
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal–organic framework-derived CeO2–ZnO catalysts for C3H6-SCR of NO: an in situ DRIFTS study
Ling Zhao *ab,
Yu Zhanga,
Sining Bia and
Qifeng Liu*a
*ab,
Yu Zhanga,
Sining Bia and
Qifeng Liu*a
aSchool of Ecology and Environment, Inner Mongolia University, China. E-mail: nmzhl@hotmail.com
bCenter for Environmental and Human Toxicology, Department of Physiological Sciences, College of Veterinary Medicine, University of Florida, USA
First published on 18th June 2019
Abstract
Metal–organic framework (MOF)-based derivatives have attracted an increasing interest in various research fields. Here, we synthesized CeO2–ZnO catalysts through the complete thermal decomposition of the Ce/MOF-5 precursor. The catalysts were characterized using XRD, FTIR, TG-DSC, SEM and H2-TPR. It is found that the as-prepared CeO2–ZnO is favorable for strengthening the interaction between Ce4+ and Zn2+. A significant improvement in the catalytic performance for C3H6-SCR of NO was found over the Ce-doped catalysts with the highest N2 yield of 69.1% achieved over 5% CeO2–ZnO. In situ DRIFTS and NO-TPD experiments demonstrated the formation of monodentate nitrates, bidentate nitrates, chelating nitrite, nitro compounds, nitrosyl and CxHyOz species (enolic species and acetate) on the surface, followed by the formation of hydrocarbonate or carbonate as intermediates to directly generate N2, CO2 and H2O.
1. Introduction
It is particularly notorious that the sheer quantity of gasoline and diesel powered vehicles grows with each passing day. The nitrogen oxides (NOx) from automobile exhaust gases have aroused significant concern among the public and government officials because these gases can cause a great threat to the environment and public health, including the greenhouse effect, acidification, photochemical smog, and ozone depletion.1,2 Owing to the ground-breaking work of Iwamoto et al., hydrocarbon-selective catalytic reduction (HC-SCR) has become a promising technology for clearing NOx away from the automobile exhausts, taking into account the cost and gas components, as well as the hydrocarbons coexisting with NO in the exhaust streams.3,4 Different kinds of hydrocarbons have been employed to improve the performance of HC-SCR, including propene,5 methane,6 ethanol,7 ethylene,8 and propane.9 More importantly, catalysts are the core of this catalytic technology. Thus, huge efforts are dedicated to develop catalysts by both the academic community and catalyst manufacturers.Metal–organic frameworks (MOFs) are a kind of crystalline porous materials with a periodic network structure formed by the self-assembly of inorganic metals (metal ions or clusters) and bridged organic ligands.10–14 On account of their low-density structures, tunable cavities, high surface area, tailorable chemistry and facile synthesis,10,11 MOFs are good candidates for applications in gas separation and storage, as well as catalysis.12–14 Recently, researchers gradually shifted their focus from MOFs to MOF derivatives.15,16 During the calcination process, MOFs are completely decomposed and gradually converted to metal oxides from the outer to inner part. Small cavities and open channels within the MOF-derived metal oxides provide pathways for the reactant species to diffuse in and out, thus enabling these metal oxides to be suitable candidates for catalysts or catalyst supports. By way of illustration, Ce–MOF was used to prepare the CeO2 catalyst for toluene combustion,16 and Cu-based MOFs were applied as the precursors to attain carbon-based catalysts for low-temperature DeNOx.15 Therefore, the development of efficient functional MOF-derived metal oxides for further exploration of their practical applications has great significance.17 More importantly, the composite materials usually exhibit excellent performances for environmental catalytic reactions. For example, Yang et al. prepared different functional composites and studied their outstanding performance of catalytic oxidative desulfurization and atrazine degradation.18,19
Cerium oxide is an extensively used promoter or catalyst support for the SCR reaction due to its superior oxygen storage capability and excellent redox properties.15 The oxygen vacancies are produced during the electron transfer between Ce3+ and Ce4+. The reduction of Ce4+ to Ce3+ and enhanced oxygen transfer could facilitate the oxidation process of NO to NO2, leading to excellent HC-SCR performance.20 In addition, it can not only highly disperse the metals but also strengthen the thermal stability in reaction with other metal oxides.21
In the present study, we synthesized ZnO and CeO2–ZnO catalysts through the complete thermal decomposition of MOF-5 and Ce/MOF-5 precursor, and then compared their activity in HC-SCR. C3H6 was used as the reducing agent due to it being the major hydrocarbon component during the process of automobile exhaust emissions. First, the crystal structure, morphology and textural properties of the samples were investigated via XRD, TG-DSC, SEM, FTIR and H2-TPR. Second, the adsorption and activation abilities of NO were studied using temperature-programmed desorption (TPD). In addition, the in situ DRIFTS experiments were performed to illuminate the C3H6-SCR process as well as the possible reaction pathways.
