
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhytochemical composition and the cholinesterase and xanthine oxidase inhibitory properties of seed extracts from the Washingtonia filifera palm fruit†
Sonia Florisa,
Antonella Fais *a,
Antonella Rosab,
Alessandra Pirasc,
Hanen Marzoukid,
Rosaria Meddaa,
Ana M. González-Paramáse,
Amit Kumarf,
Celestino Santos-Buelgae and
Benedetta Eraa
*a,
Antonella Rosab,
Alessandra Pirasc,
Hanen Marzoukid,
Rosaria Meddaa,
Ana M. González-Paramáse,
Amit Kumarf,
Celestino Santos-Buelgae and
Benedetta Eraa
aDepartment of Life and Environmental Sciences, University of Cagliari, Monserrato, CA, Italy. E-mail: fais@unica.it; Tel: +39 0706754506
bDepartment of Biomedical Sciences, University of Cagliari, Monserrato, CA, Italy
cDepartment of Chemical and Geological Sciences, University of Cagliari, Monserrato, CA, Italy
dLaboratory of Transmissible Diseases and Biologically Active Substances, Faculty of Pharmacy, University of Monastir, Tunisia
eGrupo de Investigación en Polifenoles (GIP-USAL), Universidad de Salamanca, Spain
fDepartment of Electrical and Electronic Engineering, University of Cagliari, Cagliari, Italy
First published on 8th July 2019
Abstract
The chemical composition and biological properties of palm Washingtonia filifera (Lindl.) H. Wendl. seeds are seldom studied. Bearing this in mind, the seeds of W. filifera fruits were analysed for their fatty acid and phenolic composition and their antioxidant activity in addition to their cholinesterase and xanthine oxidase inhibitory activities. Seed extracts were revealed as a good source of phenolics with significant antioxidant activity. The phenolic profile mainly consisted of proanthocyanidins or procyanidin dimers B1–B4 among the major compounds. The highest butyrylcholinesterase inhibitory activity was found in the ethanolic extracts of seeds, with IC50 values of 13.73 ± 1.31 μg mL−1. Seed alcoholic extracts also displayed interesting xanthine oxidase inhibitory activity, with IC50 values ranging between 75.2 ± 17.0 μg mL−1 and 95.8 ± 5.9 μg mL−1. Procyanidin B1, a major component in the extracts, could be an important contributor to that activity, as it was found to possess good xanthine oxidase inhibition capacity (IC50 value of 53.51 ± 6.03 μg mL−1). Docking studies were also performed to predict the binding sites of procyanidins B1 and B2 within the xanthine oxidase structure. In all, W. filifera seeds appear as a promising natural source for the extraction of bioactive compounds with antioxidant and butyrylcholinesterase as well as xanthine oxidase inhibitory potential.
Introduction
In recent years, researchers have been attempting to better valorise flora as a natural source of bioactive products.1,2 Fruits or underutilized fruit species are receiving more attention, as they contain different phytochemicals that manifest many biological activities,3 including the ability to inhibit several enzymatic targets.4,5 Moreover, compared to metabolites in the edible portion of fruits, those present in seeds have an added value and good commercial potential as promising phytochemicals/antioxidants for use in food.6The palm family includes a range of plant species with wide application in human food, some of which may also be of pharmacological interest.7 Few studies, however, exist on Washingtonia palms, a genus belonging to the Coryphoideae subfamily that includes two species: W. filifera and W. robusta. Fruits, including the seeds, of W. filifera have been analysed for their nutritional composition, with the conclusion that they possess a higher concentration of carbohydrates than proteins.8,9 W. filifera fruits and seeds are also relevant sources of dietary oils.8 As for phytochemicals, previously authors10 have studied the antioxidant activities of the aerial part of W. filifera and reported the presence of eight known flavonoids, including various luteolin and C-glycosyl derivatives, together with two newly described compounds, luteolin 7-O-glucoside 4′′-sulfate and 8-hydroxyisoscoparin (i.e., 8-hydroxychrysoeriol 6-C-glucoside).
Flavonoids are a major class of the secondary metabolites of plants that occur ubiquitously in foods of plant origin.11–13 The greatest antioxidant activity appears to be exhibited by the flavanol class, the procyanidin group.14,15
Experimental findings suggest that these molecules can act simultaneously as antioxidants, cholinesterase and xanthine oxidase inhibitors, and anti-fibril agents.16,17 Different biological activities have also been reported for C-glycosylated derivatives, such as anti-inflammatory, antioxidant and anticholinesterase properties.18
Alzheimer's disease (AD) is a neurodegenerative disease that results from the synaptic dysfunction and death of neurons in specific brain regions and circuits, specifically the populations of nerve cells sub-serving memory and cognition.19 Cholinesterase inhibitors are in the first line of pharmacotherapy for mild to moderate AD, delaying the breakdown of acetylcholine released into synaptic clefts and enhancing cholinergic neurotransmission. Several studies have been conducted to discover new substances based on plant products that can inhibit the action of cholinesterase and mitigate the effects of AD, while also with fewer side effects than the drugs currently available.20,21 The phenomena related to AD are mainly initiated and enhanced by oxidative stress, a process referring to an imbalance between antioxidants and oxidants in favour of oxidants. Several medicinal plants have been shown to assist in mitigating dementia; indeed, different medicinal plants can produce a therapeutic effect owing to different properties, including cholinesterase inhibition and antioxidant activity.22
An important enzyme that has been reported to proliferate during oxidative stress is xanthine oxidase (XO), which catalyses the reaction of hypoxanthine to xanthine and xanthine to uric acid.23 In both steps, molecular oxygen is reduced, forming the superoxide anion, followed by the generation of hydrogen peroxide. Therefore, compounds that can inhibit xanthine oxidase may reduce both the circulating levels of uric acid and the production of reactive oxygen species (ROS). The overactivity of XO has been associated with the development of gout.24
In this context, in search of novel sources of bioactive molecules with potential beneficial effects on AD, the phenolic composition and total polyphenol and flavonoid contents, as well as the antioxidant, anti-cholinesterase and anti-XO properties, have been analysed in pulp and seed extracts from W. filifera from two different geographical areas of Tunisia.
