
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and biological evaluation of 4-(1H-1,2,4-triazol-1-yl)benzoic acid hybrids as anticancer agents†
Hatem A. Abuelizz a,
Hanem M. Awadb,
Mohamed Marzoukc,
Fahd A. Nasrd,
Ali S. Alqahtanie,
Ahmed H. Bakheitaf,
Ahmed M. Naglahgh and
Rashad Al-Salahi*a
a,
Hanem M. Awadb,
Mohamed Marzoukc,
Fahd A. Nasrd,
Ali S. Alqahtanie,
Ahmed H. Bakheitaf,
Ahmed M. Naglahgh and
Rashad Al-Salahi*a
aDepartment of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, PO Box 2457, Riyadh 11451, Saudi Arabia. E-mail: ralsalahi@ksu.edu.sa
bDepartment of Tanning Materials and Leather Technology, National Research Centre, 33 El-Bohouth St. (Former El-Tahrir St.), Dokki, Cairo 12622, Egypt
cChemistry of Natural Products Group, Center of Excellence for Advanced Sciences, National Research Centre, 33 El-Bohouth St. (Former El-Tahrir St.), Dokki, Cairo 12622, Egypt
dMedicinal Aromatic, and Poisonous Plants Research Center, College of Pharmacy, King Saud University, PO Box 2457, Riyadh, 11451, Saudi Arabia
ePharmacognosy Department, College of Pharmacy, King Saud University, PO Box 2457, Riyadh, 11451, Saudi Arabia
fDepartment of Chemistry, Faculty of Science and Technology, El-Neelain University, PO Box 12702, Khartoum 11121, Sudan
gDepartment of Pharmaceutical Chemistry, Drug Exploration and Development Chair (DEDC), College of Pharmacy, King Saud University, Riyadh 11451, Saudi Arabia
hPeptide Chemistry Department, Chemical Industries Research Division, National Research Centre, 33 El-Bohouth St. (Former El-Tahrir St.), Dokki, Cairo 12622, Egypt
First published on 17th June 2019
Abstract
A series of 4-(1H-1,2,4-triazol-1-yl)benzoic acid hybrids (1–17) was successfully synthesized and their structures were established by NMR and MS analysis. In vitro cytotoxic evaluation indicated that some of the hybrids exhibited potent inhibitory activities against MCF-7 and HCT-116 cancer cell lines, with IC50 values ranging from 15.6 to 23.9 µM, compared with reference drug doxorubicin (19.7 and 22.6 µM, respectively). Notably, the most potent compounds, 2, 5, 14, and 15, not only exhibited an obvious improvement in IC50 values, but demonstrated very weak cytotoxic effects toward normal cells (RPE-1) compared with doxorubicin. A further investigation showed that compounds 2 and 14 clearly inhibited the proliferation of MCF-7 cancer cells by inducing apoptosis. In addition, these hybrids showed acceptable correlation with bioassay results in regression plots generated by 2D QSAR models. Our results indicated that 1,2,4-triazole benzoic acid hybrids could be used as a structural optimization platform for the design and development of more selective and potent anticancer molecules.
Introduction
Cancer has affected our ancestors throughout history. Today, cancer remains a major health concern worldwide, and is considered a life-threatening disease, representing the second most frequent cause of mortality after cardiovascular diseases, particularly in developed countries.Although many advances have been made in understanding cancer biology, the ultimate goal of controlling this malignancy remains a distant possibility. Cancer cells closely resemble normal cells because they result from the uncontrolled growth of otherwise normal cells. Most currently available chemotherapeutic agents for cancer act against all rapidly proliferating cells. Therefore, their activities against carcinoma cells are nonselective, killing both cancer and some normal cells, specifically those dividing rapidly. Furthermore, potent and effective compounds developed for use as treatments are often associated with undesired toxic side effects. Therefore, strategic therapies for cancer continues to be a fascinating challenge and remain among the most urgent areas of research in medicinal chemistry.1–3
The exploration of new heterocyclic compounds that exhibit potency for multiple biological purposes remains an intriguing scientific effort. For instance, triazoles and their annulated heterocycles have received great interest owing to their synthetic and pharmacophore features, and potential applicability to a range of bioactivities, such as anticancer, antibacterial, antiviral, and anti-inflammation activities. Furthermore, these compounds can be attractive moieties for the combination of two pharmacophore fragments into one compound to generate innovative multifunctional molecules.4–14
Recently, we reported that triazoloquinazolines, which are derived from the combination of triazole and quinazoline, constitute an interesting antitumor class of compounds. For instance, 3-pyridyl-bis-[1,2,4]triazoloquinazoline and 5-ethoxy-triazoloquinazoline have shown the highest cytotoxicity against Hep-G2 and colon HCT-116 carcinoma cells.15 Furthermore, 5-chloro-[1,2,4] triazoloquinazolines have been found to possess promising anticancer activities against medulloblastoma (Daoy) and melanoma (SK-MEL28) cell lines.16–18
Letrozole and anastrozole containing a 1,2,4-triazole ring have been approved by the FDA and used as first-line therapies for the treatment of breast cancer in postmenopausal women owing to their apparent superiority to estrogen receptor antagonists, such as tamoxifen. In aromatase inhibitors (AIs), which are responsible for inhibiting estrogen production, the triazole ring plays a pivotal role in the chelation with heme iron.6
Based on the above information and our interest in determining the anticancer pharmacological activities of triazole compounds, we incorporated benzoic acid and the triazole pharmacophore into one molecule to generate novel 1,2,4-triazole benzoic acid hybrid scaffolds. Herein, we aim to describe the synthesis of seventeen hybrids (1–17) and evaluate their in vitro antiproliferative activity against HCT-116 and MCF-7 human cancer cell lines, and normal human RPE-1 cells.
Results and discussion
Chemistry
Synthetic routes for the formation of 1,2,4-triazole benzoic acid hybrids (1–17) are shown in Scheme 1. Briefly, 4-hydrazinobenzoic acid was reacted with dialkyl-N-cyanoimido(dithio)carbonate to derive 1,2,4-triazole benzoic acids 1 and 2 in good yields. The structures of 1 and 2 were confirmed by their physical properties, and by NMR and MS analyses. In the target products, the presence of the benzoic acid moiety was confirmed by the two ortho doublets in the range of 8.20–7.60 ppm (J ≈ 8.6 Hz) and the COOH-singlet signal at ∼13.10–13.20 in the 1H NMR spectra, and their five 13C-resonances, especially that of the carboxyl group at ∼167.0 ppm. Similarly, the –SCH3 functional group of 1 was validated by its typical resonances at ∼2.6 (s) and 13.9 in the 1H and 13C NMR spectra, respectively. Furthermore, the 1,2,4-triazolo ring was established by assigning its two intrinsic 13C-signals (C-3′ and C-5′), which were highly deshielded according to the type of functional group attached in each case. The amino group protons were assigned as broad singlets ranging from 6.80 to 7.00 ppm. Treating compound 1 or 2 with the appropriate aldehydes in ethanol for 24–36 h under reflux