presented data in the graphs are the averages of the replicates ± the standard deviation. Differences were considered statistically significant if the p value from t-test (MS Excel 2010) for two samples with equal variance was p < 0.05.Results and discussion
Fermentation characteristics and growth behavior of M-Z18 and SS-62
Bioassays including ε-PL concentration quantification, DCW and ε-PL/DCW analysis of culture supernatants from the two strains were performed at 96 h. As shown in Fig. 1, the ε-PL concentration detected in the culture supernatants from the SS-62 strain attained 3.04 g L−1 in shake-flask fermentation, which was a 1.79-fold increase compared to the production in S. albulus M-Z18 culture supernatants. Meanwhile, the DCW and ε-PL/DCW of SS-62 were enhanced 1.25-fold and 1.55-fold, respectively.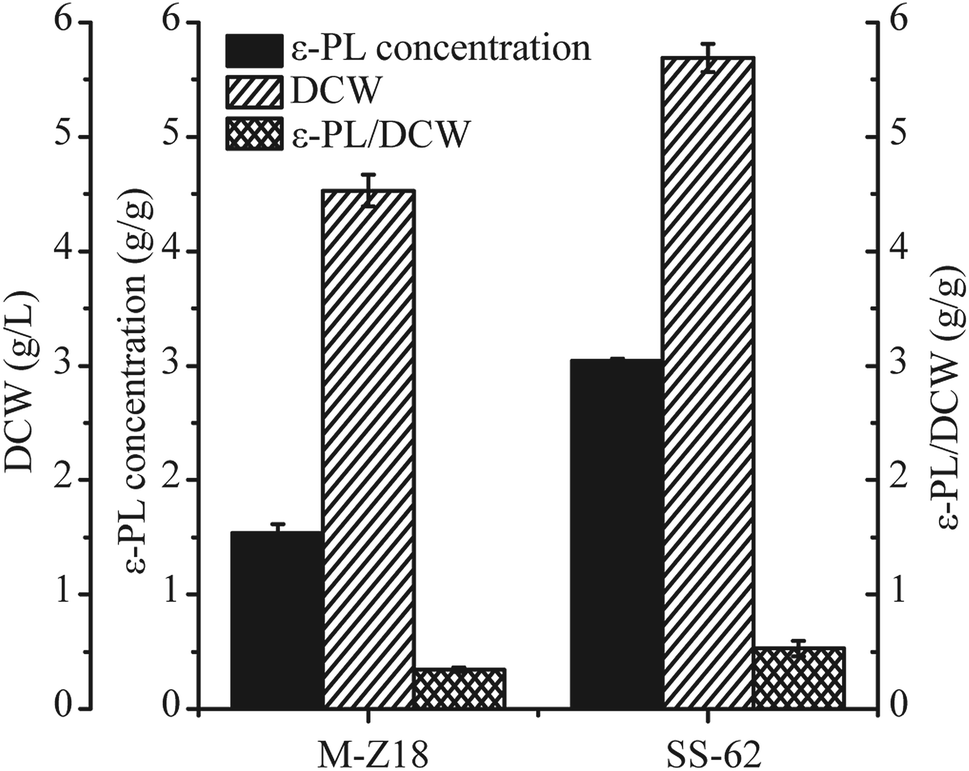 | ||
| Fig. 1 Fermentation parameters of parent strain S. albulus M-Z18 and mutant strain SS-62 in Erlenmeyer flask. Data represents the means of three independent experiments, and error bars represent the standard deviation. | ||
It is of great value to recognize changes in the cultivation characteristics of high-yielding strain for industrial application.7,20 ESI File 3† shows the macroscopic and microscopic photographs of M-Z18 and SS-62 in solid and liquid culture. M-Z18 colonies were smooth and pure white, while SS-62 colonies had a volcanic rock shape with a central depression and were surrounded by folds. Further, SEM analysis showed the hyphae of SS-62 appeared full and abundant as the parent strain M-Z18, but no vesicles and the existence of mycelial helixes were observed in SS-62 (ESI Files 3A-1 and B-4†). When the colonies matured, M-Z18 colonies had an abundance of spores, but SS-62 colonies were flat with some white dots spread in the spores. In addition, M-Z18 spores were brown, while SS-62 spores were greyish green. From the SEM analysis, it can be found that SS-62's spores were bigger and had more spines than the parent strain M-Z18 (ESI Files 3A-2, and B-5†). In contrast, there was not much difference between mycelium morphology of M-Z18 and SS-62 in liquid culture (ESI Files 3A-3, and B-6†).
Protein synthesis activity in vivo
Luzhetskyy's group has developed a versatile and sensitive reporter system for actinomycetes which is based on gusA encoding β-glucuronidase, and has shown the utility of the GUS reporter system for gene expression study at the translation level in actinomycetes.17 GFP was used for the evaluation of protein synthesis in recombinant strains in an earlier study,21 but its reliability when investigating protein synthesis in actinomycetes is unclear. Therefore, to determine the relationship between protein synthesis and ε-PL production in the parental strain and mutants, a reporter gene assay based on glucuronidase gene (GUS) was performed. The glucuronidase activity was measured at 24, 48 and 72 h after inoculation. As shown in Fig. 2, mutant SS-62 exhibited higher glucuronidase activity compared to that of parental strain M-Z18 at 48 and 72 h. It indicated that the high-level ε-PL production may be caused by accelerated protein synthesis.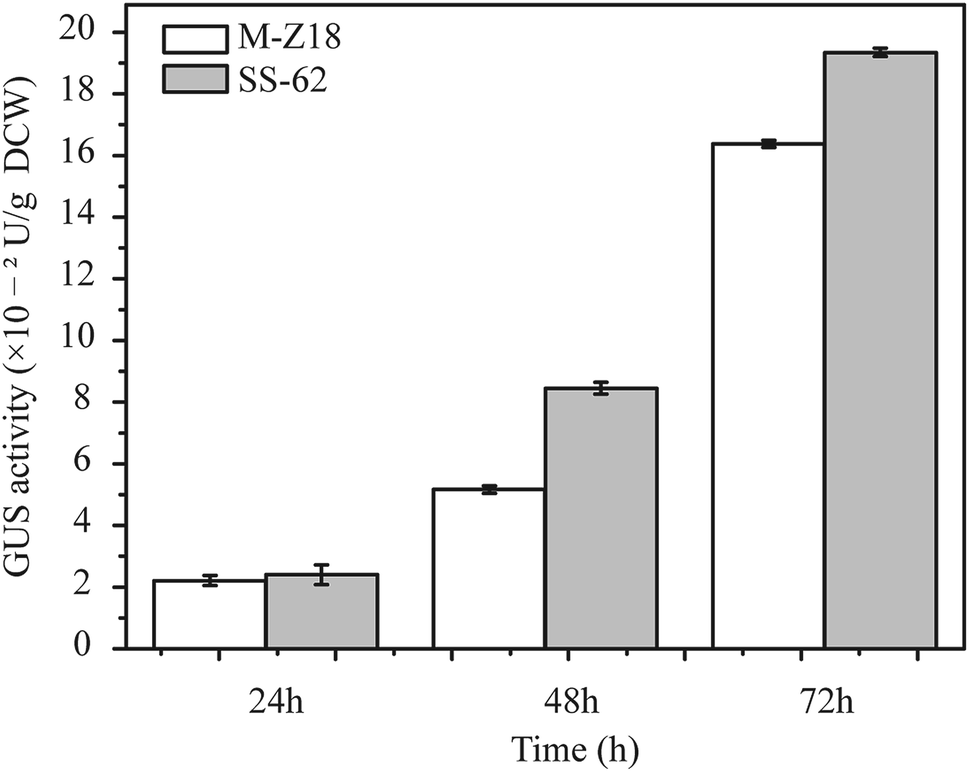 | ||