Experimental
Chemicals
All chemicals were obtained as pure commercial products and used without further purification. Standards of fatty acids and fatty acid methyl esters, Desferal (deferoxamine mesylate salt), Trolox, Folin-Ciocalteau's phenol reagent, 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS), allopurinol, xanthine oxidase from cow's milk (XO), acetylcholinesterase (AChE) from Electrophorus electricus, butyrylcholinesterase (BChE) from equine serum, xanthine, 5,5′-dithiobis-(2-nitrobenzoic) acid (DTNB), acetylthiocholine iodide (ATCI), S-butyrylthiocholine iodide (BTCI), (−)-epicatechin-(4β,8)-(+)-catechin (procyanidin B1) and all solvents used, of the highest available purity, were obtained from Sigma-Aldrich (Milan, Italy). Methanolic HCl (3 N) was purchased from Supelco (Bellefonte, PA). The phenolic compound standards were obtained from Extrasynthese (Genay, France). HPLC-grade acetonitrile was obtained from Merck KgaA (Darmstadt, Germany), and formic acid was purchased from Prolabo (VWR International, France). Water was treated in a Milli-Q water purification system (TGI Pure Water Systems, USA).Plant material
The fruits of W. filifera were collected in Tunisia in the areas of Gabès (G) (33.880444 N, 10.082222 E) and Sousse (S) (35.855727 N, 10.567089 E) in August 2013. The plant was identified by Dr Marzouki Hanene, Laboratory of Transmissible Diseases and Biologically Active Substances, Faculty of Pharmacy, University of Monastir, Tunisia.The plant materials were washed with deionized water, frozen at −20 °C and then lyophilized. Lyophilization was carried out overnight, using an LIO-5P Freeze Dryer apparatus. The dried material was stored at −20 °C until required.
The fruits of W. filifera, separated as pulp and seeds, were crushed separately and then macerated in different solvent systems to compare the bioactivity of the extracts. The lyophilized plant materials (25 g) were extracted in 100 mL of water (AE, aqueous extract), ethanol (EE, ethanol extract) or methanol (ME, methanol extract) for 72 h at room temperature under continuous stirring. After filtration and centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, the ethanol and methanol extracts were concentrated, using a rotary evaporator under reduced pressure at 60–70 °C. For fatty acid analysis, the seeds were also extracted with n-hexane (HE) in a conventional Soxhlet extraction apparatus, and the samples were further concentrated under vacuum on a rotary evaporator.
000 rpm, the ethanol and methanol extracts were concentrated, using a rotary evaporator under reduced pressure at 60–70 °C. For fatty acid analysis, the seeds were also extracted with n-hexane (HE) in a conventional Soxhlet extraction apparatus, and the samples were further concentrated under vacuum on a rotary evaporator.
Soxhlet extractions were performed using 15 g of each sample. The powder plant was transferred into a cellulose extraction thimble and inserted into a Soxhlet assembly fitted with a 100 mL flask. A 50 mL portion of n-hexane was added, and the whole assembly was heated for 6 h using a heating mantle at 60 °C. The extracts were concentrated using a rotary evaporator at 40 °C, and the dry extracts obtained were stored at −20 °C for chemical and biological assays.
Fatty acid analysis of n-hexane extracts
Dried aliquots of seed HE (3 mg) were dissolved in ethanol solution to be subjected to a mild saponification process at room temperature in the dark with a mixture of Desferal/ascorbic acid/10 N KOH, as previously described.25 Dried saponified fractions were injected into an Agilent Technologies 1100 liquid chromatograph equipped with a diode array detector (HPLC-DAD system) (Agilent Technologies, Palo Alto, CA) for the quantification of unsaturated fatty acids (UFAs). The separation was carried out on an Agilent Technologies XDB-C18 Eclipse column (150 × 4.6 mm, 3.5 μm particle size) (at 37 °C), equipped with a Zorbax XDB-C18 Eclipse guard column (12.5 × 4.6 mm, 5 μm particle size); a mixture of CH3CN/H2O/CH3COOH (75/25/0.12, v/v/v) was used as the mobile phase at a flow rate of 2.3 mL min−1, and detection was performed at 200 nm.25 UFAs were identified using standard compounds and conventional UV spectra. The quantification of UFAs was made from the peak area ratio, which was based on a calibration curve (in the amount range of 0.5–6 μg on the column for PUFA and 1–10 μg for MUFA, respectively) generated from standard compounds in CH3CN solution. The calibration curves of all the compounds were found to be linear, with correlation coefficients > 0.97, as previously reported.26 An Agilent OpenLAB Chromatography data system was employed to record and integrate the results.A portion of dried fatty acids after saponification was methylated with 1 mL of methanolic HCl (3 N) for 30 min at room temperature, as previously described.25 Fatty acid methyl esters (FAME) were analysed using a gas chromatograph Hewlett-Packard HP-6890 (Hewlett-Packard, Palo Alto, USA) with a flame ionisation detector (FID) and equipped with a cyanopropyl methyl-polysiloxane HP-23 FAME column (30 m × 0.32 mm × 0.25 μm) (Hewlett-Packard).25 Nitrogen was used as a carrier gas at a flow rate of 2 mL min−1. The oven temperature was set to 175 °C; the injector temperature was set to 250 °C; and the detector temperature was set to 300 °C. FAME were identified by comparing the retention times with those of standard compounds and quantified as a percentage of the total amount of fatty acids (g%) using the Hewlett-Packard software.
Determination of the total phenolics and flavonoids
The total phenolic and flavonoid content in the extracts were determined as previously reported.25 The phenolic concentration was calculated using gallic acid as a reference standard and expressed as mg of gallic acid equivalents (GAE) per g of dry weight (dw). Flavonoid concentration was expressed as mg of quercetin equivalents (QE) per g of dw.Antioxidant assays and enzyme assays
ABTS radical-scavenging activity was evaluated with the method reported by Delogu et al.,27 using Trolox as a standard. Briefly, the free radical ABTS˙+ was produced by reacting 7 mM ABTS with 2.45 mM potassium persulfate (final concentration) in aqueous solution and kept in the dark at room temperature for 24 h before use. Subsequently, an aliquot of this mixture was diluted to obtain an absorbance of approximately 0.7 ± 0.05 (mean ± SD). Samples of each extract (10 μL) were added to 1 mL of ABTS˙+ and the absorbance at 734 nm was recorded after incubation for 1 min.The results were expressed as the concentration of sample necessary to cause a 50% reduction in the original absorbance (EC50).
