
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlobal ab initio exploration of potential energy surfaces for radical generation in the initial stage of benzene oxidation†
Hai-Bei Li * and
Qingqing Jia
* and
Qingqing Jia
School of Ocean, Shandong University, Weihai 264209, People's Republic of China. E-mail: lihaibei@sdu.edu.cn
First published on 29th May 2019
Abstract
The potential energy surfaces (PESs) of benzene oxidation by molecular oxygen were explored using the anharmonic downward distortion following (ADDF) and artificial force induced reaction (AFIR) methods of the global reaction route mapping (GRRM) strategy. The reaction mechanism of benzene activation by initial molecular oxygen depends on the combustion temperature. At high temperature, the benzene molecule could be oxidized by abstracting hydrogen atoms and form the radical fragments, C6H5 and OOH. However, before reaching its auto-ignition point, the formation of a singlet bridging peroxide molecule C6H6O2 from the triplet reactants via electronic non-adiabatic transition will play a critical role in the increase of the combustion temperature by the generation of initial free radicals. Bridging peroxide C6H6O2 could isomerize to other stable isomers by a consecutive series of oxygen and hydrogen atom transfers. Importantly, these C6H6O2 isomers are vital sources of free radical generation in the initial stage of benzene oxidation. Free radicals, such as OOH, O, and OH, could be generated during the further oxidation of these oxygenated hydrocarbon species C6H6O2 due to the presence of active groups or sp3-C–H bonds.
1. Introduction
The pervasive use and importance of aromatic compounds in fuels contrast strikingly with uncertainties in their major reaction pathways and limited chemical kinetic and thermodynamic data. Benzene is one of the simplest aromatic hydrocarbons and its chemical kinetics of combustion have long been of interest as a mirror to the study of the chemical and physical processes of the formation of polycyclic aromatic hydrocarbons (PAH) and soot.Many efforts have been made experimentally and theoretically in benzene combustion over the last three decades to understand and evaluate the important reaction pathways and to validate the mechanisms.1–15 It is well known that hydrocarbon combustion follows a chain reaction mechanism, where free radicals play a dominant role in the destruction of reactant molecules. Free radicals such as OOH, OH, O, and H, are well known to be active in the benzene flame10,16–18 as well as in other hydrocarbon combustion.14,19–21 In near-sooting premixed low-pressure flames of benzene, Bittner and Howard2 have observed an abundance of the free radicals OH, O, OOH, and H in the benzene flame and suggested that the activation and decomposition of benzene are initiated by the oxidation of atomic O, while that by molecular oxygen and other free radicals would be too slow to account for their experimental observations, which is consistent with the observations by Ko et al.22 While, Madronich and Felder17 found that the hydrogen abstraction channel by OH radical plays a critical role in benzene consumption, giving an important intermediate phenyl radial. In contrast, in hydrogen-rich environments, hydrogen abstraction by atomic H is predicted to be important in the benzene oxidation, due to its low activation barrier.8
These benzene destruction mechanisms by free radical species have been well proven by a large number of kinetic modeling studies, which are used to present a quantitative description of the combustion processes.6–9,16–18,22 In addition to these combustion models, investigations of the reaction pathways of benzene activation by OH and H radicals have been undertaken using high-level ab initio methods,23,24 which provide evidence that hydrogen abstractions from benzene by these radicals are favorable with a low activation energy of around 10 kcal mol−1. All of these investigations indicated that free radicals, such as OH, O and atomic H, play crucial roles in benzene decomposition and subsequent PAH formation. A chain reaction mechanism consists of several steps, which are the initiation of radicals, then their propagation and branching, followed by a termination process. It is obvious that, until now, almost all investigations of benzene combustion have focused mainly on the reactions between free radicals and benzene or its derivatives, that is, on the propagation and branching reactions.
The significance of these free radicals in benzene combustion makes it reasonable to raise questions such as how these free radicals are generated in the initial stage of benzene oxidation. The proposed mechanism for the initiation step of benzene oxidation by molecular oxygen is the generation of a phenyl radical through hydrogen abstraction.4,25 Westbrook and coworkers16 using chemical kinetic modeling have proposed that in benzene combustion, hydrogen abstraction by molecular oxygen is the principal initiation reaction as well as that by various free radicals. However, theoretical studies of benzene oxidation, that is C6H6 + O2, are still rare, except for recent work,26 where the barrier to hydrogen abstraction by molecular oxygen was calculated to be around 250 kJ mol−1 by density functional theory calculations. The higher activation barrier implies that it is difficult to generate the free radicals via direct hydrogen abstraction by molecular oxygen in the initial stage of benzene oxidation. Despite many experimental and modeling efforts, a large number of uncertainties still exist regarding the main benzene combustion kinetics and reaction pathways, such as the initial chemical reactions of benzene oxidation, C6H6 + O2, the chemistry of free radical generation, and the fate of the oxygenated C6 and C5 species. For instance, it was reported that an oxygenated hydrocarbon compound C6H6O2 achieves a certain population in an early stage of benzene combustion.2 However, its generation mechanism and how this species decomposes into free radicals have not yet been explained well. The importance of the generation of free radicals in the initial stage of combustion could be imagined, which will induce and simultaneously determine the subsequent ignition and combustion processes. Therefore, it is necessary to systematically investigate the elementary processes of free radical production in the initial stage of benzene oxidation.
Global reaction route mapping (GRRM)27 has been confirmed as finding all local minima and transition states on single potential energy surfaces and providing a systematic elucidation of complex chemical reaction mechanisms. Thus, for free radical generation in benzene oxidation, GRRM is a powerful tool to globally explore its reaction mechanism. The present work focuses on systematically elucidating the fundamental chemical and physical processes taking place in the initial stage of benzene oxidation, with special attention on free radical generation. The lowest reaction routines for benzene activation and the generation of free radicals will be presented by systematically exploring all the possible chemical processes which involve a series of competitive reaction paths. This detailed theoretical study could provide deeper insights into the chemical kinetics and important reaction pathways involved in benzene oxidation.