2. Experimental
2.1. Materials
All of the reactants are of analytical grade and were used without further purification. Zn(NO3)2·6H2O and Ce(NO3)2·6H2O were purchased from Sinopharm Chemical Reagent Co., Ltd. 1,4-Benzenedicarboxylic acid (H2BDC), N,N-dimethylformamide (DMF) and dichloromethane (CH2Cl2) were purchased from Macklin.2.2. Catalyst preparation
MOF-5 was prepared by the solvothermal method according to the following procedures. First, 9.360 g Zn(NO3)2·6H2O and 2.540 g H2BDC were dissolved in 190 ml N,N-dimethylformamide (DMF), and the resulting suspension was sonicated for 30 min. Second, the mixture was transferred into a 250 ml Teflon-lined stainless steel reactor. Then, the autoclave was placed into an oven at 120 °C for 24 h to obtain the white crystals. Subsequently, they were repetitively washed with N,N-dimethylformamide (DMF) and dichloromethane (CH2Cl2). Finally, the homogeneous material obtained was oven-dried at 120 °C for 12 h.An impregnation technique was used to synthesize Ce/MOF-5. Namely, the predetermined amount of cerium nitrate (Ce(NO3)2·6H2O) and MOF-5 precursor were dissolved in ethyl alcohol and the mixed solution was sonicated for 30 min. Afterward, the mixture was left to equilibrate at room temperature for 24 h. Finally, the product was dried at 100 °C for 12 h, followed by calcination at 600 °C for 2 h under air to obtain the CeO2–ZnO catalysts, and were denoted as 1% CeO2–ZnO, 3% CeO2–ZnO, 5% CeO2–ZnO, and 10% CeO2–ZnO.
2.3. Catalysts characterization
In the present study, we adopted a series of methods to evaluate the crystal structure, morphology, thermal stability, reducibility and adsorption properties of the catalysts. XRD patterns were gained on the Empyrean (PANalytical B.V.) using Cu Kα radiation and the X-ray tube was operated under 40 kV and 40 mA. All the as-synthesized samples were scanned over the 2θ range between 5° and 80° at a scan speed of 10° min−1. The surface morphology of all the samples was characterized by SEM (S-4800, Japan). Thermogravimetric and differential scanning calorimetry (TG-DSC) analysis curves were obtained using a STA409pc (NETZSCH German) synchronous thermal analyzer. The samples were heated from 30 °C to 700 °C at a rate of 5 °C min−1 under a flow of nitrogen at 40 ml min−1. Hydrogen temperature-programmed reduction (H2-TPR) and NO temperature-programmed desorption (NO-TPD) were performed on a Chembet PULSAR TPR/TPD chemisorption analyzer loaded with 50 mg samples. Prior to the reduction, the sample was pretreated under pure He atmosphere at 350 °C for 30 min for the sake of removing the absorbent (e.g., H2O). In the case of H2-TPR, the sample began from room temperature to 800 °C under an H2 stream at a rate of 10 °C min−1. For NO-TPD, the sample was treated in a flow of NO at 100 °C for 60 min, followed by heating from 100 °C to 300 °C for desorption at a rate of 10 °C min−1. Fourier transform infrared spectroscopy (FT-IR) was performed on a VERTEX German infrared spectrometer with the KBr pellet technique at room temperature. Spectra were collected in the range of 4000–400 cm−1 at a resolution of 4 cm−1.2.4. Catalytic performance test
The C3H6-SCR catalytic tests were performed in a fixed-bed quartz tube reactor. Prior to an experiment, a catalyst (200 mg) was pretreated at 300 °C for 1 h in an Ar stream, and the activity measurement was carried out from 150 °C to 350 °C at a heating rate of 10 °C min−1. The reaction conditions were as follows: 1000 ppm NO, 1000 ppm C3H6, 5 vol% O2 and Ar as balance; the total gas flow rate was 100 ml min−1 and the GHSV was 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. The concentrations of N2 in the effluent gas were analyzed on-line using a gas chromatograph. The conversion of NO to N2 was calculated as follows:
000 h−1. The concentrations of N2 in the effluent gas were analyzed on-line using a gas chromatograph. The conversion of NO to N2 was calculated as follows:| NO conversion to N2 = 2[N2]out/[NO]in × 100% | (1) |
2.5. In situ DRIFTS measurements
NO and C3H6 adsorption on the catalysts were recorded using a FT-IR spectrometer (VERTEX German). Concentrations of the reactants were 1000 ppm of NO, 1000 ppm of C3H6, 5 vol% oxygen. The sample (20 mg) was mounted in a quartz DRIFTS cell and activated by calcination in Ar stream (30 ml min−1) at 300 °C for 30 min before introducing the reaction mixtures, followed by cooling to the desired temperature, and then the spectrum was collected and used as the background. All spectra were recorded at a resolution of 4 cm−1 (number of scans, 32) in the range of 4000–400 cm−1.3. Results and discussion
3.1. Physical and texture characterizations
From the X-ray diffraction profiles, we can identify the transformation in the phase texture on the MOF-5, MOF-5-derived ZnO and CeO2–ZnO samples. Fig. 1(a) exhibits the previous diffraction peaks for MOF-5; the 2θ angles of 6.85°, 9.62°, 13.69°, 15.39° correspond to the (200), (220), (400), (420) planes.11 A sharp peak appeared at 6.85°, which proved that we successfully synthesized the MOF-5 materials with high crystallinity.11 From the MOF-5-derived composite characterization, it can be observed that the primary structure of the framework collapsed and the characteristic diffraction peaks completely disappeared, with the main peaks of the wurtzite ZnO structure appearing at 31.7° (100), 34.4° (002), 36.3° (101), 47.4° (102), 56.6° (110), 62.8° (103), 67.9° (112) and 69.1° (201) (JCPDS No. 36-1451).22 The sample with Ce loading also displays an additional XRD peak corresponding to the structure of CeO2, indicating that the doped-Ce species exists in the form of CeO2 (Fig. 1(b)).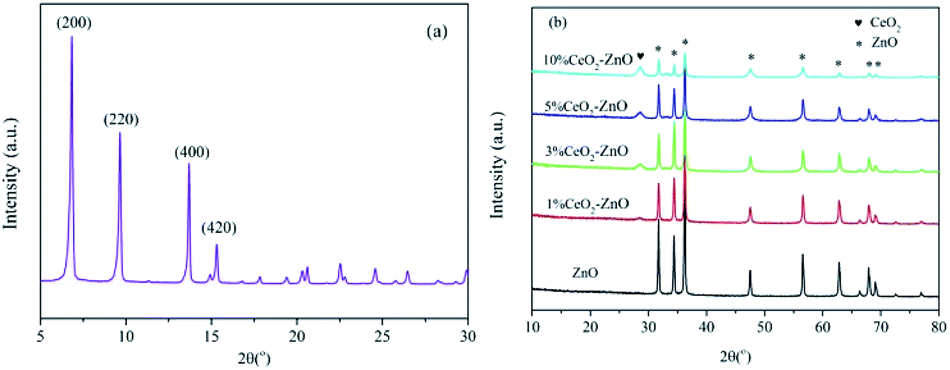 | ||
| Fig. 1 XRD pattern of (a) MOF-5 and (b) ZnO and CeO2–ZnO. | ||
The thermogravimetric and differential scanning calorimetry (TG-DSC) curves of the fresh MOF-5 are shown in Fig. 2. Three noticeable weight loss events were evident. The continuous weight loss of about 4% at the temperature range between 50 °C and 100 °C was caused by the evaporation of water from the surface of the sample. The second major loss in the temperature range of 150–320 °C was about 27%, which was related to the evaporation and decomposition of residual organic components (DMF).23 The most prominent weight loss appeared at a temperature over 420 °C, and the weight loss rapidly reached almost 60%, demonstrating that the MOF-5 framework had collapsed. According to the previous study, for MOF-5, the breakdown of the carboxylic bridges between the benzene rings and Zn4O clusters results in the decomposition of the organic ligand molecules to form CO2 and benzene.11 The DSC curve also shows a sharp exothermic peak between 420 °C and 485 °C with a maximum peak temperature at 480 °C due to the decomposition of the organic components. The decomposition process was completed after 550 °C. The evidence from the thermogravimetric analysis verified that the MOF-5 crystal transformed into ZnO completely above 550 °C. In view of these results, we chose 600 °C as the calcination temperature to attain the oxides.