Cholinesterase assay
Kinetic assays of cholinesterase activity were performed via Ellman's method28 with slight modifications29 using a plate reader. Briefly, the reaction mixture containing 70 μL of phosphate buffer (0.1 M, pH 8.0), enzyme solution (AChE, 4 μL or BChE, 6 μL), 100 μL of DTNB (1.5 mM final concentration) and inhibitor was dissolved in DMSO at the desired concentrations or in DMSO alone (2%). After, ATCI or BTCI (20 μL) was added as the substrate to the reaction mixture, and the absorbance was monitored at 405 nm (37 °C) for 4 minutes. Each extract was evaluated at six concentrations (ranging from 5 to 50 μg mL−1). Galantamine was used as the standard cholinesterase inhibitor.Xanthine oxidase assay
The inhibitory effect of W. filifera extracts on xanthine oxidase activity was determined spectrophotometrically by monitoring the formation of uric acid at 295 nm. XO activity was measured according to the method previously reported.5 The reaction mixture contained 879 μL of 100 mM phosphate buffer pH 7.5, 50 μL of an aqueous solution of XO (0.5 U mL−1) and 10 μL of the extract sample solution or the control sample solution. After mixing, 61 μL of 0.82 mM xanthine solution was added, and the enzyme activity was determined at 295 nm for 3 min at 25 °C. Allopurinol was used as the standard XO inhibitor. The inhibition potency for ChEs and XO was expressed as the IC50 values, which represent the inhibitor concentration need to cause 50% inhibition of enzyme activity. The IC50 values were calculated by interpolation in dose–response curves. The IC50 values displayed represented the mean ± standard deviation for the three independent assays.Spectrophotometric determinations were made in an Ultrospec 2100 spectrophotometer (Biochrom Ltd, Cambridge, England) using 1 cm path cells and with a FLUOstar OPTIMA microplate reader (BMG Labtech, Offenburg, Germany).
HPLC-DAD-ESI/MS analyses
The methanolic and ethanolic extracts of W. filifera fruits were analysed using a Hewlett-Packard 1200 chromatograph (Agilent Technologies, Waldbronn, Germany) equipped with a quaternary pump and a diode array detector (DAD) coupled to an HP Chem Station (rev. A.05.04) data-processing station. The HPLC system was connected via the DAD cell outlet to an API 3200 Qtrap (Applied Biosystems, Darmstadt, Germany) mass spectrometer (MS) consisting of an ESI source and a triple quadrupole-ion trap mass analyzer, which was controlled using Analyst 5.1 software.The phenolic compounds present in the samples were identified according to their UV and mass spectra and by comparison with commercial standards when available.
Molecular docking
The crystal structure of the XO (PDB ID: 1FIQ) enzyme was considered as the modeled protein structure for computational investigation. The protein–ligand binding sites were predicted using a COACH-D server, which is an enhanced version of the COACH server.30 The COACH algorithm predicts ligand poses by using a consensus of five methods. The first four are template-based methods: TM-SITE,31 S-SITE,31 COFACTOR32 and FINDSITE.33 The fifth method is template-free and performs binding site prediction by examining both sequence conservation and the structural geometry of the cavity (region).34 The results obtained from the five individual methods were then combined for consensus predictions by the COACH algorithm.31 These ligands were then clustered based on the spatial distance between their geometric centres (average linkage clustering algorithm with a cut-off distance 4 Å). The final step in the protocol consists of docking the ligand from the user input or the templates into the predicted binding pockets to build their complex structures, employing the molecular docking algorithm AutoDock Vina.35 For each predicted binding pocket, up to 10 binding poses are generated, and the one that best matches the consensus prediction of the binding residues is selected.Statistical analyses
Statistical differences were evaluated using GraphPad Prism software version 8 (San Diego, CA, USA). Comparison between groups was assessed using the Student's unpaired t-test with Welch's correction and by the one-way analysis of variance (one-way ANOVA) followed by the Bonferroni Multiple Comparisons Test. The values with p < 0.05 were considered significant.Results and discussion
Quali-quantitative information on the individual fatty acids (FA) that compose the W. filifera n-hexane extracts was obtained by GC-FID and HPLC-DAD analyses. Table 1 shows the FA composition by the GC-FID analysis (expressed as % of total FA, g/100 g) of HE obtained from the seeds of W. filifera collected in the areas of Sousse and Gabès. The HE Sousse (HES) showed proportions of approximately 60% saturated FA (SFA) (mainly lauric acid 12:0, myristic acid 14:0, and palmitic acid 16:0, respectively 36, 12, and 6%), 28% monounsaturated FA (MUFA) (mainly oleic acid 18:1n − 9 and palmitoleic acid 16:1n − 7, 25 and 2%, respectively), and 8% polyunsaturated FA (PUFA), essentially constituted by the essential FA linoleic acid (18:2n − 6), with traces (0.04%) of α-linolenic acid (18:3n − 3). The absolute content of the main UFA was determined by HPLC as follows (Table 2): 304.3 ± 10.9 mg g−1, 102.3 ± 4.2 mg g−1 and 0.8 ± 0.04 mg g−1 of n-hexane extract; for the acids 18:1n − 9, 18:2n − 6 and 18:3n − 3, respectively.
