
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSecondary structure of end group functionalized oligomeric-L-lysines: investigations of solvent and structure dependent helicity†
Merve Basak Canalpa,
Annette Meisterb and
Wolfgang H. Binder *a
*a
aFaculty of Natural Science II (Chemistry, Physics and Mathematics), Martin Luther University Halle-Wittenberg, von-Danckelmann-Platz 4, Halle (Saale) D-06120, Germany. E-mail: wolfgang.binder@chemie.uni-halle.de
bInstitute of Biochemistry and Biotechnology, Martin Luther University Halle-Wittenberg, Kurt-Mothes-Straße 3a, Halle (Saale) D-06120, Germany
First published on 12th July 2019
Abstract
Fibrillation of supramolecular building blocks represents an important model system for complex proteins and peptides, such as amyloidogenic proteins, displaying aggregation and subsequent collapse of their biological functions. In this work, we synthesized narrow-dispersed, end group-telechelic, oligomeric-(L-lysine(carboxybenzyl (Z)/trifluoroacetyl (TFA)))ns (n = 3–33) as a model system for studying assembly and secondary structure formation, prepared via ring opening polymerization (ROP) of N-carboxyanhydrides (NCA). Our primary goal was to understand the influence of amino acid chain length and end group-modification on the secondary structure and fibrillation of the oligo-Z/TFA-protected lysines. Synthesis was accomplished by initiation of ROP with 11-amino-undecene, followed by complete chain end functionalization reactions of the N-terminus by 10-undecenoyl-chloride. The so obtained oligomeric-(L-lysine(Z/TFA))ns were fractionated according to their number of repeating units (n) with preparative GPC using DMF as the eluent. As proven by MALDI-ToF MS, 1H-NMR-spectroscopy and analytical GPC, they were separated into fractions with low polydispersity (Đ) values, ranging from 1.02–1.08. Secondary structural investigations of these narrowly-dispersed oligomeric-(L-lysine(Z/TFA))ns (n = 33 ± 6, n = 18 ± 6, n = 12 ± 4, n = 5 ± 2) were accomplished by CD spectroscopy in TFE and HFIP, indicating that TFE was able to induce/stabilize the formation of α-helicity. Fibril formation of oligomeric-(L-lysine(Z/TFA))ns with shorter chain lengths (n = 7 and n = 3) were chosen to investigate the effect of the number of repeating units' role on the self-assembly of the oligomers in TFE. TEM images of these selected fractions, f19 with n = 7 and f28 with n = 3, showed that fibrillization occured and the formation of a dense fibrillar mesh was observed when the amino acid chain length is equal to 7. Therefore, the influences of the number of repeating units (n), end-group functionalities (mono- or bis-functional) and the choice of solvents (TFE or HFIP) on the propensity to form helical structure allowed us to calibrate their secondary structure.
Introduction
Homopolymers and copolymers of amino acids,1 which are able to adopt the main secondary structural features such as alpha helices and beta sheets, hold importance for understanding the formation of the 3D structures of proteins.2 Not only protein folding, but more importantly protein and peptide assemblies3 are within the spotlight of interest4 of researchers, who seek to shed light on the pathogenesis of neurodegenerative disorders such as, e.g., in Alzheimer's and Parkinson's diseases.5–7 Quite a range of different peptides have been reported to form fibrils via mechanisms not largely dissimilar to those of amyloid-beta 1–42 peptide (Aβ42).8 Especially amphiphilic peptide-conjugates,9 that are prone to aggregation, display fibrillation via nucleation dependent aggregation mechanisms. Poly-L-lysine (PLL) in particular has received significant attention in the field of biomedicine,10–16 as it possesses inter- and intra-molecular folding behaviour, where its subsequent assembly can lead to complex, often even fibrillar-like structures. Thus, dynamic conformational changes of PLL depends on various external chemical and physical conditions having been studied both in solution and in the solid state, with an unclear contribution of end groups and chain length.17,18 In solution, high molecular weight PLL(Z) (n > 5000) has shown transitions from helical conformations to random coil in 35–40% of dichloroacetic acid/chloroform mixture as measured by its optical rotatory dispersion.19 Secondary structural changes of PLL·HCl (n = 200) have been analysed by CD spectroscopy, indicating a random coil at low pH, an α-helix at a pH > 10.6 and a β-sheet at 60 °C.20 There has been a pronounced solvent-influence on the conformation of PLL-oligomers, stimulating investigations on the conformational distributions of PLL·HCl (31 kDa) by IR spectroscopy in D2O, DMSO, TFE and ethylene glycol. Mirtič et al. have suggested that an increase in temperature (T = 80 °C) induced the formation of β structures, while TFE and DMSO stabilized α-helices.21 Solid state investigations of PLL(Z) films (DP: 18), cast from DMF as a solvent, reveal a 3.6 nm α-helix (5 turns) within a simple hexagonal unit cell.18 Huesmann et al.22 have investigated poly-L-lysine(Z)s with n = 24, 57, 87, 196 and Đs around 1.70 as well as poly-L-lysine(TFA)s (n = 20, 65, 90, 143) with Đs up to 1.95 via CD, showing an increase in helicity with increasing number of repeating units (n) for poly-L-lysine(Z)s in 1,1,1,3,3,3-hexafluoroisopropanol (HFIP). In these investigations, poly-L-lysine(Z) with n = 5–15 did not display an ordered secondary conformation as proven via CD, whereas with a chain length of n ≈ 60 α-helicity was observed in HFIP.Our study aims to investigate the secondary structural behaviour of: (i) the chain end effects introduced by C11 alkyl groups on each end of the peptide, similar to lipid/peptide hybrids in e.g. lipopeptides23 or peptide amphiphiles24,25 and (ii) investigate the influence of the side chain effects of the protecting groups, Z(Cbz) and TFA, exerted on the oligomeric lysines in solution. In order to get a deeper understanding of the effects of the number of repeating units (n) on the formation of helices and their fibrillation behaviour, we have fractionated the N-terminus functionalized oligomers, enabling us to perform investigations by CD in two different solvents (HFIP and TFE) with the aim to understand to what extent TFE can stabilize α-helix formation. We therefore have prepared oligomeric L-lysines with low molecular weights (Mn = 2–3 kDa) via ROP of NCA monomers using 11-aminoundecene as the primary amine initiator, followed by an additional chain end functionalization via amidation reaction of the amino group of the N-terminus with 10-undecenoyl chloride. Subsequently, fractionated samples of N-terminus functionalized oligomers obtained by preparative GPC were investigated by MALDI-ToF MS, analytical GPC and CD spectroscopy to study their conformational behaviour at precise chain lengths (n).