 | ||
| Fig. 2 TG profile for the MOF-5 precursor. | ||
The FTIR spectra of the as-prepared MOF-5-derived ZnO and CeO2–ZnO catalysts are shown in Fig. 3. A significant peak at 430 cm−1 may be attributed to the Zn–O stretching vibrations.23 The band at 1620 cm−1 is ascribed to the bond stretching of the carboxyl (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–OH) functional groups on the surface of the samples.24
C–OH) functional groups on the surface of the samples.24
 | ||
| Fig. 3 FT-IR spectra of ZnO and CeO2–ZnO catalysts. | ||
3.2. Morphology
Scanning electron microscopy is a technique that enables the study of the microstructure of nanoparticles. The scanning electron micrographs of MOF-5, and the MOF-5-derived ZnO and CeO2–ZnO catalysts are presented in Fig. 4. The MOF-5 samples clearly exhibit the cubic morphology with smooth facets and the particle size ranged from 200 nm to 500 nm (Fig. 4(a)). After thermal treatment at 600 °C, the MOF-5-derived ZnO and CeO2–ZnO catalysts (Fig. 4(b) and (c)) mainly maintain the cubic structure, but the edge becomes smooth and the surface appears porous. In addition, compared to MOF-5, the particle sizes of the ZnO and CeO2–ZnO catalysts show a slight decrease. During the heat-treatment of MOF-5 and Ce/MOF-5, as expected, they gradually decompose with time, resulting in the smaller and porous particles with cubic structure. | ||
| Fig. 4 SEM images of (a) MOF-5, (b) ZnO and (c) CeO2–ZnO. | ||
3.3. H2-TPR measurements
H2-TPR analysis was performed to determine the reducibility of the MOF-5-derived ZnO and CeO2–ZnO catalysts. As shown in Fig. 5, one obvious hydrogen consumption peak centered at 600 °C exists in each sample. Compared with the ZnO catalyst, the CeO2–ZnO catalyst presented an obvious shift towards lower temperatures with the increasing amount of Ce, suggesting that the redox properties of the catalysts were enhanced by the interaction between CeO2 and ZnO. The enhanced redox properties could lead to a better dispersion, promoting oxygen mobility and the oxidation process of NO to NO2. | ||
| Fig. 5 H2-TPR spectra of the ZnO and CeO2–ZnO catalysts. | ||
3.4. Catalytic test
The N2 yield as a function of temperature over the MOF-5-derived ZnO and CeO2–ZnO catalysts is shown in Fig. 6. The MOF-5-derived ZnO achieved the maximum N2 yield of 31.5% at 250 °C, while the activity of the Ce-doped catalysts was enhanced significantly, though the temperature corresponding to the maximum N2 yield shifted to a higher temperature at 270 °C. Among the tested catalysts, 3% CeO2–ZnO and 5% CeO2–ZnO exhibited outstanding catalytic performances, with a maximum N2 yield of 68.3% and 69.1%, respectively. The unsatisfactory performance of the 10% CeO2–ZnO catalyst (55.7%) may be attributed to the agglomeration of excess Ce. | ||
| Fig. 6 N2 yield as a function of temperature for the ZnO and CeO2–ZnO catalysts. | ||
3.5. NO-TPD measurements
To evaluate the NO adsorption ability of the as-synthesized catalyst, NO-TPD measurements were conducted to explore the interaction between NO and the samples (Fig. 7). For the MOF-5-derived ZnO catalyst, two NO desorption peaks at 176 and 219 °C could be observed. The two peaks shifted to higher temperatures after Ce was added to the catalysts, indicating that Ce can largely strengthen the bonding of NO with the catalyst surface. Moreover, the intensities of the two NO desorption peaks enhanced after the addition, which indicated that adding Ce can result in the redistribution of the adsorbed NOx species, thus facilitating the NO oxidation. It is worth noting that the promotion effect enhanced very slowly after Ce loading, reaching over 10%. This might be caused by agglomeration of the excess Ce. | ||