| Fatty acid | g/100 g | |
|---|---|---|
| HES | HEG | |
| a Abbreviations: SFA, saturated fatty acids; MUFA, monounsaturated fatty acids; PUFA, polyunsaturated fatty acids. Oil analysis was performed in quadruplicate, and all data are expressed as mean values ± standard deviations (SD); (n = 4). Evaluation of the statistical significance of differences between the two groups was performed using the Student's unpaired t-test with Welch's correction; ap < 0.01; bp < 0.05. | ||
| 8:0 |
1.06 ± 0.17 | 1.01 ± 0.23 |
| 10:0 |
1.55 ± 0.24 | 1.61 ± 0.25 |
| 12:0 |
36.11 ± 4.23 | 33.50 ± 2.70 |
| 14:0 |
12.26 ± 0.58 | 10.40 ± 0.45a |
| 16:0 |
6.23 ± 0.29 | 6.32 ± 0.37 |
| 16:1 |
2.23 ± 0.42 | 3.07 ± 0.33b |
| 18:0 |
2.62 ± 0.45 | 3.05 ± 0.05 |
| 18:1n − 9 |
25.09 ± 2.17 | 25.47 ± 1.39 |
| 18:2n − 6 |
8.40 ± 0.63 | 9.95 ± 1.36 |
| 18:3n − 3 |
0.04 ± 0.01 | 0.06 ± 0.00 |
| 18:3n − 6 |
0.02 ± 0.01 | 0.05 ± 0.00 |
| 20:0 |
0.33 ± 0.43 | 0.08 ± 0.01 |
| 20:1 |
0.31 ± 0.06 | 0.26 ± 0.19 |
| SFA | 60.16 ± 4.03 | 55.97 ± 2.92 |
| MUFA | 27.63 ± 2.45 | 28.80 ± 1.58 |
| PUFA | 8.47 ± 0.62 | 10.06 ± 1.35 |
| Fatty acids | HES | HEG |
|---|---|---|
| a Oil analysis was performed in quadruplicate and all data are expressed as mean values ± standard deviations (SD); (n = 4). | ||
| 18:1n − 9 |
304.33 ± 10.93 | 275.41 ± 13.26 |
| 18:2n − 6 |
102.31 ± 4.21 | 108.19 ± 6.26 |
| 18:3n − 3 |
0.84 ± 0.04 | 0.88 ± 0.06 |
The HE Gabès (HEG) was characterized by a similar FA profile, with a high level of SFA (56%, with 34% of 12:0), followed by MUFA (29%) and PUFA (10%). The HEG showed a slightly lower level of SFA and higher amounts of UFA than HES. Significant differences were only observed in the levels of myristic acid (14:0), with 12% and 10% for HES and HEG, respectively (p < 0.01), and palmitoleic acid (16:1n − 7), with 2% and 3% for HES and HEG, respectively (p < 0.05). The absolute values of the main UFA determined by HPLC (Table 2) for the HEG were 275.4 ± 13.3 mg g−1, 108.2 ± 6.3 mg g−1 and 0.9 ± 0.1 mg g; for acids 18:1n − 9, 18:2n − 6 and 18:3n − 3, respectively. Both HES and HEG contained lauric acid (12:0) as the main fatty acid (34–36%) but also exhibited a high content of oleic acid (18:1n − 9; 25%). Thus, W. filifera seed oil can be regarded as a lauric-oleic oil because of the abundance of these two fatty acids.8
The FA composition of the HEG and HES oil extracts determined herein was slightly different from that of W. filifera seed oil obtained previously from a Tunisian sample.8 Specifically, like the HEG and HES, the major FA were SFA (43%), followed by MUFA (41%) and PUFA (16%); however, Tunisian seed oil showed oleic acid as the most abundant fatty acid (41%), followed by lauric acid (18%), linoleic acid (16%), myristic acid (11%) and palmitic acid (9%). This result could be ascribable to several factors, e.g., differences in FA metabolism due to the impact of the harvesting location such as climate, soil, and water availability.
The total phenolic and flavonoid contents in the analysed seed extracts are shown in Table 3. The highest total phenolic content was found in ME, followed by EE and AE. MEG showed a total phenolic content two times higher than the corresponding EEG. Very low amounts of phenolic compounds were also detected in the pulp extracts (data not shown). A positive correlation was found between total phenolic content versus flavonoid content (r = 0.98, r2 = 97%), determined in the alcoholic seed extracts, whereas hardly any flavonoids were found in the aqueous extracts.
| Total Phenolic mg GAE per g dw | Flavonoid mg QE per g dw | ABTS EC50 μg mL−1 | |
|---|---|---|---|
| Each value is the mean ± SD of three independent measurements (n = 3). §Below limit of detection. a,b,c,dDifferent letters within the same column denote statistically significant differences between extracts (p < 0.05). *Values of EC50 of EEG, EES, MES, AEG and AES compared to Trolox are significantly different (p < 0.01). | |||
| EEG | 325.96 ± 32.20a,b | 215.43 ± 98.61a | 11.11 ± 1.15a,* |
| EES | 412.30 ± 115.78a,b | 308.33 ± 137.23a | 9.06 ± 0.35a,* |
| MEG | 708.83 ± 169.10 a | 591.98 ± 386.14a | 5.52 ± 0.84b |
| MES | 637.4 ± 275.11a,c | 462.60 ± 294.20a | 9.71 ± 1.21a,* |
| AEG | 133.54 ± 30.0b | § | 22.64 ± 0.14c,* |
| AES | 233.06 ± 33.68b,c | § | 17.78 ± 0.45d,* |
| Trolox | 3.4 ± 0.3 |
Phenolic compounds have redox properties, which allow them to act as antioxidants.36 The antioxidant activity of the extracts was assessed by their ability to scavenge the ABTS radical. The results obtained for the seed extracts are included in Table 3. As for the total phenolic content, the aqueous extracts showed lower antioxidant capacity (higher EC50 values) than the alcoholic extracts. The correlation of the total phenol content and ABTS radical scavenging activity was also shown in Fig. S1 (ESI†). This correlation seems logical considering that the Folin–Ciocalteu reagent measures the reducing capacity of a sample, lacking specificity for phenolics. Pulp extracts presented much lower antioxidant capacity than seeds, with the EC50 value for ABTS radical scavenging ability was higher than 150 μg mL−1, which is in line with their low levels of phenolic compounds.
