
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRoom temperature oxidative desulfurization with MoO3 subnanoclusters supported on MCM-41
Qianfan Yang,
Jiasheng Wang *,
Wan-Hui Wang and
Ming Bao*
*,
Wan-Hui Wang and
Ming Bao*
State Key Laboratory of Fine Chemicals, School of Petroleum and Chemical Engineering, Dalian University of Technology, Panjin, 124221, China. E-mail: jswang@dlut.edu.cn; mingbao@dlut.edu.cn
First published on 9th July 2019
Abstract
Subnano MoO3/MCM-41 was successfully prepared through doping (NH4)6Mo7O24 in the synthesis process of MCM-41. The morphology of MoO3/MCM-41 was visually observed by TEM and HADDF-STEM. N2 sorption, XPS and Raman were further applied to investigate the structure of the material. MoO3/MCM-41 was used in the oxidative desulfurization process with tert-butyl hydroperoxide as oxidant. MoO3/MCM-41 showed outstanding catalytic activity and recycling ability at room temperature.
1. Introduction
Sulfur compounds in fuel are the main sources of air pollution.1 Sulfur compounds are also harmful to industrial catalysts such as the catalysts of catalytic converters2 and fuel-cells.3 So it is necessary to reduce the sulfur content in fuel oil. Hydrodesulfurization (HDS) is the traditional technology for sulfur removal,4 but it is not efficient for gaining ultralow diesel under mild conditions. Because refractory sulfur compounds like dibenzothiophene (DBT) and its derivatives do not undergo hydrogenation easily, harsh operating conditions are inevitable when using HDS to deal with these compounds. But DBT and its derivatives can be easily oxidized to corresponding sulfoxides and sulfones because of their high electron density.5 So oxidative desulfurization (ODS) is an alternative technology and has been investigated widely.A series of oxidants have been studied in the ODS like H2O2 and tert-butyl hydroperoxide (TBHP). TBHP has attracted great attention for high oxidizability. Compared to H2O2, TBHP is a kind of oil-soluble oxidant, so no subsequent separatory process is needed when using TBHP as the oxidant of ODS.6 TBHP is thermodynamically stable below 80 °C, so most experiments are tested at 50–80 °C5,7–13 for keeping acceptable catalytic activity. Different kinds of supported catalysts have been investigated for improving the ODS capacity. Lots of transition metal oxides (e.g. MoO3, WO3, V2O5, MnO2) supported on oxides (e.g. SiO2, Al2O3, TiO2) show great catalytic activity at 50–80 °C. Though the mild conditions have been achieved, a heating unit is still indispensable. Du et al. has achieved room temperature ODS, where DBT was removed within 2 h at 30 °C.14 In this study, we want to achieve room temperature ODS within a shorter reaction time.
In our previous study, MoO3 subnanoclusters supported on disordered mesoporous silica (MoO3/SiO2) was prepared by a microemulsion method. MoO3 subnanoclusters can catalyze the DBT oxidation in the model diesel at 70 °C within 15 min.13 MoO3 subnanoclusters show high catalytic activity. But because of the low Mo content (0.2 wt%), the productivity15 (activity per catalyst mass, rather than per Mo) was limited. MCM-41 is a kind of ordered mesoporous silica with large surface and suitable pore size for ODS. During the synthesis processes of disordered mesoporous silica and MCM-41, anion precursor of activity phase can combine with the normal template cationic surfactant cetyltrimethyl ammonium bromide (CTAB). Compared to SiO2 prepared by microemulsion method, more water is added for synthesizing MCM-41, which causes the lower local concentration of Mo7O246− on the micelle surface. MoO3 supported on MCM-41 can be more homogeneous and stable. So MCM-41 is a kind of suitable silica support for MoO3 subnanoclusters.
In this study, MoO3/MCM-41 was successfully prepared by soft template method. Mo7O246− can be dispersed on the surface of template during the synthesis process and converted to MoO3 in situ.16 The Mo content of MoO3/MCM-41 tested in this study was 2.0 wt% (noted as MoM-2.0). The size of MoO3 was kept lower than 1 nm while the Mo content increased by an order of magnitude compared with disordered mesoporous silica supported MoO3.13 MoO3/MCM-41 showed superiority ODS capacity at 20 °C. So the heating unit was not necessary which means greener and more economical. The recycle ability was also tested in this study.
2. Experimental
2.1 Materials
Decalin was purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. DBT and biphenyl were purchased from Shanghai Macklin Biochemical Co., Ltd. CTAB, ammonia solution (28%), ammonium molybdate ((NH4)6Mo7O24·4H2O), tetraethyl orthosilicate (TEOS), and TBHP (65%) were all purchased from Sinopharm Chemical Reagent Co., Ltd. All chemicals were used as received without further purification.2.2 Synthesis of MoO3/MCM-41
The method of preparing MCM-41 was referred to the literature with some adjustments.16 In a typical synthesis process, 0.0675 g of CTAB and 30 mL of deionized water were added into a 50 mL flask with 900 rpm stirring rate at 30 °C.When CTAB was dissolved, 5 mL of ammonia solution (28 wt%) was added. After stirring for 10 min, 1.2 mL of 0.05 mol L−1 (NH4)6Mo7O24 aqueous solution was added during the process to dope MoO3. 20 min later, 1 mL of TEOS was added drop by drop and continued to react for another 2 h. After the reaction was finished, the product was separated by ultrasound and centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm). And then, this product was washed one time by ethanol. The obtained product was dried at 100 °C for 10 h. Finally, the sample was calcined in air at 550 °C for 6 h to get the MoO3/MCM-41.
000 rpm). And then, this product was washed one time by ethanol. The obtained product was dried at 100 °C for 10 h. Finally, the sample was calcined in air at 550 °C for 6 h to get the MoO3/MCM-41.
2.3 Catalysis of oxidative desulfurization
The model diesel investigated in this study was made by dissolving DBT (1000 ppm) in decalin. The oxidant was prepared by solving TBHP in decalin (1.88 mg mL−1), there was no obvious enhancement when the molar ratio of TBHP to DBT is higher than 2 (the stoichiometry of the reaction) in our previous study. In the present experiment, the molar ratio was set to be 2.5 to make sure that DBT can be totally oxidized. In a ODS reaction process, 1 mL of model diesel was added into a 10 mL reaction tube under stirring at 20 °C. Then 19 mg of catalyst was added to the model diesel. After effective mixing, 1 mL of oxidant was added and continued to stir for 15 min (500 ppm DBT in the reaction system). The DBT conversion was analyzed by GC-FID; biphenyl was used as the internal standard substance. The molar ratio of activity phase (MoO3) to the substrate (DBT) were abbreviated as [CAT]/[S].2.4 Recycling of catalyst
The recycling performance of the catalyst was tested as following: after the reaction was finished, the reaction system was transferred to a centrifuge tube. After centrifuging for 10 min, the liquid phase was separated by decantation and the catalyst was washed by acetone. Subsequently, the catalyst was dried for 1 h at 100 °C. After cooling down to room temperature, the refreshed catalyst was transferred to a reaction tube. Then, the fresh model oil and oxidant were added to the reaction system for the next run.2.5 Characterization
TEM was performed at room temperature on a JEOL JEM-2000 EX transmission electron microscope using an accelerating voltage of 120 kV. Aberration-corrected high-angle annual dark-filed scanning transmission electron microscopy (HAADF-STEM) images were obtained on a Titan cubed 60-300STEM. The pore channel information was gained from N2 sorption by automatic physical adsorption analyzer (Quantachrome Instruments, Autosorb-iQ-C) at 77 K. The pore size was studied by Barrett–Joyner–Halenda (BJH) method. The composition was detected by X-ray photoelectron spectroscopy (XPS, Thermo Fisher, ESCALAB™250Xi) with Al-Kα X-ray source. The Raman spectrum was got from Renishaw InVia Raman. The content of Mo was detected by an inductively coupled plasma mass spectrometry (ICP-MS, Agilent, 7900).3. Results and discussion
3.1 Characterization of MoO3/MCM-41 nanoparticles
Fig. 1a was the TEM image of pure MCM-41; the size of the spheroid silica was around 300 nm. Fig. 1b was the TEM image of MoM-2.0; the pore channel can be seen clearly in the image which confirmed that the doping of MoO3 didn't influence the structure of MCM-41 obviously. Fig. 1c was the HADDF-STEM image of MoM-2.0, and the red circles marked MoO3 subnanoclusters with the size around 0.9 nm (Fig. 1d).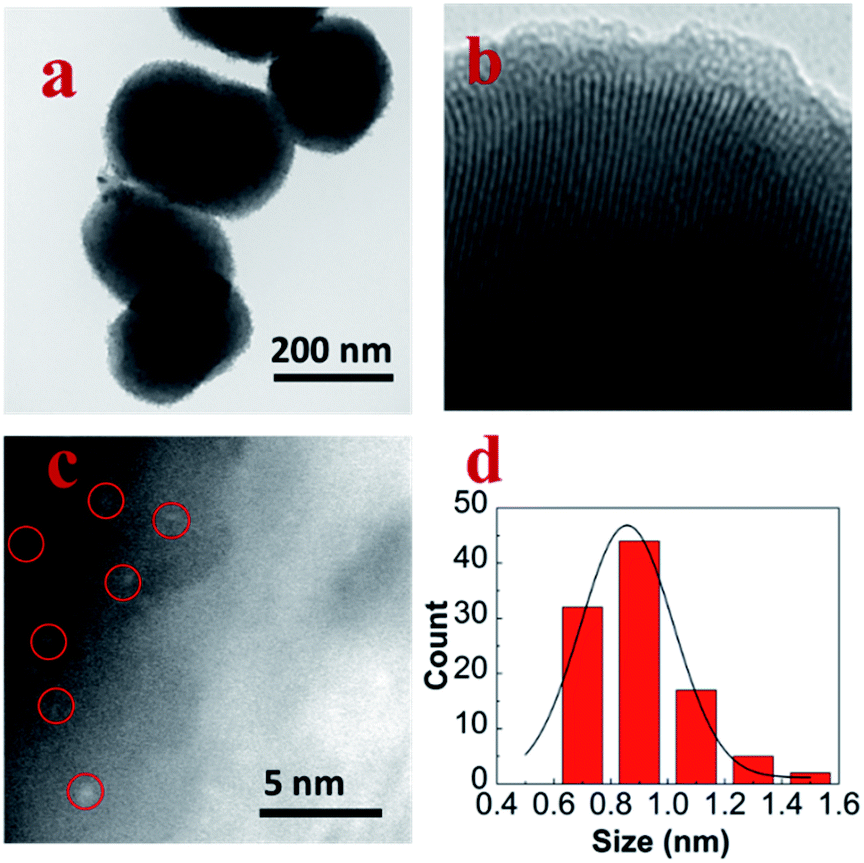 | ||
| Fig. 1 (a) TEM image of pure MCM-41, (b) TEM image of MoM-2.0, (c) HADDF-STEM image of MoM-2.0, and (d) size distribution histogram of MoO3 in MoM-2.0. | ||
The surface area and pore size of the prepared materials were analyzed by nitrogen sorption. Both pure MCM-41 and MoM-2.0 showed a typical type IV adsorption isotherm with an H1 hysteresis loop of MCM-41 (Fig. 2a and c), which was consistent with the results of TEM. Pure MCM-41 possesses high surface area (976 m2 g−1), and doping of MoO3 reduces the surface area to 734 m2 g−1 (MoM-2.0). The most probable pore diameters were similar for both pure MCM-41 and MoM-2.0 (2.7 nm, Fig. 2b and d), which confirmed that no obvious influence was caused to the structure by doping MoO3.
 | ||
| Fig. 2 N2 sorption curve and pore size distribution of MCM-41 (a and b) and MoM-2.0 (c and d). | ||
XPS results were shown in Fig. 3. Mo, O and Si were detected in the survey spectrum (Fig. 3a). The existence of Mo6+ in MoM-2.0 was evidenced by the shoulders observed on the peaks 236.3 eV and 233.1 eV, which were fitted to the binding energy of Mo 3d3/2 and 3d5/2 of MoO3 (Fig. 3b), respectively. Fig. 3c showed the spectrum of Si 2p with 103.8 eV belonging to the binding energy of SiO2. There were three peaks observed in the O 1s spectrum (Fig. 3d). 533.2 eV and 533.9 eV which could be assigned to the peaks of Si–O–Si and Si–OH, respectively.17 No obvious peak around 531 eV fitted to MoO3 was shown.18 However, a peak at 532.6 eV was observed, which could be assigned to Si–O–Mo. The signal of Mo–O shifted from 531 eV to 532.6 eV due to the connection with Si, which could confirm the formation of Si–O–Mo bonds.
 | ||
| Fig. 3 XPS spectrum of MoM-2.0: (a) survey spectrum; (b) Mo 3d; (c) Si 2p; (d) O 1s. | ||
Raman spectroscopic investigation was also conducted to analyze the surface structure of MoM-2.0. Fig. 4 was the Raman spectrum of pure MCM-41 and MoM-2.0. Compared with pure MCM-41, one higher peak (976 cm−1) and three new peaks (670 cm−1, 395 cm−1 and 235 cm−1) were shown in the spectrum of MoM-2.0. The peak at 976 cm−1 was attributed to asymmetric stretching vibration of Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. The peak at 235 cm−1 was due to twist vibration of MoO. The peaks at 670 cm−1 and 395 cm−1 were attributed to asymmetric stretching and scissor vibration of O–Mo–O, respectively.19,20
O. The peak at 235 cm−1 was due to twist vibration of MoO. The peaks at 670 cm−1 and 395 cm−1 were attributed to asymmetric stretching and scissor vibration of O–Mo–O, respectively.19,20
 | ||
| Fig. 4 Raman spectrum of (a) pure MCM-41 and (b) MoM-2.0. | ||
Combining the above results of HADDF-STEM, XPS and Raman, MoO3 was highly dispersed on MCM-41. MoO3 subnanoclusters connect with SiO2 through Si–O–Mo, and lots of terminal MoO existed on the surface of MoM-2.0. The formation process and model structure of MoM-2.0 are showed in Scheme 1. After the self-assembling of CTAB, Mo7O246− connects with CTAB through electrostatic interaction. Then, the hydrolysis of TEOS makes up a net of SiO2, and the Mo7O246− was covered by the net. Subsequently, the CTAB is removed by calcining and Si–O–Mo is generated on the surface of SiO2.
 | ||
| Scheme 1 Formation and structure model of MoO3/MCM-41. | ||
3.2 Catalysis of oxidative desulfurization
The results of ODS were shown in Table 1. Subnanocluster MoO3 shows high catalytic activities in our previous study and the TOF of ODS can attain 50 h−1 (entry 1). Because of the high Mo content of MoM-2.0, only 1.9 mg catalyst per mL of model diesel was added into the reaction. The DBT conversion and TOF of entry 1 and entry 2 were similar. The subnanocluster MoO3 possessed similar catalytic activity whether supported on ordered or disordered mesoporous silica. Due to the higher content, the ODS productivity of MoM-2.0 was higher than MoO3/SiO2, which only need 1.9 mg of catalyst to get a comparable conversion. Due to the high ODS ability of MoM-2.0 at 70 °C, we further checked the catalytic performance at 20 °C (entry 3–5). When 1.9 mg catalyst was added per mL model diesel for entry 4, DBT conversion is 42.4% after 15 min reaction with a TOF = 23 h−1. When the catalyst amount was increased to 19 mg per mL model diesel (entry 4–5), the DBT conversion was 94.4% after 10 min reaction and can be totally removed within 15 min. The catalyst showed outstanding activity at room temperature (20 °C).| Entry | DBT initial concentration/ppm | Catalyst | [Cat]/[S] | Cat/mg mL−1 | T/°C | t/min | DBT conversion% | TOF/h−1 |
|---|---|---|---|---|---|---|---|---|
| 1 (ref. 13) | 500 | MoO3/SiO2 | 0.075 | 19 | 70 | 15 | 94.1 | 50 |
| 2 | 500 | MoM-2.0 | 0.075 | 1.9 | 70 | 15 | 89.6 | 48 |
| 3 | 500 | MoM-2.0 | 0.075 | 1.9 | 20 | 15 | 42.4 | 23 |
| 4 | 500 | MoM-2.0 | 0.75 | 19 | 20 | 10 | 94.4 | 8 |
| 5 | 500 | MoM-2.0 | 0.75 | 19 | 20 | 15 | 100 | 5 |
The results of recycling test were shown in Fig. 5. The catalyst was refreshed by acetone washing. No obvious inactivation was found during the recycling process. The DBT conversion can get 94.8% in the 5 run reaction. And the recycling result confirms that MoO3 was settled on the surface of MCM-41.
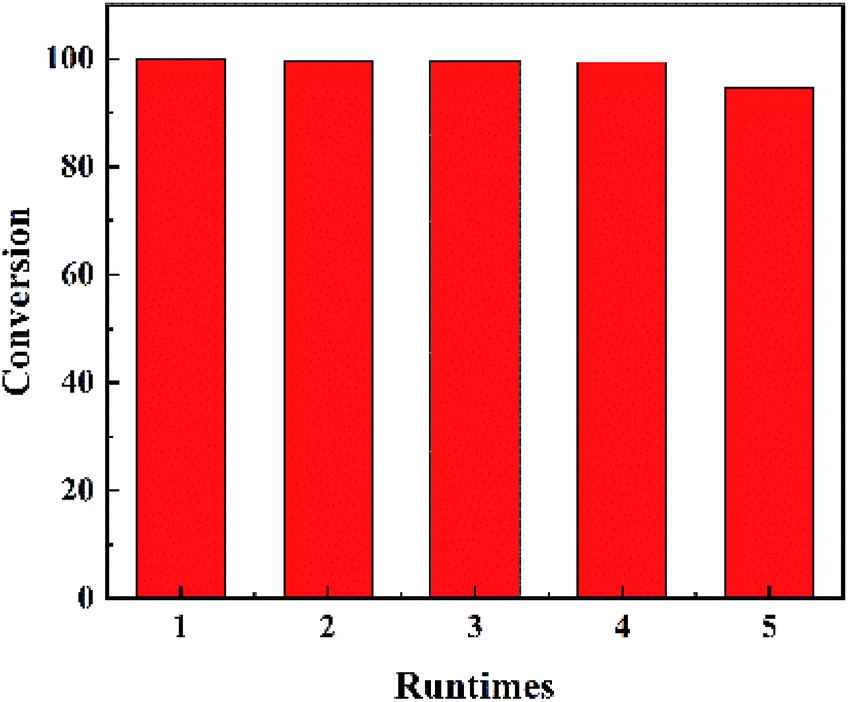 | ||
| Fig. 5 Recycling tests for MoM-2.0. | ||
4. Conclusions
In this study, MoO3 supported on MCM-41 was successfully synthesized. The structure of prepared catalyst was investigated by TEM, HADDF-STEM, N2 sorption, XPS and Raman. MoO3 subnanoclusters were highly dispersed on MCM-41, and the MoO3 connected with SiO2 through Si–O–Mo. So the Mo content was successfully increased 10 times by changing the support from disordered mesoporous silica to MCM-41. MoO3/MCM-41 showed high catalytic ability at 20 °C, DBT in the model diesel could be totally removed within 15 min. The recycle ability at room temperature was also outstanding, no obvious decrease was observed after 5 runs reaction within 15 min.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge the financial support of the National Natural Science Foundation of China (No. 21506026) and the China Postdoctoral Science Foundation (No. 2016M601311).Notes and references
- S. K. Guttikunda, G. R. Carmichael, G. Calori, C. Eck and J.-H. Woo, Atmos. Environ., 2003, 37, 11–22 CrossRef CAS.
- I. Mejia-Centeno, A. Martinez-Hernandez and G. A. Fuentes, Top. Catal., 2007, 42–43, 381–385 CrossRef.
- V. A. Sethuraman and J. W. Weidner, Electrochim. Acta, 2010, 55, 5683–5694 CrossRef CAS.
- L. Yang, X. Li, A. Wang, R. Prins, Y. Chen and X. Duan, J. Catal., 2015, 330, 330–343 CrossRef CAS.
- D. Wang, E. W. Qian, H. Amano, K. Okata, A. Ishihara and T. Kabe, Appl. Catal., A, 2003, 253, 91–99 CrossRef CAS.
- Z. Jiang, H. LÜ, Y. Zhang and C. Li, Chin. J. Catal., 2011, 32, 707–715 CrossRef CAS.
- A. Chica, A. Corma and M. E. Dómine, J. Catal., 2006, 242, 299–308 CrossRef CAS.
- V. V. D. N. Prasad, K.-E. Jeong, H.-J. Chae, C.-U. Kim and S.-Y. Jeong, Catal. Commun., 2008, 9, 1966–1969 CrossRef CAS.
- W. A. W. A. Bakar, R. Ali, A. A. A. Kadir and W. N. A. W. Mokhtar, Fuel Process. Technol., 2012, 101, 78–84 CrossRef CAS.
- Q. Tang, S. Lin, Y. Cheng, S. Liu and J.-R. Xiong, Ultrason. Sonochem., 2013, 20, 1168–1175 CrossRef CAS.
- D. H. Wang, N. Liu, J. Y. Zhang, X. Zhao, W. H. Zhang and M. H. Zhang, J. Mol. Catal. A: Chem., 2014, 393, 47–55 CrossRef CAS.
- A. Bazyari, A. A. Khodadadi, A. Haghighat Mamaghani, J. Beheshtian, L. T. Thompson and Y. Mortazavi, Appl. Catal., B, 2016, 180, 65–77 CrossRef CAS.
- J. Wang, W. Wu, H. Ye, Y. Zhao, W.-H. Wang and M. Bao, RSC Adv., 2017, 7, 44827–44833 RSC.
- D. Yue, L. Jiaheng, Z. Lina, G. Z. ran, D. Xiaodi and L. Junsheng, Mater. Res. Bull., 2018, 105, 210–219 CrossRef CAS.
- L. Zhang, Y. Ren, W. Liu, A. Wang and T. Zhang, Natl. Sci. Rev., 2018, 5, 653–672 CrossRef.
- X.-J. Lin, A.-Z. Zhong, Y.-B. Sun, X. Zhang, W.-G. Song, R.-W. Lu, A.-M. Cao and L.-J. Wan, Chem. Commun., 2015, 51, 7482–7485 RSC.
- M. E. Simonsen, C. Sonderby, Z. Li and E. G. Sogaard, J. Mater. Sci., 2009, 44, 2079–2088 CrossRef CAS.
- M. Anwar, C. A. Hogarth and R. Bulpett, J. Mater. Sci., 1989, 24, 3087–3090 CrossRef CAS.
- M. Dieterle, G. Weinberg and G. Mestl, Phys. Chem. Chem. Phys., 2002, 4, 812–821 RSC.
- M. Dieterle and G. Mestl, Phys. Chem. Chem. Phys., 2002, 4, 822–826 RSC.
| This journal is © The Royal Society of Chemistry 2019 |