Experimental section
Materials and methods
DMF was dried over CaH2 and freshly distilled by applying vacuum (20 mbar) at 50 °C. Ethyl acetate was dried at 100 °C with P2O5. N-heptane was refluxed over sodium/benzophenone at 120 °C. All of the dried solvents were flushed with N2 gas before usage. All of the reagents were purchased from Sigma-Aldrich. All NMR-spectra were recorded on a Varian spectrometer (Gemini 200, Gemini 2000 and Unity 500) at 400 or 500 MHz at 27 °C. Trimethylsilane was used as internal standard. Deuterated chloroform (CDCl3) and dimethyl sulfoxide (DMSO-d6) were used as solvent. In the case of all polymer samples trifluoroacetic acid (TFA) (15% of volume) was added to the deuterated chloroform (CDCl3). All chemical shifts (δ) were reported in parts per million (ppm) and the coupling constant (J) in Hertz. For the interpretation of the spectra MestReNova v. 6.0.2 5475 was used. For analytical GPC measurements a Viscotek GPCmax VE 2001 with an HHR-H Guard-17369 and a GMHHR-N-18055 column in DMF at 60 °C was used. The sample concentration was 5 mg mL−1, the injection volume was 100 μL. The detection was carried out via the refractive index with a VE 3580 RI detector of Viscotek at a temperature of 35 °C and a flow rate of 1 mL min−1. Polystyrene standards with molecular weights from 1000, g mol−1 to 115![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 were used for external calibration. Preparative GPC was performed by a KD-2002.5 column from Shodex company attached on a VWR HITACHI Chromaster instrument using DMF (HPLC graded) as the eluent at 55 °C with a flow rate of 0.70 mL min−1 injecting sample with the concentration of 15 mg mL−1 where refractive index detector from VWR at 50 °C was employed as the detector. The obtained data were analysed by using EZChrom Elite (version 3.3.2 SP2) software. ESI-ToF measurements were performed on a Focus micro ToF by Bruker Daltonics. The sample (1.00 mg) was dissolved in methanol (1.00 mL, HPLC grade) and directly infused (180.00 μL h−1, positive or negative mode). MALDI-TOF MS measurements were carried out in reflector mode on a Bruker Autoflex III Smart beam equipped with a nitrogen laser source (λ = 337 nm). The samples were dissolved in DMF (HPLC grade) (c = 10 mg mL−1) with a ratio of 100:10:1 (matrix:analyte:salt). The matrix dithranol with c = 10 mg mL−1 in THF, the salt potassium trifluoroacetate (KTFA) with c = 5 mg mL−1 in THF were used. For the data evaluations and simulation of the mass spectra of the polymer samples the computer programme Flex analysis (version 3.0) was used. CD spectroscopy measurements were performed with the instrument, JASCO Corp., J-810, Rev. 1.00, at a constant temperature (20 °C). The UV absorption was measured in CD units of millidegrees in the wavelength range of 260–190 nm. The cuvette cell used had a diameter of 0.1 cm. The correction of the measurements was done by subtraction of the absorption of the pure solvents e.g. 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) and 2,2,2-trifluoroethanol (TFE) from the absorbance of the sample. The negatively stained samples for Transmission Electron Microscopy (TEM) were prepared by spreading 5 μL of the dispersion (0.2 mg mL−1 in TFE) onto a Cu grid coated with a Carbon-film (PLANO, Wetzlar, D). After 1 min excess liquid was blotted off with filter paper and 5 μL of 1% aqueous uranyl acetate solution were placed onto the grid and drained off after 1 min. The dried specimens were examined with an EM 900 transmission electron microscope (Carl Zeiss Microscopy GmbH, Oberkochen, Germany). Micrographs were taken with a SSCCD SM-1k-120 camera (TRS, Moorenweis, Germany).
000 g mol−1 were used for external calibration. Preparative GPC was performed by a KD-2002.5 column from Shodex company attached on a VWR HITACHI Chromaster instrument using DMF (HPLC graded) as the eluent at 55 °C with a flow rate of 0.70 mL min−1 injecting sample with the concentration of 15 mg mL−1 where refractive index detector from VWR at 50 °C was employed as the detector. The obtained data were analysed by using EZChrom Elite (version 3.3.2 SP2) software. ESI-ToF measurements were performed on a Focus micro ToF by Bruker Daltonics. The sample (1.00 mg) was dissolved in methanol (1.00 mL, HPLC grade) and directly infused (180.00 μL h−1, positive or negative mode). MALDI-TOF MS measurements were carried out in reflector mode on a Bruker Autoflex III Smart beam equipped with a nitrogen laser source (λ = 337 nm). The samples were dissolved in DMF (HPLC grade) (c = 10 mg mL−1) with a ratio of 100:10:1 (matrix:analyte:salt). The matrix dithranol with c = 10 mg mL−1 in THF, the salt potassium trifluoroacetate (KTFA) with c = 5 mg mL−1 in THF were used. For the data evaluations and simulation of the mass spectra of the polymer samples the computer programme Flex analysis (version 3.0) was used. CD spectroscopy measurements were performed with the instrument, JASCO Corp., J-810, Rev. 1.00, at a constant temperature (20 °C). The UV absorption was measured in CD units of millidegrees in the wavelength range of 260–190 nm. The cuvette cell used had a diameter of 0.1 cm. The correction of the measurements was done by subtraction of the absorption of the pure solvents e.g. 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) and 2,2,2-trifluoroethanol (TFE) from the absorbance of the sample. The negatively stained samples for Transmission Electron Microscopy (TEM) were prepared by spreading 5 μL of the dispersion (0.2 mg mL−1 in TFE) onto a Cu grid coated with a Carbon-film (PLANO, Wetzlar, D). After 1 min excess liquid was blotted off with filter paper and 5 μL of 1% aqueous uranyl acetate solution were placed onto the grid and drained off after 1 min. The dried specimens were examined with an EM 900 transmission electron microscope (Carl Zeiss Microscopy GmbH, Oberkochen, Germany). Micrographs were taken with a SSCCD SM-1k-120 camera (TRS, Moorenweis, Germany).
Synthesis of N-carboxyanhydride (NCA) monomers
Ring opening polymerization (ROP) of NCA monomers
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CH2, 3,3,3JH,H = 13.5, 10.3, 6.7 Hz), 5.26–4.78 (m, 25H, CH2CC, CH2), 4.42 (d, 9H, CH, 3JH,H = 6.7 Hz), 3.39–2.99 (m, 25H, CH2), 2.30–1.15 (m, 94H, CH2). MS (MALDI-ToF, Dithranol/KTFA): m/z calc. = 1518.8 [M + K]+, m/z exp. = 1519.0 [M + K]+.CH–, 3,3JH,H = 16.6, 6.6 Hz), 4.95 (dd, 2H, CH2CH–, 2,3JH,H = 30.2, 13.6 Hz), 4.79 (dd, 1H, CH, 3,3JH,H = 9.9, 5.4 Hz), 4.40 (m, 4H, CH), 3.43 (d, 18H, CH2, 3JH,H = 6.6 Hz), 2.14–1.15 (m, 51H, CH2). MS (MALDI-ToF, Dithranol/KTFA): m/z calc. = 1328.5 [M + K]+, m/z exp. = 1328.6 [M + K]+.
CH–CH2, 3,3,3JH,H = 13.5, 10.3, 6.7 Hz), 5.26–4.78 (m, 25H, CH2CC, CH2), 4.42 (d, 9H, CH, 3JH,H = 6.7 Hz), 3.39–2.99 (m, 25H, CH2), 2.30–1.15 (m, 94H, CH2). MS (MALDI-ToF, Dithranol/KTFA): m/z calc. = 1518.8 [M + K]+, m/z exp. = 1519.0 [M + K]+.CH–, 3,3JH,H = 16.6, 6.6 Hz), 4.95 (dd, 2H, CH2CH–, 2,3JH,H = 30.2, 13.6 Hz), 4.79 (dd, 1H, CH, 3,3JH,H = 9.9, 5.4 Hz), 4.40 (m, 4H, CH), 3.43 (d, 18H, CH2, 3JH,H = 6.6 Hz), 2.14–1.15 (m, 51H, CH2). MS (MALDI-ToF, Dithranol/KTFA): m/z calc. = 1328.5 [M + K]+, m/z exp. = 1328.6 [M + K]+.N-terminus functionalization of oligomers
CH–, 3,3JH,H = 10.2, 6.8 Hz), 5.32–4.84 (m, 74H, H2CC, CH2), 4.43 (s, 7H, CH), 3.24 (d, 2H, CH2, 3JH,H = 5.9 Hz), 2.48–1.05 (m, 340H, CH2). MS (MALDI-ToF, Dithranol/KTFA): m/z calc. = 1669.0 [M + K]+, m/z exp. = 1668.5 [M + K]+.CH–, 3,3JH,H = 10.2, 6.8 Hz), 4.95 (dd, H2CC, CH2), 4.43 (s, 7H, CH), 3.24 (d, 2H, Hd, i, CH2, 3JH,H = 5.9 Hz), 2.48–1.05 (m, 340H, CH2). MS (MALDI-ToF, Dithranol/KTFA): m/z calc. = 1494.7 [M + K]+, m/z exp. = 1494.7 [M + K]+.Results and discussion
The synthesis of defined oligo-/poly-L-lysines is conventionally accomplished by ring opening polymerization of N-carboxyanhydrides (NCA)28,29 bearing either carboxybenzyl (Cbz, Z)-L-lysine (NCA(Cbz, Z)) or the trifluoroacetyl (TFA)-L-lysine (NCA(TFA)), initiated by primary and secondary amines such as hexylamine or diethylamine in DMF.22,30–33 As livingness and end group fidelity during ROP of NCAs can be achieved via the “Normal Amine Mechanism (NAM)” using primary or secondary amines as initiators,34,35 we have polymerized the two NCA-monomers of carboxybenzyl-L-lysine, NCA(Z), and trifluoroacetyl-L-lysine, NCA(TFA) via the primary amine, 11-aminoundecene to obtain the mono-functionalized oligomers OK(Z) and OK(TFA) in the first step. Subsequently, we have functionalized the N-terminus end groups of the oligomers OK(Z) and OK(TFA) by direct amidation with 10-undecenoyl chloride in situ to achieve the respective bis-functionalized oligomers N-OK(Z) and N-OK(TFA) (see Scheme 1). Subsequently, the bis-functionalized oligomers N-OK(Z) and N-OK(TFA) were fractionated by preparative GPC and then investigated by MALDI-ToF MS, analytical GPC, 1H-NMR and CD spectroscopy. | ||
| Scheme 1 Synthetic pathway of mono- and bis-functionalized oligo-L-lysines. | ||
Fractionation by preparative GPC
The bis-functionalized oligo-L-lysines N-OK(Z) and N-OK(TFA) were separated into fractions according to their molecular weights (the number of repeating units (n)) displaying lower polydispersity (Đ) values, obtained via preparative GPC in DMF as the eluent at a column temperature of 55 °C.As presented in Fig. 1 and 2, both N-OK(Z) and N-OK(TFA) were fractionated successfully, see Fig. 1A and B, collecting the different fractions, labelled as fx. As can be seen from Fig. 1A, for N-OK(Z) 11 fractions (f) f12–f28 were collected, whereas the fractionation of N-OK(TFA) resulted only in 4 fractions which were referred to as f2 (n = 6), f3 (n = 5), f4 (n = 4) and f5 (n = 3) (Fig. 1B). After fractionation, all fractions were analysed by analytical GPC as shown in Fig. 1C (for N-OK(Z)) and Fig. 1D (for N-OK(TFA)), displaying significantly narrow polydispersity values as compared to the unfractionated samples, N-OK(Z) and N-OK(TFA). Subsequently, all selected fractions were also analysed by MALDI-ToF MS (see Fig. 2 and Table 1) as well as 1H-NMR spectroscopy (see Fig. 3).
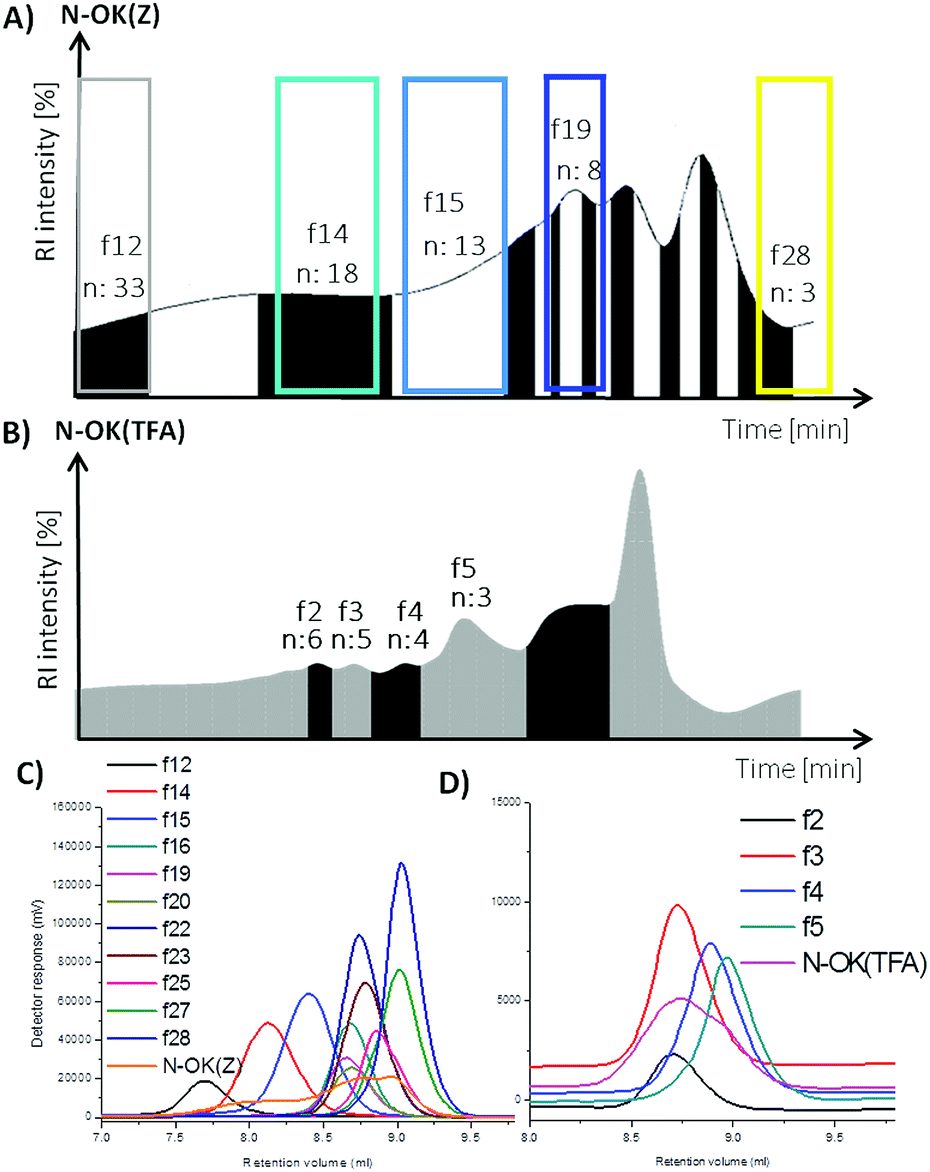 | ||
| Fig. 1 Preparative GPC graphs of (A) N-OK(Z), (B) N-OK(TFA) and analytical GPC graphs of (C) N-OK(Z), (D) N-OK(TFA) along with the selected fractions. | ||
 | ||
| Fig. 2 MALDI-ToF MS of (a) N-OK(Z), (b–f) fractions of N-OK(Z), (g) N-OK(TFA) and (h–k) fractions of N-OK(TFA). | ||
| Entry | Sample | n(MALDI) | Mn(MALDI) | Mn(GPC) | Đ | CD-spec. (HFIP) | CD-spec. (TFE) |
|---|---|---|---|---|---|---|---|
| I | OK(Z) | 15 ± 10 | 4.1 | 2.5 | 1.36 | 22% α-helicity | 32% α-helicity |
| II | N-OK(Z) | 15 ± 10 | 4.2 | 1.4 | 1.36 | 16% α-helicity | 19% α-helicity |
| III | N-OK(Z)-2 | 8 ± 2 | 2.1 | 1.9 | 1.02 | β II turn | 18% α-helicity |
| IV | N-OK(Z)-3 | 24 ± 8 | 5.6 | 8.0 | 1.33 | 40% α-helicity | 53% α-helicity |
| V | f12 | 33 ± 6 | 8.9 | 9.1 | 1.08 | 19% α-helicity | 32% α-helicity |
| VI | f14 | 18 ± 6 | 4.9 | 4.9 | 1.07 | 15% α-helicity | 25% α-helicity |
| VII | f15 | 12 ± 4 | 3.4 | 3.7 | 1.07 | β II turn | 18% α-helicity |
| VIII | f16 | 8 ± 2 | 2.3 | 2.6 | 1.02 | β II turn | 16% α-helicity |
| IX | f19 | 6 & 7 | 2.1 | 2.6 | 1.03 | β II turn | 16% α-helicity |
| X | f20 | 6 ± 2 | 2.1 | 2.6 | 1.03 | β II turn | 16% α-helicity |
| XI | f23 | 5 ± 1 | 1.8 | 2.3 | 1.03 | β II turn | 15% α-helicity |
| XII | f25 | 4 ± 1 | 1.5 | 2.0 | 1.03 | β II turn | 12% α-helicity |
| XIII | f28 | 3 | 1.1 | 1.8 | 1.02 | β II turn | 12% α-helicity |
| XIV | OK(TFA) | 6 ± 3 | 1.5 | 1.0 | 1.09 | 16% α-helicity | 31% α-helicity |
| XV | N-OK(TFA) | 6 ± 3 | 1.6 | 1.2 | 1.05 | β II turn | 15% α-helicity |
| XVI | f2 | 6 ± 1 | 1.4 | 1.3 | 1.02 | β II turn | 19% α-helicity |
| XVII | f3 | 5 ± 1 | 1.2 | 1.2 | 1.02 | β II turn | 15% α-helicity |
| XVIII | f4 | 4 ± 1 | 1.0 | 1.0 | 1.02 | β II turn | β II turn |
| XIX | f5 | 3 & 4 | 0.90 | 0.96 | 1.02 | β II turn | β II turn |
 | ||
| Fig. 3 1H-NMR spectra of fractions of N-OK(Z), f28 and f12 measured in CDCl3 (15 vol% TFA). | ||
In Fig. 2, the MALDI spectra of both fractionated oligomers f28, f19, f15, f14, f12 of N-OK(Z) (b–f) and f5, f4, f3, f2 of N-OK(TFA) (h–k) along with the oligomers (a and g) before fractionation are shown. MALDI spectra prove that the fractions were well separated, with defined end group functionalities on both sides of the respective oligomers, as shown by the excellent match between the simulated and the measured isotopic patterns of the samples (see details in the ESI, Fig. 19S–29S and 31S–33S†). The analytical GPC results also support (see Fig. 2 and Table 1) the very narrow Đ values for each fraction with small variations in the numbers of repeating units (n). Thus, at n = 3 to 8 only a chain variation of ±2 was obtained, and at n = 12–33 the variation was ±6.
As shown in Fig. 3, we have investigated the fractions f12 and f28 of N-OK(Z) by 1H-NMR spectroscopy in CDCl3 (15% volume TFA added), proving their chemical identity and a good match to the data obtained by GPC and MALDI.
All analytical data of the samples are summarized in Table 1, along with the respective n, Mn and Đ values.
Secondary structural analyses by CD spectroscopy
Circular dichroism (CD) is an excellent tool for the rapid determination of the secondary structure and folding properties of proteins.36 In this work, the analysis of the secondary structures of the oligomers was conducted in 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) and 2,2,2-trifluoroethanol (TFE). We chose HFIP and TFE as solvents, as they both act as the good solvents24 for our side chain protected oligomers for the CD measurements. The measured CD spectroscopy data were reported as ellipticity (θ) [mdeg]. The percentage values of α-helicity of the samples were calculated according to the eqn (1), used for the estimation of the helicity of the peptide chains by Krannig and Sun et al.:37|
α-helix (%) = (−[θ]222 + 3000)/39000 × 100%
| (1) |
The summary of all CD spectroscopy measurements along with analytical GPC results of oligomers and their fractions are presented in Table 1. The secondary structural investigations of the fractions of N-OK(Z); f28, f19, f15, f14, f12 and of N-OK(TFA); f4, f3, f2 in TFE are depicted in Fig. 4A and B respectively (also see Table 1).
 | ||
| Fig. 4 CD spectra of the fractions of (A) N-OK(Z) and (B) N-OK(TFA) in TFE (c = 0.2 mg mL−1). | ||
Firstly, we investigated the effect of the number of repeating units (n) on formation of secondary structures by CD spectroscopy, aiming to understand changes in α-helicity with increasing chain length n of the bis-substituted N-OK(Z) oligomers (Fig. 4A and Table 1, entries V–XIII). In Fig. 4A, one can see an increase in α-helicity of the oligomers with increasing n via enhancement of the [θ]222 signal of the fractions of N-OK(Z) f12, f14, f15, f19 and f28 (Table 1, entries V–VII, IX, XIII) in TFE. Because of their low number of repeating units (n = 3–7), fractions of N-OK(TFA) f2, f3, f4 (Table 1, entries XVI–XIX) do not display helicity for f4, with only 19% helicity for f2. Therefore, as shown in Fig. 5, chain length dependence of α-helicity in TFE is demonstrated for the bis-functional fractionated samples with the Z-group protected moieties possessing low Đ values (<1.07), e.g. fractions of N-OK(Z): f28, f19, f15, f14, f12), indicating an almost linear increase in α-helicity (from 12% up to 33%) with increasing number of repeating units (n). It should be noted that this trend is also visible in the unfractionated samples: thus the unfractionated bis-functional oligomer N-OK(Z)-2, (n = 8) (Table 1, entry III) displays a lower α-helicity (18% in TFE) compared to the unfractionated bis-functional oligomer N-OK(Z)-3, (n = 24) (53% in TFE, 40% in HFIP) (Table 1, entry IV), yet confirming the increasing helicity behaviour of oligomers with increasing n as depicted with the “linear” line in Fig. 5.
 | ||
| Fig. 5 Depiction of increase in α-helicity [%] with respect to increasing number of repeating units (n) of fractions of N-OK(Z) in TFE (line is a guide for the eye only). | ||
The chain end effects introduced by alkyl groups on one end (mono-functional) e.g. OK(Z) and OK(TFA), or on both ends (bis-functional) e.g. N-OK(Z) and N-OK(TFA), of the oligomer chains were examined by CD spectroscopy in HFIP and in TFE (see Table 1). Mono-functional oligomers, OK(Z) (Table 1, entry I) and OK(TFA) (Table 1, entry XIV) with equal chain length show higher α-helicity in comparison to their bis-functional derivatives, N-OK(Z) (Table 1, entry II) and N-OK(TFA) (Table 1, entry XV), suggesting that the alkyl end groups do not favour the helix formation. Furthermore, side chain effects stemming from amino-protecting groups, Z(Cbz) and TFA on conformational changes were also studied. We found that there was no distinctive difference in their α-helicity values of N-OK(Z) and N-OK(TFA) oligomers induced by their side chains (Table 1, entries II and XV respectively). Thus for instance, fractions f19 (with Z) (entry IX, n = 6, 7) and f2 (with TFA) (entry XVI, n = 6 ± 1) possess similar α-helicity values. However, as shown in Table 1, there is a slight difference in α-helicity values for the fractions with lower n, e.g. f5 (entry XIX, n = 3–4) does not display α-helicity, whereas f25 (entry XII, n = 4 ± 1) and f28 (entry XIII, n = 3) with similar ns show 12% helicity in TFE.
From Table 1 it can be also seen that TFE promotes helicity38 compared to HFIP. Fraction f12 of N-OK(Z) (entry V) displays 32% α-helicity in TFE whereas only 19% α-helicity is observed in HFIP. In the case of the fractions of N-OK(Z) measured in TFE (entries V–XIII) α-helicity of more than 16% is observed with a number of repeating units higher than 8 (n > 8) starting from fractions f19 and f20 (entries IX and X respectively). TFE increases the stability and formation of the α-helices over HFIP, explained by comparing polarities of both solvents, where TFE is more polar than HFIP. Pengo and Pasquato et al.39 have indicated a correlation between the polarities of different solvents and the equilibrium of the formation of α-helix structures, proving that the more polar the solvent is e.g. TFE, the equilibrium shifts to the formation of α-helical structures, whereas the less polar solvents promote the formation of 310-helix structures. It has been stated that the hydroxyl group of TFE can bind to the carbonyl group in the peptide backbone via hydrogen bonding without disrupting the hydrogen bonding interactions between the carbonyls and the amines of the peptide forming the α-helical structure.40
Influence of solvent composition and effects of number of repeating units of the peptide on the dynamics of the secondary structure formation of peptides and PEG-peptide diblock copolymers have reported that α-helicity is increasing with increasing number of repeating units of a peptide chain and with addition of TFE.41,42
Transmission electron microscopy (TEM) analyses
There has been a number of reports on the visualization of the fibril formation of synthetic peptides at different physical and chemical conditions.43–45 More complex molecular assemblies of alkyl chain grafted PLLs with n = 60, 130 and 300 with varying degree of grafting have been analysed by TEM to observe the formation of vesicles at neutral pH.46 Energy landscapes of peptide amphiphile nanofibers consisting of an alkyl tail and peptide unit with a propensity to form β-sheets have revealed that many products form monodisperse short fibres.47In our study the TEM images of the fractions of N-OK(Z), f19 and f28, obtained after preparation in TFE (c = 0.2 mg mL−1) are shown in Fig. 6. The end groups of both f19 and f28 are functionalized with two C11 vinyl chains. The repeating unit (n) of each sample is different; for f19, the repeating unit is n ≈ 7, indicating that helix formation is already favoured, whereas for f28 (n = 3) the α-helical turn could be only 1, stabilized only with the help of TFE solvent. The TEM image of f19 (n = 7) shows a dense mesh of large fibrils (see Fig. 6a), while stacks of fibrillar bundles are formed by f28 (n = 3) (see Fig. 6b).
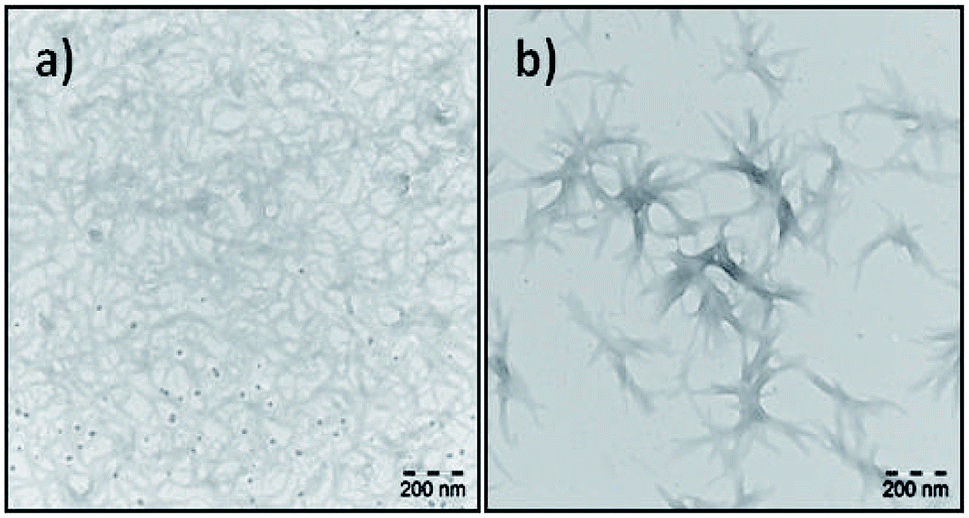 | ||
| Fig. 6 TEM images of f19 (a) and f28 (b) in TFE. | ||
As shown in Fig. 6, we chose fractions f19 and f28 to demonstrate the effect of number of repeating units (n), specifically in the case of very low Đ values, on the formation of fibrils due to their secondary structural interactions. We did also investigate fibril formation of various other samples (see Fig. 36S–40S†), where solvent of choice and the fraction between helical chain lengths have been probed. In addition to the data shown in Fig. 6 and the CD results discussed above (Table 1, Fig. 4 and 5), a higher propensity to form fibrils is accompanied with a higher degree to form alpha helices, suggesting the formed to be promoted by the latter. We propose that increasing α-helicity is crucial to form fibrils owing to their improved packing accompanied by a more precise conformational identity of the peptide with the lower entropy.
Conclusions
In this work, we successfully synthesized mono- and bis-functionalized lysine oligomers with two different protecting groups (Z) and (TFA). We separated bis-functional N-OK(Z) and N-OK(TFA) into fractions by using preparative GPC in DMF to obtain lower polydispersity values of the low molecular weight oligomers. All synthesized oligomers and selectively chosen fractionated samples were analysed by 1H-NMR, analytical GPC and MALDI-ToF MS. The secondary structural investigations were conducted with the help of CD spectroscopy in HFIP and TFE. From our results we conclude that TFE stabilizes helicity better than HFIP. Additionally, as shown in Table 1, we have observed that the functionalization of both end groups of the peptide decreases the stability of the α-helicity for the cases of O-K(Z/TFA) and N-OK(Z/TFA). The fractions of N-OK(Z) and N-OK(TFA) were selectively investigated by CD spectroscopy in HFIP and in TFE, aiming to focus on the effect of the chain length of the peptide on the formation of α-helices. We can prove that the number of repeating units of the oligomers significantly affects the ability to form helical structure, showing that higher n values result in an increased amount of α-helicity as depicted with the linear line in Fig. 5. Moreover, from TEM images we can confirm that in TFE larger fibrils form a dense mesh, when n is larger than 3 (f19), whereas oligomers with a small number of repeating unit (n = 3) (f28) form small stacks of fibrillar bundles.The so obtained data allow us to quantify the amount of secondary structure (α-helicity) present within the functionalized oligo-L-lysines of precise chain length on basis of eqn (1). It is crucial to understand peptide assemblies in view of the initial conformational interactions and their secondary structure formation, hence we here for the first time represent a rational approach to engineer future fibril forming systems, based on oligomeric-L-lysines with very low Đ values (Đ < 1.07). Last but not least, it should be also emphasized that such artificial fibril forming assemblies hold great potential applications as inhibitors for amyloidosis of complex proteins e.g. Aβ. The influence of lipidic alkyl end groups and chain length of peptide unit on the folding behaviours of peptides within a hybrid polymer can thus be quantified, prospectively reported in future work.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the grant Project A03 nr. 189853844-TRR 102 (Polymers under multiple constraints) for financial support from the German Research Foundation (DFG) via the research centre SFB/TRR 102 (Project A3 and A12). We also thank a grant from the “Leistungszentrum Chemie und Biosystemtechnik des Landes Sachsen-Anhalt” for financial support, internal project S1 (Binder). I would like to thank Prof. Hauke Lilie from the Institute of Biochemistry and Biotechnology, Martin Luther University Halle-Wittenberg for allowing me to work with the CD spectroscopy instrument and also Dr Gerd Hause from Biocentre, Martin Luther University Halle-Wittenberg for his contributions to our TEM measurements.Notes and references
- H. Lu, J. Wang, Z. Song, L. Yin, Y. Zhang, H. Tang, C. Tu, Y. Lin and J. Cheng, Chem. Commun., 2014, 50, 139–155 RSC.
- J. C. Johnson and L. T. J. Korley, Soft Matter, 2012, 8, 11431–11442 RSC.
- I. W. Hamley, Angew. Chem., Int. Ed., 2007, 46, 8128–8147 CrossRef CAS.
- B. Wang, E. H. Pilkington, Y. Sun, T. P. Davis, P. C. Ke and F. Ding, Environ. Sci.: Nano, 2017, 4, 1772–1783 RSC.
- S. Williams, T. P. Causgrove, R. Gilmanshin, K. S. Fang, R. H. Callender, W. H. Woodruff and R. B. Dyer, Biochemistry, 1996, 35, 691–697 CrossRef CAS.
- K. Pagel, S. C. Wagner, K. Samedov, H. von Berlepsch, C. Böttcher and B. Koksch, J. Am. Chem. Soc., 2006, 128, 2196–2197 CrossRef CAS.
- J. Li, X. Du, S. Hashim, A. Shy and B. Xu, J. Am. Chem. Soc., 2017, 139, 71–74 CrossRef CAS.
- I. W. Hamley, Chem. Rev., 2012, 112, 5147–5192 CrossRef CAS PubMed.
- I. W. Hamley and V. Castelletto, Bioconjugate Chem., 2017, 28, 731–739 CrossRef CAS.
- G. D. Fasman, Biopolymers, 1987, 26, S59–S79 CrossRef.
- V. Stanić, Y. Arntz, D. Richard, C. Affolter, I. Nguyen, C. Crucifix, P. Schultz, C. Baehr, B. Frisch and J. Ogier, Biomacromolecules, 2008, 9, 2048–2055 CrossRef.
- V. Incani, X. Lin, A. Lavasanifar and H. Uludağ, ACS Appl. Mater. Interfaces, 2009, 1, 841–848 CrossRef CAS.
- M. Chittchang, H. H. Alur, A. K. Mitra and T. P. Johnston, J. Pharm. Pharmacol., 2002, 54, 315–323 CrossRef CAS PubMed.
- M. Chittchang, N. Salamat-Miller, H. H. Alur, D. G. V. Velde, A. K. Mitra and T. P. Johnston, J. Pharm. Pharmacol., 2002, 54, 1497–1505 CrossRef CAS.
- Y.-C. Huang, M. Arham and J.-S. Jan, Soft Matter, 2011, 7, 3975–3983 RSC.
- S. Sinha, D. H. J. Lopes, Z. Du, E. S. Pang, A. Shanmugam, A. Lomakin, P. Talbiersky, A. Tennstaedt, K. McDaniel, R. Bakshi, P.-Y. Kuo, M. Ehrmann, G. B. Benedek, J. A. Loo, F.-G. Klärner, T. Schrader, C. Wang and G. Bitan, J. Am. Chem. Soc., 2011, 133, 16958–16969 CrossRef CAS.
- E. Katchalski, I. Grossfeld and M. Frankel, J. Am. Chem. Soc., 1948, 70, 2094–2101 CrossRef CAS.
- H. Yakel Jr, Acta Crystallogr., 1953, 6, 724–727 CrossRef.
- G. D. Fasman, M. Idelson and E. R. Blout, J. Am. Chem. Soc., 1961, 83, 709–712 CrossRef CAS.
- B. Davidson and G. D. Fasman, Biochemistry, 1967, 6, 1616–1629 CrossRef CAS.
- A. Mirtič and J. Grdadolnik, Biophys. Chem., 2013, 175–176, 47–53 CrossRef.
- D. Huesmann, A. Birke, K. Klinker, S. Türk, H. J. Räder and M. Barz, Macromolecules, 2014, 47, 928–936 CrossRef CAS.
- V. Castelletto, A. Kaur, R. M. Kowalczyk, I. W. Hamley, M. Reza and J. Ruokolainen, Biomacromolecules, 2017, 18, 2013–2023 CrossRef CAS.
- P. A. Korevaar, C. J. Newcomb, E. W. Meijer and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 8540–8543 CrossRef CAS.
- M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2017, 50, 2440–2448 CrossRef CAS.
- G. J. M. Habraken, M. Peeters, C. H. J. T. Dietz, C. E. Koning and A. Heise, Polym. Chem., 2010, 1, 514–524 RSC.
- G. J. M. Habraken, K. H. R. M. Wilsens, C. E. Koning and A. Heise, Polym. Chem., 2011, 2, 1322–1330 RSC.
- A. C. Farthing and R. J. W. Reynolds, Nature, 1950, 165, 647 CrossRef CAS.
- C. Deng, J. Wu, R. Cheng, F. Meng, H.-A. Klok and Z. Zhong, Prog. Polym. Sci., 2014, 39, 330–364 CrossRef CAS.
- H. H. James, in Ring-Opening Polymerization, American Chemical Society, 1985, ch. 5, vol. 286, pp. 67–85 Search PubMed.
- W. Zhao, Y. Gnanou and N. Hadjichristidis, Biomacromolecules, 2015, 16, 1352–1357 CrossRef CAS.
- J. Zou, J. Fan, X. He, S. Zhang, H. Wang and K. L. Wooley, Macromolecules, 2013, 46, 4223–4226 CrossRef CAS.
- W. Vayaboury, O. Giani, H. Cottet, A. Deratani and F. Schué, Macromol. Rapid Commun., 2004, 25, 1221–1224 CrossRef CAS.
- W. N. E. Van Dijk-Wolthuis, L. van de Water, P. van de Wetering, M. J. Van Steenbergen, J. J. Kettenes-van den Bosch, W. J. W. Schuyl and W. E. Hennink, Macromol. Chem. Phys., 1997, 198, 3893–3906 CrossRef CAS.
- T. Aliferis, H. Iatrou and N. Hadjichristidis, Biomacromolecules, 2004, 5, 1653–1656 CrossRef CAS.
- N. J. Greenfield, Nat. Protoc., 2007, 1, 2876–2890 CrossRef.
- K.-S. Krannig, J. Sun and H. Schlaad, Biomacromolecules, 2014, 15, 978–984 CrossRef CAS.
- A. Kentsis and T. R. Sosnick, Biochemistry, 1998, 37, 14613–14622 CrossRef CAS.
- P. Pengo, L. Pasquato, S. Moro, A. Brigo, F. Fogolari, Q. B. Broxterman, B. Kaptein and P. Scrimin, Angew. Chem., 2003, 115, 3510–3514 CrossRef.
- R. Rajan and P. Balaram, Int. J. Pept. Protein Res., 1996, 48, 328–336 CrossRef CAS.
- M. Pechar, P. Kopečková, L. Joss and J. Kopeček, Macromol. Biosci., 2002, 2, 199–206 CrossRef CAS.
- G. W. M. Vandermeulen, C. Tziatzios and H.-A. Klok, Macromolecules, 2003, 36, 4107–4114 CrossRef CAS.
- D. Punihaole, R. S. Jakubek, R. J. Workman, L. E. Marbella, P. Campbell, J. D. Madura and S. A. Asher, J. Phys. Chem. B, 2017, 121, 5953–5967 CrossRef CAS.
- I. W. Hamley, A. Dehsorkhi and V. Castelletto, Langmuir, 2013, 29, 5050–5059 CrossRef CAS.
- M. J. Krysmann, V. Castelletto and I. W. Hamley, Soft Matter, 2007, 3, 1401–1406 RSC.
- B.-Y. Chen, Y.-C. Huang and J.-S. Jan, RSC Adv., 2015, 5, 22783–22791 RSC.
- F. Tantakitti, J. Boekhoven, X. Wang, R. V. Kazantsev, T. Yu, J. Li, E. Zhuang, R. Zandi, J. H. Ortony, C. J. Newcomb, L. C. Palmer, G. S. Shekhawat, M. O. de la Cruz, G. C. Schatz and S. I. Stupp, Nat. Mater., 2016, 15, 469 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03099a |
| This journal is © The Royal Society of Chemistry 2019 |