The characterization of individual phenolic compounds was performed by HPLC-DAD/ESI-MS. Data of the retention time, λmax, pseudomolecular ions, main fragment ions in MS2, and tentative identification are presented in Table 4. As can be seen, the sample mostly consists of flavan-3-ols (i.e., catechins and proanthocyanidins). Epicatechin and procyanidin B1 were identified by comparison with standards, whereas the identities of the procyanidin dimers B2–B4 and trimer C2 were tentatively assigned by comparison with data available in our data library. The identities of the remaining compounds were established based on their molecular weights. A point to highlight is the presence of some proanthocyanidins containing possible (epi)afzelechin units as well as A-type linkages. B-type procyanidin dimers (B1–B4) were among the main phenolic compounds in the extracts of W. filifera seeds that, in a previous study, were reported for their different biological activity.37,38 Minor amounts of other flavonoids, mainly quercetin and isorhamnetin derivatives possessing sulfate residues, were also detected. Although flavonoid sulfates are not very common in plants, they have been reported to occur in species of the Palmae family.39 As far as we know, no previous reports have been published on the phenolic profile of W. filifera seeds.
| Peak | Rt (min) | λmax (nm) | Pseudomolecular ion [m – H]− (m/z) | MS2 (m/z) | Tentative identification |
|---|---|---|---|---|---|
| 1 | 12.0 | 260, 293 | 331 | Galloylglucose | |
| 2 | 16.3 | 280, 307 | 451 | (epi)Catechin glucoside | |
| 3 | 20.2 | 279 | 577 | 451, 425, 407, 289 | B-type procyanidin dimer (B3) |
| 4 | 20.7 | 279 | 577 | B-type procyanidin dimer (B1) | |
| 5 | 21.1 | 278 | 865 | 695, 577, 425, 407, 287 | B-type procyanidin trimer (C2) |
| 6 | 22.5 | 577 | B-type procyanidin dimer (B4) | ||
| 7 | 22.9 | 577 | B-type procyanidin dimer (B2) | ||
| 8 | 23.7 | 863 | A-type procyanidin trimer | ||
| 9 | 25.4 | 1153 | 849, 577, 407, 287 | B-type procyanidin tetramer | |
| 10 | 27.5 | 278 | 289 | 245, 203, 179, 109 | Epicatechin |
| 11 | 28.4 | 561 | 435, 407, 289 | (epi)Catechin–(epi)afzelechin dimer | |
| 12 | 29.4 | 283 | 449 | 287, 269 | Dihydrokaempferol hexoside |
| 13 | 30.9 | 863 | 711, 575, 423 | A-type procyanidin trimer | |
| 14 | 32.6 | 865 | B-type procyanidin trimer | ||
| 15 | 34.1 | 865 | B-type procyanidin trimer | ||
| 16 | 38.3 | 865 | B-type procyanidin trimer | ||
| 17 | 39.1 | 1153 | B-type procyanidin tetramer | ||
| 18 | 40.5 | 865 | B-type procyanidin trimer | ||
| 19 | 41.4 | 849 | 697, 577, 407, 287 | B-type proanthocyanidin trimer containing one afzelechin unit | |
| 20 | 43.3 | 254, 353 | 689 | 301 | Quercetin rutinoside sulfate |
| 21 | 43.8 | 1441 | B-type procyanidin pentamer | ||
| 22 | 45.3 | 256, 358 | 703 | 315 | Isorhamnetin rutinoside sulfate |
| 23 | 45.3 | 577 | B-type procyanidin dimer | ||
| 24 | 46.5 | 849 | B-type procyanidin trimer containing one afzelechin unit | ||
| 26 | 50.4 | 557 | 315 | Isorhamnetin glucoside sulfate | |
| 28 | 61.8 | 255, 353 | 463 | 301 | Quercetin glucoside |
| 29 | 65.1 | 577 | B-type procyanidin dimer | ||
| 30 | 865 | B-type procyanidin trimer |
The anticholinesterase activity of all the extracts at a concentration of 20 μg mL−1 was checked using AChE/BChE assays.
Table 5 shows the AChE and BChE inhibitory activities of the of W. filifera seeds extracts, compared with those of the standard inhibitor galantamine. The IC50 for AChE was not determined because the inhibition at the highest screened concentration (20 μg mL−1) was less than 40%.
| Extracts | AChE% I | AChE IC50 | BChE% I | BChE IC50 |
|---|---|---|---|---|
| n.d: not determined because the inhibition at the highest screened concentration (20 μg mL−1) was less than 40%. Values were expressed as mean ± SD (n = 3). a,b,cDifferent letters within the same column denote statistically significant differences between extracts (p < 0.05). *Values of the IC50 of EES, MES and AES compared to galantamine are significantly different (p < 0.05). | ||||
| EEG | 3.2 ± 0.5 | n.d | 65.6 ± 0.78 | 13.73 ± 1.31a |
| EES | 16.5 ± 6.22 | n.d | 53.9 ± 6.5 | 27.30 ± 5.37b,* |
| MEG | 7.7 ± 2.22 | n.d | 64.5 ± 9.26 | 15.13 ± 2.05a,c |
| MES | 20.9 ± 1.56 | n.d | 63.1 ± 1.91 | 22.6 ± 2.72b,c,* |
| AEG | 28.6 ± 7.78 | n.d | 45.6 ± 1.06 | 15.08 ± 1.05a,c |
| AES | 38.5 ± 11.6 | n.d | 48.5 ± 1.06 | 18.51 ± 0.001a,c,* |
| Galantamine | 0.895 ± 0.043 | 7.65 ± 1.78 |
The IC50 values ranged from 13.73 ± 1.31 μg mL−1 to 27.30 ± 5.37 μg mL−1 in the different seed extracts. There was no statistically significant difference between the IC50 values of EEG, MEG and AEG compared to galantamine. The results obtained revealed that EEG showed very potent BChE inhibitory activity, with IC50 values (13.73 ± 1.31 μg mL−1) close to those of the standard drug galantamine (IC50 = 7.65 ± 1.78 μg mL−1) calculated under the same experimental conditions.
This can be considered a satisfactory result, since the standard inhibitor is a single molecule, while a mixture of numerous compounds exists in the plant extracts.
The inhibitory activity against BChE of a procyanidin B1 standard at a concentration of 20 μg mL−1 was also checked, obtaining a value of 18.98 ± 2.52%.
Thus, anti-BChE activity observed in the seed extracts cannot be mainly attributed to procyanidin B1, even if it is present in high concentrations. However, our findings led us to consider that this compound could contribute to the anti-BChE effect in these extracts.
Several studies on plant AChE inhibitors have been performed;40 however, fewer BChE inhibitors have been identified.41 No ChE inhibitory activity was found for any of the pulp extracts examined (data not shown).
ChEs inhibition has been extensively used as an approach for the treatment of Alzheimer's disease (AD). BChE activity progressively increases in patients with AD, while AChE activity remains unchanged or declines. Therefore, the use of molecules selectively interacting with BChE might have a relevant role in the treatment of patients with advanced AD.
The extracts of W. filifera were proven to have great potential and should be considered in future studies to identify the constituents responsible for the selective BChE inhibitory activity.
The extracts were also evaluated for their inhibition of XO enzyme activity (Table 6).
| Extracts | % I | IC50 | Inhibitory mode |
|---|---|---|---|
| a n.d: not determined because inhibition at the highest screened concentration (150 μg mL−1) was less than 40%. Values were expressed as mean ± SD (n = 3). Values of the IC50 for alcoholic extracts compared to allopurinol were significantly different (p < 0.05). | |||
| EEG | 52.4 ± 0.8 | 95.8 ± 5.9* | Mixed |
| EES | 63.9 ± 0.1 | 87.0 ± 0.5* | Mixed |
| MEG | 72.8 ± 0.3 | 75.2 ± 17.0* | Mixed |
| MES | 74.6 ± 0.2 | 76.1 ± 5.2* | Mixed |
| AEG | 36.7 ± 0.1 | n.d | n.d |
| AES | 37.7 ± 0.1 | n.d | n.d |
| Allopurinol | 2.0 ± 0.4 | ||
It was encouraging to observe that only pulp extracts were inactive against the XO enzyme, while all seed extracts displayed inhibitory activity at 150 μg mL−1, ranging between 36.7 ± 0.1 and 74.6 ± 0.2%. The alcoholic seed extracts showed IC50 values for the XO inhibitory activity in the 75.2 ± 17.0 μg mL−1 and 95.8 ± 5.9 μg mL−1 range, higher than those of the standard drug allopurinol (IC50 = 2.0 ± 0.4 μg mL−1).
Flavan-3-ols, the major compounds in the seed extracts, have been reported to possess inhibitory XO activity.42,43 Epicatechin behaves as a good XO inhibitor.44 Procyanidin B1, one the major components detected in the seed extracts of W. filifera, also revealed good XO inhibition capacity, showing an IC50 value of 53.5 ± 6.0 μg mL−1. As far as we know, no previous reports exist on XO inhibition by this procyanidin.
Molecular docking is a powerful technique that allows the prediction and identification of the most probable binding mode of the ligand to a partner protein.44 Therefore, to predict the best ligand pose within the XO binding site, we performed the docking of ligands procyanidin B1 and B2, which consists of catechin and epicatechin units joined in a beta-configuration. For comparison, the docking of ligands catechin and epicatechin, which are the elementary flavan-3-ol units in these dimers, was also checked.45
For the procyanidin ligands (B1, B2), the two most probable binding sites (Fig. 1) was observed. Binding site 1, which is located a distance from the protein active site exhibited the best docking energy values for both ligands (Table 7). We observed a reasonable overlap in the ligand poses, with procyanidin B1 displaying favourable docking energy.
 | ||
| Fig. 1 Predicted docked positions for the ligands bound to the XO protein. The active site residues are shown as grey van der Waals spheres (grey). Two probable binding sites 1 and 2 for the ligands procyanidin B1 (red) and B2 (blue) are circled in pink, while the binding region for the ligands catechin (green) and epicatechin (yellow) are within the green circle. In the rectangular box, a zoomed representation of the binding region for the ligands close to the active site is shown. | ||
| Protein–ligand | Docking energy (kcal mol−1) | C-score | Cluster size |
|---|---|---|---|
| XO–procyanidin B1 | −4.6 (site 1) | 0.12 | 28 |
| −2.7 (site 2) | 0.22 | 41 | |
| XO–procyanidin B2 | −3.8 (site 1) | 0.12 | 28 |
| −3.0 (site 2) | 0.22 | 41 | |
| XO–catechin | −8.6 kcal mol−1 | 0.19 | 30 |
| XO–epicatechin | −9.2 kcal mol−1 | 0.15 | 28 |
On the other hand, binding site 2 (Fig. 1) for both procyanidin ligands (B1, B2) was found to be near the XO protein active site. Interestingly, the binding region for the ligands catechin and epicatechin are in the active site and are in close proximity to binding site 2 of the procyanidin ligands.
Experimental data performed on the seed extracts indicated a mixed-type inhibition against the XO-enzyme. Now, considering that the concentration of procyanidin is pronounced (among the dimers) in the seed extracts, a plausible explanation for the mixed-type inhibition can be established from the spatial location of the predicted binding sites (different from the active site) for the procyanidin ligands. Since experiments revealed very low inhibitory activity for procyanidin B1 against BChE, we do not discuss the docking results of procyanidin B1 and the BChE protein. However, for completeness, we have provided the data in the ESI.†
To delve into the binding mode of the ligands with the XO protein, further examination was made using Ligplot software,46 which revealed a conserved interaction image regarding XO binding for both procyanidins B1 and B2 (Fig. 2) and for catechin and epicatechin (Fig. 3).