| Fig. 2 Detection of β-glucuronidase activity. Glucuronidase activity was measured in cell lysates of parental strain S. albulus M-Z18 and mutant SS-62 from fermentation broth at 24, 48 and 72 h, respectively. The data shown are the mean values of three replicates ± standard deviation (SD), which is indicated by an error bar. | ||
Sampling conditions and summary of proteomics analysis
To understand the potential elements involved in controlling ε-PL production in S. albulus, comparative proteomics based on iTRAQ analysis was performed. It is well known that the synthesis of ε-PL is closely related to pH.22 To exclude the impact of pH, 30 h shake-flask fermentation broth was chosen as sample to obtain a proteomic profile at the beginning of ε-PL synthesis. A list of amino acid sequences of all proteins predicted to be encoded by Streptomyces albulus PD-1 chromosome and plasmids was compiled based on previously reported genome sequences (ESI File 4†).23iTRAQ experiments identified 3486 proteins from S. albulus according to UniProt database. Overall 401 proteins (11.50%) in SS-62 were significantly differentially expressed compared to expression in M-Z18. Of these proteins, 188 proteins (46.88%) were up-regulated and 213 proteins (53.12%) were down-regulated (Fig. 3A, ESI File 5†). GO analysis revealed the first 20 significant biological functional pathways of the differential proteins (Fig. 3B).
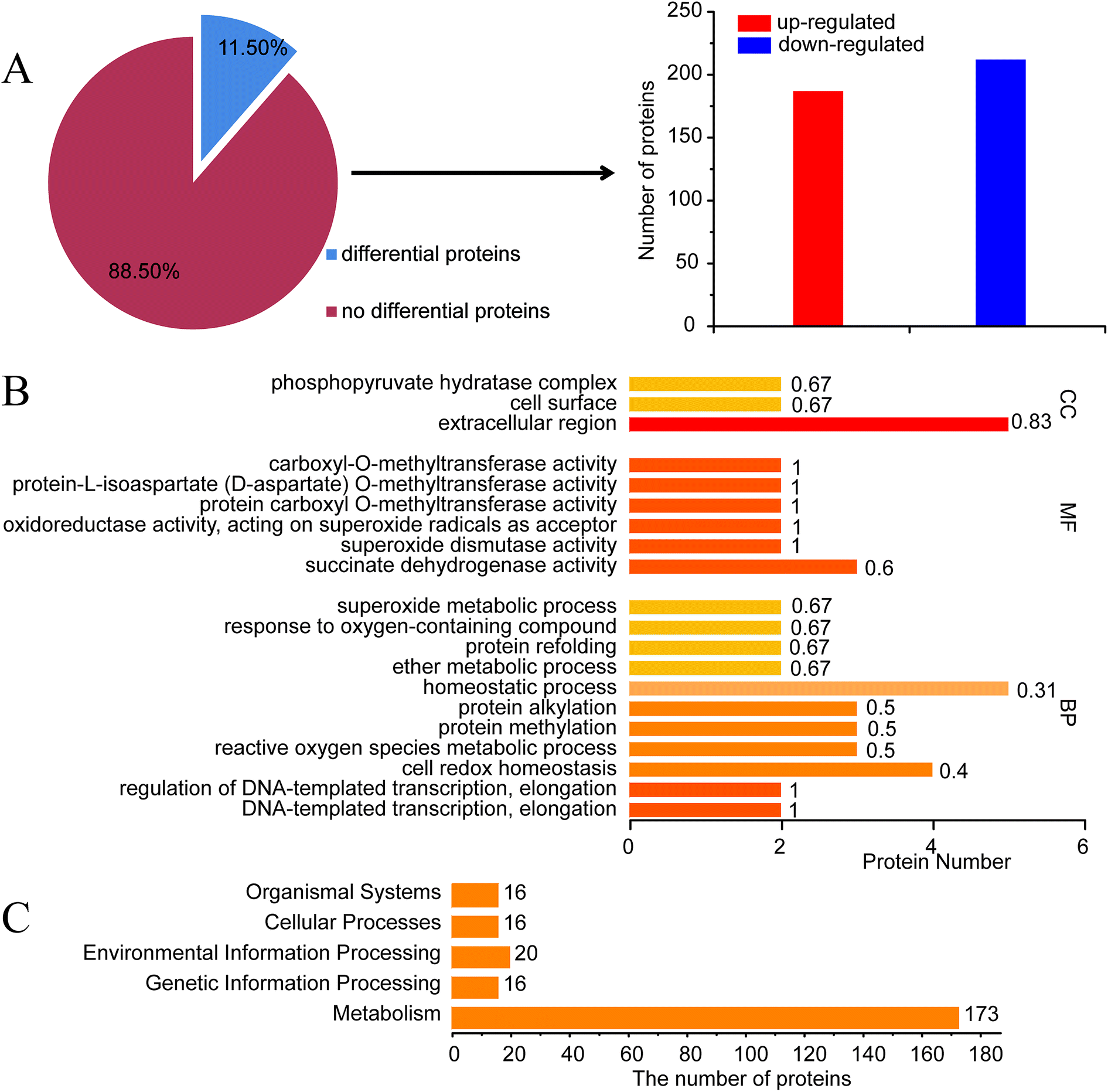 | ||
| Fig. 3 Proteins identified by searching UniProt and GO enrichment analysis. Numbers of the detected proteins (3486) of S. albulus, of which 11.50% are differentially expressed proteins (401) compared with the patent strain (A). The first 20 significant biological functional pathways of GO analysis related to the percentage of the differential proteins (B). Differential proteins related to the different biological functions by KEGG analysis (C). | ||
KEGG analysis results showed some different biological functional properties (Fig. 3C) on the basis of differential proteins, including organismal systems, cellular processes, environmental information processing, genetic information processing and metabolism. Of particular interest in this study was the identification of metabolic and regulatory proteins that may be directly or indirectly regulation ε-PL biosynthesis in SS-62. This includes carbohydrate metabolism, amino acid metabolism, energy metabolism and other transcriptional regulators.
Changes of carbohydrate metabolism, amino acid metabolism and energy metabolism in SS-62
Carbohydrate metabolism, amino acid metabolism and energy metabolism are essential for ε-PL synthesis. ε-PL synthesis was strengthened from carbon metabolism and energy metabolism with 0.5% n-dodecane in broth.24 Results indicated that most of the differential proteins were mainly related to metabolism and genetic information processing (Fig. 4A). However, the proteins associated with carbohydrate metabolism, amino acid metabolism and energy metabolism accounted for most of the differential proteins. Carbohydrate metabolism is a crucial important pathway for cellular activities.25 The pathway mainly includes PPP, EMP, TCA, DAP and anaplerotic pathway. Differentially expressed proteins from iTRAQ results were displayed in Fig. 4B and ESI File 6.† G6PDH (A0A059VZG9) is the rate-limiting enzyme in the pentose phosphate pathway, which is responsible for providing NADPH and pentoses used for cell growth and metabolites biosynthesis.26 The expression of G6PDH in SS-62 was increased 1.21-fold. PEPC (A0A059W8H1) and PYC (X0MXA7) catalyze conversion of phosphoenolpyruvate and pyruvate into oxaloacetic acid in the anaplerotic pathway, respectively, as a supplementary to tricarboxylic acid cycle. Results showed that the expression levels of both proteins were enhanced 1.22- and 1.3-fold, respectively. It is reported that L-lysine, the precursor to ε-PL, is synthesized through diaminopimelate pathway in bacteria.2 This pathway is initiated from the phosphorylation of aspartate by the catalytic activity of Ask. As shown in Fig. 4B, the expression of ASK (Q0KJ33) was up-regulated by 1.28-fold. pls is a nonribosomal peptide synthetase that catalyzes the polymerization of L-lysine to produce ε-PL with consumption of ATP.27,28 Results showed that the expression of Pls (B5BR95) in SS-62 was significantly increased, about 1.55-fold. These data show iterative introduction of streptomycin resistance significantly increased the protein expression related to cell growth and ε-PL synthesis in mutant SS-62.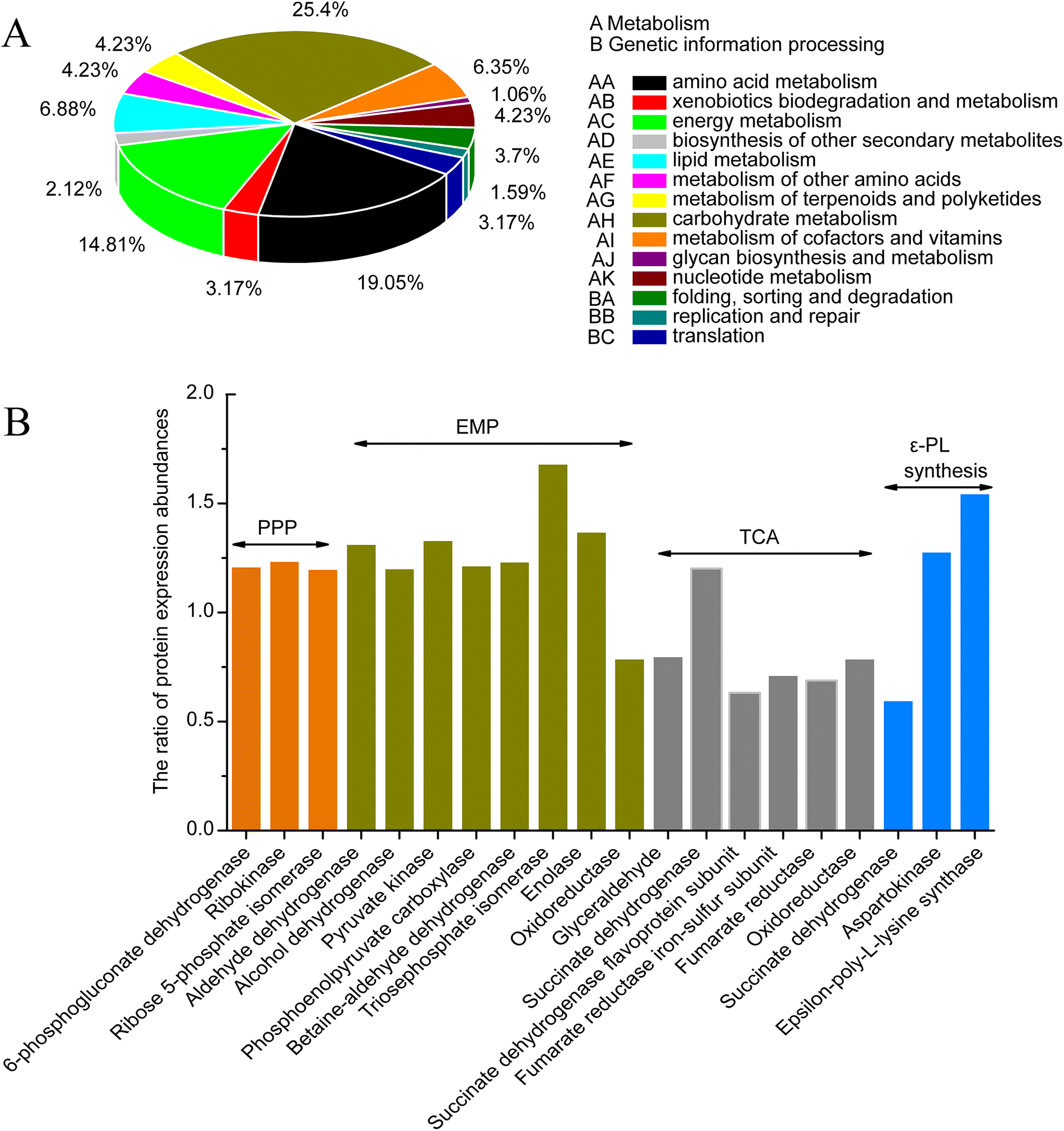 | ||
| Fig. 4 Differential proteins related to the metabolism and genetic information processing by KEGG analysis. | ||
Amino acid metabolism and energy metabolism were compared between SS-62 and M-Z18 (ESI File 6†). The metabolism of histidine (X0N9B5, X0MQF6, X0MXT6, X0N359) and arginine (X0NZK5, X0N514) were strengthened in SS-62. It is possible that after continuous stimulation with streptomycin, basic amino acids were more readily synthesized and the cells had a stronger ε-PL synthesis capacity. However, lysine degradation ability was enhanced, and the reduced lysine might be used for cell growth and ε-PL synthesis. Phenylalanine (A0A059W7X8, X0MY85), tyrosine and tryptophan biosynthesis (X0MZG0, X0N525), glycine, serine and threonine metabolism (A0A059WCN5, X0MW61, A0A059W2E1), alanine, glutamic acid (X0N4C0, A0A059W6Z3) and other metabolic byproducts had been weakened. This result indicated that the synthesis of byproducts decreased in the process of ε-PL synthesis in SS-62. Moreover, some increased expression of proteins associated with oxidative phosphorylation (X0NA06, A0A1A9QKT3, X0MJ64), carbon fixation pathways in prokaryotes (A0A059VUK9, X0NA06), and methane metabolism (A0A059W0D7, X0MSW1, A0A059WCI8, A0A059VUK9, A0A1A9QG52) also suggest that SS-62 synthesizes more energy and may promote the synthesis of ε-PL.
Translation and related proteins
Several proteins that are known or may be involved in regulating ε-PL production were detected in the dataset (Table 3). Expression of one glutamyl-tRNAGln amidotransferase family protein (X0N7H0, A0A059W8F5/GatA) was increased 1.22-fold in SS-62 compared with the parental strain. X0N571 was increased 1.36-fold. The decrease of 30S ribosomal protein S1 (A0A1A9QG37) expression in SS-62 mutant was not expected as it has a role in the ribosome dynamic properties to initiate translation of a large set of mRNAs with diverse structural features.29 However, 30S ribosomal protein S6 (A0A059W5Y0) in mutant SS-62 was up-regulated 1.89-fold. Interestingly, three elongation factors (X0P1T9, A0A1A9QQA8, X0P2R5) that promote polypeptide chain in mRNA translation were also up-regulated significantly in mutant SS-62, which might be due to the increase in expression of proteins related to ε-PL synthesis. Protein biosynthesis, which occurs on the ribosome, consists of four steps: initiation, elongation, termination, and ribosome recycling. The subsequent translation cycle requires breakdown of the ribosome–mRNA–tRNA posttermination complex and recycling of the ribosomal subunits. The ribosome recycling factor (RRF), together with the elongation factor-G (EF-G), is involved in catalyzing the disassembly of the post-termination complex and recycling the ribosome.30 Furthermore, RRF, a product of frr gene responsible for the dissociation of ribosomes from messenger RNA after the termination of translation, was up-regulated in SS-62. frr gene encoding RRF is widely distributed in prokaryotes and eukaryotic organelles, and is present in all prokaryotes examined to date. RRF has been shown to strongly stimulate in vitro protein synthetic activity in Escherichia coli, suggesting that the efficiency of protein synthesis is increased by recycling components of the post-termination complex.31 An increased expression of RRF in Streptomyces coelicolor was recently shown to cause enhanced protein synthesis during the late growth phase and subsequent overproduction of the antibiotic.32| Accession | Putative function | Protein | Gene | Unique peptides | Peptides | Fold changea | p Value |
|---|---|---|---|---|---|---|---|
| a Fold change between two strains (mutant SS-62/parent M-Z18), fold change > 1.2 represented upregulation, fold change < 0.83 represented downregulation.b Function unknown. | |||||||
| Translation | |||||||
| X0N7H0 | Translation | Glutamyl-tRNA (Gln) amidotransferase subunit A | gatA | 16 | 17 | 1.22 | 0.0212 |
| X0N571 | Regulation of translational fidelity | Isoleucine-tRNA ligase | ileS | 25 | 25 | 1.36 | 0.0004 |
| A0A059W8F5 | Regulation of translational fidelity | Aspartyl/glutamyl-tRNA (Asn/Gln) amidotransferase subunit C | gatC | 2 | 2 | 1.53 | 0.0225 |
| A0A059W5Y0 | Translation | 30S ribosomal protein S6 | rpsF | 5 | 5 | 1.89 | 0.0005 |
| A0A1A9QQ8 | —b | Peptidase | A4V12_17360 | 1 | 1 | 1.20 | 0.0104 |
| A0A1A9QG37 | Translation | 30S ribosomal protein S1 | rpsA | 1 | 26 | 0.71 | 0.0269 |
|
|||||||
| Others | |||||||
| X0P1T9 | Translational elongation | Elongation factor Tu | tuf | 14 | 24 | 1.35 | 0.0293 |
| A0A1A9QQA8 | Translational elongation | Elongation factor Ts | tsf | 19 | 20 | 1.36 | 0.0019 |
| X0P2R5 | Translational elongation | Transcription elongation factor GreA | greA | 6 | 6 | 1.27 | 0.0367 |
| X0MS37 | Translational termination | Ribosome-recycling factor | frr | 15 | 15 | 1.21 | 0.0415 |
Transcriptional regulators
The expression of secondary metabolic gene clusters is controlled by many different families of regulatory proteins, some of which are found only in actinomycetes, and is elicited by both extracellular and intracellular signaling molecules.33 A total of 33 transcriptional regulators were detected in iTRAQ dataset (Table 4), which were categorized into 12 significant families and one other cluster. The MarR-family transcriptional regulators (A0A059WCR4, A0A059W106) were up-regulated in mutant SS-62 compared to in the parent strain. In addition, Crp/Fnr family protein (X0MIN4), which is a pleiotropic transcriptional regulator that controls a broad range of cellular functions,34 was up-regulated in SS-62. Further analysis indicated that the LysR-family transcriptional regulators (X0N4A8, A0A059WFL3) and one single IclR transcriptional regulator detected in dataset were down-regulated in SS-62. And disruption or deletion of orf10 (LysR-type transcriptional regulators) in Streptomyces lividans causes actinorhodin overproduction.35| Accession | Putative function | Protein | Gene | Unique peptides | Peptides | Fold changea | p Value |
|---|---|---|---|---|---|---|---|
| a Fold change between two strains (mutant SS-62/parent M-Z18), fold change > 1.5 represented upregulation, fold change < 0.83 represented downregulation.b Function unknown. | |||||||
| X0MIN4 | —b | Crp/Fnr family transcriptional regulator | P354_08015 | 19 | 20 | 1.28 | 0.0010 |
| A0A059WCR4 | Regulation of transcription, DNA-templated | MarR family transcriptional regulator | DC74_5136 | 9 | 9 | 1.21 | 0.0124 |
| X0MS01 | — | Transcriptional regulator | P354_28905 | 3 | 3 | 1.40 | 0.0050 |
| X0MWP7 | Regulation of transcription, DNA-templated | Transcriptional regulator | P354_18815 | 6 | 6 | 1.74 | 0.0001 |
| X0MRL4 | Sequence-specific DNA binding | DNA-binding protein | P354_30280 | 1 | 1 | 1.30 | 0.0161 |
| X0N564 | Integral component of membrane | Transcriptional regulator | P354_27980 | 1 | 1 | 1.25 | 0.0116 |
| X0N2N8 | Oxidation–reduction process | DNA-binding protein | P354_25910 | 2 | 2 | 1.23 | 0.0024 |
| A0A059WA88 | Regulation of transcription, DNA-templated | AsnC family transcriptional regulator | DC74_4159 | 1 | 1 | 1.23 | 0.0194 |
| A0A059W106 | — | MarR family transcriptional regulator | DC74_1054 | 1 | 1 | 1.22 | 0.0090 |
| A0A059VZR2 | Biosynthetic process | Putative GntR family transcriptional regulator | DC74_566 | 5 | 5 | 1.21 | 0.0145 |
| A0A059WBX8 | — | Transcriptional regulator | DC74_4870 | 5 | 5 | 1.22 | 0.0276 |
| X0N4Z4 | — | Transcriptional regulator | P354_28185 | 9 | 9 | 0.78 | 0.0130 |
| X0NBV0 | Regulation of transcription, DNA-templated | TetR family transcriptional regulator | P354_12810 | 3 | 3 | 0.78 | 0.0212 |
| A0A059WBQ9 | Regulation of transcription, DNA-templated | DeoR family transcriptional regulator | DC74_6728 | 4 | 4 | 0.79 | 0.0335 |
| X0N4A8 | Regulation of transcription, DNA-templated | LysR family transcriptional regulator | P354_31270 | 4 | 4 | 0.76 | 0.0049 |
| X0N5W5 | Regulation of transcription, DNA-templated | TetR family transcriptional regulator | P354_27365 | 2 | 2 | 0.81 | 0.0343 |