2. Computational methods
All the potential energy surfaces (PESs) were explored by the global reaction route mapping (GRRM) program28 in which energies, gradient, and Hessian were calculated using the Gaussian 09 program29 as an external subroutine. The reaction pathways between two molecules were searched using the artificial force induced reaction (AFIR) method.27 The isomerization and dissociation pathways starting from stable intermediates were explored by the GRRM method,30,31 where the l-ADDF option of the GRRM method, which searches for the 10 lowest barrier paths around each intermediate, was adopted in this study.32 These automated searches, performed at the UB3LYP/6-31+G(d) level, gave several reaction pathways starting from each intermediate. The pathways with barriers higher than Elowest + 50 kJ mol−1 were omitted, where Elowest is the barrier height for the most preferred path among them. All obtained structures were re-optimized at the UB3LYP/cc-pVTZ level, and frequency analysis calculations were performed to confirm the local minima (with zero imaginary frequency) and the transition states (TSs, possessing one and only one imaginary frequency) on the potential energy surfaces. The intrinsic reaction coordinate (IRC) method33 was also performed to ensure that the TS connects two appropriate local minima in the reaction pathway. Zero-point energy (ZPE) corrections were taken into account during the calculation of barrier heights. The Gibbs free energies, which are presented in the ESI,† were calculated including ZPE and thermal correction at 1000.0 K. Some minimum energy structures on seam-of-crossing hyperspaces (MESXs) were searched as critical points of non-adiabatic transitions between the ground singlet and triplet PESs at the same level. The UB3LYP method might result in significant error due to the absence of dispersion or significant spin contamination. For MESXs searching, Maeda and coworkers26 have confirmed that, for the oxidation of hydrocarbon, the UB3LYP method in combination with the aug-cc-pVDZ basis set could give similar results and correct energy trends which were obtained at CASPT2/aug-cc-pVDZ. Thus, considering the high requirement for computing resources in this work, we applied UB3LYP/cc-pVTZ to investigate the potential energy surfaces of benzene oxidation.Based on the thermodynamic parameters obtained from the above calculation, we calculated the reaction rate constant for the first step of benzene oxidation, utilizing the well-known Rice–Ramsperger–Kassel–Marcus (RRKM) theory with the expression as follows:
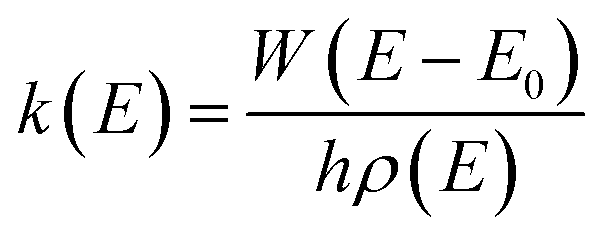 | (1) |
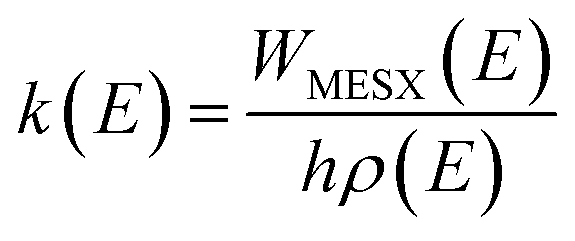 | (2) |
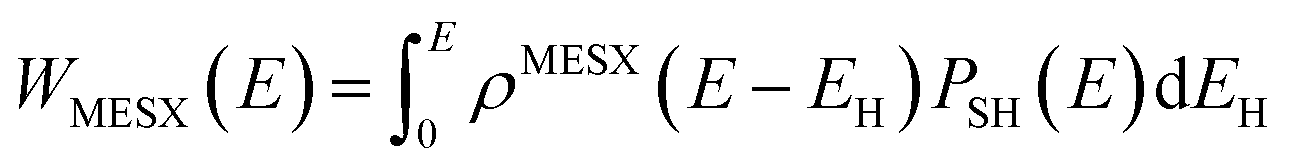 | (3) |
| PSH(EH) = (1 + P)(1 − P) | (4) |
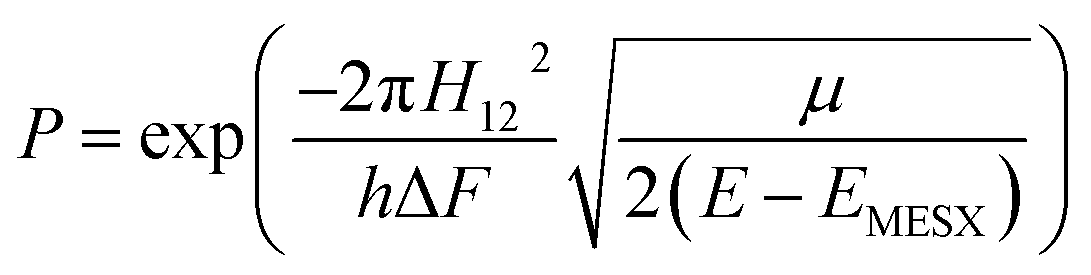 | (5) |
3. Results and discussion
3.1 Activation of benzene
Fig. 1 shows all of the optimized structures of intermediates, transition states, and MESXs with their energies for the considered reaction pathways during benzene interacting with the first molecular oxygen. The singlet and triplet PESs for all possible reaction pathways of benzene activation are presented in Fig. 2. The corresponding Gibbs free energies are supplied in the ESI (Fig. S1–S4†). In the initial stage of benzene oxidation, ground state triplet molecular oxygen instead of excited singlet state would have a high population. Interestingly, for the triplet reaction pathways, two well-known radicals, phenyl and phenoxy, are generated through the reaction pathways 1′ → TS1′/9′ → 9′ → C6H5 + OOH, and 1′ → TS1′/11′ → 11′ → TS11′/12′ → 12′→ C6H5O + OH, respectively. The generation of a phenyl radical is directly by the hydrogen abstraction process (C6H6 + 3O2 → C6H5 + OOH). This hydrogen abstraction mechanism has been proposed for the activation of benzene in previous studies.4,25 Direct hydrogen abstraction from benzene by triplet molecular oxygen is quite endothermic, requiring around 260.0 kJ mol−1 in order to generate the radical fragments C6H5 and OOH. This high barrier makes it difficult to activate benzene through direct hydrogen abstraction by initial molecular oxygen at a low temperature. This is consistent with a theoretical study concerning the initial chemical events of benzene oxidation by molecular dynamic simulation using a reactive force field,37 where there is no C–H bond cleavage by molecular oxygen. Compared to hydrogen abstraction, the generation of a phenoxy radical is much more difficult. The initial molecular oxygen inserts into the C–H bond forming phenyl hydroperoxide, followed by the dissociation of O–O peroxide group generating phenoxy, C6H5O and OH. It has been proposed from kinetic modeling that species C6H5O is highly abundant and is considered to play critical roles in the subsequent formation of small fragments.18 While here in the former insertion process, transition state TS1′/11′ lies 332.1 kJ mol−1 higher than the reactants. This higher barrier indicates that species C6H5O is not produced by initial molecular oxygen, but might be by atomic oxygen, as suggested by Richter et al.18 On the other hand, the higher barrier of this path also demonstrates that the formation of phenyl hydroperoxide is highly unlikely by the addition of initial molecular oxygen.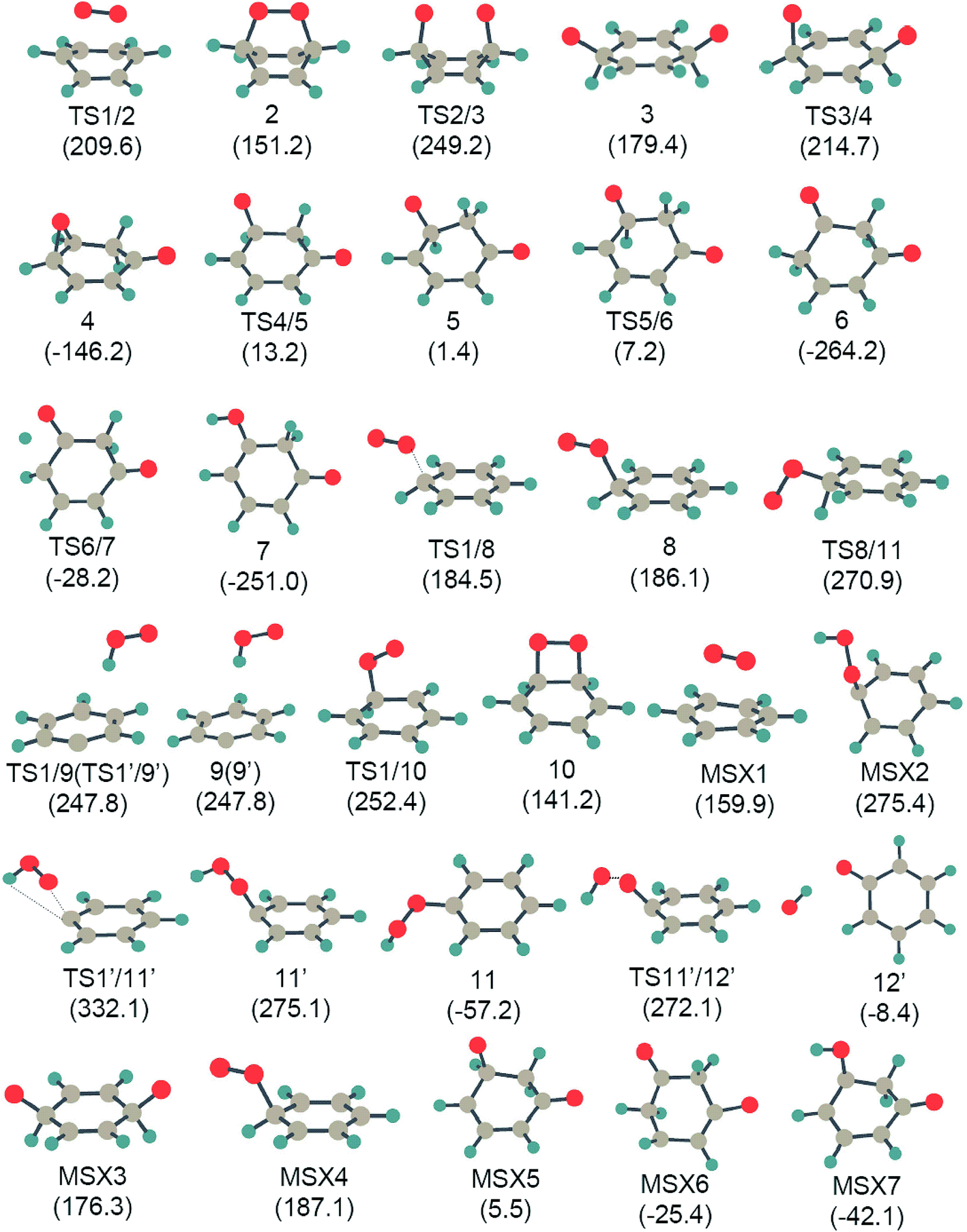 | ||
| Fig. 1 The optimized structures for all intermediates, transition states, and MESX, with their energies (in parenthesis) existing in Fig. 2 at the UB3LYP/cc-pVTZ level. Energy values (in kJ mol−1) are relative to the sum of the energy of triplet molecular oxygen plus benzene. | ||
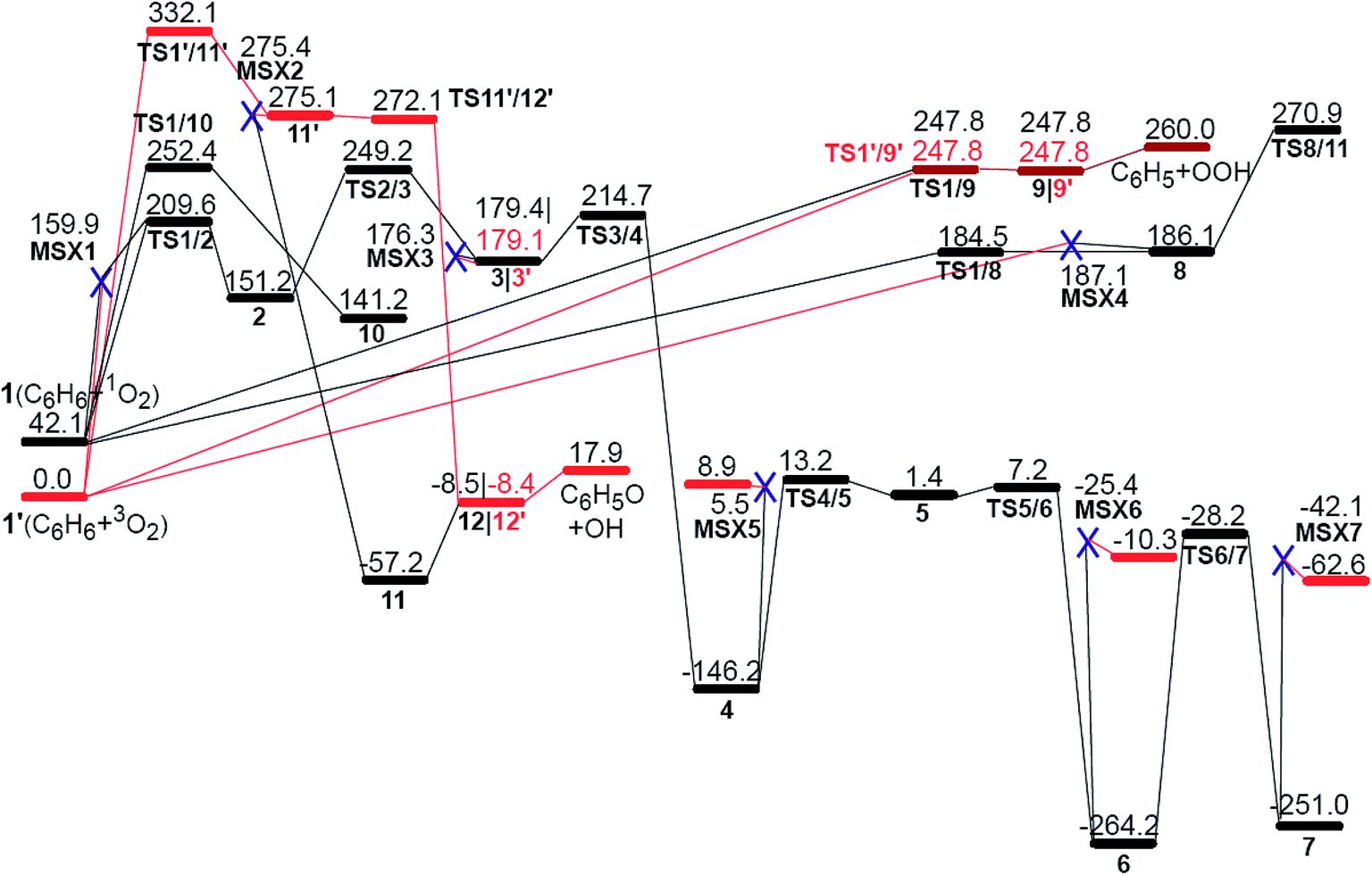 | ||
| Fig. 2 The potential energy surfaces (red for triplet and black for singlet) for benzene activation at UB3LYP/cc-pVTZ level. Energy values (in kJ mol−1) are relative to the sum of the energy of triplet molecular oxygen plus benzene. The MESX points are shown with a blue X. The corresponding structures are illustrated in Fig. 1. | ||
The other likely pathway for benzene activation, i.e., production of 2, 8, 9, or 10, involves a singlet molecular oxygen generated by the non-adiabatic transition from triplet to singlet state PES through the lowest MESX1 (see Fig. 2). The generation of singlet molecular oxygen has been discussed in detail in ref. 26. The spin-orbital coupling for MESX1 is calculated with a value of 159.9 cm−1 using the state-averaged CASSCF method and the cc-pVTZ basis set with an active space including 10 electrons and 8 orbitals. Unsurprisingly, the generated singlet molecular oxygen could react further with another benzene molecule to produce intermediates 8, 9, or 10 as well as intermediate 2. Thermodynamically, intermediate 8 with one trans-·C–C–O–O· group is not stable. Instead of transforming into phenyl hydroperoxide via a high barrier transition state TS8/11, it could quickly reverse to the reactants, C6H6 + 1O2, through TS1/8 or C6H6 + 3O2 via MESX4. Correspondingly, the molecular oxygen could also attack one carbon atom, generating a cis-·C–C–O–O· group in TS1/10, producing intermediate 10 with 4/6-membered rings. The barrier for this path is comparable to the direct hydrogen abstraction process of benzene on the singlet state PES, 1 → TS1/9 → 9 → C6H5 + OOH, both requiring around 205 kJ mol−1 ∼ 210 kJ mol−1. In contrast to all these reaction paths starting from reactants C6H6 + 1O2 (3O2), the reaction pathway, 1′ → MESX1 → TS1/2 → 2, is the most favorable way in benzene activation, which requires around 209.6 kJ mol−1 to produce the bridging peroxide molecule 2. This is in good agreement with the experimental results,2 where oxygenated hydrocarbon compound C6H6O2 has been observed early in the benzene flame. In order to assess the relative importance of reaction channels under different temperature conditions, we performed a calculation of the reaction rate constant (k) based on the well-known RRKM theory, as shown in Fig. 3. At a lower temperature, the production of bridging peroxide molecule 2 is predominant due to the easier non-adiabatic transition from triplet to singlet molecular oxygen. With an increase in combustion temperature, the hydrogen abstraction processes 1 (1′) → TS1/9(TS1′/9′) → 9(9′) → C6H5 + OOH become more important. At a temperature higher than 1200 K, which is much higher than its auto-ignition temperature, the reaction rate of hydrogen abstraction by molecular oxygen has exceeded that of the formation of the bridging peroxide molecule 2. This illustrates that at high temperature, the benzene molecule could be easily oxidized by abstracting the hydrogen atoms and forming radical fragments C6H5 and OOH. However, before it reaches its auto-ignition point, the production of bridging peroxide molecule 2 will play a critical role in the increase of the combustion temperature by the generation of initial free radicals.
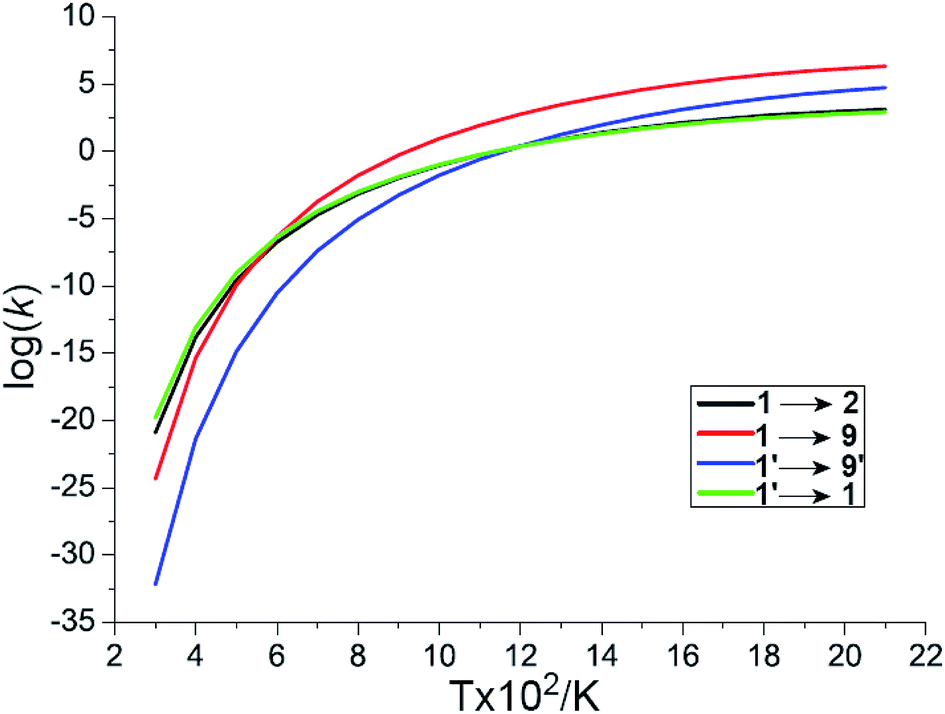 | ||
| Fig. 3 The reaction rate constants of four reaction channels for the activation of benzene. | ||
Obviously, the bridging peroxide structure of intermediate 2 is not stable kinetically and thermodynamically, especially at high temperature. The unstable feature of structure 2 is characterized by the approach of another triplet molecular oxygen, which results in the scission of the peroxy O–O bond forming complex 3 + O2 with a barrier height of 97.8 kcal mol−1 (the IRC calculation with the TS and product are presented in Fig. S5†). This indicates that the entropy effect at high temperature has a significant influence on the stability of intermediate 2. This characteristic of intermediate 2 might be the reason why some research groups preferred the hydrogen abstraction mechanism in the initiation step,4,25 instead of the formation of a bridging peroxide molecule. We stepwisely explored the global minimum isomerization processes for the oxygenated hydrocarbon compound C6H6O2 based on the structure of intermediate 2. The first isomerization step 2 → 3, the scission of the bridging O–O peroxy bond and the formation of two sp3-C atoms and two radical sites C–O·, was found to have a barrier height of 98.0 kJ mol−1, and this reaction is endothermic by 28.2 kJ mol−1. Compared to bridging peroxide structure 2, intermediate 3 is more active with two radical sites. It is liable to transform into structure 4 by oxygen transfer, forming one epoxide group. Step 3 → 4 has a barrier height of only 35.5 kJ mol−1 and is quite exothermic by 325.6 kJ mol−1 (see Fig. 2). The active epoxide group in structure 4 induces further isomerization, 4 → TS4/5 → 5 → TS5/6 → 6, corresponding to oxygen and hydrogen transfer, respectively, generating more stable intermediates 6. The high stability of 4 and 6 provides direct evidence to explain the long-term existence of the species C6H6O2 early in the benzene flame.2 Further carbonyl-phenol tautomerization from 6 to 7 requires a higher barrier height with a value of 236.0 kJ mol−1, which implies that this isomerization has a low probability of occurrence. The small mole fraction of species C6H6O2 in the benzene flame, which is shown in Fig. 7 in ref. 2, indicates that, in addition to these consecutive isomerization processes, 2 → 3 → 4 → 5 → 6, it is highly likely there will be other competitive or more favorable reaction pathways for the consumption of the species C6H6O2. Therefore, we will consider the further oxidation of intermediates 3, 4, and 6 by adding another molecular oxygen in the following section.
3.2 Generation of radicals
The computed PESs for the interactions between intermediates 3, 4, 6 and molecular oxygen are depicted in Fig. 4–6. As discussed above, bridging peroxide intermediate 2 could automatically transform into isomer 3 following the approach of the second molecular oxygen. Thus, instead of considering the oxidation of intermediate 2, we present the PES for the oxidation of intermediate 3 in Fig. 4. Structure 3 has two H–C–O· groups which find it easier to form C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbonyl group by losing the hydrogen atoms. Compared to the isomerization step 3 → 4, the hydrogen abstraction process by the second molecular oxygen is more favorable kinetically for reaction path 13 → 14 with a calculated barrier of only 7.3 kJ mol−1. It was found that the interaction between triplet intermediate 3′ and molecular oxygen has almost the same barrier height as that in reaction 13 → 14. Step 13 → 14 is found to be 205.7 kJ mol−1 exothermic and product 14 is a complex with one carbonyl group hydrogen-bonded to an OOH radical. After this, two possible pathways could happen, (1) the OOH radical moves away from species 15 after absorbing heat of 42.0 kJ mol−1 and forms free radicals, which could play pivotal roles with other radicals in the following ignition and combustion processes, or (2) the OOH radical, roaming to the other side of fragment 15, abstracts hydrogen from the other sp3-C atom with only a small barrier of 27.9 kJ mol−1, and produces complex 19 consisting of H2O2 and C6H4O2 fragments. The production of H2O2 is another source of free radical generation. It has been found that the decomposition of H2O2 could produce OH or OOH radicals under different experimental conditions.38 Although the barriers for these two processes are similar, the OOH radical is likely to dissociate before finding the H atom to be abstracted, especially at high temperature because the migration length of the OOH in the isomerization from 14 to 18 is pretty long and also because the dissociation energy (15 + OOH) is 18.1 kJ mol−1 lower than that of TS18/19. In this case, the first free radical in the initiation step of benzene oxidation should be OOH. If there are many free radicals such as OOH around 15, it will immediately be dehydrogenated into 1,4-benzoquinone through TS like TS18/19. The produced intermediate 15, C6H5O2, has three possibilities for isomerization to structures 16, 20, and 21 by oxygen and hydrogen transfer, respectively. Thermodynamically, reaction 15 → 21 is more exothermic with an energy of 194.6 kJ mol−1, while this reaction has a higher barrier of 58.1 kJ mol−1, compared to reactions 15 → 16 and 15 → 20 with only 30.5 kJ mol−1 and 37.6 kJ mol−1, respectively. Intermediate 16 is easier to generate kinetically, which is quickly followed by the formation of a seven-membered ring isomer 17 via C–C bond cleavage of the epoxide group. The structures and their energies for isomers 15–21 have been studied by Mebel and coworkers,39 to elucidate the reaction mechanisms of the fundamental reaction, C6H5 + O2, which produces atomic hydrogen or oxygen. It could be imagined that the isomers 15 and 20 could immediately lose a hydrogen atom to form a stable conjugated structure 1,4-benzoquinone by another molecular oxygen or other radicals in a later stage of combustion.
O carbonyl group by losing the hydrogen atoms. Compared to the isomerization step 3 → 4, the hydrogen abstraction process by the second molecular oxygen is more favorable kinetically for reaction path 13 → 14 with a calculated barrier of only 7.3 kJ mol−1. It was found that the interaction between triplet intermediate 3′ and molecular oxygen has almost the same barrier height as that in reaction 13 → 14. Step 13 → 14 is found to be 205.7 kJ mol−1 exothermic and product 14 is a complex with one carbonyl group hydrogen-bonded to an OOH radical. After this, two possible pathways could happen, (1) the OOH radical moves away from species 15 after absorbing heat of 42.0 kJ mol−1 and forms free radicals, which could play pivotal roles with other radicals in the following ignition and combustion processes, or (2) the OOH radical, roaming to the other side of fragment 15, abstracts hydrogen from the other sp3-C atom with only a small barrier of 27.9 kJ mol−1, and produces complex 19 consisting of H2O2 and C6H4O2 fragments. The production of H2O2 is another source of free radical generation. It has been found that the decomposition of H2O2 could produce OH or OOH radicals under different experimental conditions.38 Although the barriers for these two processes are similar, the OOH radical is likely to dissociate before finding the H atom to be abstracted, especially at high temperature because the migration length of the OOH in the isomerization from 14 to 18 is pretty long and also because the dissociation energy (15 + OOH) is 18.1 kJ mol−1 lower than that of TS18/19. In this case, the first free radical in the initiation step of benzene oxidation should be OOH. If there are many free radicals such as OOH around 15, it will immediately be dehydrogenated into 1,4-benzoquinone through TS like TS18/19. The produced intermediate 15, C6H5O2, has three possibilities for isomerization to structures 16, 20, and 21 by oxygen and hydrogen transfer, respectively. Thermodynamically, reaction 15 → 21 is more exothermic with an energy of 194.6 kJ mol−1, while this reaction has a higher barrier of 58.1 kJ mol−1, compared to reactions 15 → 16 and 15 → 20 with only 30.5 kJ mol−1 and 37.6 kJ mol−1, respectively. Intermediate 16 is easier to generate kinetically, which is quickly followed by the formation of a seven-membered ring isomer 17 via C–C bond cleavage of the epoxide group. The structures and their energies for isomers 15–21 have been studied by Mebel and coworkers,39 to elucidate the reaction mechanisms of the fundamental reaction, C6H5 + O2, which produces atomic hydrogen or oxygen. It could be imagined that the isomers 15 and 20 could immediately lose a hydrogen atom to form a stable conjugated structure 1,4-benzoquinone by another molecular oxygen or other radicals in a later stage of combustion.
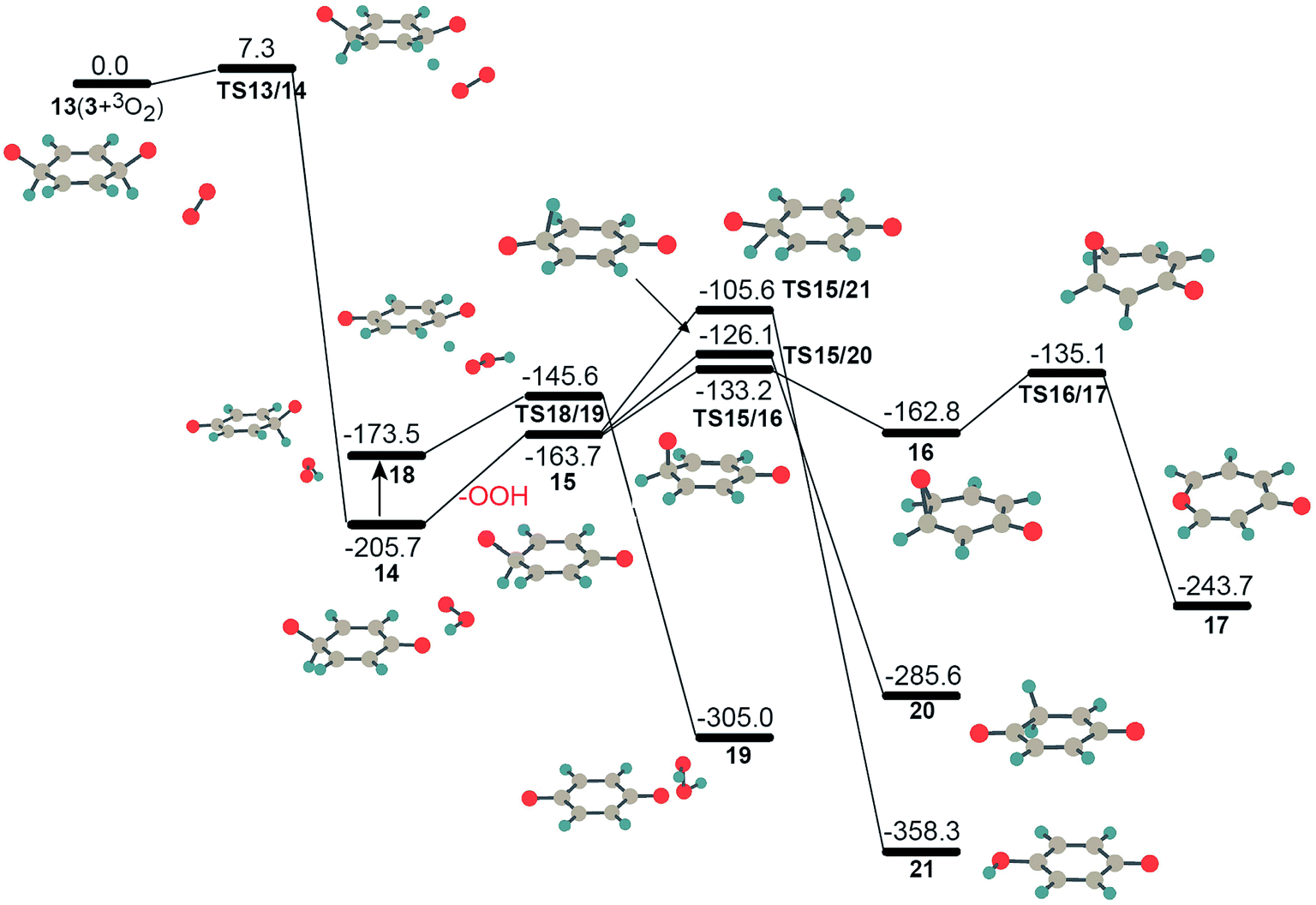 | ||
| Fig. 4 The potential energy surfaces for the oxidation of intermediate 3 by triplet molecular oxygen at UB3LYP/cc-pVTZ level. Energy values (in kJ mol−1) are relative to the sum of the energy of triplet molecular oxygen plus intermediate 3. | ||
In addition to isomerization to 5, there are two alternative reaction pathways for intermediate 4, which are further oxidation by another molecular oxygen, as illustrated in Fig. 5. Unlike the active structure 3 with its radical sites, intermediate 4 has one epoxy group and two sp3-C–H bonds, which could result in (1) a ring-opening in the epoxy group and the subsequent production of a chain structure with a terminal peroxide radical, 22 → TS22/23 → 23 → 24, or (2) hydrogen atom abstraction from an sp3-C atom, generating an OOH radical and intermediate 16, 22 → TS22/34 → 34 → 16. The ring-opening exhibits a barrier of 157.8 kJ mol−1, which is close to the corresponding values of 160.2 kJ mol−1 (TS22/34) and 159.4 kJ mol−1 (TS4/5) for the parallel hydrogen abstraction and isomerization processes, respectively, indicating that these three reaction pathways for intermediate 4 are competitive. On the one hand, hydrogen abstraction of intermediate 4 with molecular oxygen could produce an OOH radical. On the other hand, chain intermediate 24 is not stable due to its active peroxide radical site, which is liable to lose the terminal atomic 3O, forming a stable intermediate 31 with double-ended aldehyde groups, as shown in Fig. 5. This process requires only 50.0 kJ mol−1. In addition, the generated atomic 3O atom could also actively abstract an adjacent hydrogen atom with only 11.9 kJ mol−1 and produce stable complex 26 with an OH radical, 25 → TS25/26 → 26. Complex 26 is likely to lose an OH radical, and then chain structure 27 proceeds through several rearrangements to stable conjugated structure 30 with one radical site. A further reaction between the OH radical and the remaining fragment in 26 forming 32 has a higher barrier (TS26/32) than OH radical dissociation, and thus is a minor channel. In short, the further oxidation of intermediate 4, to some extent, could result in the generation of OOH, O and OH radicals.
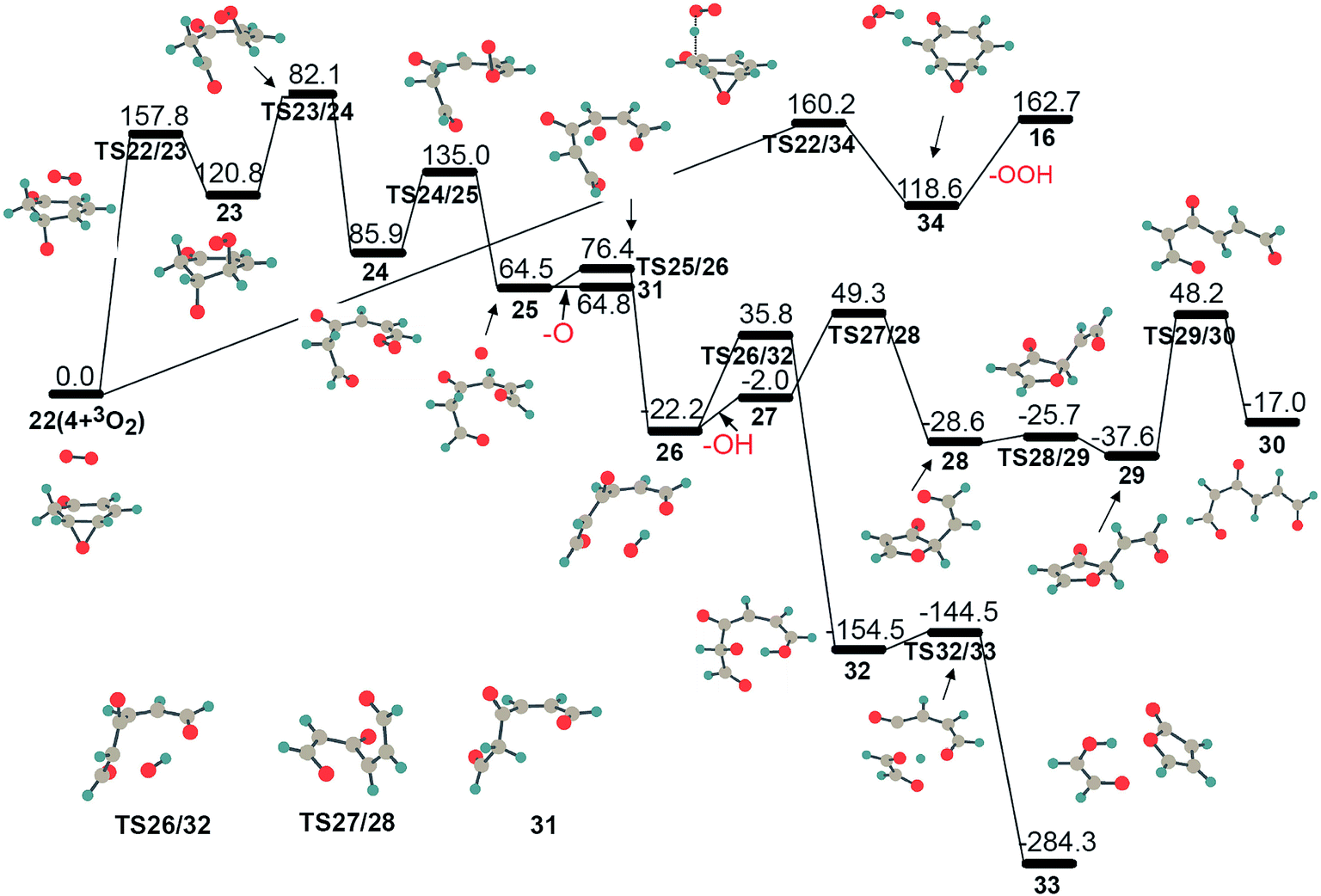 | ||
| Fig. 5 The potential energy surfaces for the oxidation of intermediate 4 by triplet molecular oxygen at UB3LYP/cc-pVTZ level. Energy values (in kJ mol−1) are relative to the sum of the energy of triplet molecular oxygen plus intermediate 4. | ||
As mentioned above, an alternative reaction pathway for intermediate 4 is isomerization to 6 via the sequential migration of oxygen and hydrogen atoms. In Fig. 2, intermediate 6 is located in a deep well on the PES of C6H5O2. It needs to overcome a barrier height of 236.0 kJ mol−1 to isomerize into structure 7 through hydrogen transfer. However, structure 6 has two sp3-C atoms with two sp3-C–H bonds, respectively, which has a higher possibility for further oxidization by another molecular oxygen. The PES for the further oxidation of intermediate 6 is shown in Fig. 6. Two hydrogen atoms were abstracted from two sp3-C atoms with reaction barriers of 120.9 kJ mol−1 and 152.9 kJ mol−1, respectively, giving complexes 36 and 41 with an OOH radical through hydrogen-bonding interactions. Obviously, complex 36 is more stable due to the higher delocalization of the lone electron within the π network of the oxygenated hydrocarbon fragment. Thus, the reaction 35 → TS35/36 → 36 has a higher possibility of proceeding kinetically and thermodynamically, due to its lower barrier height and being less endothermic. Complex 36 could produce intermediate 37 after absorbing heat of 40.6 kJ mol−1, giving free radical OOH. Alternatively, the generated OOH radical, wandering around 37, might abstract the second hydrogen atom from the sp3-C atom, giving intermediate 40 and an active H2O2 molecule, 36 → 38 → TS38/39 → 39 → 40. Undoubtedly, the latter path requires a higher barrier than the former one, which prefers to happen in the following ignition and combustion step.
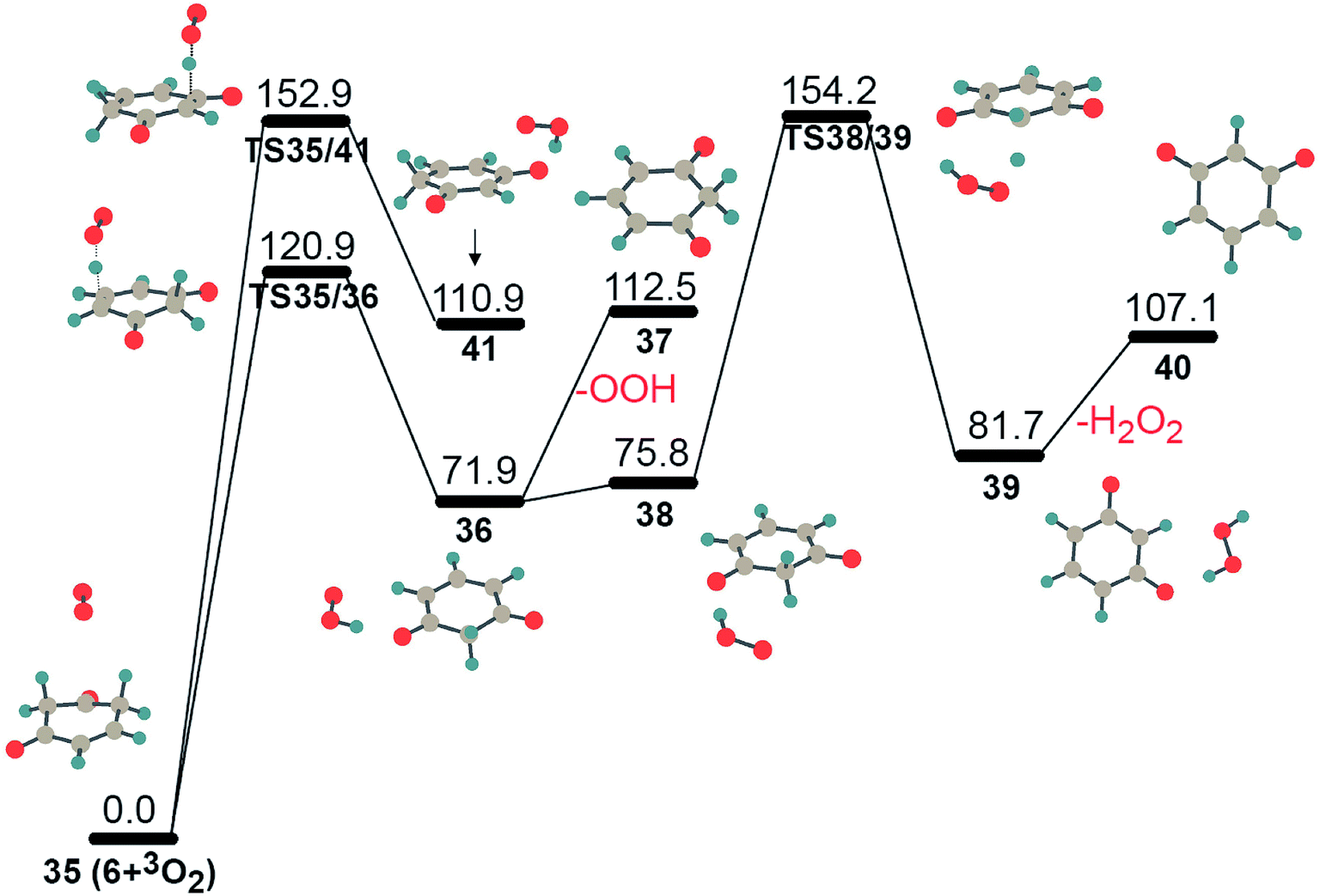 | ||
| Fig. 6 The potential energy surfaces for the oxidation of intermediate 6 by triplet molecular oxygen at UB3LYP/cc-pVTZ level. Energy values (in kJ mol−1) are relative to the sum of the energy of triplet molecular oxygen plus intermediate 6. | ||
It is worth noting that the oxygenated hydrocarbon species C6H6O2 can be vital sources of the initial free radical generation, such as OOH, OH, and atomic 3O. Free radical OOH is likely to be generated by the further oxidation of C6H6O2 through TS13/14. The presence of a terminal peroxide radical group in chain structure 24 could easily split into stable aldehyde and atomic 3O, which subsequently abstracts hydrogen atom, giving radical OH through TS25/26. Up to now, this is the first time the reaction mechanisms for the initial generation of free radicals in benzene oxidation have been systematically studied.
3.3 Role of radicals on PAH growth and small fragment dissociation
As mentioned in the above discussion, free radicals OOH, OH, and O could be produced from the oxidation of the species C6H6O2. The importance of these free radicals including H2O2 in the combustion chemistry has been studied in detail earlier,16,18–21,40 which mainly focused on small aliphatic hydrocarbons. They could abstract hydrogen atoms from fuel and generate hydrocarbon radical species. Importantly, they could induce chain reactions and introduce more and more free radicals into the combustion system.18 These chain-propagation processes in the combustion account to a considerable extent for the combustion kinetics, including not only the oxidation of hydrocarbon fuels to final products, such as CO2, H2O, and CO, but also the growth of PAH fragments. The oxidation of benzene by free radicals has also been studied,6,22,24,39 and was found to be much easier compared with oxidation by molecular oxygen. It only requires around 40 kJ mol−1 to abstract a hydrogen atom by OH24 or atomic H,23 generating the hydrocarbon radical C6H5. In this way, free radicals will play a leading role in the activation of most benzene molecules by abstracting hydrogen atoms, which could explain why the mole fraction of C6H6O2 is not large in experiments.2 In addition to the radical C6H5, species C6H6O and C5H6 were also observed as having large mole fractions early in the flame,2 but their formation mechanisms have not yet been investigated. It would be interesting to study thoroughly how these free radicals, such as OH, O, OOH, and H, oxidize benzene molecules using ADDF and AFIR methods and find the most favorable reaction pathway for benzene oxidation in combustion and explain the experimental observations. Furthermore, the generated species C6H5 could not only be further oxidized41 by molecular or atomic oxygen via radical–molecule or radical–radical reaction mechanisms producing CO2, and CO, but also could react with other hydrocarbon radical or benzene molecules forming PAH fragments. It is well known that PAH fragments are widespread organic pollutants, while their formation mechanisms are still in a period of exploration.42,43 Therefore, one of the most urgent tasks is to demonstrate clearly the reaction mechanism for PAH growth. Using ADDF and AFIR methods could be expected to supply deeper insights into the formation mechanism of PAH fragments in the near future. These works are currently in progress within our group.4. Conclusions
We have explored the various potential energy surfaces (PESs) of the oxidation of benzene by molecular oxygen using the powerful anharmonic downward distortion following (ADDF) and artificial force induced reaction (AFIR) methods of the global reaction route mapping (GRRM) strategy. Before reaching the auto-ignition point, the most favorable reaction route of benzene activation is the addition of singlet molecular oxygen, forming a singlet bridging peroxide molecule C6H6O2. The system undergoes non-adiabatic transition from triplet to singlet PES during the initiation step of benzene oxidation. The formed bridging peroxide molecule could isomerize to three stable isomers by stepwise oxygen and hydrogen transfer. The further oxidations of these oxygenated hydrocarbon species were studied in detail by the addition of another triplet molecular oxygen. Interestingly, it was found that species C6H6O2 are important sources for free radical generation in the initial stage of benzene oxidation. Free radicals, such as OH, OOH and O, could be generated when the above isomers C6H6O2 interact with molecular oxygen. The molecular oxygen could easily abstract hydrogen atoms from the sp3-C atom of the isomers of C6H6O2, generating OOH radicals. Alternatively, it could also attack one C atom, resulting in ring opening and the formation of a ring structure with a terminal peroxide radical group, which could further dissociate into atomic O. These initial free radical species could propagate and branch into more free radicals through chain-propagation processes. They would play critical roles in the following PAH growth and production of small molecules, such as CO2, CO and H2O.In the present study, the reaction mechanism of free radical generation in the initial stage of benzene oxidation have been systematically studied theoretically. Due to the high requirement for computing resources, we have to admit that the UB3LYP/cc-pVTZ level applied in this work is not the best computational method to study the non-adiabatic transitions and the chemical kinetics, although it has been confirmed to give the correct energy trends compared with the CASPT2/aug-cc-pVDZ level [J. Phys. Chem. Lett., 2011, 2, 852]. A higher computational level is needed for a further and more accurate description of the reaction kinetics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (21403127) and the Natural Science Foundation of Shandong Province, China (ZR2014BQ015). Hai-Bei Li thanks the fund of Young Scholars Program of Shandong University (Weihai), YSPSDUWH, and the supercomputing system at the Supercomputing Center, Shandong University, Weihai.References
- N. Fujii and T. Asaba, Symposium (International) on Combustion, 1973, 14, 433–442 CrossRef.
- J. D. Bittner and J. B. Howard, Symposium (International) on Combustion, 1981, 18, 1105–1116 CrossRef.
- R. D. Kern, H. J. Singh, M. A. Esslinger and P. W. Winkeler, Symposium (International) on Combustion, 1982, 19, 1351–1358 CrossRef.
- C. Venkat, K. Brezinsky and I. Glassman, Symposium (International) on Combustion, 1982, 19, 143–152 CrossRef.
- H. J. Singh and R. D. Kern, Combust. Flame, 1983, 54, 49–59 CrossRef CAS.
- J. H. Kiefer, L. J. Mizerka, M. R. Patel and H. C. Wei, J. Phys. Chem., 1985, 89, 2013–2019 CrossRef CAS.
- R. P. Lindstedt and G. Skevis, Combust. Flame, 1994, 99, 551–561 CrossRef CAS.
- R. A. Shandross, J. P. Longwell and J. B. Howard, Symposium (International) on Combustion, 1996, 26, 711–719 CrossRef.
- Y. Tan and P. Frank, Symposium (International) on Combustion, 1996, 26, 677–684 CrossRef.
- A. Schöbel, A. G. Class, L. Krebs, M. Braun-Unkhoff, C. Wahl and P. Frank, Chemosphere, 2001, 42, 591–599 CrossRef.
- F. Defoeux, V. Dias, C. Renard, P. J. Van Tiggelen and J. Vandooren, Proc. Combust. Inst., 2005, 30, 1407–1415 CrossRef.
- H. Richter, S. Granata, W. H. Green and J. B. Howard, Proc. Combust. Inst., 2005, 30, 1397–1405 CrossRef.
- S.-I. Shih, T.-C. Lin and M. Shih, J. Hazard. Mater., 2005, 117, 149–159 CrossRef CAS PubMed.
- J. M. Simmie, Prog. Energy Combust. Sci., 2003, 29, 599–634 CrossRef CAS.
- M. Frenklach, Z. Liu, R. I. Singh, G. R. Galimova, V. N. Azyazov and A. M. Mebel, Combust. Flame, 2018, 188, 284–306 CrossRef CAS.
- C. K. Westbrook and F. L. Dryer, Prog. Energy Combust. Sci., 1984, 10, 1–57 CrossRef CAS.
- S. Madronich and W. Felder, J. Phys. Chem., 1985, 89, 3556–3561 CrossRef CAS.
- H. Richter and J. B. Howard, Phys. Chem. Chem. Phys., 2002, 4, 2038–2055 RSC.
- D. E. Hoare and M. Patel, Trans. Faraday Soc., 1969, 65, 1325–1333 RSC.
- V. N. Kondratiev and V. V. Azatyan, Symposium (International) on Combustion, 1973, 14, 37–44 CrossRef.
- X. Zhong and J. W. Bozzelli, J. Phys. Chem. A, 1998, 102, 3537–3555 CrossRef CAS.
- T. Ko, G. Y. Adusei and A. Fontijn, J. Phys. Chem., 1991, 95, 8745–8748 CrossRef CAS.
- A. M. Mebel, M. C. Lin, T. Yu and K. Morokuma, J. Phys. Chem. A, 1997, 101, 3189–3196 CrossRef CAS.
- D. S. Hollman, A. C. Simmonett and H. F. Schaefer, Phys. Chem. Chem. Phys., 2011, 13, 2214–2221 RSC.
- G. Bermudez and L. Pfefferle, Combust. Flame, 1995, 100, 41–51 CrossRef CAS.
- S. Maeda, R. Saito and K. Morokuma, J. Phys. Chem. Lett., 2011, 2, 852–857 CrossRef CAS PubMed.
- S. Maeda and K. Morokuma, J. Chem. Phys., 2010, 132, 241102–241104 CrossRef PubMed.
- S. Maeda, Y. Osada, K. Morokuma and K. Ohno, GRRM, a developmental version, 2011 Search PubMed.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. ratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- K. Ohno and S. Maeda, Chem. Phys. Lett., 2004, 384, 277–282 CrossRef CAS.
- S. Maeda and K. Ohno, J. Phys. Chem. A, 2005, 109, 5742–5753 CrossRef CAS PubMed.
- S. Maeda and K. Ohno, J. Phys. Chem. A, 2007, 111, 4527–4534 CrossRef CAS PubMed.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- J. N. Harvey, Phys. Chem. Chem. Phys., 2007, 9, 331–343 RSC.
- A. W. Jasper, C. Zhu, S. Nangia and D. G. Truhlar, Faraday Discuss., 2004, 127, 1–22 RSC.
- D. R. Glowacki, C.-H. Liang, C. Morley, M. J. Pilling and S. H. Robertson, J. Phys. Chem. A, 2012, 116, 9545–9560 CrossRef CAS PubMed.
- K. Chenoweth, A. C. T. van Duin and W. A. Goddard, J. Phys. Chem. A, 2008, 112, 1040–1053 CrossRef CAS PubMed.
- H. Kijewski and J. Troe, Int. J. Chem. Kinet., 1971, 3, 223–235 CrossRef CAS.
- A. M. Mebel and M. C. Lin, J. Am. Chem. Soc., 1994, 116, 9577–9584 CrossRef CAS.
- R. W. Walker, Symposium (International) on Combustion, 1989, 22, 883–892 CrossRef.
- P. Frank, J. Herzler, T. Just and C. Wahl, Symposium (International) on Combustion, 1994, 25, 833–840 CrossRef.
- H. Richter and J. B. Howard, Prog. Energy Combust. Sci., 2000, 26, 565–608 CrossRef CAS.
- D. Golea, Y. Rezgui, M. Guemini and S. Hamdane, J. Phys. Chem. A, 2012, 116, 3625–3642 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The Gibbs free energy surfaces for all of the corresponding PESs. See DOI: 10.1039/c9ra03048d |
| This journal is © The Royal Society of Chemistry 2019 |