 | ||
| Fig. 2 Procyanidin ligands B1 and B2 bound to the XO protein. In (a) and (b), the ligand poses in the binding site 1, while in (c) and (d), it does so in the binding site 2. Hydrophobic interactions are represented by red spokes radiating towards the interacting ligand atoms, while hydrogen-bonded interactions are shown with a dashed green line. | ||
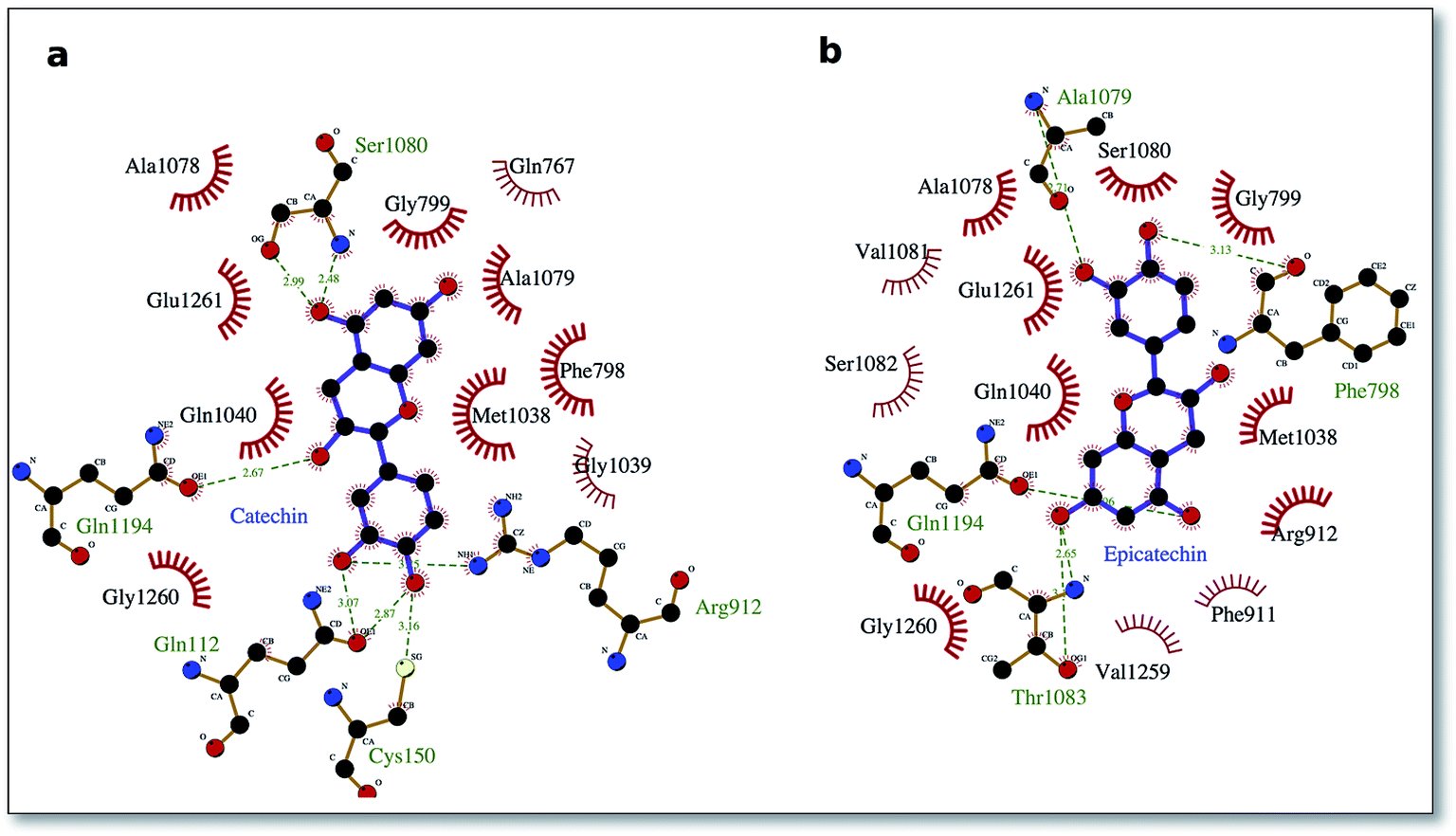 | ||
| Fig. 3 Interaction picture of the ligands catechin and epicatechin bound to the XO protein. Hydrophobic interactions are represented by red spokes radiating towards the interacting ligand atoms, while hydrogen-bonded interactions are shown with a dashed green line. | ||
For binding site 1 (Fig. 2a and b), we note two additional interactions involving residues Leu 147, Ile 1229 and Pro 1230 and procyanidin B1, thus confirming the better docking energy value with respect to procyanidin B2. On the other hand, for binding site 2, a good overlap between the ligands poses was confirmed by a conserved interaction picture (Fig. 2c and d).
The well-conserved binding regions for the ligands catechin and epicatechin (Fig. 3), involving interactions with amino acid residues Gly799, Glu802, Phe914, Ala1078, Ala1079 and Glu1261 in the active site, confirmed the significantly better docking energy with respect to the procyanidin ligands (B1, B2).
Conclusion
Natural products from plants provide unlimited opportunities for new drugs because their chemical diversity shows a range of biological activities. W. filifera seeds have been revealed to possess components with antioxidant and also butyrylcholinesterase and xanthine oxidase inhibition properties, which is of interest, considering that they are an inedible part of the fruit and are usually discarded. Molecular docking studies predicted the XO protein binding regions of procyanidin ligands and provided a plausible explanation to the mixed-type inhibition characteristic found for the seed extracts against the XO enzyme. These findings should contribute to valorise W. filifera seeds as a source for the extraction of bioactive compounds with nutraceutical and therapeutic potential.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Dr Giorgia Sarais (University of Cagliari) for her support. This work was supported by the University of Cagliari. The GIP-USAL was financially supported by MINECO (Spanish National Projects AGL2015-64522-C2).References
- M. G. Arvizu-Espinosa, G. L. von Poser, A. T. Henriques, A. Mendoza-Ruiz, A. Cardador-Martínez, R. Gesto-Borroto, P. N. Núñez-Aragón, M. L. Villarreal-Ortega, A. Sharma and A. Cardoso-Taketa, J. Nat. Prod., 2019, 82, 785–791 CrossRef CAS.
- B. David, J.-L. Wolfender and D. A. Dias, Phytochem. Rev., 2014, 14, 299–315 CrossRef.
- R. K. Firdose, Afr. J. Pharm. Pharmacol., 2011, 5, 2067–2072 Search PubMed.
- A. Di Petrillo, A. M. González-Paramás, B. Era, R. Medda, F. Pintus, C. Santos-Buelga and A. Fais, BMC Complement Altern. Med., 2016, 16, 453 CrossRef.
- A. Fais, B. Era, A. Di Petrillo, S. Floris, D. Piano, P. Montoro, C. I. G. Tuberoso, R. Medda and F. Pintus, BioMed Res. Int., 2018, 2018, 1–9 CrossRef.
- Y. Xu, M. Fan, J. Ran, T. Zhang, H. Sun, M. Dong, Z. Zhang and H. Zheng, Saudi J. Biol. Sci., 2016, 23, 379–388 CrossRef CAS.
- P. Geavlete, R. Multescu and B. Geavlete, Ther. Adv. Urol., 2011, 3, 193–198 CrossRef.
- I. A. Nehdi, Food Chem., 2011, 126, 197–202 CrossRef CAS.
- J. W. Cornett, Principes, 1987, 31, 159–161 Search PubMed.
- N. H. El-Sayed, N. M. Ammar, S. Y. Al-Okbi, L. T. A. El-Kassem and T. J. Mabry, Nat. Prod. Res., 2006, 20, 57–61 CrossRef CAS.
- P. C. Hollman and M. B. Katan, Biomed. Pharmacother., 1997, 51, 305–310 CrossRef CAS.
- T. R. Dias, M. G. Alves, S. Casal, P. F. Oliveira and B. M. Silva, Curr. Med. Chem., 2017, 24, 334–354 CrossRef.
- A. Tresserra-Rimbau, R. M. Lamuela-Raventos and J. J. Moreno, Biochem. Pharmacol., 2018, 156, 186–195 CrossRef CAS.
- C. A. Rice-Evans and N. J. Miller, Biochem. Soc. Trans., 1996, 24, 790–795 CrossRef CAS.
- J. E. Wood, S. T. Senthilmohan and A. V. Peskin, Food Chem., 2002, 77, 155–161 CrossRef CAS.
- D. Szwajgier, Pol. J. Food Nutr. Sci., 2014, 64, 59–64 CrossRef CAS.
- D. Di Majo, M. La Guardia, G. Leto, M. Crescimanno, C. Flandina and M. Giammanco, Int. J. Food Sci. Nutr., 2014, 65, 886–892 CrossRef CAS.
- J. S. Choi, M. N. Islam, M. Y. Ali, Y. M. Kim, H. J. Park, H. S. Sohn and H. A. Jung, Arch. Pharmacal Res., 2014, 37, 1354–1363 CrossRef CAS.
- G. M. Shankar and D. M. Walsh, Mol. Neurodegener., 2009, 4, 48 CrossRef.
- N. SatheeshKumar, P. K. Mukherjee, S. Bhadra and B. P. Saha, Phytomedicine, 2010, 17, 292–295 CrossRef CAS.
- M. Herrera-Ruiz, G. García-Morales, A. Zamilpa, M. González-Cortazar, J. Tortoriello, E. Ventura-Zapata and E. Jimenez-Ferrer, Bol. Latinoam. Caribe Plantas Med. Aromat., 2012, 11, 526–541 CAS.
- T. S. Anekonda and P. H. Reddy, Brain Res. Rev., 2005, 50, 361–376 CrossRef.
- R. Harrison, Drug Metab. Rev., 2004, 36, 363–375 CrossRef CAS.
- B. T. Emmerson and A. J. J. Wood, N. Engl. J. Med., 1996, 334, 445–451 CrossRef CAS.
- A. Rosa, A. Maxia, D. Putzu, A. Atzeri, B. Era, A. Fais, C. Sanna and A. Piras, Food Chem., 2017, 230, 82–90 CrossRef CAS.
- A. Rosa, D. Caprioglio, R. Isola, M. Nieddu, G. Appendino and A. M. Falchi, Food Funct., 2019, 10, 1629–1642 RSC.
- G. L. Delogu, M. J. Matos, M. Fanti, B. Era, R. Medda, E. Pieroni, A. Fais, A. Kumar and F. Pintus, Bioorg. Med. Chem. Lett., 2016, 26, 2308–2313 CrossRef CAS.
- G. L. Ellman, K. D. Courtney, V. Andres and R. M. Featherstone, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS.
- A. Kumar, F. Pintus, A. Di Petrillo, R. Medda, P. Caria, M. J. Matos, D. Vina, E. Pieroni, F. Delogu, B. Era, G. L. Delogu and A. Fais, Sci. Rep., 2018, 8, 4424 CrossRef.
- Q. Wu, Z. Peng, Y. Zhang and J. Yang, Nucleic Acids Res., 2018, 46, W438–W442 CrossRef CAS.
- J. Yang, A. Roy and Y. Zhang, Bioinformatics, 2013, 29, 2588–2595 CrossRef CAS.
- A. Roy, J. Yang and Y. Zhang, Nucleic Acids Res., 2012, 40, W471–W477 CrossRef CAS.
- M. Brylinski and J. Skolnick, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 129–134 CrossRef CAS.
- J. A. Capra, R. A. Laskowski, J. M. Thornton, M. Singh and T. A. Funkhouser, PLoS Comput. Biol., 2009, 5, e1000585 CrossRef.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- M. A. Soobrattee, V. S. Neergheen, A. Luximon-Ramma, O. I. Aruoma and T. Bahorun, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2005, 579, 200–213 CrossRef CAS.
- Z.-Q. Ling, B.-J. Xie and E.-L. Yang, J. Agric. Food Chem., 2005, 53, 2441–2445 CrossRef CAS PubMed.
- C. Ao, T. Higa, H. Ming, Y.-t. Ding and S. Tawata, J. Enzyme Inhib. Med. Chem., 2010, 25, 406–413 CrossRef CAS.
- J. B. Harborne, Phytochemistry, 1975, 14, 1147–1155 CrossRef CAS.
- A. Murray, M. Faraoni, M. Castro, N. Alza and V. Cavallaro, Curr. Neuropharmacol., 2013, 11, 388–413 CrossRef CAS PubMed.
- J. H. Kim, S.-H. Lee, H. W. Lee, Y. N. Sun, W.-H. Jang, S.-Y. Yang, H.-D. Jang and Y. H. Kim, Int. J. Biol. Macromol., 2016, 91, 1033–1039 CrossRef CAS.
- H. Moini, Q. Guo and L. Packer, J. Agric. Food Chem., 2000, 48, 5630–5639 CrossRef CAS.
- M. Ozyurek, B. Bektasoglu, K. Guclu and R. Apak, Anal. Chim. Acta, 2009, 636, 42–50 CrossRef CAS.
- A. Fais, B. Era, S. Asthana, V. Sogos, R. Medda, L. Santana, E. Uriarte, M. J. Matos, F. Delogu and A. Kumar, Int. J. Biol. Macromol., 2018, 120, 1286–1293 CrossRef CAS.
- A. Grosdidier, V. Zoete and O. Michielin, Nucleic Acids Res., 2011, 39, W270–W277 CrossRef CAS.
- R. A. Laskowski and M. B. Swindells, J. Chem. Inf. Model., 2011, 51, 2778–2786 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02928a |
| This journal is © The Royal Society of Chemistry 2019 |