| Fig. 7 NO-TPD profiles of the ZnO and CeO2–ZnO catalysts. | ||
3.6. DRIFTS studies
 | ||
| Fig. 8 In situ DRIFTS of (a) ZnO, (b) 1% CeO2–ZnO, (c) 3% CeO2–ZnO (d) 5% CeO2–ZnO (e) 10% CeO2–ZnO exposed to the SCR reaction at 250 °C for various times under NO + O2 adsorption and during C3H6 interaction with the NO + O2 gas mixture. | ||
In the case of the 1% CeO2–ZnO catalyst (Fig. 8(b)), the formation of monodentate nitrate, chelating nitrite and gas-phase NO (1028 cm−1, 1180 cm−1 and 1634 cm−1, respectively) occurred on the surface after 10 min and was slightly enhanced with increasing time. Switching to the C3H6/O2 mixture, several new bands were detected. The bands at 1666 cm−1 and 1444 cm−1 are assigned to the enolic species (RCHCH–O−) and acetic acid, respectively.27,28 The IR bands centered at 2950 cm−1 and 3091 cm−1 are attributed to the asymmetric and symmetric CH stretch (vCH) of the CH3 group, which were proposed by Efstathiou et al. to indicate the –CH3 adsorbed species produced from the interaction of C3H6 with the catalyst surface.30 In addition, a large amount of gas phase CO2 was observed at 2343 cm−1 and 2358 cm−1.
For the 3% CeO2–ZnO catalyst (Fig. 8(c)), NO/O2 flushing after 1 min produced the monodentate nitrate (at 1028 and 1137 cm−1), chelating nitrite (at 1160 and 1189 cm−1), nitrosyl (at 1845 cm−1 and 1910 cm−1), and gas-phase NO (at 1634 cm−1), indicating that the 3% CeO2–ZnO catalyst exhibited better adsorption performance in comparison with the 1% CeO2–ZnO catalyst. In addition, the peaks at 2343 and 2358 cm−1 belong to the gas phase CO2 and are attributed to the CO bonds on the carbon surface. The intensities of these peaks were markedly increased by prolonging the adsorption time. Similar to the 1% CeO2–ZnO catalyst, the chelating nitrite and nitrosyl group clearly decreased after purging with C3H6/O2, accompanied by an appearance of the enolic species (1666 cm−1) and acetate (1443 cm−1) over 3% CeO2–ZnO.
In the case of the 5% CeO2–ZnO catalyst (Fig. 8(d)), excluding the similar characteristic peaks with the 1% CeO2–ZnO and 3% CeO2–ZnO catalysts, several new bands such as the bidentate nitrate (1569 and 1540 cm−1) and nitro compounds (1280 cm−1) were observed at the same time, suggesting the enhanced adsorption ability of NO on the 5% CeO2–ZnO catalyst. For 10% CeO2–ZnO (Fig. 8(e)), the peaks were significantly weakened, revealing that the doping amount of cerium should be appropriate for the ceria–zinc mixed oxides.
On the basis of these facts, we can reach the following conclusion. The nitrates (monodentate nitrates, bidentate nitrates), chelating nitrite, nitro compounds and nitrosyl groups are formed on the surface of the catalysts from the interaction of NO and O2. After introducing C3H6, these species decreased or diminished, accompanied by hydrocarbonate, enolic species and acetate. In addition, significant amounts of gas phase CO2 and H2O (the products of C3H6 oxidation) were also observed. All of the bands were significantly enhanced by adding ceria.