conditions yielded targets 3–12. Their identities were verified by the characteristic resonances of the benzylidene functional group at ∼9.00–9.50 (s) and 160.0–166.0 ppm in the 1H and 13C NMR, respectively. Reacting benzyl(phenethyl) isothiocyanate with parents 1 and 2 produced compounds 13–15, whose structures were confirmed by assignment of the thiocarbonyl group peak at 188–189 ppm. Esterification of 1 yielded ester 16, which did not have a –COOH proton signal. Furthermore, the carboxyl carbon was slightly shifted upfield (166.0 ppm) owing to ester formation, with –CH3 resonances recorded at 3.88 and 52.8 ppm by 1H and 13C NMR, respectively. Oxidation of –SCH3 to an –SO2CH3 group was successfully achieved by reacting 1 with hydrogen peroxide in boiling ethanol, yielding product 17. This –SO2CH3 group was deduced from the singlet at 2.94 ppm in the 1H NMR spectrum, and a corresponding 13C resonance at 38.7 ppm. For the 3′-phenoxy products (2, 10–12, and 15), a characteristic coupling system of three 1H signals was recorded at ∼7.40 (br t, H-3/5), 7.27 (br d, 2/6), and 7.19 ppm (br t, H-4). Unambiguous confirmation of all products was finally achieved by clear identification of the 1H and 13C resonances of the accompanying functional groups or moieties, such as –SCH3, –SO2CH3, –OPh, benzyl, and phenethyl, and through comparison with data of structurally related compounds.19–24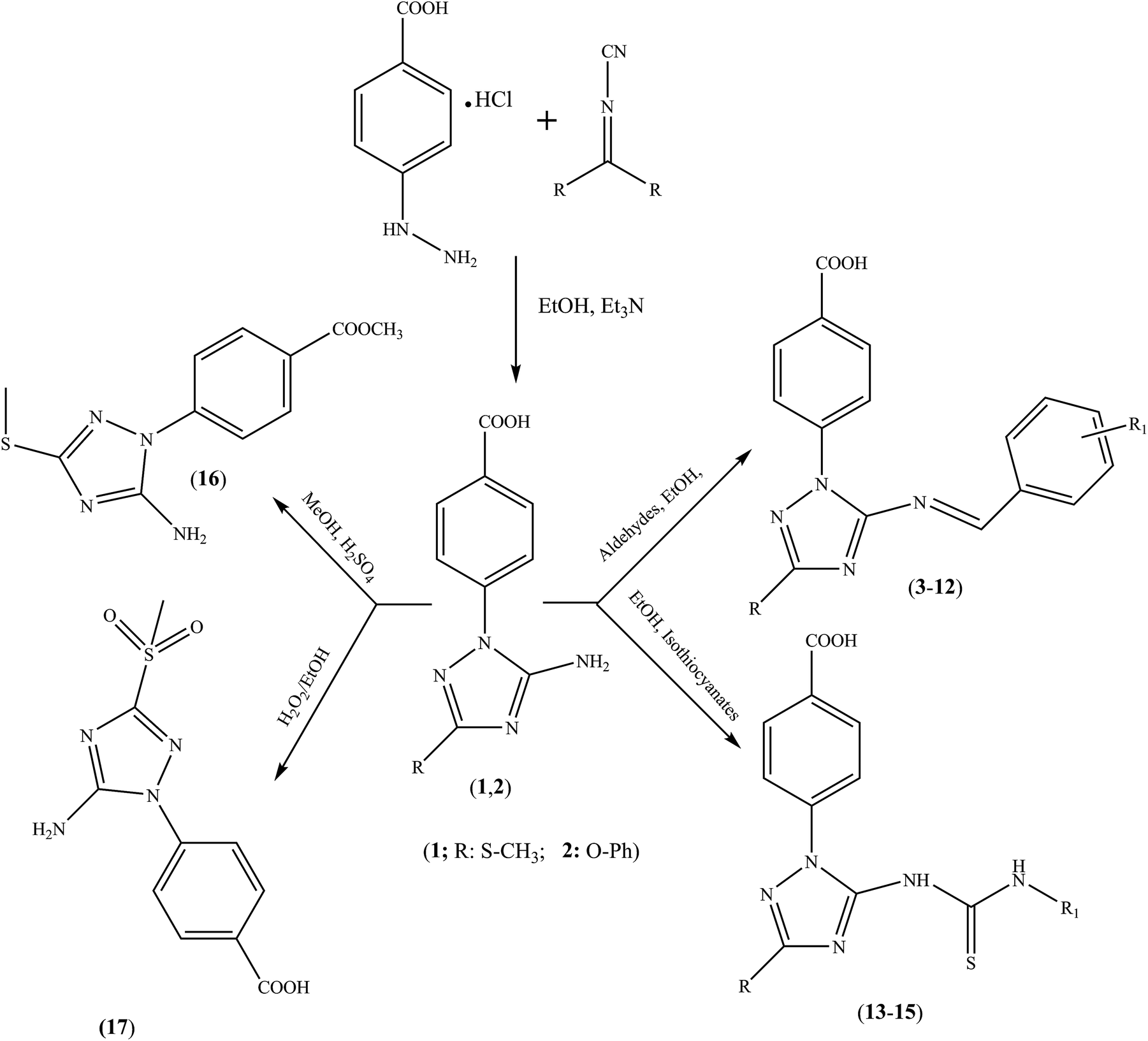 | ||
| Scheme 1 Synthetic routes to triazole benzoic acid compounds 1–17. | ||
Biology
From the afore-mentioned synthetic routes, we obtained a series of novel 1,2,4-triazole-benzoic acid hybrids 1–17 (Table 1). To investigate whether substituents affected their cytotoxic effects, in vitro cytotoxic studies against HCT-116 and MCF-7 human cancer cells, and normal human RPE-1 cells, were performed using the MTT assay. Doxorubicin was used as the reference drug.| Compound | R | R1 | Compound | R | R1 |
|---|---|---|---|---|---|
| 3 | S-CH3 | 2-OH-5-methyl | 10 | O-Ph | 2-OH-5-methyl |
| 4 | S-CH3 | 2-OH-5-methoxy | 11 | O-Ph | 2-OH-5-methoxy |
| 5 | S-CH3 | 2-OH-5-nitro | 12 | O-Ph | 2-OH-5-nitro |
| 6 | S-CH3 | 2-OH-5-Br | 13 | S-CH3 | Benzyl |
| 7 | S-CH3 | 4-OH-3-methoxy | 14 | S-CH3 | Phenethyl |
| 8 | S-CH3 | 3-OH | 15 | O-Ph | Benzyl |
| 9 | S-CH3 | 4-OH |
The percentages of viable cells were calculated and compared with those treated with doxorubicin. The activities of compounds 1–17 against the two cell lines were compared with those of doxorubicin. All compounds suppressed the human cell lines in a dose-dependent manner (see ESI†). To determine the efficacy of the synthesized 1,2,4-triazole benzoic acid hybrids, the cytotoxic effect of each compound was compared with that of the reference drug. Table 2 shows that compounds 2, 5, 12, 14, and 15 had significantly more potent cytotoxic activities in relation to doxorubicin against the two examined cells. The remaining compounds had significantly lower cytotoxic effects. For MCF-7 cells, 2, 5, 12, 14, and 17 exhibited the highest activities compared with that of doxorubicin. Among them, compounds 2 and 14 appeared more active than doxorubicin, while compounds 5, 12, 13, 15 and 17 showed comparable activities. Compounds 1, 3, 4, 6–11, and 16 had slightly lower anticancer activities. Against HCT-116 cells, compounds 2, 5, 12, 14, and 15 were found to possess good cytotoxicity compared with doxorubicin, while the remaining compounds showed less activity. Concerning RPE-1 cells, compounds 3, 4, 11, 14, 16, and 17 were more cytotoxic than the reference drug, while 12 and 13 had comparable effects. Furthermore, products 1, 2, 5, 7, and 9 had slightly weaker cytotoxic effects and the remaining compounds showed significantly lower cytotoxicity (Table 2 and ESI†).
| Compound code | IC50 (µM) ± SD | Therapeutic index (TI) | |||
|---|---|---|---|---|---|
| HCT-116 | MCF-7 | RPE-1 | HCT-116 | MCF-7 | |
| 1 | 41.8 ± 6.5 | 32.9 ± 5.3 | 61.0 ± 13.9 | 1.5 | 1.9 |
| 2 | 25.7 ± 4.1 | 18.7 ± 3.5 | 61.2 ± 10.9 | 2.4 | 3.3 |
| 3 | 36.0 ± 6.1 | 23.9 ± 1.6 | 43.9 ± 10.5 | 1.2 | 1.8 |
| 4 | 35.2 ± 5.2 | 27.3 ± 4.2 | 55.3 ± 11.1 | 1.6 | 2.0 |
| 5 | 24.0 ± 4.5 | 20.0 ± 4.1 | 60.2 ± 12.8 | 2.5 | 3.0 |
| 6 | 37.6 ± 6.4 | 31.3 ± 5.1 | 59.4 ± 11.7 | 1.6 | 1.9 |
| 7 | 29.8 ± 4.2 | 29.4 ± 5.3 | 61.1 ± 12.5 | 2.1 | 2.1 |
| 8 | 34.6 ± 6.1 | 31.2 ± 5.6 | 70.1 ± 15.3 | 2.0 | 2.2 |
| 9 | 39.1 ± 5.6 | 34.8 ± 5.5 | 68.0 ± 16.7 | 1.7 | 2.0 |
| 10 | 33.7 ± 5.2 | 30.3 ± 5.3 | 74.0 ± 16.5 | 2.2 | 2.4 |
| 11 | 33.1 ± 4.4 | 27.5 ± 4.1 | 58.5 ± 10.7 | 1.8 | 2.1 |
| 12 | 26.8 ± 4.5 | 21.6 ± 3.1 | 62.8 ± 13.9 | 2.3 | 2.9 |
| 13 | 30.1 ± 4.1 | 23.4 ± 4.6 | 63.9 ± 13.5 | 2.1 | 2.7 |
| 14 | 28.3 ± 5.2 | 15.6 ± 2.2 | 45.8 ± 8.9 | 1.6 | 2.9 |
| 15 | 23.9 ± 4.5 | 24.0 ± 4.3 | 91.6 ± 15.9 | 3.8 | 3.8 |
| 16 | 37.3 ± 6.1 | 23.4 ± 3.5 | 39.1 ± 9.9 | 1.0 | 1.7 |
| 17 | 34.2 ± 5.2 | 21.9 ± 4.2 | 48.5 ± 9.8 | 1.4 | 2.2 |
| Doxorubicin | 22.6 ± 3.9 | 19.7 ± 3.1 | 64.0 ± 13.2 | 2.8 | 3.3 |
To determine the safety of each compound, we calculated their respective therapeutic indexes (TIs) using the following equation:
| TI = IC50 against normal cells/IC50 against cancer cells |
Table 2 shows that, for HCT-116, compound 15 had a wider TI than doxorubicin. Compounds 2, 5, 8, 10, 12, and 13 had slightly lower TIs, while the remaining compounds had a significantly lower TI than doxorubicin.
Similarly, compared with doxorubicin, compound 15 had wider TI against MCF-7 cells, while 2 had the same TI, compounds 5 and 12–14 had slightly lower TIs, and the remaining compounds had significantly lower TIs. Regarding compound safety and efficacy, the results suggested that 2, 5, 14, and 15 are potent anticancer candidate drugs in human HCT-116 and MCF-7 cancer cell lines.
The resulting findings showed that 1,2,4-triazole hybrids (1–17) had weak to high cytotoxic activities against the two tumor cell lines, with IC50 value ranges of 15.6–39.8 and 23.9–41.8 µM for MCF-7 and HCT-116 cells, respectively (Table 2). Generally, introducing an electron-rich group significantly increased the compound cytotoxicity, as exemplified by target 2, which demonstrated promising IC50 values of 18.7 and 25.7 µM against MCF-7 and HCT-116, respectively.
Different substituents at the 5-position of the 1,2,4-triazole moiety in 2 contributed to the cytotoxicity profile. Furthermore, SAR studies showed that introducing isothiocyanate (15) and nitrobenzylidene (12) substituents was crucial for the potent cytotoxic activity.
Although target 1 was less active, its transformed product 5 showed the highest activity among all test compounds. Compound 7 showed a moderate effect against both cancer cell lines. Through this work, we found that aldehyde substituents at the 5-position showed varying cytotoxicities, as indicated by compounds 3–12.
Furthermore, the selectivity of chemotherapeutic agents remains a major impediment to successful cancer therapy. Therefore, the discovery and design of new chemical entities that can exert selectivity towards carcinoma cells are likely to improve the therapeutic index.