| X0N9V3 | Regulation of transcription, DNA-templated | LuxR family transcriptional regulator | P354_18990 | 1 | 1 | 0.79 | 0.0227 |
| A0A059WD02 | Regulation of transcription, DNA-templated | TetR family transcriptional regulator | DC74_6856 | 2 | 2 | 0.77 | 0.018 |
| A0A059WE02 | Regulation of transcription, DNA-templated | TetR family transcriptional regulator | DC74_7592 | 1 | 1 | 0.63 | 0.0142 |
| X0MHH6 | DNA binding | DNA-binding protein | P354_13010 | 3 | 3 | 0.82 | 0.0474 |
| X0MQA0 | Regulation of transcription, DNA-templated | GntR family transcriptional regulator | P354_33540 | 1 | 1 | 0.75 | 0.0028 |
| X0NHJ | Regulation of transcription, DNA-templated | LuxR family transcriptional regulator | P354_00025 | 2 | 2 | 0.79 | 0.0391 |
| A0A059WFL3 | Regulation of transcription, DNA-templated | LysR family transcriptional regulator | DC74_7800 | 1 | 1 | 0.27 | 0.0079 |
| X0MNE4 | Sequence-specific DNA binding | Cro/Cl family transcriptional regulator | P354_37150 | 2 | 2 | 0.76 | 0.0045 |
| A0A059WH51 | Regulation of transcription, DNA-templated | TetR family transcriptional regulator | DC74_6720 | 1 | 1 | 0.78 | 0.0067 |
| X0N092 | Regulation of transcription, DNA-templated | TetR family transcriptional regulator | P354_05815 | 1 | 1 | 0.71 | 0.0266 |
| A0A1A9QJ90 | — | IclR family transcriptional regulator | A4V12_27770 | 1 | 1 | 0.78 | 0.0098 |
| A0A059VZH0 | Regulation of transcription, DNA-templated | MarR family transcriptional regulator | DC74_486 | 1 | 1 | 0.78 | 0.0067 |
| A0A059W3M0 | Regulation of transcription, DNA-templated | Cold shock protein | DC74_3899 | 1 | 3 | 1.68 | 0.0361 |
| X0NAN3 | Regulation of transcription, DNA-templated | Cold shock protein | P354_16170 | 4 | 5 | 1.40 | 0.0075 |
| X0P2W9 | Regulation of transcription, DNA-templated | Cold shock protein | P354_13760 | 2 | 3 | 1.78 | 0.0091 |
| X0MYX2 | Regulation of transcription, DNA-templated | Cold shock protein | P354_42355 | 2 | 4 | 1.79 | 0.0296 |
| A0A059VY86 | Regulation of transcription, DNA-templated | Cold shock protein | DC74_24 | 2 | 3 | 1.28 | 0.0009 |
Six additional regulators were differentially expressed in the comparison, which included proteins belonging to the AsnC-family (A0A059WA88), Cro/Cl-family (X0MNE4), LuxR-family (X0N9V3, X0NHJ), NmrA-family (497683061) and DeoR-family (490052578). AsnC (A0A059WA88) was up-regulated 1.23-fold than in M-Z18. It is reported that deletion of SCO3361, an Lrp/AsnC family protein from Streptomyces coelicolor, led to dramatic reduction in actinorhodin production.36 A single GntR-family transcriptional regulator (490058535), which has various functions including gluconate repression, and multiple TetR-family transcriptional regulators were all found to be under-expressed in SS-62. TetR-family members are involved in the transcriptional regulation of multidrug efflux, antibiotic biosynthesis pathways, osmotic stress response, catabolic pathways and differentiation processes. Importantly, it has been proven that the TetR family contains transcriptional repressors by Ramos et al.37 The down-regulation of TetR transcriptional regulators may be beneficial to ε-PL synthesis. In addition, five cold shock proteins (A0A059W3M0, X0NAN3, X0P2W9, X0MYX2, A0A059VY86) were significantly over-expressed in the mutant SS-62. Cold shock proteins are small nucleic acid-binding proteins ranging from 65 to 75 amino acids in length, which are not only produced during cold stress but have a much wider role in the stress response of bacteria.38 It has a function as RNA chaperones by destabilizing secondary structures in target RNA at low temperature so that the single-stranded state of target RNA is maintained, this enables efficient transcription and translation.39
qRT-PCR analysis of related genes and overexpression of frr and pls
We performed gene expression analysis of some genes, viz., B5BR95 (pls), X0MS37 (frr), A0A059VZL5 (relA), and HrdD (hrdD), based on the criteria of fold change observed in the proteomic analysis and involvement in important physiological pathways. As shown in Fig. 5, qRT-PCR results showed that the transcription level of pls, frr, and hrdD in SS-62 were increased at different levels. Especially, the transcription level of frr was enhanced by 6.3-fold. Moreover, the transcription levels of pls and hrdD in the SS-62 mutant were all increased by 4.6-fold, which may be responsible for enhancing ε-PL production. It is reported that HrdD can specifically bind to the pls-gene promoter, regulate the pls expression, and initiate ε-PL synthesis.40,41 However, there was no difference in the expression of relA at transcriptional and protein levels. This was surprising as ppGpp (relA gene encoding ppGpp synthetase) is necessary for antibiotic production under conditions of nitrogen limitation in Streptomyces coelicolor A3(2)42 and for cephamycin C production in Streptomyces clavuligerus.43,44 Therefore, whether ppGpp was directly involved in promoting transcription of ε-PL biosynthetic genes was unclear. | ||
| Fig. 5 mRNA expression quantitation of related genes in S. albulus M-Z18 and SS-62 upon iTRAQ analysis. | ||
Proteomics and qRT-PCR results showed that the expression of Pls, RRF and HrdD in SS-62 were all increased. Herein, we investigated the positive role of Pls, RRF and HrdD in ε-PL production through overexpression of pls, frr and hrdD genes in S. albulus M-Z18, respectively. As shown in Fig. 6, the results indicated that there is no difference in the consumption rate of glucose between M-Z18 and M-Z18-pls (Fig. 6A). But the ε-PL production and the synthesis of ε-PL per unit cell of M-Z18-pls increased by 7.2% and 20.3% compared to M-Z18, respectively (Fig. 6C and D). However, the consumption rate of glucose and the DCW by S. albulus M-Z18-frr was slower than M-Z18 (Fig. 6A and B). Although the ε-PL production of S. albulus M-Z18-frr increased slightly, the ε-PL synthesis of M-Z18-frr was always higher than that of M-Z18 before 30 h, and the synthesis of ε-PL per unit cell increased significantly (Fig. 6C and D). Hosaka et al.32 and Ma et al.45 showed that an increased expression of RRF in S. coelicolor and S. diastatochromogenes 1628 can enhance the production of antibiotics, which were consistent to our results. However, the detailed mechanism of up-regulated frr to increase antibiotic production remains to be clarified. Unfortunately, the overexpression of hrdD in S. albulus M-Z18 was not made difference in terms of DCW, ε-PL production and the synthesis of ε-PL per unit cell than the S. albulus M-Z18 (ESI File 2†).
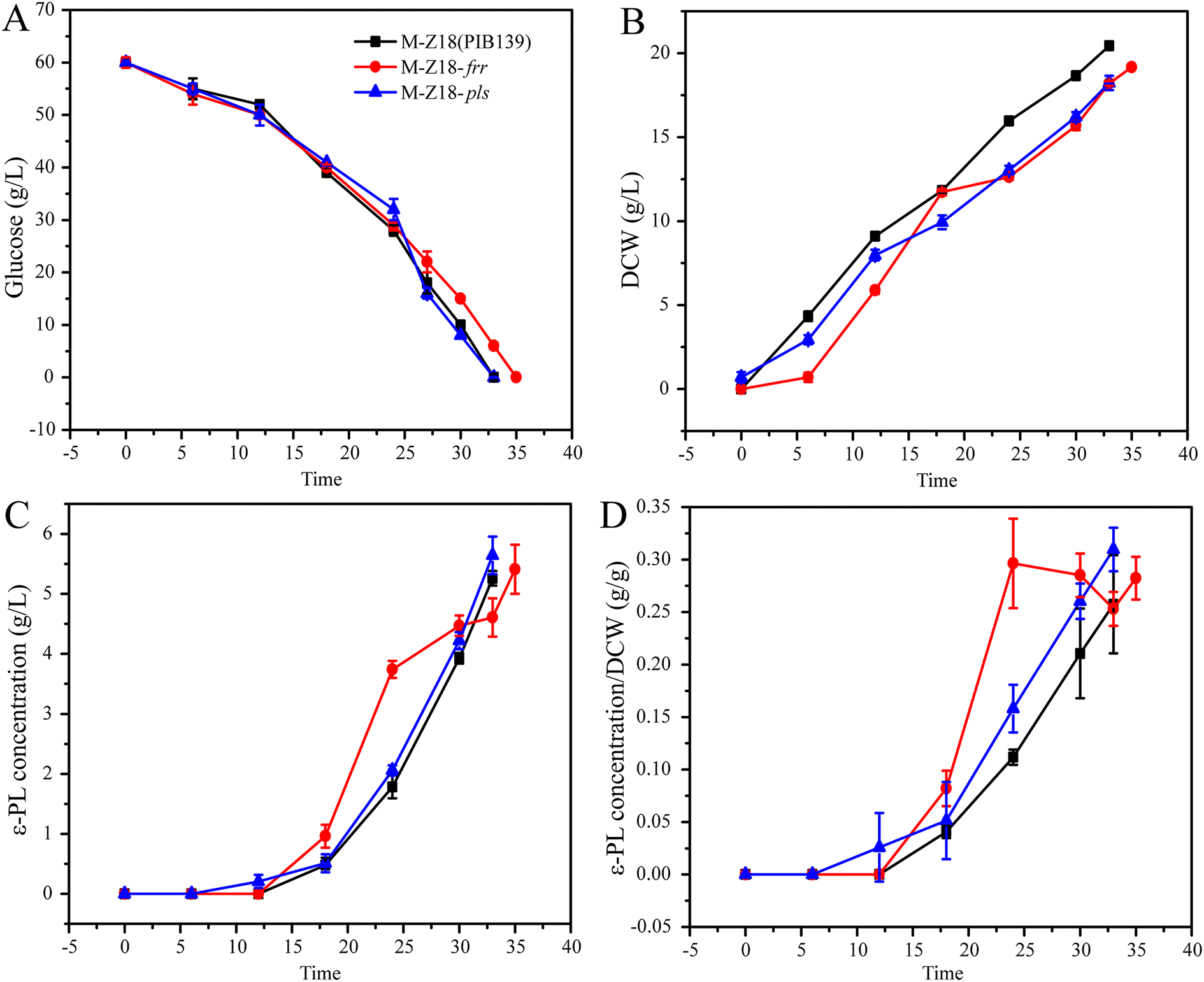 | ||
| Fig. 6 Fermentation parameters of the parent strain S. albulus M-Z18 and overexpressed strain S. albulus M-Z18-frr and S. albulus M-Z18-pls in 1 L multi-bioreactor system. | ||
Conclusions
In this study, iTRAQ-based proteomic analysis was used to explain the high-yield mechanism of streptomycin-resistant ε-PL-producing mutant in Streptomyces albulus at the level of proteomics. The dramatic changes were related to the carbohydrate metabolism, amino acid metabolism and energy metabolism based on the GO and KEGG analysis. Analysis of the proteome indicated that mutant SS-62 could not only strengthen the ε-PL precursor and energy metabolism but also tune the pathways related to transcriptional regulation and translation, resulting in a better intracellular metabolic environment for the synthesis of ε-PL. Specifically, the strain showed up-regulated expression levels of Pls and RRF in the pathway of L-lysine and ε-PL synthesis, which led to a significant increase of ε-PL biosynthesis capacity in the mutant strain.Funding
This work was financially supported by the National Natural Science Foundation of China (31671846) and the National First-Class Discipline Program of Light Industry Technology and Engineering (LITE2018-27).Conflicts of interest
The authors declare that they have no conflict of interest.Abbreviations
| ε-PL | ε-Poly-L-lysine |
| iTRAQ | Isobaric tags for relative and absolute quantification |
| UniProt | Universal protein |
| GO | Gene ontology |
| KEGG | Kyoto encyclopedia of genes and genomes |
| qRT-PCR | Real time quantitative PCR |
| EMP | Embden-meyerhof-parnas |
| PPP | Pentose phosphate pathway |
| TCA | Tricarboxylic acid cycle |
| DAP | Diaminopimelate pathway |
| G6PDH | Glucose-6-phosphate dehydrogenase |
| PEPC | Phosphoenolpyruvate carboxylase |
| PYC | Pyruvate carboxylase |
| ASK | Aspartokinase |
| Pls | ε-PL synthetase |
| RRF | Ribosome recycling factor |
References
- S. Shima and H. Sakai, Polylysine produced by Streptomyces, Agric. Biol. Chem., 1977, 41, 1807–1809 CAS.
- S. B. Bankar and R. S. Singhal, Panorama of poly-ε-lysine, RSC Adv., 2013, 3, 8586–8603 RSC.
- J. Hiraki, M. Hatakeyama, H. Morita and Y. Izumi, Improved epsilon-poly-L-lysine production of an S-(2-aminoethyl)-L-cysteine resistant mutant of Streptomyces albulus, Seibutsu Kogaku Kaishi, 1998, 76, 487–493 CAS.
- H. Zong, Y. Zhan, X. Li, L. Peng, F. Feng and D. Li, A new mutation breeding method for Streptomyces albulus by an atmospheric and room temperature plasma, Afr. J. Microbiol. Res., 2012, 6, 3154–3158 CAS.
- L. Wang, X. Chen, G. Wu, S. Li, X. Zeng, X. Ren, L. Tang and Z. Mao, Improved ε-poly-l-lysine production of Streptomyces sp. FEEL-1 by atmospheric and room temperature plasma mutagenesis and streptomycin resistance screening, Ann. Microbiol., 2015, 65, 2009–2017 CrossRef CAS.
- S. Li, F. Li, X. Chen, L. Wang, J. Xu, L. Tang and Z. Mao, Genome shuffling enhanced ε-poly-l-lysine production by improving glucose tolerance of Streptomyces graminearus, Appl. Biochem. Biotechnol., 2012, 166, 414–423 CrossRef CAS PubMed.
- L. Wang, X. Chen, G. Wu, X. Zeng, X. Ren, S. Li, L. Tang and Z. Mao, Genome Shuffling and Gentamicin-Resistance to Improve ε-Poly-l-Lysine Productivity of Streptomyces albulus W-156, Appl. Biochem. Biotechnol., 2016, 180, 1601–1617 CrossRef CAS PubMed.
- W. Geng, C. Yang, Y. Gu, R. Liu, W. Guo, X. Wang, C. Song and S. Wang, Cloning of ε-poly-L-lysine (ε-PL) synthetase gene from a newly isolated ε-PL-producing Streptomyces albulus NK660 and its heterologous expression in Streptomyces lividans, Microb. Biotechnol., 2014, 7, 155–164 CrossRef CAS PubMed.
- Y. Gu, X. Wang, C. Yang, W. Geng, J. Feng, Y. Wang, S. Wang and C. Song, Effects of Chromosomal Integration of the Vitreoscilla Hemoglobin Gene (vgb) and S-Adenosylmethionine Synthetase Gene (metK) on [epsilon]-Poly-l-Lysine Synthesis in Streptomyces albulus NK660, Appl. Biochem. Biotechnol., 2016, 178, 1445 CrossRef CAS PubMed.
- D. Xu, H. Yao, C. Cao, Z. Xu, S. Li, Z. Xu, J. Zhou, X. Feng and H. Xu, Enhancement of epsilon-poly-L-lysine production by overexpressing the ammonium transporter gene in Streptomyces albulus PD-1, Bioprocess Biosyst. Eng., 2018, 41, 1337–1345 CrossRef CAS PubMed.
- K. Ochi, S. Okamoto, Y. Tozawa, T. Inaoka, T. Hosaka, J. Xu and K. Kurosawa, Ribosome engineering and secondary metabolite production, Adv. Appl. Microbiol., 2004, 56, 155–184 CAS.
- N. Tamehiro, T. Hosaka, J. Xu, H. Hu, N. Otake and K. Ochi, Innovative approach for improvement of an antibiotic-overproducing industrial strain of Streptomyces albus, Appl. Environ. Microbiol., 2003, 69, 6412–6417 CrossRef CAS PubMed.
- G. Wang, T. Hosaka and K. Ochi, Dramatic activation of antibiotic production in Streptomyces coelicolor by cumulative drug resistance mutations, Appl. Environ. Microbiol., 2008, 74, 2834–2840 CrossRef CAS PubMed.
- Y. Liu, X. Chen, J. Zhao, Q. Li and Z. Mao, Improvement of ε-poly-L-lysine production of Streptomyces albulus by continuous introduction of streptomycin resistance, Process Biochem., 2019, 82, 10–18 CrossRef CAS.
- M. Nishikawa and K. Ogawa, Distribution of Microbes Producing Antimicrobial-Poly-L-Lysine Polymers in Soil Microflora Determined by a Novel Method, Appl. Environ. Microbiol., 2002, 68, 3575–3581 CrossRef CAS PubMed.
- J. Sambrook, D. W. Russell, and D. W. Russell, Molecular cloning: a laboratory manual (3-volume set), Cold spring harbor laboratory press, New York, 2001 Search PubMed.
- M. Myronovskyi, E. Welle, V. Fedorenko and A. Luzhetskyy, β-Glucuronidase as a sensitive and versatile reporter in actinomycetes, Appl. Environ. Microbiol., 2011, 77, 5370–5383 CrossRef CAS PubMed.
- K. J. Livak and T. D. Schmittgen, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- R. F. Itzhaki, Colorimetric method for estimating polylysine and polyarginine, Anal. Biochem., 1972, 50, 569–574 CrossRef CAS PubMed.
- X. Ren, Y. Xu, X. Zeng, X. Chen, L. Tang and Z. Mao, Microparticle-enhanced production of ε-poly-l-lysine in fed-batch fermentation, RSC Adv., 2015, 5, 82138–82143 RSC.
- Z. Ma, L. Tao, A. Bechthold, X. Shentu, Y. Bian and X. Yu, Overexpression of ribosome recycling factor is responsible for improvement of nucleotide antibiotic-toyocamycin in Streptomyces diastatochromogenes 1628, Appl. Microbiol. Biotechnol., 2014, 98, 5051 CrossRef CAS PubMed.
- X. Ren, X. Chen, X. Zeng, L. Wang, L. Tang and Z. Mao, Acidic pH shock induced overproduction of ε-poly-L-lysine in fed-batch fermentation by Streptomyces sp. M-Z18 from agro-industrial by-products, Bioprocess Biosyst. Eng., 2015, 38, 1113–1125 CrossRef CAS PubMed.
- Z. Xu, X. Feng, S. Li, H. Xu, F. Bo and Z. Sun, Genome Sequence of Streptomyces albulus PD-1, a Productive Strain for Epsilon-Poly-L-Lysine and Poly-L-Diaminopropionic Acid, Microbiology Resource Announcements, 2014, 2, e00297 Search PubMed.
- Z. Xu, F. Bo, J. Xia, Z. Sun, S. Li, X. Feng and H. Xu, Effects of oxygen-vectors on the synthesis of epsilon-poly-lysine and the metabolic characterization of Streptomyces albulus PD-1, Biochem. Eng. J., 2015, 94, 58–64 CrossRef CAS.
- H. Hayrapetyan, M. Tempelaars, M. Nierop Groot and T. Abee, Bacillus cereus ATCC 14579 RpoN (Sigma 54) Is a Pleiotropic Regulator of Growth, Carbohydrate Metabolism, Motility, Biofilm Formation and Toxin Production, PLoS One, 2015, 10, e0134872 CrossRef PubMed.
- X. Zeng, X. Chen, X. Ren, Q. Liu, L. Wang, Q. Sun, L. Tang and Z. Mao, Insights into the role of glucose and glycerol as a mixed carbon source in the improvement of ε-poly-l-lysine productivity, Appl. Biochem. Biotechnol., 2014, 173, 2211–2224 CrossRef CAS PubMed.
- K. Yamanaka, N. Kito, Y. Imokawa, C. Maruyama, T. Utagawa and Y. Hamano, Mechanism of ε-poly-l-lysine production and accumulation revealed by identification and analysis of an ε-poly-l-lysine-degrading enzyme, Appl. Environ. Microbiol., 2010, 76, 5669–5675 CrossRef CAS PubMed.
- K. Yamanaka, C. Maruyama, H. Takagi and Y. Hamano, Epsilon-poly-L-lysine dispersity is controlled by a highly unusual nonribosomal peptide synthetase, Nat. Chem. Biol., 2008, 4, 766–772 CrossRef CAS PubMed.
- M. Duval, A. Korepanov, O. Fuchsbauer, P. Fechter, A. Haller, A. Fabbretti, L. Choulier, R. Micura, B. P. Klaholz, P. Romby, M. Springer and S. Marzi, Escherichia coli ribosomal protein S1 unfolds structured mRNAs onto the ribosome for active translation initiation, PLoS Biol., 2013, 11, e1001731 CrossRef PubMed.
- L. S. I. Janosi and A. Kaji, Ribosome recycling factor (ribosome releasing factor) is essential for bacterial growth, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 4249–4253 CrossRef CAS PubMed.
- M. K. J. Ryoji and A. Kaji, Further characterization of ribosome releasing factor and evidence that it prevents ribosomes from reading through a termination codon, J. Biol. Chem., 1981, 256, 5798–5801 CAS.
- T. Hosaka, J. Xu and K. Ochi, Increased expression of ribosome recycling factor is responsible for the enhanced protein synthesis during the late growth phase in an antibiotic-overproducing Streptomyces coelicolor ribosomal rpsL mutant, Mol. Microbiol., 2006, 61, 883–897 CrossRef CAS PubMed.
- M. J. Bibb, Regulation of secondary metabolism in streptomycetes, Curr. Opin. Microbiol., 2005, 8, 208–215 CrossRef CAS PubMed.
- Y. Huang, D. Yang, G. Pan, G. L. Tang and B. Shen, Characterization of LnmO as a pathway-specific Crp/Fnr-type positive regulator for leinamycin biosynthesis in Streptomyces atroolivaceus and its application for titer improvement, Appl. Microbiol. Biotechnol., 2016, 100, 10555–10562 CrossRef CAS PubMed.
- O. H. Martínez-Costa AJM-T, E. Martínez, A. Miguel, Fernández-Moreno and F. Malpartida, An Additional Regulatory Gene for Actinorhodin Production in Streptomyces lividans Involves a LysR-Type Transcriptional Regulator, J. Bacteriol., 1999, 181, 4353–4364 Search PubMed.
- J. Liu, J. Li, H. Dong, Y. Chen, Y. Wang, H. Wu, C. Li, D. T. Weaver, L. Zhang and B. Zhang, Characterization of an Lrp/AsnC family regulator SCO3361, controlling actinorhodin production and morphological development in Streptomyces coelicolor, Appl. Microbiol. Biotechnol., 2017, 101, 5773–5783 CrossRef CAS PubMed.
- J. L. Ramos, M. Martinez-Bueno, A. J. Molina-Henares, W. Teran, K. Watanabe, X. Zhang, M. T. Gallegos, R. Brennan and R. Tobes, The TetR family of transcriptional repressors, Microbiol. Mol. Biol. Rev., 2005, 69, 326–356 CrossRef CAS PubMed.
- R. Keto-Timonen, N. Hietala, E. Palonen, A. Hakakorpi, M. Lindstrom and H. Korkeala, Cold Shock Proteins: A Minireview with Special Emphasis on Csp-family of Enteropathogenic Yersinia, Front. Microbiol., 2016, 7, 1151 Search PubMed.
- W. H. Y. Jiang and M. Inouye, CspA, CspA, the Major Cold-shock Protein of Escherichia coli, Is an RNA Chaperone, J. Biol. Chem., 1997, 272, 196–202 CrossRef CAS PubMed.
- L. Wang, C. Gao, N. Tang, S. Hu and Q. Wu, Identification of genetic variations associated with epsilon-poly-lysine biosynthesis in Streptomyces albulus ZPM by genome sequencing, Sci. Rep., 2015, 5(1), 9201 CrossRef CAS PubMed.
- L. Wang, X. Chen, G. Wu, S. Li, X. Zeng, X. Ren, L. Tang and Z. Mao, Enhanced ε-poly-l-lysine production by inducing double antibiotic-resistant mutations in Streptomyces albulus, Bioprocess Biosyst. Eng., 2017, 40, 271–283 CrossRef CAS PubMed.
- BIBB* RCAM, The ppGpp synthetase gene (relA) of Streptomyces coelicolor A3(2) plays a conditional role in antibiotic production and morphological differentiation, J. Bacteriol., 1997, 179, 5854–5861 CrossRef PubMed.
- W. Jin, H. K. Kim, J. Y. Kim, S. G. Kang, S. H. Lee and K. J. Lee, Cephamycin C production is regulated by relA and rsh genes in Streptomyces clavuligerus ATCC27064, J. Biotechnol., 2004, 114, 81–87 CrossRef CAS PubMed.
- W. Jin, Y. G. Ryu, S. G. Kang, S. K. Kim, N. Saito, K. Ochi, S. H. Lee and K. J. Lee, Two relA/spoT homologous genes are involved in the morphological and physiological differentiation of Streptomyces clavuligerus, Microbiology, 2004, 150, 1485–1493 CrossRef CAS PubMed.
- Z. Ma, L. Tao, A. Bechthold, X. Shentu, Y. Bian and X. Yu, Overexpression of ribosome recycling factor is responsible for improvement of nucleotide antibiotic-toyocamycin in Streptomyces diastatochromogenes 1628, Appl. Microbiol. Biotechnol., 2014, 98, 5051 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03156a |
| This journal is © The Royal Society of Chemistry 2019 |