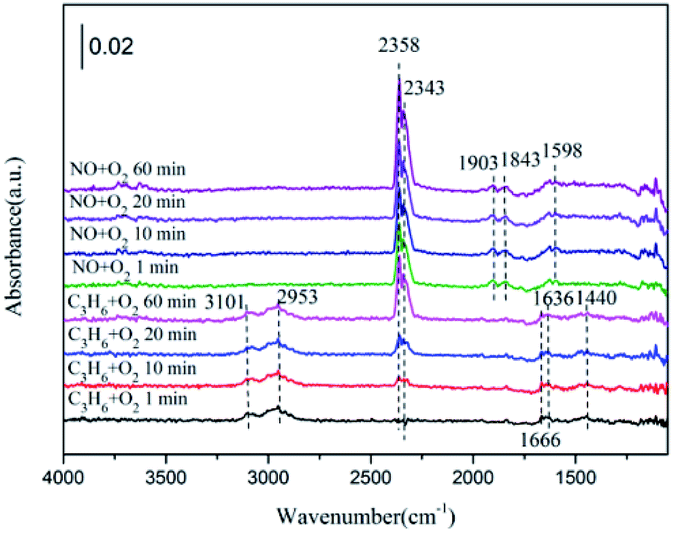 | ||
| Fig. 9 In situ DRIFTS of 5% CeO2–ZnO exposed to the SCR reaction at 250 °C for various times under C3H6 + O2 adsorption and during NO + O2 interaction with the C3H6 + O2 gas mixture. | ||
Exposure to NO resulted in the surface CxHyOz, hydrocarbonate and C3H6 gas fade away, combined with an enhancement in the bidentate nitrate (at 1598 cm−1) and nitrosyl (at 1845 cm−1 and 1910 cm−1) species. More importantly, the formation of CO2 (2358 and 2343 cm−1) was increased by the flow of NO. The CxHyOz amounts decreased by the reaction with NO, although the hydrocarbonate species were more likely to decompose directly to CO2 and O2.
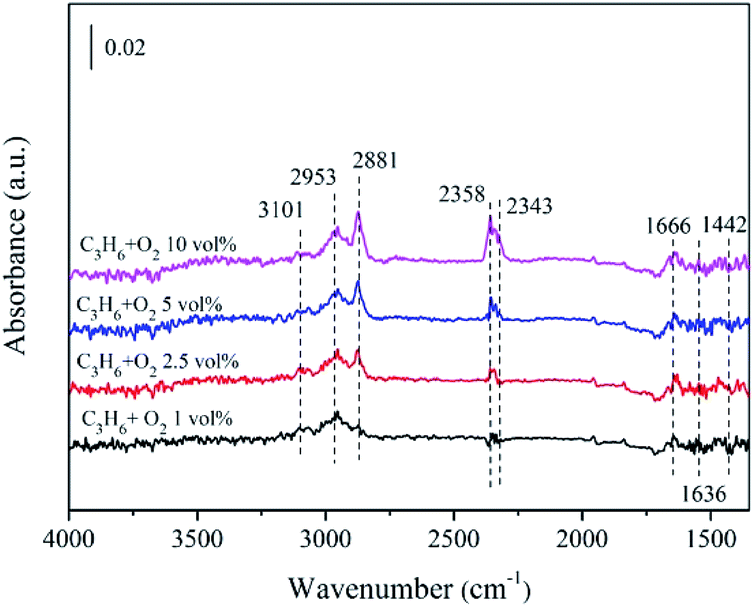 | ||
| Fig. 10 In situ DRIFTS of 5% CeO2–ZnO exposed to C3H6 + O2 with different O2 content after 30 min. | ||
3.7. Reaction pathway
Many literature reports have confirmed that NO reduction undergoes different reaction pathways, depending on the type of hydrocarbon reductants and SCR catalysts, along with the reaction temperature.32 In general, two schemes are proposed for the process. One is known as the “decomposition” mechanism, involving the dissociation and adsorption of NO on the metal active sites, followed by the recombination of the adsorbed N to generate the gas-phase N2. The other is the “reduction” mechanism, which consists of complex surface interactions between the adsorbed NOx and surface hydrocarbon fragments.33,34In this study, the reaction mechanism of the HC-SCR over the MOF-5-derived CeO2–ZnO catalysts conforms to the “reduction” mechanism scheme. It can be understood through an analysis of the adsorbed species by means of in situ DRIFTS. The scheme of reaction mechanism is shown in Fig. 11. First, during the adsorption and subsequent oxidation of NO process, the nitrates (monodentate nitrates, bidentate nitrates), chelating nitrite, nitro compounds and nitrosyl are generated on the active sites (Ce4+ and Zn2+). In addition, an abundance of CxHyOz (enolic species and acetate) is formed and activated on the active sites (Ce4+ or Zn2+). Subsequently, in the typical C3H6-SCR, the two kinds of surface species above are able to react with each other through the formation of a hydrocarbonate or carbonate species, and then directly produce N2, CO2 and H2O. Based on our present study, these results demonstrate that the Ce addition has a significant positive effect on the formation of more adsorbed NOx species due to the increase in the available active sites, and contributes to the improvement in the C3H6-SCR activity.