Apoptosis detection
MCF-7 cells were treated with 2 and 14 at 18.7 and 15.6 µM, respectively, for 24 h, stained with fluorescence DNA-binding dye Hoechst 33258. As shown in (Fig. 1) DNA fragmentation and typical marks of apoptosis are evident after compounds treatment. In contrast, control or vehicle-treated cells retained their cellular morphology, confluency and uniformly stained. To further confirm the apoptosis detection, MCF-7 treated cells were analyzed by flow cytometry. This method is based on the binding of Annexin V to phosphatidylserine on the plasma membrane of cells that undergoes apoptosis. In contrast, 7-amino-actinomycin (7-AAD) bind to the nucleic acids of cells in the later stages of apoptosis or necrosis. The results indicated that 2 and 14 induced cell apoptosis in MCF-7 cells. The MCF-7 control gave apoptosis percentage of 1.84 ± 0.63 and 2.94 ± 0.5% in early and late apoptosis, respectively, while treated cells showed an increase of 17.82 ± 1.30 and 24.91 ± 1.98% with compound 2 and 29.11 ± 1.44 and 45.47 ± 2.10 % with compound 14 in early and late apoptosis, respectively. As shown in Fig. 2, there was a significant difference (p < 0.05) in the early and late apoptosis of the control and MCF-7 treated cells. Apparently, compound 2 and 14 can mediate apoptosis in cancer cells, which contributed, at least in part, to their cytotoxic effects. | ||
| Fig. 1 Nuclear morphological changes in MCF-7 cell lines. Cells were fixed and stained with Hoechst 33258, and the images were captured by fluorescence microscopy (EVOS, USA); magnification: 200×. (A) Control (without treatment); (B and C) treatment with compounds 2 and 14 at the IC50 (18.7 and 15.6 µM, respectively) for 24 h. Arrows indicate apoptotic cells. | ||
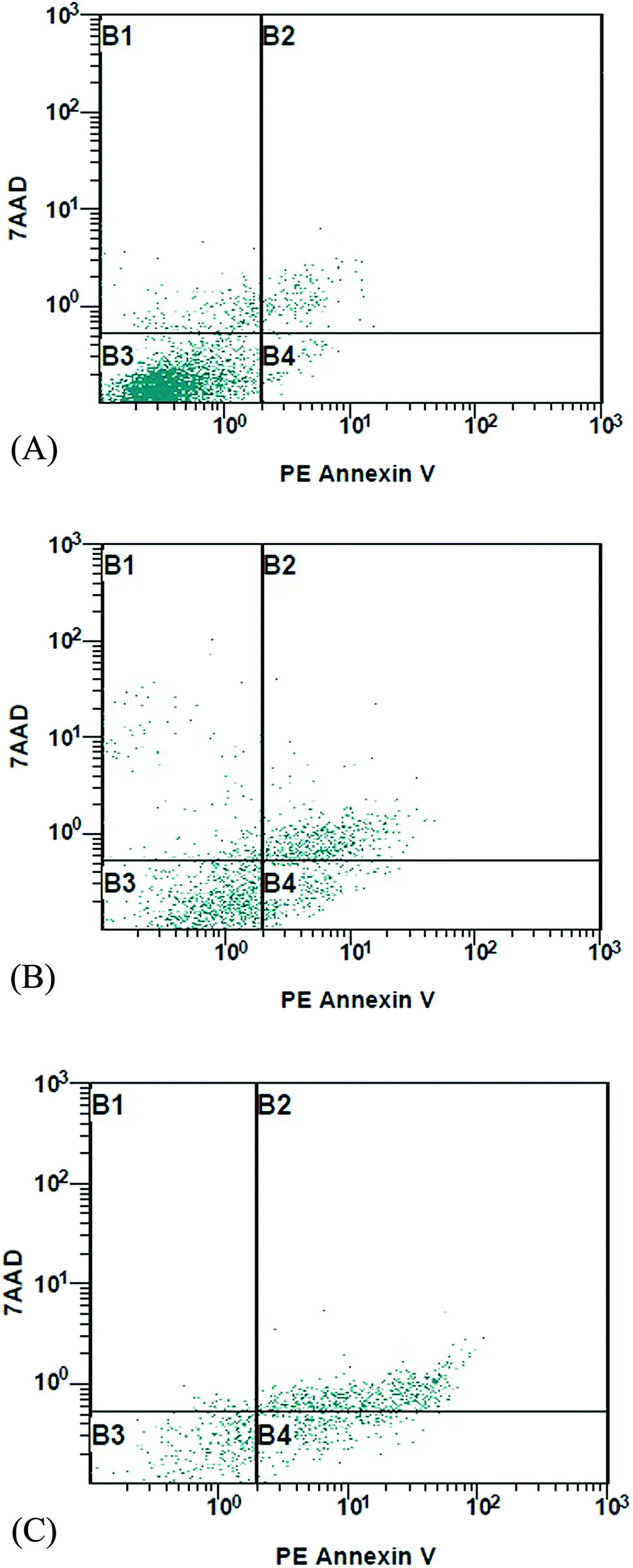 | ||
| Fig. 2 Effects of compounds 2 and 14 on apoptosis induction in MCF-7 cells. MCF-7 tumor cells were grown, treated, and stained with PE Annexin V and 7-AAD dye prior to flow cytometry analysis. Lower left quadrants (B3) show live cells; lower right quadrants (B4) represent early apoptotic cells; upper left and right quadrants (B1 and B2) contain necrotic or late apoptotic cells. (A) Control (without treatment); (B and C) treatment with compounds 2 and 14 at the IC50 (18.7 and 15.6 µM, respectively) for 24 h. | ||
2D QSAR analysis
QSAR is a method used to analyze the molecular descriptors (molecular weight, SlogP, HBA, HBD, rotatable bonds, and TPSA) of ligands/drugs and their correlation with in vitro/in vivo biological characteristics. The QuaSAR module was employed in the MOE to perform 2D QSAR for compounds 1–17. The calculated pIC50 values for compounds 1–17 against MCF-7 and HCT-116 cancer cells were input manually. Correlation plots were generated by plotting pIC50 values on the x-axis and predicted values ($PRED) on the y-axis for compounds 1–17.6 From the resulting pIC50 and $PRED values, a 2D QSAR model for compounds 1–17 was created to investigate potential ligand compounds. Regression plots were built for pIC50 vs. $PRED for the two cancer cells and the normal cells. A linear correlation resulted from regression analysis of the target compounds against MCF-7 cancer cells, exhibiting excellent linearity (Fig. 3). The Z-score of compounds 1–17 was predicted for the two cancer cells and normal cells. All compounds, except 3 and 8, showed good Z-score values. Compounds 3 and 8 showed Z-score values (absolute difference between the value of the model and the activity field, divided by the square root of the mean square error of the data set) beyond the limit of 2.5 (2.985 and 2.803). Compounds 2, 5, 14, and 15 showed more significant Z-scores of 0.175, 0.0779, 0.0998, and 0.208, respectively, against the MCF-7 cells. Compound 15 had a significant Z score of 0.733 against HCT-116 cells. Furthermore, principal component analysis was performed using the three eigenvectors, namely, PCA1, PCA2, and PCA3, and 3D graphical plots were created that included 98% of variance. In the plot, all values were found to lie in the range of +3 to −3 and are shown as different colored spots in Fig. 4, with each colored spot representing one compound.6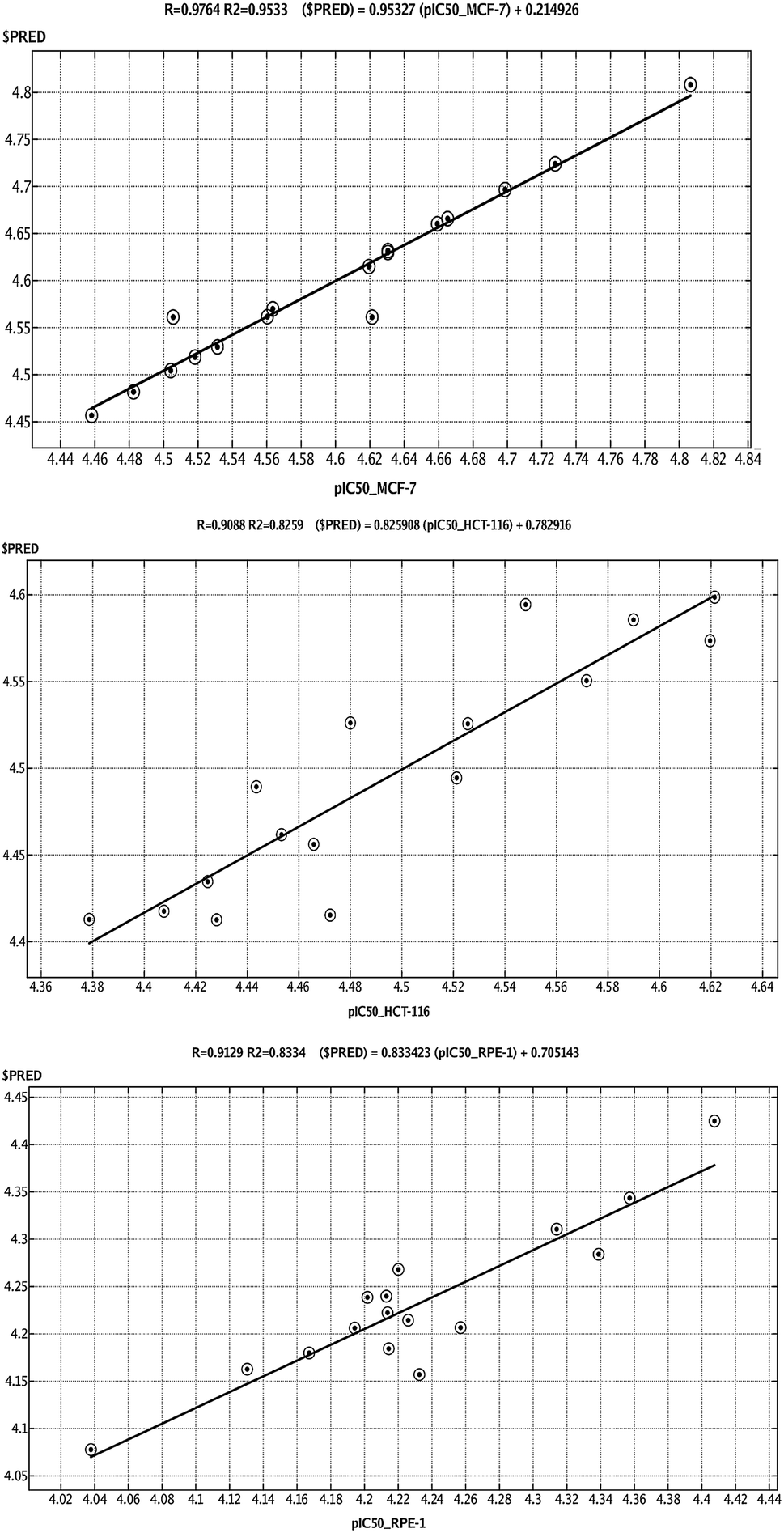 | ||
| Fig. 3 Linear correlation graph of experimentally measured pIC50 values of compounds 1–17 against (A) breast and (B) colon cancer cell lines, and (C) RPE-1 cell lines with predicted pIC50 values based on the calculated 2D QSAR model. The linearity of the 2D QSAR models are shown with error (RMSE) and correlation factor (R2) values. | ||
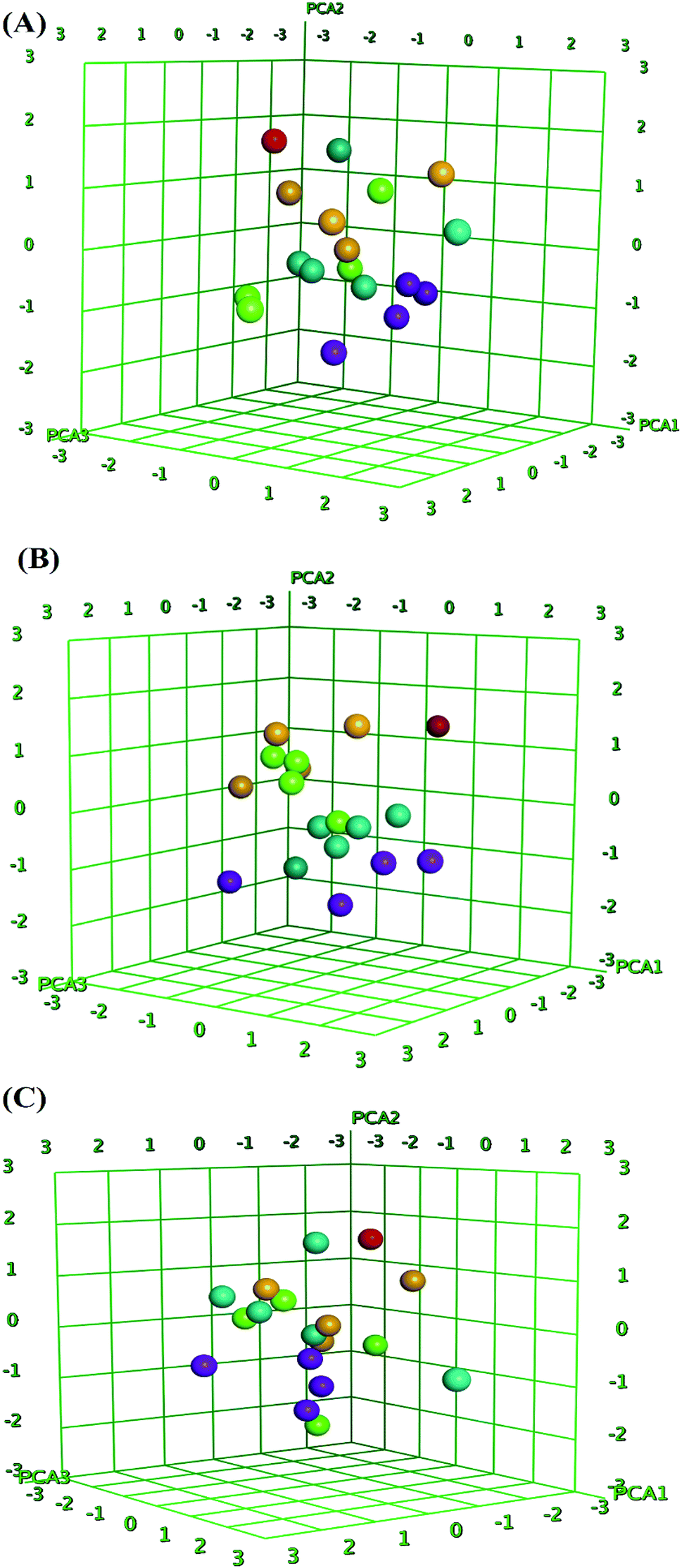 | ||
| Fig. 4 Principal component analysis (PCA) plots of compounds 1–17 for data obtained for breast (A), colon cancer cells (B) and (C) RPE-1 cell line. The 3D graphical plot was constructed with three eigenvectors PCA1, PCA2 and PCA3 in the range of +3 to −3 and coordinate value for each compound has represented with colored spots. | ||
Conclusions
A series of novel 1,2,4-triazole benzoic acid derivatives were synthesized, and their potential in vitro cytotoxicity against MCF-7 and HCT-116 human cancer cell lines was determined. Through this investigation, we showed that hybrids 2, 5, 14, and 15 exhibited potent antiproliferative activities. The SAR study also showed that the incorporation of isothiocyanate and nitrobenzylidene moieties into the 1,2,4-triazole scaffold would be beneficial to its promising cytotoxic effects. Furthermore, 2 and 15 not only demonstrated promising cytotoxic effects against the examined tumor cells, but exhibited very weak cytotoxicity against normal cells, implying that a therapeutic window exists for their use. A confirmation test also indicated that compounds 2 and 14 induce apoptosis in MCF-7 cancer cells and are, therefore, worthy of further research to develop novel anticancer drugs. In 2D QSAR analysis, the predicted pIC50 values of active compounds showed significant correlations with the experimental values from the generated regression and principal component analysis plots. These results strongly support the cytotoxicity results of compounds 2, 5, 14, and 15.Materials and methods
Chemistry
General procedure for the preparation of compounds 1 and 2
Through dropwise addition, 2-hydrazinobenzoic acid (10 mmol) dissolved in EtOH (10 mL) and Et3N (20 mmol) was added to a solution of dimethyl(phenyl)-N-cyanoimido(dithio)carbonate at room temperature. The mixture was refluxed for 3–5 h and then poured into ice/water. The obtained solid was collected, washed with water, and dried.![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ), 8.05 (d, J = 8.7 Hz, 2H, H-2/6), 7.68 (d, J = 8.7 Hz, 2H, H-3/5), 6.79 (br s, 2H,–N2), 2.49 (s, 3H, –SC3); 13C NMR (175 MHz, DMSO-d6): δ 167.1 (C-7), 159.3 (C-3′), 155.9 (C-5′), 140.9 (C-4), 131.1 (C-2/6), 128.8 (C-1), 121.9 (C-3/5), 13.8 (–S
), 8.05 (d, J = 8.7 Hz, 2H, H-2/6), 7.68 (d, J = 8.7 Hz, 2H, H-3/5), 6.79 (br s, 2H,–N2), 2.49 (s, 3H, –SC3); 13C NMR (175 MHz, DMSO-d6): δ 167.1 (C-7), 159.3 (C-3′), 155.9 (C-5′), 140.9 (C-4), 131.1 (C-2/6), 128.8 (C-1), 121.9 (C-3/5), 13.8 (–S![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3); HRMS (EI): m/z calcd for C10H10N4O2S (M)˙+ 250.0524, found 250.0563.), 8.04 (d, J = 8.6 Hz, 2H, H-2/6), 7.66 (d, J = 8.6 Hz, 2H, H-3/5), 7.41 (br t, J = 7.5 Hz, 2H, H-3′/5′), 7.27 (br d, J = 7.8 Hz, 2H, H-2′/6′), 7.19 (br t, J = 7.5 Hz, 1H, H-4′), 6.92 (br s, 2H, –N2); 13C NMR (175 MHz, DMSO-d6): δ 167.1 (C-7), 164.8 (C-3′), 155.0 (C-5′), 154.9 (C-1″), 140.9 (C-4), 131.1 (C-2/6), 130.1 (C-3″/5″ & 1), 124.8 (C-4″), 121.9 (C-3/5), 119.8 (C-2″/6″); HRMS (EI): m/z calcd for C15H12N4O3 (M)˙+ 296.0909, found 296.0947.
H3); HRMS (EI): m/z calcd for C10H10N4O2S (M)˙+ 250.0524, found 250.0563.), 8.04 (d, J = 8.6 Hz, 2H, H-2/6), 7.66 (d, J = 8.6 Hz, 2H, H-3/5), 7.41 (br t, J = 7.5 Hz, 2H, H-3′/5′), 7.27 (br d, J = 7.8 Hz, 2H, H-2′/6′), 7.19 (br t, J = 7.5 Hz, 1H, H-4′), 6.92 (br s, 2H, –N2); 13C NMR (175 MHz, DMSO-d6): δ 167.1 (C-7), 164.8 (C-3′), 155.0 (C-5′), 154.9 (C-1″), 140.9 (C-4), 131.1 (C-2/6), 130.1 (C-3″/5″ & 1), 124.8 (C-4″), 121.9 (C-3/5), 119.8 (C-2″/6″); HRMS (EI): m/z calcd for C15H12N4O3 (M)˙+ 296.0909, found 296.0947.General procedure for the preparation of compounds 3–15
A mixture of aldehyde (1.1 mmol) or benzyl (phenethyl)isothiocyanate (1.1 mmol) with compounds 1 and 2 (1 mmol) was heated under reflux in ethanol (15 mL) for 24–36 h. After cooling, the precipitate was collected by filtration, washed with water, and dried.), 10.94 (br s, 1H, –O), 9.51 (s, 1H, H-7″), 8.13 (d, J = 8.6 Hz, 2H, H-2/6), 7.95 (d, J = 8.6 Hz, 2H, H-3/5), 7.71 (d, J = 1.3 Hz, 1H, H-6″), 7.33 (dd, J = 8.4, 1.3 Hz, 1H, H-4″), 6.91 (d, J = 8.4 Hz, 1H, H-3″), 2.63 (s, 3H, –SC3), 2.27 (s, 3H, –ArC3); 13C NMR (175 MHz, DMSO-d6): δ 167.0 (C-7), 166.1 (C-7″), 160.9 (C-3′), 159.0 (C-2″), 157.7 (C-5′), 140.3 (C-4), 137.2 (C-4″), 130.9 (C-2/6), 130.2 (C-5″), 130.1 (C-6″), 129.0 (C-1), 123.5 (C-3/5), 120.2 (C-1″), 117.3 (C-3″), 20.4 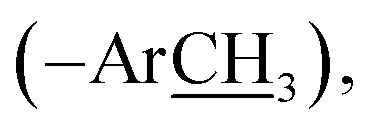 13.9 (–SH3); HRMS (EI): m/z calcd for C18H16N4O3S (M)˙+ 368.0943, found 368.0980.), 10.72 (br s, 1H, –O), 9.52 (s, 1H, H-7″), 8.12 (d, J = 8.4 Hz, 2H, H-2/6), 7.97 (d, J = 8.4 Hz, 2H, H-3/5), 7.44 (d, J = 2.8 Hz, 1H, H-6″), 7.15 (dd, J = 8.9, 2.8 Hz, 1H, H-4″), 6.95 (d, J = 8.9 Hz, 1H, H-3″), 3.75 (s, 3H, –OC3), 2.64 (s, 3H, –SC3); 13C NMR (175 MHz, DMSO-d6): δ 167.0 (C-7), 165.5 (C-7″), 161.0 (C-3′), 157.6 (C-5′), 155.5 (C-2″), 152.7 (C-5″), 140.3 (C-4), 130.8 (C-2/6), 130.2 (C-1), 124.2 (C-4″), 123.5 (C-3/5), 120.5 (C-1″), 118.6 (C-3″), 111.8 (C-6″), 55.9
13.9 (–SH3); HRMS (EI): m/z calcd for C18H16N4O3S (M)˙+ 368.0943, found 368.0980.), 10.72 (br s, 1H, –O), 9.52 (s, 1H, H-7″), 8.12 (d, J = 8.4 Hz, 2H, H-2/6), 7.97 (d, J = 8.4 Hz, 2H, H-3/5), 7.44 (d, J = 2.8 Hz, 1H, H-6″), 7.15 (dd, J = 8.9, 2.8 Hz, 1H, H-4″), 6.95 (d, J = 8.9 Hz, 1H, H-3″), 3.75 (s, 3H, –OC3), 2.64 (s, 3H, –SC3); 13C NMR (175 MHz, DMSO-d6): δ 167.0 (C-7), 165.5 (C-7″), 161.0 (C-3′), 157.6 (C-5′), 155.5 (C-2″), 152.7 (C-5″), 140.3 (C-4), 130.8 (C-2/6), 130.2 (C-1), 124.2 (C-4″), 123.5 (C-3/5), 120.5 (C-1″), 118.6 (C-3″), 111.8 (C-6″), 55.9  13.9 (–SH3); HRMS (EI): m/z calcd for C18H16N4O4S (M)˙+ 384.0892, found 384.0912.), 10.31 (br s, 1H, –O), 9.55 (s, 1H, H-7″), 8.75 (d, J = 2.6 Hz, 1H, H-6″), 8.37 (dd, J = 9.1, 2.6 Hz, 1H, H-4″), 8.05 (d, J = 8.5 Hz, 2H, H-2/6), 7.68 (d, J = 8.4 Hz, 2H, H-3/5), 7.19 (d, J = 9.1 Hz, 1H, H-3″), 2.65 (s, 3H, –SC3); 13C NMR (175 MHz, DMSO-d6): δ 167.1 (C-7), 166.1 (C-7″), 161.2 (C-3′), 157.7 (C-5′), 156.0 (C-2″), 140.5 (C-4), 140.2 (C-5″), 131.1 (C-2/6), 130.5 (C-1), 122.7 (C-4″), 121.9 (C-3/5), 121.5 (C-1″), 118.9 (C-3″), 118.3 (C-6″), 13.8 (–SH3); HRMS (EI): m/z calcd for C17H13N5O5S (M)˙+ 399.0637, found 399.0686.), 10.22 (br s, 1H, –O), 9.49 (s, 1H, H-7″), 8.12 (d, J = 8.6 Hz, 2H, H-2/6), 7.93 (d, J = 8.6 Hz, 2H, H-3/5), 7.72 (d, J = 2.6 Hz, 1H, H-6″), 7.49 (dd, J = 8.4, 2.6 Hz, 1H, H-4″), 6.95 (d, J = 8.4 Hz, 1H, H-3″), 2.64 (s, 3H, –SC3); 13C NMR (175 MHz, DMSO-d6): δ 167.0 (C-7), 164.2 (C-7″), 161.1 (C-3′), 159.9 (C-2″), 157.4 (C-5′), 140.1 (C-4), 134.6 (C-4″), 131.1 (C-6″), 130.8 (C-2/6), 130.3 (C-1), 123.7 (C-3/5), 122.0 (C-1″), 119.2 (C-3″), 111.2 (C-5″), 13.9 (–SH3); HRMS (EI): m/z calcd for C17H13BrN4O3S (M)˙+431.9892, found 431.9924.), 9.77 (br s, 1H, –O), 9.17 (s, 1H, H-7″), 8.13 (d, J = 8.7 Hz, 2H, H-2/6), 8.04 (d, J = 8.7 Hz, 2H, H-3/5), 7.63 (d, J = 1.8 Hz, 1H, H-2″), 7.60 (dd, J = 8.1, 1.8 Hz, 1H, H-6″), 6.97 (d, J = 8.1 Hz, 1H, H-5″), 3.87 (s, 3H, –OC3), 2.63 (s, 3H, –SC3); 13C NMR (175 MHz, DMSO-d6): δ 167.8 (C-7), 160.7 (C-7″), 159.3 (C-3′), 155.9 (C-5′), 153.3 (C-4″), 148.6 (C-3″), 140.6 (C-4), 131.1 (C-2/6), 129.6 (C-1″), 129.2 (C-1), 126.7 (C-6″), 122.8 (C-3/5), 113.1 (C-2″), 56.0
13.9 (–SH3); HRMS (EI): m/z calcd for C18H16N4O4S (M)˙+ 384.0892, found 384.0912.), 10.31 (br s, 1H, –O), 9.55 (s, 1H, H-7″), 8.75 (d, J = 2.6 Hz, 1H, H-6″), 8.37 (dd, J = 9.1, 2.6 Hz, 1H, H-4″), 8.05 (d, J = 8.5 Hz, 2H, H-2/6), 7.68 (d, J = 8.4 Hz, 2H, H-3/5), 7.19 (d, J = 9.1 Hz, 1H, H-3″), 2.65 (s, 3H, –SC3); 13C NMR (175 MHz, DMSO-d6): δ 167.1 (C-7), 166.1 (C-7″), 161.2 (C-3′), 157.7 (C-5′), 156.0 (C-2″), 140.5 (C-4), 140.2 (C-5″), 131.1 (C-2/6), 130.5 (C-1), 122.7 (C-4″), 121.9 (C-3/5), 121.5 (C-1″), 118.9 (C-3″), 118.3 (C-6″), 13.8 (–SH3); HRMS (EI): m/z calcd for C17H13N5O5S (M)˙+ 399.0637, found 399.0686.), 10.22 (br s, 1H, –O), 9.49 (s, 1H, H-7″), 8.12 (d, J = 8.6 Hz, 2H, H-2/6), 7.93 (d, J = 8.6 Hz, 2H, H-3/5), 7.72 (d, J = 2.6 Hz, 1H, H-6″), 7.49 (dd, J = 8.4, 2.6 Hz, 1H, H-4″), 6.95 (d, J = 8.4 Hz, 1H, H-3″), 2.64 (s, 3H, –SC3); 13C NMR (175 MHz, DMSO-d6): δ 167.0 (C-7), 164.2 (C-7″), 161.1 (C-3′), 159.9 (C-2″), 157.4 (C-5′), 140.1 (C-4), 134.6 (C-4″), 131.1 (C-6″), 130.8 (C-2/6), 130.3 (C-1), 123.7 (C-3/5), 122.0 (C-1″), 119.2 (C-3″), 111.2 (C-5″), 13.9 (–SH3); HRMS (EI): m/z calcd for C17H13BrN4O3S (M)˙+431.9892, found 431.9924.), 9.77 (br s, 1H, –O), 9.17 (s, 1H, H-7″), 8.13 (d, J = 8.7 Hz, 2H, H-2/6), 8.04 (d, J = 8.7 Hz, 2H, H-3/5), 7.63 (d, J = 1.8 Hz, 1H, H-2″), 7.60 (dd, J = 8.1, 1.8 Hz, 1H, H-6″), 6.97 (d, J = 8.1 Hz, 1H, H-5″), 3.87 (s, 3H, –OC3), 2.63 (s, 3H, –SC3); 13C NMR (175 MHz, DMSO-d6): δ 167.8 (C-7), 160.7 (C-7″), 159.3 (C-3′), 155.9 (C-5′), 153.3 (C-4″), 148.6 (C-3″), 140.6 (C-4), 131.1 (C-2/6), 129.6 (C-1″), 129.2 (C-1), 126.7 (C-6″), 122.8 (C-3/5), 113.1 (C-2″), 56.0  13.9 (–SH3); HRMS (EI): m/z calcd for C18H16N4O4S (M)˙+ 384.0892, found 384.0923.), 9.92 (br s, 1H, –O), 9.24 (s, 1H, H-7″), 8.14 (d, J = 8.3 Hz, 2H, H-2/6), 7.99 (d, J = 8.3 Hz, 2H, H-3/5), 7.51 (br d, J = 7.5 Hz, 1H, H-6″), 7.47 (br s, 1H, H-2″), 7.40 (t-like, J = 7.9 Hz, 1H, H-5″), 7.21 (br d, J = 7.5 Hz, 1H, H-4″), 2.65 (s, 3H, –SC3); 13C NMR (175 MHz, DMSO-d6): δ 167.1 (C-7), 160.9 (C-7″), 159.3 (C-3′), 158.3 (C-3″), 155.9 (C-5′), 140.9 (C-4), 136.3 (C-1″), 130.8 (C-2/6), 130.0 (C-1), 123.4 (C-3/5), 122.9 (C-6″), 121.6 (C-4″), 115.4 (C-2″), 13.8 (–SH3); HRMS (EI): m/z calcd for C17H14N4O3S (M)˙+ 354.0787, found 354.0819.), 9.19 (s, 1H, H-7″), 8.13 (d, J = 8.6 Hz, 2H, H-2/6), 8.03 (d, J = 8.6 Hz, 2H, H-3/5), 7.67 (d, J = 8.5 Hz, 2H, H-2″/6″), 6.95 (d, J = 8.5 Hz, 2H, H-3″/5″), 2.63 (s, 3H, –SC3); 13C NMR (175 MHz, DMSO-d6): δ 167.8 (C-7), 163.5 (C-4″), 160.7 (C-7″), 159.3 (C-3′), 155.9 (C-5′), 140.9 (C-4), 133.4 (C-1″), 131.1 (C-2/6), 130.8 (C-2″/6″), 129.7 (C-1), 122.9 (C-3/5), 116.7 (C-3″/5″), 13.9 (–SH3); HRMS (EI): m/z calcd for C17H14N4O3S (M)˙+ 354.0787, found 354.0819.), 10.23 (br s, 1H, –O), 9.42 (s, 1H, H-7″), 8.12 (d, J = 8.6 Hz, 2H, H-2/6), 7.96 (d, J = 8.6 Hz, 2H, H-3/5), 7.72 (br s, 1H, H-6‴), 7.40 (br t, J = 7.4 Hz, 2H, H-3″/5″), 7.34 (br d, J = 8.4 Hz, 1H, H-4‴), 7.26 (br d, J = 7.8 Hz, 2H, H-2″/6″), 7.19 (br t, J = 7.5 Hz, 1H, H-4″), 6.91 (d, J = 8.4 Hz, 1H, H-3‴), 2.25 (s, 3H, –ArC3); 13C NMR (175 MHz, DMSO-d6): δ 167.1 (C-7), 166.9 (C-7‴), 164.8 (C-3′), 159.2 (C-2‴), 154.9 (C-5′/1″), 141.0 (C-4), 137.7 (C-4‴), 131.1 (C-2/6), 130.3 (C-5‴), 130.1 (C-3″/5″), 129.2 (C-1), 128.7 (C-6‴), 124.8 (C-4″), 121.9 (C-3/5), 119.8 (C-2″/6″), 119.7 (C-1‴), 117.3 (C-3‴), 20.3
13.9 (–SH3); HRMS (EI): m/z calcd for C18H16N4O4S (M)˙+ 384.0892, found 384.0923.), 9.92 (br s, 1H, –O), 9.24 (s, 1H, H-7″), 8.14 (d, J = 8.3 Hz, 2H, H-2/6), 7.99 (d, J = 8.3 Hz, 2H, H-3/5), 7.51 (br d, J = 7.5 Hz, 1H, H-6″), 7.47 (br s, 1H, H-2″), 7.40 (t-like, J = 7.9 Hz, 1H, H-5″), 7.21 (br d, J = 7.5 Hz, 1H, H-4″), 2.65 (s, 3H, –SC3); 13C NMR (175 MHz, DMSO-d6): δ 167.1 (C-7), 160.9 (C-7″), 159.3 (C-3′), 158.3 (C-3″), 155.9 (C-5′), 140.9 (C-4), 136.3 (C-1″), 130.8 (C-2/6), 130.0 (C-1), 123.4 (C-3/5), 122.9 (C-6″), 121.6 (C-4″), 115.4 (C-2″), 13.8 (–SH3); HRMS (EI): m/z calcd for C17H14N4O3S (M)˙+ 354.0787, found 354.0819.), 9.19 (s, 1H, H-7″), 8.13 (d, J = 8.6 Hz, 2H, H-2/6), 8.03 (d, J = 8.6 Hz, 2H, H-3/5), 7.67 (d, J = 8.5 Hz, 2H, H-2″/6″), 6.95 (d, J = 8.5 Hz, 2H, H-3″/5″), 2.63 (s, 3H, –SC3); 13C NMR (175 MHz, DMSO-d6): δ 167.8 (C-7), 163.5 (C-4″), 160.7 (C-7″), 159.3 (C-3′), 155.9 (C-5′), 140.9 (C-4), 133.4 (C-1″), 131.1 (C-2/6), 130.8 (C-2″/6″), 129.7 (C-1), 122.9 (C-3/5), 116.7 (C-3″/5″), 13.9 (–SH3); HRMS (EI): m/z calcd for C17H14N4O3S (M)˙+ 354.0787, found 354.0819.), 10.23 (br s, 1H, –O), 9.42 (s, 1H, H-7″), 8.12 (d, J = 8.6 Hz, 2H, H-2/6), 7.96 (d, J = 8.6 Hz, 2H, H-3/5), 7.72 (br s, 1H, H-6‴), 7.40 (br t, J = 7.4 Hz, 2H, H-3″/5″), 7.34 (br d, J = 8.4 Hz, 1H, H-4‴), 7.26 (br d, J = 7.8 Hz, 2H, H-2″/6″), 7.19 (br t, J = 7.5 Hz, 1H, H-4″), 6.91 (d, J = 8.4 Hz, 1H, H-3‴), 2.25 (s, 3H, –ArC3); 13C NMR (175 MHz, DMSO-d6): δ 167.1 (C-7), 166.9 (C-7‴), 164.8 (C-3′), 159.2 (C-2‴), 154.9 (C-5′/1″), 141.0 (C-4), 137.7 (C-4‴), 131.1 (C-2/6), 130.3 (C-5‴), 130.1 (C-3″/5″), 129.2 (C-1), 128.7 (C-6‴), 124.8 (C-4″), 121.9 (C-3/5), 119.8 (C-2″/6″), 119.7 (C-1‴), 117.3 (C-3‴), 20.3 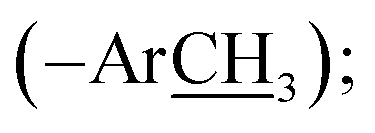 HRMS (EI): m/z calcd for C23H18N4O4 (M)˙+ 414.1328, found 414.1357.), 10.26 (br s, 1H, –O), 9.42 (s, 1H, H-7‴), 8.13 (d, J = 8.4 Hz, 2H, H-2/6), 7.96 (d, J = 8.4 Hz, 2H, H-3/5), 7.43 (br s, 1H, H-6‴), 7.41 (br t, J = 7.6 Hz, 2H, H-3″/5″), 7.26 (br d, J = 7.7 Hz, 2H, H-2″/6″), 7.15 (br d, 1H, H-4‴), 7.19 (br t, J = 7.3 Hz, 1H, H-4″), 6.95 (d, J = 8.8 Hz, 1H, H-3‴), 3.73 (s, 3H, –OC3); 13C NMR (175 MHz, DMSO-d6): δ 167.1 (C-7), 164.8 (C-7‴), 161.0 (C-3′), 155.8 (C-2‴), 154.9 (C-5′/1″), 152.6 (C-5‴), 141.0 (C-4), 131.1 (C-2/6), 130.1 (C-3″/5″), 128.7 (C-1), 124.8 (C-4″/4‴), 121.9 (C-3/5), 119.8 (C-2″/6″), 119.7 (C-1‴), 119.1 (C-3‴), 110.5 (C-6‴), 55.9
HRMS (EI): m/z calcd for C23H18N4O4 (M)˙+ 414.1328, found 414.1357.), 10.26 (br s, 1H, –O), 9.42 (s, 1H, H-7‴), 8.13 (d, J = 8.4 Hz, 2H, H-2/6), 7.96 (d, J = 8.4 Hz, 2H, H-3/5), 7.43 (br s, 1H, H-6‴), 7.41 (br t, J = 7.6 Hz, 2H, H-3″/5″), 7.26 (br d, J = 7.7 Hz, 2H, H-2″/6″), 7.15 (br d, 1H, H-4‴), 7.19 (br t, J = 7.3 Hz, 1H, H-4″), 6.95 (d, J = 8.8 Hz, 1H, H-3‴), 3.73 (s, 3H, –OC3); 13C NMR (175 MHz, DMSO-d6): δ 167.1 (C-7), 164.8 (C-7‴), 161.0 (C-3′), 155.8 (C-2‴), 154.9 (C-5′/1″), 152.6 (C-5‴), 141.0 (C-4), 131.1 (C-2/6), 130.1 (C-3″/5″), 128.7 (C-1), 124.8 (C-4″/4‴), 121.9 (C-3/5), 119.8 (C-2″/6″), 119.7 (C-1‴), 119.1 (C-3‴), 110.5 (C-6‴), 55.9  HRMS (EI): m/z calcd for C23H18N4O5 (M)˙+ 430.1277, found 430.1304.), 10.31 (br s, 1H, –O), 9.55 (s, 1H, H-7‴), 8.75 (d, J = 2.6 Hz, 1H, H-6‴), 8.37 (dd, J = 9.1, 2.6 Hz, 1H, H-4‴), 8.05 (d, J = 8.5 Hz, 2H, H-2/6), 7.68 (d, J = 8.4 Hz, 2H, H-3/5), 7.41 (br t, J = 7.6 Hz, 2H, H-3″/5″), 7.26 (br d, J = 7.7 Hz, 2H, H-2″/6″), 7.19 (br t, J = 7.3 Hz, 1H, H-4″), 7.19 (d, J = 9.1 Hz, 1H, H-3‴); 13C NMR (175 MHz, DMSO-d6): δ 167.1 (C-7), 166.1 (C-7‴), 161.2 (C-3′), 157.7 (C-5′), 156.0 (C-2‴), 154.9 (C-1″), 140.5 (C-4), 140.2 (C-5‴), 131.1 (C-2/6), 130.1 (C-3″/5″), 130.5 (C-1), 124.8 (C-4″), 122.7 (C-4‴), 121.9 (C-3/5), 121.5 (C-1‴), 119.8 (C-2″/6″), 118.9 (C-3‴), 118.3 (C-6‴); HRMS (EI): m/z calcd for C22H15N5O6 (M)˙+ 445.1022, found 445.1059.), 9.63 (br s, 1H, –NH–), 8.05 (d, J = 8.6 Hz, 2H, H-2/6), 7.68 (d, J = 8.6 Hz, 2H, H-3/5), 7.33 (br t, J = 7.5 Hz, 2H, H-3″/5″), 7.28 (br d, J = 7.3 Hz, 2H, H-2″/6″), 7.24 (br t, J = 7.5 Hz, 1H, H-4″), 4.95 (s, 2H, H-7″), 2.49 (s, 3H, –SC3); 13C NMR (175 MHz, DMSO-d6): δ 188.2 (