 | ||
| Fig. 11 Proposed reaction mechanism of NOx reduction by propene over CeO2–ZnO catalysts. | ||
4. Conclusions
In summary, the MOF-based derivative x% CeO2–ZnO (x = 1, 3, 5, 10) catalysts were facilely synthesized using a complete thermal decomposition of the Ce/MOF-5 precursor. Combined with XRD, FTIR, TG-DSC, SEM and H2-TPR experiments, it was found that the as-prepared CeO2–ZnO catalysts were favorable for strengthening the interaction between Ce4+ and Zn2+ and exhibited excellent catalytic performance for the C3H6-SCR of NO. A significant improvement in the catalytic activity was found over the Ce-doped catalysts with the highest N2 yield of 69.1% achieved over 5% CeO2–ZnO. In situ DRIFTS and NO-TPD experiments demonstrated the formation of monodentate nitrates, bidentate nitrates, chelating nitrite, nitro compounds, nitrosyl and CxHyOz species (enolic species and acetate) on the surface, and the additional formation of hydrocarbonate or carbonate as intermediates to directly generate N2, CO2 and H2O. Thus, this research highlights new perspectives on the application of MOF materials in the field of deNOx.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study is supported by the National Natural Science Foundation of China (No. 21866022, 21567018, 51868054), Inner Mongolia Natural Science Foundation (No. 2017MS0214, 2017MS0522), Science and Technology Major Project on Lakes of Inner Mongolia (No. ZDZX2018054), and Inner Mongolia Engineering Research Center of Coal Chemical Wastewater Treatment & Resourcelization.References
- G. Pekridis, N. Kaklidis, V. Komvokis, C. Athanasiou, M. Konsolakis, I. V. Yentekakis and G. E. Marnellos, J. Phys. Chem. A, 2010, 114, 3969–3980 CrossRef CAS PubMed.
- P. Granger and V. I. Parvulescu, Chem. Rev., 2011, 111, 3155–3207 CrossRef CAS PubMed.
- N. A. S. Amin, E. F. Tan and Z. A. Manan, J. Catal., 2004, 222, 100–106 CrossRef CAS.
- X. Y. Liu, Z. Jiang, M. X. Chen, J. W. Shi, Z. X. Zhang and W. F. Shangguan, Ind. Eng. Chem. Res., 2011, 50, 7866–7873 CrossRef CAS.
- H. A. Habib, R. Basner, R. Brandenburg, U. Armbruster and A. Martin, ACS Catal., 2014, 2479–2491 CrossRef CAS.
- H. Pan, Y. H. Guo, Y. F. Jian and C. He, Energy Fuels, 2015, 29, 5282–5289 CrossRef CAS.
- G. Y. Xu, Y. B. Yu and H. He, ACS Catal., 2018, 8, 2699–2708 CrossRef CAS.
- Y. H. Hu and K. Griffiths, Appl. Surf. Sci., 2008, 254, 5048–5054 CrossRef CAS.
- K. Köhler and C. H. He, J. Phys. Chem. C, 2011, 115, 1248–1254 CrossRef.
- H. X. Jiang, Q. Y. Wang, H. Q. Wang, Y. F. Chen and M. H. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 26817–26826 CrossRef CAS PubMed.
- L. Zhang and Y. H. Hu, Mater. Sci. Eng., B, 2011, 176, 573–578 CrossRef CAS.
- W. Y. Huang, X. Zhou, Q. B. Xia, J. H. Peng, H. H. Wang and Z. Li, Ind. Eng. Chem. Res., 2014, 53, 11176–11184 CrossRef CAS.
- Z. J. Li, Y. L. Xiao, W. J. Xue, Q. Y. Yang and C. L. Zhong, J. Phys. Chem. C, 2015, 119, 3674–3683 CrossRef CAS.