HRMS (EI): m/z calcd for C23H18N4O5 (M)˙+ 430.1277, found 430.1304.), 10.31 (br s, 1H, –O), 9.55 (s, 1H, H-7‴), 8.75 (d, J = 2.6 Hz, 1H, H-6‴), 8.37 (dd, J = 9.1, 2.6 Hz, 1H, H-4‴), 8.05 (d, J = 8.5 Hz, 2H, H-2/6), 7.68 (d, J = 8.4 Hz, 2H, H-3/5), 7.41 (br t, J = 7.6 Hz, 2H, H-3″/5″), 7.26 (br d, J = 7.7 Hz, 2H, H-2″/6″), 7.19 (br t, J = 7.3 Hz, 1H, H-4″), 7.19 (d, J = 9.1 Hz, 1H, H-3‴); 13C NMR (175 MHz, DMSO-d6): δ 167.1 (C-7), 166.1 (C-7‴), 161.2 (C-3′), 157.7 (C-5′), 156.0 (C-2‴), 154.9 (C-1″), 140.5 (C-4), 140.2 (C-5‴), 131.1 (C-2/6), 130.1 (C-3″/5″), 130.5 (C-1), 124.8 (C-4″), 122.7 (C-4‴), 121.9 (C-3/5), 121.5 (C-1‴), 119.8 (C-2″/6″), 118.9 (C-3‴), 118.3 (C-6‴); HRMS (EI): m/z calcd for C22H15N5O6 (M)˙+ 445.1022, found 445.1059.), 9.63 (br s, 1H, –NH–), 8.05 (d, J = 8.6 Hz, 2H, H-2/6), 7.68 (d, J = 8.6 Hz, 2H, H-3/5), 7.33 (br t, J = 7.5 Hz, 2H, H-3″/5″), 7.28 (br d, J = 7.3 Hz, 2H, H-2″/6″), 7.24 (br t, J = 7.5 Hz, 1H, H-4″), 4.95 (s, 2H, H-7″), 2.49 (s, 3H, –SC3); 13C NMR (175 MHz, DMSO-d6): δ 188.2 (![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S), 167.1 (C-7), 159.4 (C-3′), 155.9 (C-5′), 140.9 (C-4), 138.6 (C-1″), 131.1 (C-2/6), 129.3 (C-3″/5″), 128.7 (C-2″/6″), 128.6 (C-1), 127.8 (C-4″), 121.9 (C-3/5), 48.5 (C-7″), 13.9 (–SH3); HRMS (EI): m/z calcd for C18H17N5O2S2 (M)˙+ 399.0824, found 399.0857.), 8.05 (d, J = 8.5 Hz, 2H, H-2/6), 7.68 (d, J = 8.6 Hz, 2H, H-3/5), 7.34 (br t, J = 7.5 Hz, 2H, H-3″/5″), 7.29 (br d, J = 7.3 Hz, 2H, H-2″/6″), 7.27 (br t, J = 7.5 Hz, 1H, H-4″), 3.92 (t, J = 6.9 Hz, 2H, H-8″), 2.96 (t, J = 6.9 Hz, 2H, H-7″), 2.49 (s, 3H, –SC3); 13C NMR (175 MHz, DMSO-d6): δ 189.9 (CS), 167.8 (C-7), 159.4 (C-3′), 155.9 (C-5′), 140.9 (C-4), 138.1 (C-1″), 131.1 (C-2/6), 129.4 (C-3″/5″), 128.9 (C-2″/6″), 128.8 (C-1), 127.3 (C-4″), 122.0 (C-3/5), 46.4 (C-8″), 35.8 (C-7″), 13.8 (–SH3); HRMS (EI): m/z calcd for C19H19N5O2S2 (M)˙+ 413.0980, found 413.1028.), 9.64 (br s, 1H, –NH–), 8.03 (d, J = 7.9 Hz, 2H, H-2/6), 7.66 (d, J = 7.9 Hz, 2H, H-3/5), 7.41 (br t, J = 7.6 Hz, 2H, H-3″/5″), 7.34 (br t, J = 7.5 Hz, 2H, H-3‴/5‴), 7.29 (br d, J = 7.5 Hz, 2H, H-2‴/6‴), 7.26 (m, 3H, H-2″/6″,4‴), 7.18 (br t, J = 7.7 Hz, 1H, H-4″), 4.95 (s, 2H, H-7‴); 13C NMR (175 MHz, DMSO-d6): δ 188.0 (CS), 167.1 (C-7), 164.8 (C-3′), 154.9 (C-5′,1″), 141.0 (C-4), 138.5 (C-1‴), 131.1 (C-2/6), 130.1 (C-3″/5″), 129.4 (C-3‴/5‴), 128.7 (C-1), 128.6 (C-2‴/6‴), 127.8 (C-4‴), 124.8 (C-4″), 121.9 (C-3/5), 119.8 (C-2″/6″), 48.5 (C-7‴); HRMS (EI): m/z calcd for C23H19N5O3S (M)˙+ 445.1209, found 445.1243.
S), 167.1 (C-7), 159.4 (C-3′), 155.9 (C-5′), 140.9 (C-4), 138.6 (C-1″), 131.1 (C-2/6), 129.3 (C-3″/5″), 128.7 (C-2″/6″), 128.6 (C-1), 127.8 (C-4″), 121.9 (C-3/5), 48.5 (C-7″), 13.9 (–SH3); HRMS (EI): m/z calcd for C18H17N5O2S2 (M)˙+ 399.0824, found 399.0857.), 8.05 (d, J = 8.5 Hz, 2H, H-2/6), 7.68 (d, J = 8.6 Hz, 2H, H-3/5), 7.34 (br t, J = 7.5 Hz, 2H, H-3″/5″), 7.29 (br d, J = 7.3 Hz, 2H, H-2″/6″), 7.27 (br t, J = 7.5 Hz, 1H, H-4″), 3.92 (t, J = 6.9 Hz, 2H, H-8″), 2.96 (t, J = 6.9 Hz, 2H, H-7″), 2.49 (s, 3H, –SC3); 13C NMR (175 MHz, DMSO-d6): δ 189.9 (CS), 167.8 (C-7), 159.4 (C-3′), 155.9 (C-5′), 140.9 (C-4), 138.1 (C-1″), 131.1 (C-2/6), 129.4 (C-3″/5″), 128.9 (C-2″/6″), 128.8 (C-1), 127.3 (C-4″), 122.0 (C-3/5), 46.4 (C-8″), 35.8 (C-7″), 13.8 (–SH3); HRMS (EI): m/z calcd for C19H19N5O2S2 (M)˙+ 413.0980, found 413.1028.), 9.64 (br s, 1H, –NH–), 8.03 (d, J = 7.9 Hz, 2H, H-2/6), 7.66 (d, J = 7.9 Hz, 2H, H-3/5), 7.41 (br t, J = 7.6 Hz, 2H, H-3″/5″), 7.34 (br t, J = 7.5 Hz, 2H, H-3‴/5‴), 7.29 (br d, J = 7.5 Hz, 2H, H-2‴/6‴), 7.26 (m, 3H, H-2″/6″,4‴), 7.18 (br t, J = 7.7 Hz, 1H, H-4″), 4.95 (s, 2H, H-7‴); 13C NMR (175 MHz, DMSO-d6): δ 188.0 (CS), 167.1 (C-7), 164.8 (C-3′), 154.9 (C-5′,1″), 141.0 (C-4), 138.5 (C-1‴), 131.1 (C-2/6), 130.1 (C-3″/5″), 129.4 (C-3‴/5‴), 128.7 (C-1), 128.6 (C-2‴/6‴), 127.8 (C-4‴), 124.8 (C-4″), 121.9 (C-3/5), 119.8 (C-2″/6″), 48.5 (C-7‴); HRMS (EI): m/z calcd for C23H19N5O3S (M)˙+ 445.1209, found 445.1243.General procedure for the preparation of methyl 4-(5-amino-3-(methylthio)-1H-1,2,4-triazol-1-yl)benzoate (16)
A mixture of compound 1 (1 mmol), methanol (10 mL), and concentrated H2SO4 (0.4 mL) was heated under reflux for 5 h. The mixture was then cooled to room temperature, concentrated under reduced pressure, and treated with equal volumes (20 mL) of water and CH2Cl2. The aqueous phase was further extracted with CH2Cl2, which was combined with the previous CH2Cl2 layer. The organic phase was washed with aqueous Na2CO3 (20 mL) and water (20 mL), dried over Na2SO4, and evaporated to obtain 16 as a pale yellow amorphous powder. Yield (42%); mp 150–152 °C; 1H NMR (700 MHz, DMSO-d6): δ 8.06 (d, J = 8.7 Hz, 2H, H-2/6), 7.71 (d, J = 8.7 Hz, 2H, H-3/5), 6.82 (br s, 2H, –N2), 3.88 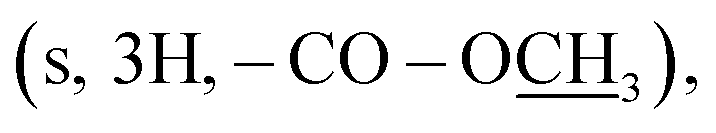 2.49 (s, 3H, –SC3); 13C NMR (175 MHz, DMSO-d6): δ 166.0 (C-7), 159.5 (C-3′), 155.9 (C-5′), 141.3 (C-4), 130.9 (C-2/6), 127.5 (C-1), 121.9 (C-3/5), 52.8
2.49 (s, 3H, –SC3); 13C NMR (175 MHz, DMSO-d6): δ 166.0 (C-7), 159.5 (C-3′), 155.9 (C-5′), 141.3 (C-4), 130.9 (C-2/6), 127.5 (C-1), 121.9 (C-3/5), 52.8 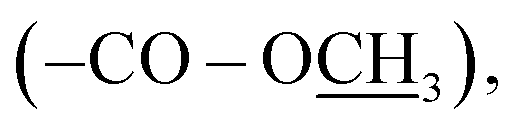 13.8 (–SH3); HRMS (EI): m/z calcd for C11H12N4O2S (M)˙+ 264.0681, found 264.0727.
13.8 (–SH3); HRMS (EI): m/z calcd for C11H12N4O2S (M)˙+ 264.0681, found 264.0727.
General procedure for the preparation of 4-(5-amino-3-(methylsulfonyl)-1H-1,2,4-triazol-1-yl) benzoic acid (17)
Compound 1 (1 mmol) was dissolved in boiling ethanol (10 mL). H2O2 (2 mL) was then added dropwise over 5 min, followed by further heating of the mixture for 3 h. The solvent was evaporated, and the remaining residue was treated with hot water and left to stand at room temperature. The resulting solid was filtered, washed with water, and dried.White amorphous powder, yield (69%); mp 249–251 °C; 1H NMR (700 MHz, DMSO-d6): δ 13.11 (br s, 1H, –COO), 8.11 (d, J = 8.5 Hz, 2H, H-2/6), 7.72 (d, J = 8.5 Hz, 2H, H-3/5), 7.12 (br s, 2H, –N2), 2.94 (s, 3H, –SO2C3); 13C NMR (175 MHz, DMSO-d6): δ 167.0 (C-7), 164.1 (C-3′), 156.6 (C-5′), 140.3 (C-4), 131.2 (C-2/6), 130.2 (C-1), 123.3 (C-3/5), 38.7 (–SO2H3); HRMS (EI): m/z calcd for C10H10N4O4S (M)˙+ 282.0432, found 282.0472.
Biology
Conflicts of interest
The authors declare that they have no competing interests.Note added after first publication
This article replaces the version published on 17 Jun 2019, which contained errors in Fig. 4. The correct version of this figure is now shown.Acknowledgements
The authors wish to thank the Deanship of Scientific Research at King Saud University for funding this work through the research group No. RG-1435-068.References
- R. Al-Salahi, I. Alswaidan and M. Marzouk, Int. J. Mol. Sci., 2014, 15, 22483–22491 CrossRef CAS PubMed
.
- H. A. Abuelizz, M. Marzouk, H. Ghabbour and R. Al-Salahi, Saudi Pharm. J., 2017, 25, 1047–1054 CrossRef PubMed
- R. Al-Salahi, R. A. El Dib and M. Marzouk, Heterocycles, 2015, 91, 1735–1751 CrossRef CAS
- H. A. M. El-Sherief, B. G. M. Youssif, S. N. Abbas-Bukhari, A. H. Abdelazeem, M. Abdel-Aziz and H. M. Abdel-Rahman, Eur. J. Med. Chem., 2018, 156, 774–789 CrossRef CAS PubMed
- R. R. Ruddarraju, A. C. Murugulla, R. Kotla, T. M. Chandra Babu, R. Wudayagiri, S. Donthabakthuni, R. Maroju, K. Baburao and L. S. Parasa, Eur. J. Med. Chem., 2016, 123, 379–396 CrossRef CAS PubMed
- R. Pingaew, V. Prachayasittikul, P. Mandi, C. Nantasenamat, S. Prachayasittikul, S. Ruchirawat and V. Prachayasittikul, Bioorg. Med. Chem., 2015, 23, 3472–3480 CrossRef CAS PubMed
- W. Hou, Z. Luo, G. Zhang, D. Cao, D. Li, H. Ruan, B. H. Ruan, L. Su and H. Xu, Eur. J. Med. Chem., 2017, 138, 1042–1052 CrossRef CAS PubMed
- T. G. Kraljević, A. Harej, M. Sedić, S. K. Pavelić, V. Stepanić, D. Drenjančević, J. Talapko and S. Raić-Malić, Eur. J. Med. Chem., 2016, 124, 794–808 CrossRef PubMed
- M. Huang, Z. Deng, J. Tian and T. Liu, Eur. J. Med. Chem., 2017, 127, 900–908 CrossRef CAS PubMed
- L. Y. Ma, L. P. Pang, B. Wang, M. Zhang, B. Hu, D. Q. Xue, K. P. Shao, B. L. Zhang, Y. Liu, E. Zhang and H. M. Liu, Eur. J. Med. Chem., 2014, 86, 368–380 CrossRef CAS PubMed
- H. N. Li, H. Wang, Z. P. Wang, H. N. Yan, M. Zhang, Y. Liu and M. S. Cheng, Bioorg. Med. Chem., 2018, 26, 4025–4033 CrossRef CAS PubMed
- L. Zhao, L. Mao, G. Hong, X. Yang and T. Liu, Bioorg. Med. Chem. Lett., 2015, 25, 2540–2544 CrossRef CAS PubMed
- S. Narsimha, K. N. Satheesh, S. B. Kumara, R. M. Vasudeva, H. S. K. Althaf and R. M. Srinivasa, Bioorg. Med. Chem. Lett., 2016, 26, 1639–1644 CrossRef CAS PubMed
- P. V. Chavan, U. V. Desai, P. P. Wadgaonkar, S. R. Tapase, K. M. A. Choudhari and D. Sarkar, Bioorg. Chem., 2019, 85, 475–486 CrossRef CAS PubMed
- R. Al-Salahi, M. Marzouk, A. Gamal Eldeen, A. Alanazi, M. Al-Omar and M. Fouda, J. Pure Appl. Microbiol., 2013, 7, 189–198 CAS
- R. Al-Salahi, A. E. Elsayed, R. E. M. Wadaan, E. Ezzeldin and M. Marzouk, Lat. Am. J. Pharm., 2016, 35, 58–65 CAS
- R. Al-Salahi, A. E. Elsayed, R. E. M. Wadaan, E. Ezzeldin and M. Marzouk, Lat. Am. J. Pharm., 2016, 35, 66–73 CAS
- R. Al-Salahi, M. Marzouk, A. Ashour and I. Alswiadan, Asian J. Chem., 2014, 26, 2173–2176 CrossRef CAS
- R. Al-Salahi, R. Ahmad, E. Anouar, N. N. I. Iwana, M. Marzouk and H. A. Abuelizz, Future Med. Chem., 2018, 10, 1889–1905 CrossRef CAS PubMed
- H. Abuelizz, R. Al-Salahi, J. Al-Asri, J. Mortier, M. Marzouk, E. Ezzeldin, A. A. Ali, M. G. Khalil, G. Wolber, H. A. Ghabbour, A. A. Almehizia and G. A. Abdel Jaleel, Chem. Cent. J., 2017, 11, 103 CrossRef PubMed
- R. Al-Salahi, K. E. Tahir, N. Lolak, M. Hamidaddin, I. Alswaidan and M. Marzouk, Chem. Cent. J., 2014, 8, 3 CrossRef PubMed
- R. Al-Salahi, I. Alswaidan, M. Al-Omar and M. Marzouk, Life Sci. J., 2013, 10, 2018–2028 Search PubMed
- R. Al-Salahi and D. Geffken, Synth. Commun., 2011, 41, 3512–3523 CrossRef CAS
- R. Al-Salahi and D. Geffken, Molecules, 2010, 15, 7016–7034 CrossRef CAS
- A. S. Hassan, M. F. Mady, H. M. Awad and T. S. Hafez, Chin. Chem. Lett., 2017, 28, 388–393 CrossRef CAS
- A. N. Emam, S. A. Loutfy, A. A. Mostafa, H. M. Awad and M. B. Mohamed, RSC Adv., 2017, 7, 23502–23514 RSC
- E. M. Flefel, W. A. El-Sayed, A. M. Mohamed, W. I. El-Sofany and H. M. Awad, Molecules, 2017, 22(170), 1–13 Search PubMed
- R. Gerl and D. L. Vaux, Carcinogenesis, 2005, 2, 263–270 Search PubMed
- M. Khan, A. Maryam, J. I. Qazi and T. Ma, Int. J. Biol. Sci., 2015, 16, 1100–1112 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03151k |
| This journal is © The Royal Society of Chemistry 2019 |