- Q. Naddaf, M. Mansour, H. Thakkar and F. Rezaei, Ind. Eng. Chem. Res., 2018, 57, 17470–17479 CrossRef.
- L. Zhang, L. Huang, Y. H. Qin and B. Z. Chen, Trans. Nonferrous Met. Soc. China, 2018, 28, 980–988 CrossRef CAS.
- X. Chen, X. Chen, E. Q. Yu, S. C. Cai, H. P. Jia, J. Chen and P. Liang, Chem. Eng. J., 2018, 344, 469–479 CrossRef CAS.
- K. Cendrowski, P. Skumial, P. Spera and E. Mijowska, Mater. Des., 2016, 110, 740–748 CrossRef CAS.
- S. H. Wu, H. J. He, X. Lia, C. P. Yang, G. M. Zeng, B. Wu, S. Y. He and L. Lu, Chem. Eng. J., 2018, 341, 126–136 CrossRef CAS.
- L. Qiu, Y. Cheng, C. P. Yang, G. M. Zeng, Z. Y. Long, S. N. Wei, K. Zhao and L. Luo, RSC Adv., 2016, 6, 17036–17045 RSC.
- Q. L. Zhang, J. Fan, P. Ning, Z. X. Song, X. Liu, L. Y. Wang, J. Wang, H. M. Wang and K. X. Long, Appl. Surf. Sci., 2018, 435, 1037–1045 CrossRef CAS.
- Q. Ye, L. N. Yan, H. P. Wang, S. Y. Cheng, D. Wang, T. Kang and H. Dai, Appl. Catal., A, 2012, 431–432, 42–48 CrossRef CAS.
- A. V. Rajgure, N. L. Tarwal, J. Y. Patil, L. P. Chikhale, R. C. Pawar, C. S. Lee, I. S. Mulla and S. S. Suryavanshi, Ceram. Int., 2014, 40, 5837–5842 CrossRef CAS.
- M. Z. Hussain, G. S. Pawar, Z. Huang, A. A. Tahir, R. A. Fischer, Y. Q. Zhu and Y. D. Xia, Carbon, 2019, 146, 348–363 CrossRef CAS.
- R. Atchudan, T. N. J. I. Edison, S. Perumal, D. Karthikeyan and Y. R. Lee, J. Photochem. Photobiol., B, 2016, 162, 500–510 CrossRef CAS.
- J. Liu, X. Y. Li, Q. D. Zhao, C. Hao and D. K. Zhang, Environ. Sci. Technol., 2013, 47, 4528–4535 CrossRef CAS.
- K. Ueda, J. Y. Ohyama and A. Satsuma, ACS Omega, 2017, 2, 3135–3143 CrossRef CAS.
- W. Yang, R. D. Zhang, B. H. Chen, D. Duprez and S. Royer, Environ. Sci. Technol., 2012, 46, 11280–11288 CrossRef CAS PubMed.
- J. Liu, X. Y. Li, Q. D. Zhao, C. Hao, S. B. Wang and M. Tade, ACS Catal., 2014, 4, 2426–2436 CrossRef CAS.
- X. X. Cheng, Y. R. Cheng, Z. Q. Wang and C. Y. Ma, Fuel, 2018, 214, 230–241 CrossRef CAS.
- C. M. Kalamaras, G. G. Olympiou, V. I. Pârvulescu, B. Cojocaru and A. M. Efstathiou, Appl. Catal., B, 2017, 206, 308–318 CrossRef CAS.
- L. Q. Nguyen, C. Salim and H. Hinode, Appl. Catal., B, 2010, 96, 299–306 CrossRef CAS.
- J. L. Long, Z. Z. Zhang, Z. X. Ding, R. S. Ruan, Z. H. Li and X. X. Wang, J. Phys. Chem. C, 2010, 114, 15713–15727 CrossRef CAS.
- M. Haneda, N. Bion, M. Daturi, J. Saussey, J. C. Lavalley, D. Duprez and H. Hamada, J. Catal., 2002, 206, 114–124 CrossRef CAS.
- R. Burch, J. P. Breen and F. C. Meunier, Appl. Catal., B, 2002, 39, 283–303 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |