
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA highly sensitive and selective spectrofluorimetric method for the determination of cerium at pico-trace levels in some real, environmental, biological, soil, food and bone samples using 2-(α-pyridyl)-thioquinaldinamide
M. Jamaluddin Ahmed *,
M. Tazul Islam and
Fahima Farhana
*,
M. Tazul Islam and
Fahima Farhana
Laboratory of Analytical Chemistry, Department of Chemistry, University of Chittagong, Chittagong-4331, Bangladesh. E-mail: pmjahmed55@gmail.com; tazul78j@gmail.com; fahimafislam@gmail.com
First published on 15th August 2019
Abstract
A very simple, ultra-sensitive and highly selective non-extractive new spectrofluorimetric method is presented for the determination of cerium at pico-trace levels using 2-(α-pyridyl)-thioquinaldinamide (PTQA). PTQA has been proposed as a new analytical reagent for the direct non-extractive spectrofluorimetric determination of cerium(IV). This novel fluorimetric reagent, PTQA becomes oxidized in a slightly acidic (0.0005–0.0015 M H2SO4) solution with cerium(IV) in absolute ethanol to produce a highly fluorescent oxidized product (λex = 303 nm; λem = 370 nm). Constant and maximum fluorescence intensities were observed over a wide range of acidity (0.0005–0.0015 M H2SO4) for the period between 5 min and 24 h. Linear calibration graphs were obtained for 0.001–600 μg L−1 of Ce, having a detection limit of 0.1 ng L−1; the quantification limit of the reaction system was found to be 1 ng L−1 and the RSD was 0–2%. A large excess of over 60 cations, anions and complexing agents (like, chloride, phosphate, azide, tartrate, oxalate, SCN− etc.) do not interfere in the determination. The developed method was successfully used in the determination of cerium in several certified reference materials (alloys, steels, noodles, ores and sediments) as well as in some environmental waters (potable and polluted), biological fluids (human blood, urine and milk), soil samples, food samples (vegetable, rice, corn and wheat), bone samples (human, cow, bull, fish, hen, goat, sheep), solutions containing both cerium(III) and cerium(IV) and complex. The results of the proposed method for assessing biological, food and vegetables samples were comparable with ICP-OES and were found to be in excellent agreement.
Introduction
Cerium is one of the most fascinating elements in the periodic table1 and continues to attract a lot of attention. Among the rare Earth elements, Ce3+ is the most widely distributed in the Earth's crust.2–4 Cerium is widely used in luminescence,5 agriculture,6 textiles7 and photographic industries, catalysis,8 nuclear energy,9 metallurgy,10 microelectronics,11 therapeutic applications,12 magnetism, glass and ceramics,13 lighting,14 solar cells,15 fuel cells,16 abrasives for chemical mechanical planarization,17 and as one of the active components of catalytic converters18 in vehicles. Cerium is especially interesting because of its variable electronic structure,19 which gives rise to dual valency states. Cerium is an iron-gray lustrous metal. Cerium in its tetravalent state, Ce(IV), differs significantly from all other lanthanides due to its immense oxidizing ability in acidic solution especially in sulfuric acid solution.20 Cerium is industrially important and is used in nuclear reactors; in alloys with nickel and chromium; and in microwave devices, lasers, and in television sets.20,21 Cerium is also used in agriculture, forestry and animal husbandry, and much attention is now being paid to the study of cerium in the environment.22,23 Addition of small amounts of the cerium to steel significantly modified its properties.22–24 Therefore, the accurate determination of cerium at trace and ultra-trace levels is important industrially.Cerium is considered to be biologically non-essential. Cerium, like all rare Earth metals, is of low to moderate toxicity.25 Fumes from cerium fires are toxic. Workers exposed to cerium have experienced itching, sensitivity to heat and skin lesions.26 Animals injected with large doses of cerium have died due to cardiovascular collapse. Cerium can, however, replace calcium in the bone structure of the human body, which may cause skeletal problems.27 The element has also been implicated as a cause of pneumoconiosis due to inhalation of cerium-bearing dust and linked to myocardial heart disease.28 The oxalate salts of the metal have been used to remedy vomiting during pregnancy,29 while other salts of the metal have been used as depressants of the central nervous system, astringents, and antiseptics. The increasing industrial use of cerium and reports on cerium toxicity make it essential to have analytical procedures suitable for monitoring cerium in the environment and related fields.14 Analytically, cerium in its tetravalent state differs significantly from all other lanthanides due to its oxidizing ability in acidic solutions.30 This property of Ce(IV) stands in the way of its determination by organic reagents. This necessitated the development of convenient and reliable analytical methods for the determination of cerium. Analytical techniques such as X-ray fluorescence,31 ICP-AES,32 electrothermal atomic absorption,33 ICP-OES,34 ICP-MS,35,36 spectrophotometry37–39 and stripping voltammetry40,41 have been used to determine cerium(III) ion. Atomic absorption spectrophotometry based method lack sensitivity, while those on flame or flameless atomization or fuel rich or fuel deficient flames have proven equally ineffective.42 Spectrofluorimetric methods43–68 were also reported for the determination of cerium. However, interference from coexist ions, especially for some rare Earth ions, Fe3+ and PO43− are always faced in the existing methods.
The goal of the present work was to develop a simpler direct spectrofluorimetric method for the pico-trace determination of cerium. In the search for a more sensitive reagent in this work a new reagent was synthesized according to the method of Porter69 and a oxidation reaction of 2-(α-pyridyl)-thioquinaldinamide (PTQA); with Ce(IV) and forms an intensely fluorescent oxidized product. The method possesses distinct advantages over existing methods43–68 (Table 1) with respect to sensitivity, selectivity, range of determination, simplicity, speed, pH/acidity range, thermal stability, accuracy, precision and ease of operation. The method is based on the oxidative reaction of non-fluorescent PTQA in a slightly acidic (0.0005–0.0015 M H2SO4) solution with Ce(IV) in presence of ethanol to produce a highly fluorescent oxidized product, followed by a direct measurement of the fluorescence intensity in an aqueous solution at room temperature. Oxidation is very rapid and no extraction is required. With suitable masking, the reaction can be made to be highly selective and the reagent blank solutions do not show any fluorescence.
| Reagent | Nature of reaction | Solvent | Medium aqueous/surfactant/organic | Acidity/pH | λex![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :λem (nm) :λem (nm) |
Beer's law (μg L−1) | Detection limit (μg L−1) | RSD (%) | Interference | Remarks | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2-Ethoxy-6,9-diaminoacridine lactate | Catalytic oxidation | Water | Sulfuric acid | 0.1 M | 265:485 |
50–500 | 12 | 6 | Pb2+, Fe3+, Sn2+, Cr3+, etc. | (i) Solvent extractive | 43 |
| (ii) Less selective due to much interference | |||||||||||
| (iii) Less sensitive | |||||||||||
| (iv) Lengthy and time consuming | |||||||||||
| Sodium 4,8-diamino-l,5-dihydroxyanthraquinone-2,6-disulphonate | Catalytic oxidation | Water | Water | 0.00 | 525:585 |
20–370 | 50 | 5 | Many | (i) Less sensitive | 44 |
| (ii) Time consuming | |||||||||||
| (iii) Less selective due to much interference | |||||||||||
| 1,5-Diphenyl-3-(2-stiryl)-Δ2-pyrazoline | Oxidation | Ethanol | HCl | 0.50 | 360–510 | 40–200 | 65 | 7 | V(V), AuIII, Pt(IV) & Fe(III) | (i) Time consuming | 45 |
| (ii) Less sensitive | |||||||||||
| (iii) Less selective due to much interference | |||||||||||
| Citric acid | Catalytic oxidation | Na2SO4 | Sulfuric acid | 0.22 | 256:360 |
8.5–300 | 5 | 3 | Many | (i) Solvent extractive | 49 |
| (ii) pH dependent | |||||||||||
| (iii) Less sensitive | |||||||||||
| (iv) Less selective due to much interference | |||||||||||
| Sodium peroxodisulfate | Fluorescence compound formation | Ethanol | Perchloric acid | 0.3 | 260:350 |
1–100 | 10 | 5 | Many | (i) Lengthy and time consuming | 53 |
| (ii) Less selective due to much interference | |||||||||||
| (iii) Less sensitive | |||||||||||
| (iv) pH dependent | |||||||||||
| Sodium dodecyl benzene sulfonate (SDBS) | Chemiluminescence | CCl4 | Acidic | 2.0 | 290–365 | 0.05–73 | 30 | 9 | Many | (i) Less sensitive | 54 |
| (ii) Time consuming | |||||||||||
| (iii) Solvent extractive | |||||||||||
| Calcein | Fluorimetric titration | KOH | HCl | 7–8.9 | 495:520 |
25–300 | 20 | 7 | Fe(II + III), Cr(VI), Mo(VI), Ce(IV) | (i) Solvent extractive | 55 |
| (ii) pH dependent | |||||||||||
| (iii) Less sensitive | |||||||||||
| (iv) Less selective due to much interference | |||||||||||
| Rhodamine 6G | Fluorimetric oxidation | Organic | Sulfuric acid | 0.45 | 530:550 |
60–240 | 15.0 | 5 | Many | (i) Solvent extractive | 58 |
| (ii) pH dependent | |||||||||||
| (iii) Less sensitive | |||||||||||
| (iv) Less selective due to much interference | |||||||||||
| Phosphoric acid | Catalytic oxidation | Water | H3PO4 acid | 3.5 | 297:346 |
0.5–100 | 12 | 3 | Fe(III), Cr(VI), Mn(VII), V(V), etc. | (i) pH dependent method | 64 |
| (ii) Less selective due to much interference | |||||||||||
| (iii) Less sensitive | |||||||||||
| KCl | Fluorescence compound formation | Ethanol | Weak acid medium | 6.2 | 252–354 | 0–420 | 84 | 4 | Many | (i) Time consuming | 65 |
| (ii) Less sensitive | |||||||||||
| (iii) Less selective due to much interference | |||||||||||
| 2-(α-Pyridyl)-thio quinaldinamide (PTQA) (present method) | Oxidation reaction | Water | Slight H2SO4 medium | 0.001 M | 303–370 | 0.001–600 | 0.1 pg mL−1 | 2 | Nil, using suitable masking agents | (i) Ultra sensitive | Present method |
| (ii) Highly selective | |||||||||||
| (iii) Aqueous medium | |||||||||||
| (1) Pico-trace levels | |||||||||||
| (2) Fluorescence is stable for over 24 h at roomtemp. 25 ± 5 °C | |||||||||||
| (3) Very simple, rapid and non-extractive | |||||||||||
| (4) Carcinogenic solvents are avoided |
Experimental section
Apparatus
A Shimadzu (Kyoto, Japan) (Model-RF-5301PC) spectrofluorophotometer with 1 cm quartz cells were used and a Jenway (England, UK) (Model-3010) pH meter with combination of electrodes were used for measurements of the fluorescence intensity and pH. The calibration and linearity of the instrument were frequently checked with standard quinine sulphate (10 mg L−1). A PerkinElmer (Toronto, Canada) (Model: Optima 5300) Inductively Coupled Plasma-Optical Emission Spectrometer (ICP-OES), [λ = 418 nm, plasma gas flow rate (L min−1) = 15, RF power (W) = 1400, nebulizer gas flow rate (L min−1) = 1–10] was used to compare of the results. The Elemental Analyzer (Exeter Analytical Inc. Model: CE 440) equipped with supersensitive thermal conductivity detector for simultaneous determination of CHN was used. Infrared spectrum was recorded with a FTIR spectrophotometer, Shimadzu (Kyoto, Japan) (Model-IR Prestige 21, Detector DTGS KBr) in the range 7500–350 cm−1 and model: JEOL 500SS, magnetic field strength: 500 MHz, solvent used: DMSO D6, standard: TMS, four channel NMR spectrometer with signal-to-noise ratio of ∼5000:1 for proton were used for characterization of the ligand.
Synthesis and characterization of the reagent
2-(α-Pyridyl)-thioquinaldinamide (PTQA, C15H11N3S) (molecular wt = 265.18) was synthesized according to the method of Porter.69 The mixture containing 2-aminopyridin (purity 98%), quinaldine (99%) and sulphur (97%) powder in the molar ratio of 2:1:1.5 were mixed and refluxed for 6 hours in 250 mL round bottom flask fitted with bulb condenser under controlled temperature (140–150) °C at 1 atm pressure over oil bath. The reaction mixture was kept over night. The thio-compound was filtered and crystallized using petroleum ether (60–80) °C to give a bright yellow crystalline (needle shaped) solid. The compound recrystallized from lime-distilled ethanol and was kept under vacuum (0.1 mm of Hg) for 24 hours. Yield of the product was 70%. The structure of the reagent is shown in Scheme 1.
 | ||
| Scheme 1 Reaction scheme of 2-(α-pyridyl)-thioquinaldinamide (PTQA). | ||
Characterization of the reagent
The reagent (PTQA) was characterized by taking the melting point, elemental analysis and an FTIR spectrum (Fig. 1) and 1H NMR spectrum (Fig. 2) and thermo-gravimetric analysis. The melting point of the synthesized compound (PTQA) was 155 ± 2 °C (lit. 155 ± 1 °C)70 which indicated the purity of PTQA.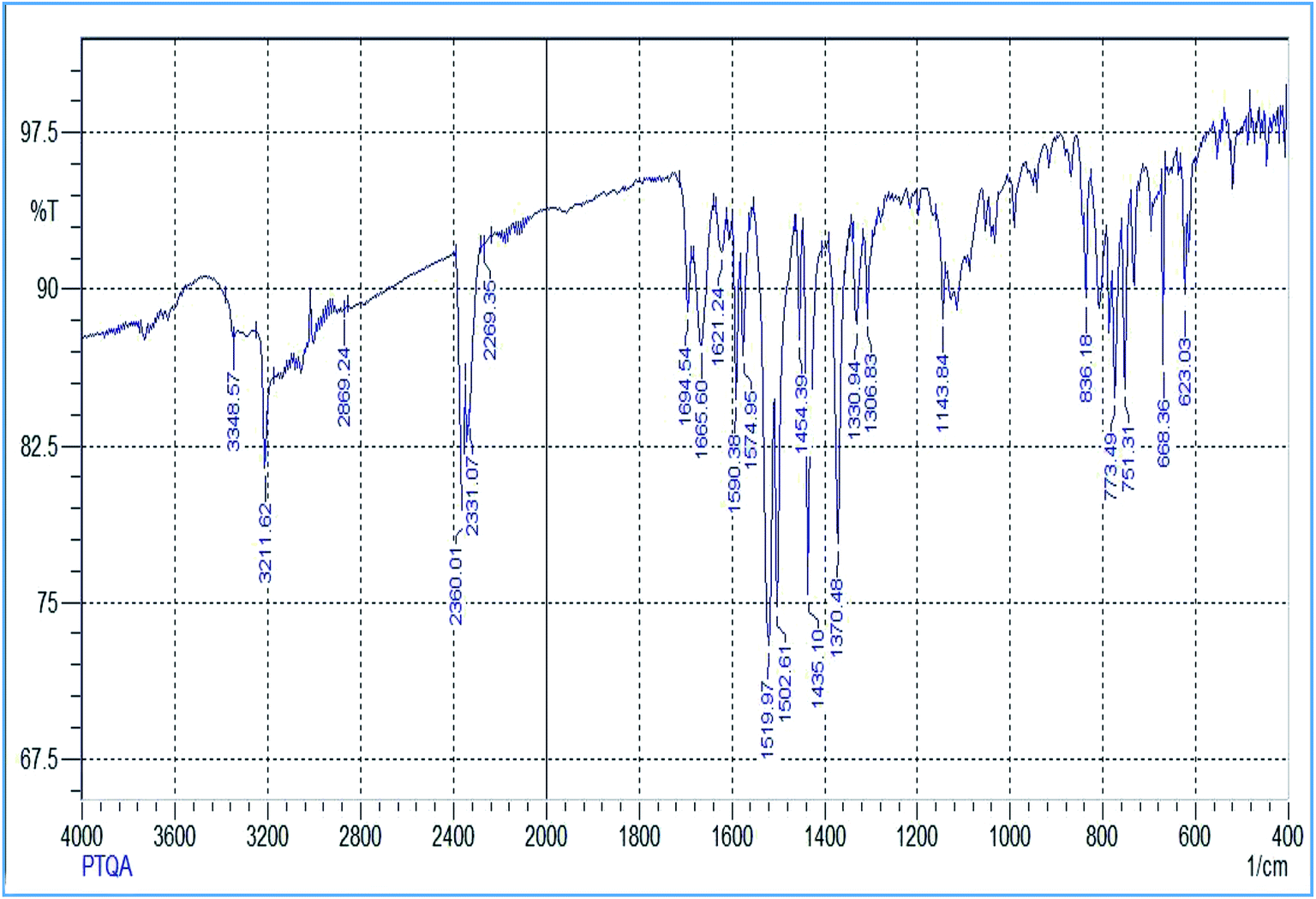 | ||
| Fig. 1 FTIR spectrum of 2-(α-pyridyl)-thioquinaldinamide. | ||
 | ||
| Fig. 2 1H NMR spectrum of 2-(α-pyridyl)-thioquinaldinamide (PTQA). | ||
The results elemental analysis (C = 72.25%, N = 13.35% and H = 4.25%) of the reagent are very in good agreement with the calculated values (C = 72.43%, N = 13.55% and H = 4.55%). The FTIR spectrum of prepared reagent (PTQA) is shown in Fig. 1. The presence of FTIR peak at 1126.43 cm−1 in Fig. 1 was due to the characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S double bond peak (νCS,1050–1200 cm−1)70 of the reagent indicating the formation of PTQA. Both FTIR spectral and elemental analysis data indicated the formation of the reagent PTQA. The formation of the reagent also tested by 1H NMR spectrum is shown in Fig. 2. The steadiness of the thermogravimetric curve obtained for about 1 g of the reagent at 80–90 °C indicated that the reagent did not contain any moisture.
S double bond peak (νCS,1050–1200 cm−1)70 of the reagent indicating the formation of PTQA. Both FTIR spectral and elemental analysis data indicated the formation of the reagent PTQA. The formation of the reagent also tested by 1H NMR spectrum is shown in Fig. 2. The steadiness of the thermogravimetric curve obtained for about 1 g of the reagent at 80–90 °C indicated that the reagent did not contain any moisture.
The elemental analysis was performed by the National Center of Excellence in Analytical Chemistry, University of Sindh, Pakistan and FTIR spectra was recorded with FTIR spectrophotometer in our laboratory and 1H NMR spectrum was recorded from University of Kanazawa of our prepared reagent.
Synthesis of the reagent
PTQA solution (9.8 × 10−4 M). The reagent solution was prepared by dissolving the requisite amount (0.0026 g) of PTQA, in a known volume (10 mL) of absolute ethanol. A freshly prepared reagent solution (9.8 × 10−4 M) was used whenever required.
Cerium(IV) standard solution (7.14 × 10−3 M). A 100 mL amount of stock solution (1 mg mL−1) of tetravalent cerium was prepared by dissolving 288.5 mg of ceric sulfate tetra-hydrate, {Ce(SO4)2·4H2O} (Sigma-Aldrich, Merck KGaA, Germany, pro-analysis grade, 99.6%) in doubly distilled de-ionized water containing 3 mL of concentrated sulfuric acid and standardized by titrimetry with ethylenediaminetetraacetic acid (EDTA) using o-phenanthroline solution (ferroin) as indicator.71 More dilute standard solutions were prepared by appropriate dilution of aliquots from the stock solution with de-ionized water as and when required. A freshly standardized solution was always used.
Cerium(III) standard solution (3.22 × 10−3 M). A 100 mL amount of stock solution (1 mg mL−1) of trivalent cerium was prepared by dissolving 309.9 mg of cerous nitrate hexahydrate {Ce(NO3)3·6H2O} (Sigma-Aldrich, Merck KGaA, pro-analysis grade, 99.5%) in doubly distilled de-ionized water and standardized by titrimetry with ethylenediaminetetraacetic acid (EDTA) using o-phenanthroline solution (ferroin) as indicator.71 More dilute standard solutions were prepared by appropriate dilution of aliquots from the stock solution with de-ionized water as and when required. A freshly standardized solution was always used.
Potassium dichromate solution. A 100 mL amount of stock solution (0.1 N) was prepared by dissolving 500 mg of finely powdered K2Cr2O7 (Merck, 99.9%) in 100 mL de-ionized water.
Ammonium persuphate solution. Ammonium persuphate solution (2% w/v) (ACS-grade 99% pure) was freshly prepared by dissolving 2 g in 100 mL of de-ionized water.
Tartrate solution. A 100 mL stock solution of tartrate (0.01% w/v) was prepared by dissolving 10 mg of ACS-grade (99%) potassium sodium tartrate tetrahydrate in (100 mL) de-ionized water.
Aqueous ammonia solution. A 100 mL solution of an aqueous ammonia solution was prepared by diluting 10 mL concentrated NH4OH (28–30%, ACS-grade) to 100 mL with de-ionized water. The solution was stored in a polypropylene bottle.
EDTA solution. A 100 mL stock solution of EDTA (0.01% w/v) was prepared by dissolving 10 mg ACS-grade (≥99%) ethylenediaminetetraacetic acid as disodium salt dehydrate in (100 mL) de-ionized water.
Other solutions. Solutions of a large number of inorganic ions and complexing agents were prepared from their annular grade or equivalent grade water-soluble salts (or the oxides and carbonates in hydrochloric acid); those of niobium, tantalum, titanium, zirconium and hafnium were specially prepared from their corresponding oxides (Specpure, Johnson Matthey) according to the recommended procedures of Mukharjee.72 In the case of insoluble substances, special dissolution methods were adopted.73
Procedure
To 0.1–1.0 mL of a neutral aqueous solution containing 0.01–6000 ng of cerium(IV) in a 10 mL calibrated flask was mixed with a 1:250–1:800 fold molar excess (preferably 1 mL) of 9.8 × 10−4 M of the 2-(α-pyridyl)-thioquinaldinamide (PTQA) reagent solution followed by the addition of 0.5–2 mL (preferably 1 mL) of 0.001 M of sulfuric acid. The solution was mixed well and allowed to stand for 5 min after which 2 mL of absolute ethanol was added and the mixture was diluted to the mark with de-ionized water. The fluorescence intensity of the system was measured at 370 nm against a corresponding reagent blank, prepared concurrently, keeping the excitation wavelength maximum at 303 nm and the instrument setting the same. The cerium content in an unknown sample was determined using a concurrently prepared calibration graph.
Environmental samples. Water and soil samples were collected in polythene bottles from different places of Bangladesh. After collection, HNO3 (1 mL−1) was added as preservative.
Blood, urine and milk. Blood and urine samples were collected in polythene bottles from effected persons of Chittagong Medical College Hospital, Bangladesh. Milk sample was collected from a Bangladeshi lactating mother. Immediately after collection they were stored in a salt-ice mixture and latter, at the laboratory, were at 20 °C.
Soil samples. Soil samples were collected from different locations of Bangladesh. Samples were dried in air and homogenized with a mortar.
Food samples. Food samples (rice, wheat, fruits and vegetables) were collected from local market of Chittagong. After collection the samples (fruits and vegetables) were stored in refrigerator for preservation. Samples (rice, wheat) were used as dry condition and homogenized with a mortar.
Bone samples. Bone samples of different animals were collected from local market of Chittagong, and that of human from the Chittagong Medical College Hospital. Dried samples were homogenized with a mortar. Bone samples were used as dry condition and homogenized with a mortar.
Results and discussion
Factors affecting the fluorescence intensity
 | ||
| Fig. 3 Spectra A and C are the excitation spectra (fluorescence collected at λem = 370 nm) and emission spectra (excitation wavelength λex = 303 nm) of CeIV–PTQA system; spectra B and D are the excitation spectra (fluorescence collected at λem = 370 nm) and emission spectra (excitation wavelength λex = 303 nm) of reagent blank, respectively in aqueous solutions. | ||
From these spectra we performed the subsequent fluorescence measurements at λex = 303 nm and λem = 370 nm.
Effect of solvent. Because PTQA is insoluble in water, an organic solvent was used for the system. Of the various solvents [chloroform, benzene, carbon tetrachloride, n-butanol, isobutanol, ethanol, 1,4-dioxane and N,N-dimethylformamide (DMF)] were tested for the system, ethanol was found to be the best solvent for the system. The effect of ethanol on the fluorescence intensity was studied and no adverse effect was observed over a wide range of ethanol concentrations. It was observed that CeIV–PTQA system with 10 μg L−1 of CeIV in absolute ethanol solution produced constant fluorescence intensity as shown in Fig. 4. A concentration of 20% v/v ethanol in the final volume was sufficient to prevent any precipitation or turbidity and to allow accurate measurements. Therefore, a 20% v/v ethanolic solution was used in the recommended procedure.
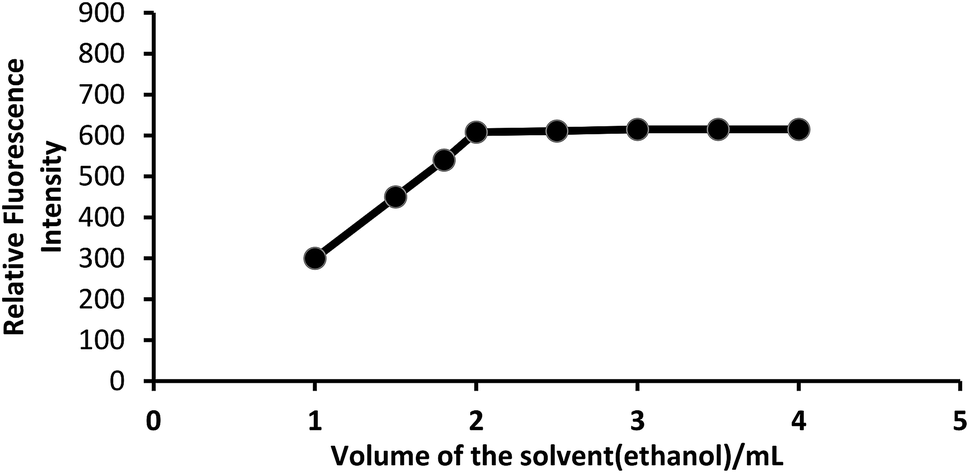 | ||
| Fig. 4 Effect of solvent (ethanol) on the fluorescence of CeIV–PTQA system. | ||
Effect of acidity or effect of H2SO4 concentration. The oxidation reaction was conducted in acid medium to avoid the precipitation of hydrated ceric oxide, CeO2·H2O. In order to determine the most suitable acid for the reaction, different acids (nitric, sulfuric, hydrochloric and phosphoric) were tested. But, sulfuric acid was found to be the best acid than any other mineral acids for the system. The fluorescence intensity was at maximum and constant when the 10 mL of solution (10 μg L−1of CeIV) contained 0.5–2 mL of 0.0005–0.0015 M sulfuric acid at room temperature (25 ± 5 °C). Outside this range of acidity, the fluorescence intensity decreased (Fig. 5). The optimum acidity range in the final solution is therefore 0.0005–0.0015 M (preferably 0.001 M) H2SO4. Therefore, 1 mL of 0.001 M sulfuric acid solution was used for all subsequent measurements.
 | ||
| Fig. 5 Effect of acidity on the fluorescence of ceriumIV–PTQA system. | ||
Effect of temperature. The influence of temperature was studied between 10–70 °C. It could be observed that temperature effect is not pronounced between 10–70 °C and so room temperature (25 ± 5) °C is recommended for all subsequent measurements.
Effect of time. The reaction is instantaneous. The CeIV–PTQA system attained maximum and constant fluorescence intensity immediately (within 5 min) after dilution of the solution to the final volume, which then remained strictly unaltered for 24 h at room temperature (25 ± 5 °C).
Effect of reagent concentration. The intensities of the fluorescence of a series of solutions containing a constant amount of Ce(IV) with varying amounts of PTQA were measured in order to establish the optimum concentration of PTQA. The change of fluorescence intensity with PTQA concentration was shown in Fig. 6 while the concentration of CeIV was kept constant. It was found that the fluorescence intensity increase at first as the PTQA concentration rises and reaches a maximum, but further addition of PTQA hardly effect the intensity even PTQA is more times concentrated than CeIV. It was observed that at 10 μg L−1 CeIV metal and the reagent molar ratios of 1
:250 to 1:800 produced a constant fluorescence intensity of the oxidized product. Outside this range of reagent, the fluorescence intensity get decreased (Fig. 6). At different CeIV concentrations (0.5 and 1 μg L−1), the effect of varying the reagent concentration was similar. For all subsequent measurements 1 mL of 9.8 × 10−4 MPTQA reagent was added.
 | ||
| Fig. 6 Effect of reagent (PTQA: ceriumIV molar concentration) on the fluorescence of ceriumIV–PTQA system. | ||
Effect of metal concentration. The effect of Ce(IV) solution was investigated by carrying out the reaction using 1 mL of Ce(IV) solution of different concentrations ranging from 0.001–1000 μg L−1. It was found that the relative fluorescence intensity increased by increasing the concentration of Ce(IV) up to 600 μg L−1, but higher concentration has negative effect on the values of the relative fluorescence intensity.
Calibration curves (Beer's law). The calibration graphs for the determination of Ce(IV) were constructed under optimum conditions. The well-known equation for spectrofluorimetric analysis in very dilute solutions derived from Beer's law. The effect of metal concentration was studied over 0.001–1000 μg L−1 distributed in six different sets (0.001–0.01, 0.01–0.1, 0.1–1, 1–10, 10–100 and 100–1000 μg L−1) for convenience of measurement. The fluorescence intensity was linear over a wide range 1 pg mL−1 to 600 μg mL−1 for 0.001–600 μg L−1 of cerium(IV) at excitation wavelength at 303 nm and emission wavelength at 370 nm. Of six calibration graphs, the one showing the limit of the linearity range (Fig. 7); the remaining five were straight-line graphs passing through the origin (R2 = 0.9998). The limit of detection and limit of quantization were found to be 0.1 ng L−1 and 1 ng L−1, respectively. Selected analytical parameters obtained with the optimization experiments are shown in Table 2.
 | ||
| Fig. 7 Calibration graph F: 100–600 μg L−1 of ceriumIV. Bandwidth: Ex. slit-1.5, Em. slit-1.5. Sensitivity: high. | ||
| Parameters | Studied range | Selected value |
|---|---|---|
| Excitation wavelength maximum/λex (nm) | 200–700 | 303 |
| Emission wavelength maximum/λem (nm) | 200–700 | 370 |
| Acidity/M H2SO4 | 0.00005–0.5 | 0.0005–0.0015 (preferably 0.001) |
| pH | 2.0–7.0 | 4.9–5.6 (preferably 5.1) |
| Time/h | 0–72 | 1 min to 24 h (preferably 5 min) |
| Temperature/°C | 5–80 | 10–70 (preferably 25 ± 5) |
| Reagent (fold molar excess, M:R) |
1:50–1:2000 |
1:250–1:800 (preferably 1:500) |
| Linear range/μg L−1 | 0.0001–1000 | 0.001–600 |
| Limit of quantization/ng L−1 | 0.01–100 | 1.0 |
| Detection limit/ng L−1 | 0.001–10.0 | 0.1 |
| Reproducibility (% RSD) | 0–10 | 0–2 |
| Regression co-efficient (R2) | 0.9996–0.9999 | 0.9998 |
Effect of foreign ions. In order to apply the proposed method to the determination of the concentration of Ce(IV) in the real sample, the effect of some co-existing species was investigated using 10 μg L−1 of cerium(IV). The tolerance limits of about 60 anions, cations and complexing agents were studied individually to investigate their effect on the determination of 10 μg L−1 of cerium(IV). The criterion for interference74 was a fluorescence intensity value varying by more than ±5% from the expected value for cerium alone. The results are summarized in Table 3. As can be seen a large number of ions have no significant effect on the determination of cerium. So the method is of good selectivity.
| Species x | Tolerance ratio x/CeIV (w/w) | Species x | Tolerance ratio x/CeIV (w/w) |
|---|---|---|---|
| a Tolerance limit was defined as ratio that causes less than ±5 percent interference.b With 10 mg L−1 tartrate.c With 10 mg L−1EDTA. | |||
| Ammonium | 1000 | Lithium | 1000 |
| Arsenic(III) | 1000 | Lead(II) | 1000 |
| Arsenic(V) | 1000 | Magnesium | 1000 |
| Aluminum | 1000 | Manganese(II) | 1000 |
| Azide | 1000 | Manganese(VII) | 1000b |
| Ascorbic acid | 5000 | Mercury(II) | 1000 |
| Antimony | 1000 | Molybdenum(VI) | 1000c |
| Bromide | 1000 | Nitrate | 1000 |
| Bismuth(III) | 1000 | Nickel | 1000 |
| Beryllium(II) | 1000 | Oxalate | 5000 |
| Barium | 1000 | Potassium | 1000 |
| Calcium | 1000 | Phosphate | 1000 |
| Chloride | 1000 | Selenium(VI) | 1000 |
| Cobalt(II) | 1000 | Selenium(IV) | 1000c |
| Cobalt(III) | 1000 | Silver | 1000 |
| Chromium(III) | 1000 | Sodium | 1000 |
| Chromium(VI) | 1000b | Strontium | 1000 |
| Cadmium | 1000 | Sulfate | 1000 |
| Carbonate | 1000 | Titanium(IV) | 1000 |
| Cesium | 1000 | Tellurium(IV) | 1000 |
| Citrate | 5000 | Tellurium(VI) | 1000 |
| Cerium(III) | 1000 | Tartrate | 5000 |
| Copper(II) | 1000 | Thiocyanate | 1000 |
| Cyanide | 1000 | Thiourea | 1000 |
| EDTA | 5000 | Tungsten(VI) | 1000 |
| Fluoride | 5000 | Tin(II) | 1000 |
| Iodide | 1000 | Tin(IV) | 1000 |
| Iron(II) | 1000 | Vanadium(V) | 1000 |
| Iron(III) | 1000 | Zinc | 1000 |
Most of the ions were tolerated over 1000 exceeds. Ascorbic acid, oxalate, citrate, tartrate, EDTA & fluoride ions etc. were tolerated over 5000 folds. In order to eliminate the interference of Se(IV), Cr(VI) and Mn(VII) ions, EDTA and tartrate can be used as masking agents, respectively.75 Strong reducing agents such as, tin(II), chloride, iron(II), sulfate, hydroxylamine, hydrochloride and sodium azide, which would otherwise reduce cerium(IV), undergo oxidation during the treatment of the cerium(III) solution with persulphate and hence are not a problem.76 The amount mentioned is not the tolerance limit but the actual amount studied. However, for those ions whose tolerance limits have been studied, their tolerance ratios are mentioned in Table 3.
| Sample | Composition of mixtures (μg L−1) | Cerium/μg L−1 | ||
|---|---|---|---|---|
| Added | Founda (n = 5) | Recovery ± SDb (%) | ||
| a Average of five analyses of each sample.b The measure of precision is the standard deviation (SD). | ||||
| A | Ce(IV) | 1.0 | 0.99 | 99.0 ± 0.5 |
| 50 | 49.98 | 99.96 ± 0.6 | ||
| B | As in A + Cr3+ (50) + As3+ (50) + Co2+ (50) + Fe3+ (50) + EDTA (50) | 1.0 | 1.0 | 100.0 ± 0.00 |
| 50 | 49.98 | 99.98 ± 0.8 | ||
| C | As in B + Pb2+ (50) + Bi3+ (50) + Hg2+ (50) + SeIV (50) + Ba2+ (50) | 1.0 | 0.98 | 98.0 ± 0.6 |
| 50 | 49.97 | 99.9 ± 1.0 | ||
| D | As in C + Sn2+ (50) + CrVI (50) + Ti2+ (50) + Mg2+ (50) + Fe2+ (50) | 1.0 | 1.0 | 100.0 ± 0.0 |
| 50 | 49.95 | 99.6 ± 1.0 | ||
| E | As in D + Ca2+ (50) + Al3+ (50) + WVI (50) + Cd2+ (50) + Ni2+ (50) | 1.0 | 0.99 | 99.0 ± 1.5 |
| 50 | 49.90 | 99.8 ± 2.0 | ||
| F | As in E + Na+ (50) + K+ (50) + Zn2+ (50) + Li+ (50) + Ce3+ (50) | 1.0 | 0.98 | 98.0 ± 0.5 |
| 50 | 49.98 | 99.96 ± 0.6 | ||
| Sample no. | Certified reference materials (composition%) | Cerium (%) | ||
|---|---|---|---|---|
| In CRM sample | Found (n = 5) | RSDb | ||
| a The standard sample was obtained from The Baotou Institute of Rare Earth, Baotou, China.b The measure of precision is the relative standard deviation (RSD).c Values in μg g−1.d Values in mg kg−1. | ||||
| 1 | GBW 01144: low alloyed steela: (C = 3.73, Si = 2.01, Mn = 0.449, Cr = 0.401, Ni = 0.63, Cu = 0.548, Ce = 0.465, Mo = 0.22, V = 0.161, Ti = 0.22, Mg = 0.059, Sn = 0.128, La = 0.13) | 0.465 | 0.461 | 1.0 |
| 2 | ECRM-688-1: iron orea: (As = 11.0, B = 5.0, Be = 1, Ce = 55.0, Cr = 21.7, Gd = 3.5, Hg = 0.5, Hf = 0.4, Ho = 0.7, La = 26.0, Lu = 0.3, Nb = 1.6, Nd = 26.0, Rb = 9.0, S = 468.0, Sc = 7, Se = 0.7, Sm = 4.6, Sn = 3.3, Sr = 19.0) | 55.0 | 54.85 | 2.0 |
| 3 | JK-37: high alloyed steela: (C = 0.013, Si = 0.14, Mn = 1.73, P = 0.016, Cr = 26.72, Ni = 30.82, Mo = 3.55, Ce = 0.123, Co = 0.058, Cu = 0.936, Al = 0.008, As = 0.004, B = 0.001) | 0.123 | 0.125 | 1.0 |
| 4 | Rare-Earth ore samplea: CeO2 = 49.21 | 49.21 | 48.32 | 1.8 |
| 5 | NIST-SRM-3110: single element CRM of Ce: Ce = 978 ± 3c | 978 ± 3 | 976.5 ± 2.5 | 2.5 |
| 6 | NIST-SRM-1575: pine noodlesc | 0.4 | 0.42 ± 1.5 | 1.3 |
| 7 | NIST-SRM-1646a: estuarine sedimentd | 80 | 78.5 | 2.2 |
| 8 | SABS-SRM-19: soild | 56 | 55.8 | 2.5 |
| 9 | GBW-09101: human haird | 1.9 | 1.88 | 1.5 |
| 10 | GBW-08503: wheat flourd | 10.1 | 9.98 | 1.3 |
| Sample | Cerium/μg L−1 | Recovery ± SD (%) | RSDb (%) | ||
|---|---|---|---|---|---|
| Added | Founda (n = 5) | ||||
| a Average of five replicate determinations of each sample.b The measure precision is the relative standard deviation (RSD).c T. S. P. Complex Ltd., Patenga, Chittagong.d Usmania Glass Sheet Factory Ltd., Chandgaon, Chittagong.e PHP Glass factory, Chittagong.f Bangladesh Steel Re-rolling Mills Ltd. (BSRM), Baizid Bosthami, Chittagong.g Karnaphuly Paper Mills, Chandraghona, Chittagong.h Eastern Cables Ltd., Patenga, Chittagong.i Berger Paints Bangladesh Limited, Kalurghat, Chittagong.j Elite Paint & Chemical Industries Ltd., Agrabad, Chittagong.k Glaxo SmithKline Bangladesh Ltd., Fouzderhat, Chittagong.l Albion Laboratories Ltd., Muradpur. | |||||
| Tap water (Chittagong city) | 0 | 6.55 | 99.8 ± 0.8 | 0.30 | |
| 10 | 14.6 | 96.0 ± 0.3 | 0.23 | ||
| 50 | 56.0 | ||||
| Rain water (Chittagong city) | 0 | 4.8 | 98.7 ± 1.2 | 0.31 | |
| 10 | 15.0 | 98.0 ± 0.5 | 0.40 | ||
| 50 | 56.0 | ||||
| Well water (Chittagong city) | 0 | 8.5 | 95.7 ± 1.5 | 0.25 | |
| 10 | 19.5 | 97.5 ± 1.3 | 0.21 | ||
| 50 | 60.5 | ||||
| River water | Karnaphully (upper) | 0 | 12.0 | 97.8 ± 1.0 | 0.22 |
| 10 | 22.5 | 97.0 ± 0.8 | 0.28 | ||
| 50 | 64.0 | ||||
| Karnaphully (lower) | 0 | 15.0 | 100.0 ± 0.0 | 0.00 | |
| 10 | 25.0 | 97.0 ± 1.0 | 0.21 | ||
| 50 | 67.0 | ||||
| Halda (upper) | 0 | 10.5 | 97.6 ± 0.8 | 0.29 | |
| 10 | 21.0 | 97.6 ± 0.6 | 0.25 | ||
| 50 | 62.0 | ||||
| Halda (lower) | 0 | 12.5 | 97.8 ± 1.0 | 0.24 | |
| 10 | 23.0 | 100.0 ± 0.0 | 0.00 | ||
| 50 | 62.5 | ||||
| Sea water | Bay of Bengal (upper) | 0 | 7.5 | 97.2 ± 1.0 | 0.37 |
| 10 | 18.0 | 98.3 ± 0.8 | 0.29 | ||
| 50 | 58.5 | ||||
| Bay of Bengal (lower) | 0 | 9.0 | 100.0 ± 0.0 | 0.00 | |
| 10 | 19.0 | 98.3 ± 0.75 | 0.19 | ||
| 50 | 60.0 | ||||
| Drain water | T. S. P. Complexc | 0 | 45.0 | 100.0 ± 0.0 | 0.00 |
| 10 | 55.0 | 98.9 ± 1.0 | 0.28 | ||
| 50 | 96.0 | ||||
| Usmania Glassd | 0 | 65.0 | 100.0 ± 0.0 | 0.00 | |
| 10 | 75.0 | 97.5 ± 0.8 | 0.27 | ||
| 50 | 118.0 | ||||
| PHP Glasse | 0 | 75.6 | 98.4 ± 0.5 | 0.65 | |
| 10 | 87.0 | 99.7 ± 0.3 | 0.45 | ||
| 50 | 126.0 | ||||
| BSRM Steelf | 0 | 85.0 | 100.0 ± 0.0 | 0.00 | |
| 10 | 95.0 | 96.4 ± 1.0 | 0.29 | ||
| 50 | 140.0 | ||||
| KPMg | 0 | 55.0 | 100.0 ± 0.0 | 0.00 | |
| 10 | 65.0 | 97.2 ± 0.8 | 0.48 | ||
| 50 | 108.0 | ||||
| Eastern Cables Ltd.h | 0 | 78.0 | 100.0 ± 0.0 | 0.00 | |
| 10 | 88.0 | 99.2 ± 1.0 | 0.26 | ||
| 50 | 129.0 | ||||
| Berger Painti | 0 | 85.30 | 100.0 ± 0.0 | 0.00 | |
| 10 | 95.0 | 97.8 ± 0.8 | 0.27 | ||
| 50 | 138.0 | ||||
| Elite Paintj | 0 | 78.0 | 100.0 ± 0.0 | 0.00 | |
| 10 | 88.0 | 98.5 ± 0.6 | 0.39 | ||
| 50 | 130.0 | ||||
| GSKk | 0 | 75.0 | 100.0 ± 0.0 | 0.00 | |
| 10 | 85.0 | 99.2 ± 0.5 | 0.18 | ||
| 50 | 126.0 | ||||
| Albion Lab. Ltd.l | 0 | 95.0 | 100.0 ± 0.0 | 0.00 | |
| 10 | 105.0 | 97.0 ± 1.2 | 0.28 | ||
| 50 | 150.0 | ||||
| Serial no. | Sample sourcea | Sample | Cerium/ng L−1 | |||
|---|---|---|---|---|---|---|
| ICP-OES (n = 5) | Proposed method (n = 5) | |||||
| Found | RSD (%) | Found | RSD (%) | |||
| a Samples were collected from Chittagong Medical College Hospital.b Values in μg g−1. | ||||||
| 1 | Brain disorder (male) | Blood | 343.5 | 2.0 | 345.8 | 2.5 |
| Urine | 86.8 | 1.5 | 88.5 | 1.8 | ||
| 2 | Liver cirrhosis patient (male) | Blood | 315.6 | 2.8 | 320.5 | 2.2 |
| Urine | 78.5 | 1.8 | 80.8 | 1.8 | ||
| 3 | Neurological problem (male) | Blood | 385.0 | 2.5 | 388.5 | 2.6 |
| Urine | 73.0 | 1.6 | 75.8 | 1.5 | ||
| 4 | Kidney disease patient (male) | Blood | 205.6 | 2.0 | 210.8 | 1.8 |
| Urine | 52.8 | 1.8 | 54.5 | 1.5 | ||
| 5 | Lung cancer patient (female) | Blood | 425.6 | 2.5 | 430.0 | 2.0 |
| Urine | 168.5 | 1.8 | 110.8 | 1.7 | ||
| 6 | Diabetic patient (female) | Blood | 150.8 | 2.6 | 152.5 | 2.5 |
| Urine | 40.8 | 1.5 | 42.8 | 1.6 | ||
| 7 | Hypertension patient (female) | Blood | 195.0 | 2.2 | 198.5 | 2.0 |
| Urine | 50.5 | 1.5 | 51.8 | 1.6 | ||
| 8 | Hepatitis B patient (male) | Blood | 220.5 | 2.0 | 225.8 | 2.1 |
| Urine | 55.8 | 1.8 | 58.5 | 2.0 | ||
| 9 | Bangladeshi lactating mother | Milk | 58.8 | 1.2 | 58.6 | 1.5 |
| 10 | Normal human hair (female) | Human hairb | 2.3 | 1.5 | 2.5 | 1.6 |
| Serial no. | Sample sourcec | Cerium (mg kg−1)a (n = 5) | RSDb (%) |
|---|---|---|---|
| a Average of five analyses of each sample.b The measure of precision is the relative standard deviation (RSD).c Composition of the soil samples: C, N, P, K, Na, Ca, Mg, Ce, Cu, Mo, Fe, Pb, V, Zn, Mn, Co, NO3, SO4 et al. | |||
| S1 | Fertilizer industrial soil (T. S. P. Complex, Chittagong) | 51.8 | 1.2 |
| S2c | Glass industrial soil (PHP Glass) | 63.5 | 1.5 |
| S3 | Steel industrial soil (Bangladesh Steel Re-rolling Mills Ltd., Chittagong, Bangladesh) | 78.5 | 2.0 |
| S4 | Paint industry soil (Berger Paint) | 85.8 | 1.6 |
| S5 | Paint industry soil (Elite Paint) | 77.5 | 1.5 |
| S6 | Industrial soil (Eastern Cables Ltd) | 75.2 | 1.8 |
| S7 | Agricultural soil (Chittagong University Campus) | 21.8 | 1.0 |
| S8 | Marine soil (sediments) (Bay of Bengal) | 66.6 | 1.5 |
| S9 | Road side soil (Chittagong to Dhaka) | 75.0 | 1.9 |
| S10 | Pharmaceutical soil (GlaxoSmithKline) | 135.8 | 1.8 |
| Serial no. | Bone sample | Cerium (μg kg−1)a (n = 5) | RSDb (%) | Sample source |
|---|---|---|---|---|
| a Average of five analyses of each sample.b The measure of precision is the relative standard deviation (RSD). | ||||
| 1 | Human (adult) | 510.5 | 1.5 | Chittagong Medical College Hospital, Chittagong |
| 2 | Human (baby) | 495.6 | 1.0 | Chittagong Medical College Hospital, Chittagong |
| 3 | Cow | 305.2 | 1.8 | Local market, Chittagong |
| 4 | Bull | 321.8 | 2.0 | Local market, Chittagong |
| 5 | Hen (deshi) | 125.8 | 1.8 | Local market, Chittagong |
| 6 | Cock (deshi) | 130.5 | 1.6 | Local market, Chittagong |
| 7 | Chicken (broiler) | 485.8 | 2.0 | Local market, Chittagong |
| 8 | Goat | 432.5 | 1.8 | Local market, Chittagong |
| 9 | Sheep | 465.8 | 1.9 | Local market, Chittagong |
| 10 | Buffalo | 498.5 | 2.5 | Local market, Chittagong |
| Serial no. | Sample | Cerium/mg kg−1, founda ± SD (n = 5) | Sample source | |||
|---|---|---|---|---|---|---|
| ICP-OES (n = 5) | Proposed method (n = 5) | |||||
| Found | RSDb | Found | RSDb | |||
| a Average of five replicate analyses of each sample.b The measure of precision is the relative standard deviation (RSD). | ||||||
| 1 | Carrot (Daucus carota) | 20.5 | 1.8 | 21.8 | 2.0 | Local market, Chittagong |
| 2 | Wheat (Trictium aestivum) | 12.5 | 1.5 | 12.8 | 1.8 | Local market, Chittagong |
| 3 | Corn (Zea mays) | 8.6 | 1.7 | 9.5 | 1.6 | Local market, Chittagong |
| 4 | Mango (Mangifera indica) | 15.6 | 2.0 | 16.8 | 2.1 | Local market, Rajshahi |
| 5 | Rice (Oryza sativa) | 9.9 | 1.8 | 10.1 | 2.0 | Local market, Chittagong |
| 6 | Arum (Arum discorides) | 14.9 | 1.8 | 15.5 | 2.0 | Local market, Chittagong |
| 7 | Radish (Raphanus sativus) | 10.5 | 1.7 | 10.9 | 1.8 | Local market, Chittagong |
| 8 | Potato (Solanum tuberosum) | 10.8 | 1.5 | 11.5 | 1.8 | Local market, Chittagong |
| 9 | Spinach (Spinacia oleracea) | 8.5 | 1.2 | 9.8 | 1.5 | Local market, Chittagong |
| 10 | Cabbage (Brassica oleracea) | 6.8 | 1.6 | 7.5 | 1.7 | Local market, Chittagong |
| Serial no. | Ce(IV): Ce(III) | Ce, taken (μg L−1) | Ce, found (μg L−1) | Error (μg L−1) | |||
|---|---|---|---|---|---|---|---|
| Ce(IV) | Ce(III) | Ce(IV) | Ce(III) | Ce(IV) | Ce(III) | ||
| 1 | 1:1 |
10 | 10 | 9.99 | 9.98 | 0.01 | 0.02 |
| 1 | 1:1 |
10 | 10 | 1.00 | 1.00 | 0.00 | 0.00 |
| 1 | 1:1 |
10 | 10 | 9.98 | 9.99 | 0.02 | 0.01 |
| Mean error: Ce(IV) = ±0.01, Ce(III) = ±0.01 | |||||||
| Standard deviation: Ce(IV) = ±0.005, Ce(III) = ±0.006 | |||||||
| 1 | 1:5 |
10 | 50 | 9.99 | 49.98 | 0.01 | 0.02 |
| 1 | 1:5 |
10 | 50 | 9.98 | 49.99 | 0.02 | 0.01 |
| 1 | 1:5 |
10 | 50 | 9.99 | 49.98 | 0.01 | 0.02 |
| Mean error: Ce(IV) = ±0.013, Ce(III) = ±0.016 | |||||||
| Standard deviation: Ce(IV) = ±0.0058, Ce(III) = ±0.006 | |||||||
| 1 | 1:10 |
10 | 100 | 9.98 | 99.99 | 0.02 | 0.01 |
| 1 | 1:10 |
10 | 100 | 10.00 | 100.00 | 0.00 | 0.00 |
| 1 | 1:10 |
10 | 100 | 9.99 | 99.98 | 0.01 | 0.02 |
| Mean error: Ce(IV) = ±0.01, Ce(III) = ±0.01 | |||||||
| Standard deviation: Ce(IV) = ±0.005, Ce(III) = ±0.006 | |||||||
| Samples | F-test resultsa | ||||
|---|---|---|---|---|---|
| (SD12/SD22)43 | (SD12/SD32)48 | (SD12/SD42)56 | (SD12/SD52)62 | (SD12/SD62)67 | |
| a Tabulated F-value for (5, 5) degrees of freedom at P(0.96) is 5.52. SD1 = standard deviation of proposed method, SD2 = standard deviation of reference method,43 SD3 = standard deviation of reference method,48 SD4 = standard deviation of reference method,56 SD5 = standard deviation of reference method,62 SD6 = standard deviation of reference method.67 | |||||
| Water | 0.107 | 0.47 | 0.17 | 0.0011 | |
| Water | 0.0416 | 0.091 | 0.023 | 0.55 | |
| Water | 0.259 | 0.200 | 0.153 | ||
| Water | 0.819 | ||||
| Blood | 0.75 | ||||
| Blood | 0.65 | ||||
| Blood | 0.93 | ||||
| Soil | 0.22 | 0.0004 | 0.0005 | ||
| Soil | 0.89 | 0.0003 | 0.0004 | ||
| Soil | 0.90 | 0.0009 | 0.0007 | ||
| Alloy | 0.72 | ||||
| Alloy | 0.09 | ||||
| Synthetic mixture | 0.21 | ||||
| Synthetic mixture | 0.69 | ||||
| Synthetic mixture | 0.67 | ||||
| Food | 0.68 | ||||
| Food | 0.72 | ||||
| Urine | 1.02 | ||||
| Urine | 0.84 | ||||
| Bone | 0.86 | ||||
| Wheat | 0.78 | ||||
 | ||
| Fig. 8 Mechanism of oxidative cyclization reaction of 2-(α-pyridyl)-thioquinaldinamide (PTQA). | ||
Applications
The procedure was applied for determination of trace amounts of cerium in some synthetic mixtures of various compositions (Table 4) and also in a number of real samples e.g. several Certified Reference Materials (CRM) (Table 5). The method was also extended to the determination of cerium in a number of environmental, biological, soil, bone, food, vegetables and fruit samples. In view of the unknown composition of environmental water samples, the same equivalent portions of each such sample were analyzed for cerium content; the recoveries in both the “spiked” (added to the samples before the mineralization or dissolution) and the “unspiked” samples are in excellent agreement (Table 6). The results of biological analyses by spectrofluorimetric method were found to be in excellent agreement with those obtained by ICP-OES (Table 7). The results of soil analyses by the spectrofluorimetric method are shown in Table 8. The results of bone samples by the spectrofluorimetric method are shown in Table 9. The results of food analyses by spectrofluorimetric method were also found to be in excellent agreement with those obtained by ICP-OES (Table 10). The results of speciation of cerium(III) and cerium(IV) in mixtures were highly reproducible (Table 11). The statistical comparison between proposed and reported existing methods for cerium (Table 12).A suitable aliquot (1–2 mL) of the abovementioned solution was taken into a 10 mL calibrated flask and the cerium(IV) content was determined; as described under procedure using tartrate or EDTA as masking agent. The proposed procedure for the spectrofluorimetric determination of cerium was applied to the analysis of single element CRM of Ce (NIST-SRM-3110), estuarine sediment (NIST-SRM-1646a), soil (SABS-SRM-19), human hair (GBW-09101), wheat flour (GBW-08503) and pine noodles (NIST-SRM-1575) CRMs obtained from The Baotou Institute of Rare Earth, Baotou, China, using tartrate or EDTA as masking agents, following a method recommended by Sun et al.79 Based on five replicate analyses, average cerium concentration determined by the spectrofluorimetric method was in an excellent agreement with the certified values. The results are given in Table 5.
Most spectrofluorimetric methods for determination of cerium in natural and sea-water require preconcentration or standard addition of cerium.81 The concentration of cerium in natural and sea water is a few ngL−1 in developed countries.81
A suitable aliquot (1–2 mL) of the final solution was pipetted out into a 10 mL calibrated flask and the cerium content was determined as described under the general procedure using tartrate or EDTA as masking agent. The cerium content was then determined by the above procedure and quantified from a calibration graph prepared concurrently. The results are shown in Table 9.
The food samples used were rice, wheat and corn and these were used under dry conditions. Each sample was first ground in a mortar. Corn and fruit samples (2 g) or rice and wheat samples (1 g) were weighed accurately and placed in a porcelain crucible and charred in an electric furnace; the sample was ashen at 555 °C in a muffle furnace in presence of excess oxidizing agent following a method recommended by Mitra.78 To it, 2.0 mL of HCl and 10 mL of water were added to the ash. The mixture of each foodstuff was heated with 2 mL of 2% (w/v) freshly prepared ammonium persulphate below the boiling point for 5–10 min below the boiling point to complete oxidation from Ce(III) to Ce(IV). The solutions were cooled and neutralized with dilute NH4OH in presence of 1–2 mL of 0.01% (w/v) EDTA solution and filtered. The resulting solution was quantitatively transferred into a 25 mL calibrated flask and mixed well and made up to the mark with de-ionized water.
A suitable aliquot (1–2 mL) of the final digested solution was pipetted into a 10 mL calibrated flask and the cerium content was determined as described under the general procedure using tartrate as masking agent. High value of cerium for Daucus carota (carrot) is probably due to the involvement of high cerium concentration in the soil. The results of food and vegetables analyses by spectrofluorimetric method were also found to be in excellent agreement with those obtained by ICP-OES. The results are shown in Table 10.
:1, 1:5, 1:10) were taken in a 25 mL Pyrex conical flask. A few drops (2–3 drops) of 2 M H2SO4, and 2 – 4 mL of 2% (w/v) freshly prepared ammonium persulphate were added to oxidize trivalent cerium to tetravalent cerium and the mixture was heated gently with further addition of 5 mL water, if necessary, for 5 minutes to drive off the excess persulphate, then the mixture was cooled to room temperature. The reaction mixture was then cooled and neutralized with dilute NH4OH in presence of 1–2 mL of 0.01% (w/v) EDTA solution. The solution was transferred quantitatively into a 10 mL volumetric flask and 1 mL of 9.8 × 10−4 M PTQA reagent solution was added followed by the addition of 1 mL of 0.001 M H2SO4. It was made up to the mark with de-ionized water. The fluorescence intensity was measured then being cooled at room temperature, (25 ± 5) °C, at 370 nm when excited at 303 nm, against a reagent blank. The total cerium content was calculated with the help of a calibration graph prepared concurrently.An equal aliquot (1–2 mL) of the above cerium(IV + III) mixture was taken into a 25 mL conical flask. The solution was neutralized with dilute NH4OH in presence of 1–2 mL of 0.01% (w/v) EDTA solution. After, the content of the beaker was transferred quantitatively into a 10 mL volumetric flask, 1 mL of 9.8 × 10−3 M PTQA reagent solution was added, followed by the addition of 1 mL of 0.001 M H2SO4. It was made up to the mark with de-ionized water. After 5 min the fluorescence intensity was measured following the general procedure at 370 nm when excited at 303 nm against a reagent blank, as before. The cerium concentration was calculated in μg L−1 or ng L−1 with the aid of a calibration graph. This gives a measure of cerium(IV) originally present in the mixture. This value was subtracted from that of the total cerium to determine the cerium(III) present in the mixture. The results of the assessment of speciation of Ce(VI) and Ce(III) were found to be highly reproducible. The occurrence of such reproducible results is also reported for different oxidation states of cerium.85 The results of a set of determination are given in Table 11.
The present method was compared with some reported methods43,48,56,62,67 statistically. It was found that present method is much superior those of the reported methods. The results are shown in Table 12.
Conclusions
A new rapid, ultra sensitive, highly selective and inexpensive spectrofluorimetric method with the cerium–PTQA system was developed for the determination of cerium in some real, environmental, biological, bone, food, and soil samples, for continuous monitoring to establish the pico-trace levels of cerium in different samples matrices. Compared with other methods, the proposed method has several remarkable analytical characteristics: firstly, the proposed method is highly sensitive that the amount, in ng L−1, of cerium can be determined without preconcentration in diluted biological solutions. Secondly, the low detection limit, 0.1 ng L−1 i.e. pg g−1 (10−12 pg g−1) levels can be measured without preconcentration or standard addition method. The proposed method is very simple, rapid, and stable. The reaction of cerium with PTQA is instantaneous, so it does not involve any stringent reaction conditions and offer the advantages of high stability of fluorescence intensity (over 24 h) at room temperature, reliable and reproducible. Thirdly, the method has added the advantages of determining individual amounts of cerium(III) and cerium(IV). With suitable masking agents, the reaction can be made highly selective and better reproducibility has been achieved (RSD = 0–2%).Finally, the proposed method using PTQA in aqueous solutions not only is one of the most sensitive methods for the determination of cerium but also is excellent in terms of selectivity, simplicity and precision (RSD: 0–2%). Therefore, this method can be successfully used in routine analysis of pico-trace amounts of cerium in real, environmental, biological, bone, food, vegetables and soil samples. It is a new method needs neither heating nor extraction to organic phase, works satisfactorily and could be an alternative method for the rapid determination of cerium in a wide variety of sample matricesand found superior to existing spectrofluorimetric methods reported in different literature.
Conflicts of interest
There are no conflicts to declare.References
- F. Riccardo, and S. Adriana, Intermetallic Chemistry, 2010, vol. 13, pp. 370–389 Search PubMed.
- T. A. Ali, G. G. Mohamed, E. M. S. Azzam and A. A. Abd-elaal, Sens. Actuators, B, 2013, 191, 192 CrossRef.
- A. Afkhami, T. Madrakian, A. Shirzadmehr, M. Tabatabaee and H. Bagheri, Sens. Actuators, B, 2012, 174, 237–248 CrossRef CAS.
- M. R. Awual, T. Yaita and H. Shiwaku, Chem. Eng. J., 2013, 228, 327–335 CrossRef CAS.
- A. Kumar, S. Babu, A. S. Karakoti, A. Schulte and S. Seal, Langmuir, 2009, 25, 10998–11009 CrossRef CAS PubMed.
- E. Diatloffa, F. W. Smith and C. J. Asher, J. Plant Nutr., 2008, 18, 1995–2009 Search PubMed.
- P. C. C. Faria, J. J. M. Orfao and M. F. R. Pereira, Appl. Catal., B, 2009, 88, 341–350 CrossRef CAS.
- T. Alessandro, C. deLeitenburg, B. Marta and D. Giuliano, Catal. Today, 1999, 50, 353–363 CrossRef.
- C. L. Melcher and J. S. Schweitzer, IEEE Trans. Nucl. Sci., 1992, 39, 502–513 CrossRef CAS.
- I. Woo and K. Nishimoto, Met. Mater. Int., 2001, 7, 241–249 CrossRef CAS.
- A. N. Shmyreva, A. V. Borisov and N. V. Maksimchuk, Nanotechnol. Russ., 2010, 5, 382–391 CrossRef.
- I. Celardo, E. Traversa and L. Ghibelli, J. Exp. Ther. Oncol., 2010, 9, 47–51 Search PubMed.
- S. Robert, H. J. Stevens and J. R. Varner, J. Non-Cryst. Solids, 1999, 249, 123–134 CrossRef.
- R. Le Toquin and A. K. Cheetham, Chem. Phys. Lett., 2006, 423, 352 CrossRef CAS.
- A. Corma, P. Atienzar, H. Garcia and J. Y. Chane-Ching, Nat. Mater., 2004, 3, 394403 CrossRef PubMed.
- E. P. Murray, T. Tsai and S. A. Barnett, Nature, 1999, 400, 649–658 CrossRef CAS.
- X. Feng, Y. S. Her, W. L. Zhang, J. Davis, E. Oswald, J. Lu, V. Bryg, S. Freeman and D. Gnizak, Mater. Res. Soc. Symp. Proc., 2003, 763, 173–181 Search PubMed.
- M. Ozawa, M. Kimura and A. Isogai, J. Alloys Compd., 1993, 193, 73–86 CrossRef CAS.
- K. H. Al-Sowdani and A. Townshend, Anal. Chim. Acta, 1986, 179, 469–476 CrossRef CAS.
- Y. Watanabe, V. Kain, T. Tonozuka, T. Shoji, T. Kondo and F. Masuyama, Scr. Mater., 2000, 42, 307–316 CrossRef CAS.
- E. K. Robert, The History and Use of Our Earth's Chemical Elements: A Reference Guide, 2006, p. 279 Search PubMed.
- L. Wang, Y. Yu, X. Huang, Z. Long and D. Cui, Chem. Eng. J., 2013, 215, 162–172 CrossRef.
- S. Abhilash, M. K. Sinha and B. D. P. Sinha, Int. J. Miner. Process., 2014, 127, 70–81 CrossRef CAS.
- S. Hirano and K. T. Suzuki, Environ. Health Perspect., 1996, 104, 85–105 CAS.
- D. I. Bleiwas, Potential for Recovery of Cerium Contained in Automotive Catalytic Converters, U.S. Department of the Interior: U.S. Geological Survey, 2013 Search PubMed.
- R. J. Lewis, Hazardous Chemicals Desk Reference, John Wiley & Sons, 6th edn, 2008, pp. 300–337 Search PubMed.
- https://www.webelements.com/cerium/chemistry.html, accessed January 2019.
- http://weppi.gtk.fi/publ/foregsatlas/text/Ce.pdf, accessed July 2004.
- M. A. Jakupec, P. Unfried and B. K. Keppler, Rev. Physiol., Biochem. Pharmacol., 2005, 153, 104–117 Search PubMed.
- P. Cukor and R. . P. Weberling, Anal. Chim. Acta, 1968, 41, 404–411 CrossRef.
- A. N. Masiand and R. A. Olsina, Talanta, 1993, 40, 931–944 CrossRef.
- M. Achilli, G. Ciceri, R. Ferraroli, D. Helati and W. Martinotti, Analyst, 1989, 114, 319–327 RSC.
- V. K. Jain, A. Handa, S. S. Sait, P. Shrivastav and Y. K. Agrawal, Anal. Chim. Acta, 2001, 429, 237–243 CrossRef CAS.
- F. Shemirani and S. Reza Yousefi, Microchim. Acta, 2007, 153, 223–229 CrossRef.
- B. Li, Y. Zhang and M. Yin, Analyst, 1997, 122, 543–549 RSC.
- B. Li, Y. Sun and M. Yin, J. Anal. At. Spectrom., 1999, 14, 1843–1851 RSC.
- H. L. Greenhaust, M. Feibush and L. Gordon, Anal. Chem., 1957, 29, 1531–1538 CrossRef.
- L. Gordon and A. M. Feibush, Anal. Chem., 1955, 27, 1051–1062 Search PubMed.
- P. L. Sharma and L. H. Dieter, Talanta, 1966, 13, 347–356 CrossRef.
- J. Wang, P. Farias and J. S. Mahmoud, Anal. Chim. Acta, 1985, 171, 215–221 CrossRef CAS.
- S. B. Khooand and J. Zhu, Electroanalysis, 1999, 11, 546–552 CrossRef.
- J. G. Sen Gupta, Talanta, 1984, 31, 1053–1560 CrossRef CAS PubMed.
- T. I. Ivkova, R. P. Pantaler and Y. A. Povrozin, Anal. Chem. Lett., 2012, 2, 220–229 CrossRef CAS.
- A. Navas, F. S. Rojas and F. G. Sanchez, Mikrochim. Acta, 1982, 1, 175–184 CrossRef CAS.
- F. Grases, C. Genestarand and J. G. March, Microchem. J., 1984, 29, 237–249 CrossRef CAS.
- B. K. Pal, F. Toneguzzo and A. Corsini, Anal. Chim. Acta, 1977, 88, 353–365 CrossRef CAS.
- N. Jie, J. Yang and T. Liu, Talanta, 1994, 41, 415–423 CrossRef CAS.
- H. Tavallali and R. Nejabat, Asian J. Chem., 2009, 21, 2441–2449 CAS.
- D. Ozyurt, B. Demirata and R. Apak, J. Fluoresc., 2011, 21, 2069–2077 CrossRef CAS PubMed.
- I. A. Darwish, A. S. Khedr, H. F. Askal and R. M. Mahmoud, Il Farmaco, 2005, 60, 555–563 CrossRef CAS PubMed.
- A. Akseli and Y. Rakicioğlu, Fresenius. J. Anal. Chem., 1996, 354, 424–431 CAS.
- F. Salinas, C. Genestar and F. Grases, Microchem. J., 1982, 27, 32 CrossRef CAS.
- K. H. Al-Sowdani and A. Townshend, Anal. Chim. Acta, 1986, 179, 469–477 CrossRef CAS.
- F. Zhao and W. Zhao, J. Fluoresc., 2012, 22, 529–536 CrossRef CAS.
- I. Berregi, D. J. Senén and C. J. Alfonso, Talanta, 1999, 48, 719–725 CrossRef CAS.
- J. X. Meng, H. J. Wu and D. X. Feng, Spectrochim. Acta, Part A, 2000, 56, 1925–1933 CrossRef.
- M. M. Karim, S. H. Lee, Y. S. Kim, H. S. Bae and S. B. Hong, J. Fluoresc., 2006, 16, 17 CrossRef CAS.
- N. Jie, Z. Si, J. Yang, Q. Zhang, X. Huang and D. Yang, Mikrochim. Acta, 1997, 126, 93–99 CrossRef CAS.
- D. T. Burns, C. D. P. Dangolle and M. Harriott, Mikrochim. Acta, 1995, 119, 49–56 CrossRef CAS.
- Y. Rakicioğlu and A. Akseli, J. Fluoresc., 1998, 8, 1–8 CrossRef.
- J. Nianqin, Y. Jinghe and G. Jian, Fenxi Huaxue, 1992, 20, 847–853 Search PubMed.
- A. Akseli and Y. Rakicioğlu, Talanta, 1996, 43, 1983–1998 CrossRef CAS.
- P. Cukor and R. P. Weberling, Anal. Chim. Acta, 1968, 41, 404–409 CrossRef.
- Z. Holzbecher, Collect. Czech. Chem. Commun., 1989, 54, 616–627 CrossRef CAS.
- C. Huang and X. Chi, J. Beijing Normal Univ. (Nat. Sci.), 1993, 03, 235–243 Search PubMed.
- Y. Takayama, T. Ujihara, K. Morishige, Y. Nishikawa and B. Kagaku, Bunseki Kagacu, 1986, 35, 713–719 CrossRef CAS.
- A. Feng, Q. Hu and W. Wang, J. Food Sci. Technol., 2011, 48(4), 489–494 CrossRef PubMed.
- M. K. Rofouei, N. Tajarrod, M. Masteri-Farahani and R. Zadmard, J. Fluoresc., 2015, 25, 1855–1864 CrossRef CAS PubMed.
- H. D. Porter, J. Am. Chem. Soc., 1954, 76, 127–136 CrossRef CAS.
- M. Jamaluddin Ahmed, M. Tazul Islam and F. Hossain, RSC Adv., 2018, 8, 5509–5522 RSC.
- Vogel's Textbook of Quantitative Chemical Analysis, ed. G. H. Jeffery, J. Bassett, J. Mendham and R. C. Denney, ELBS, Bath Press Ltd., London, 5th edn, 1994, pp. 380–382 Search PubMed.
- A. K. Mukharjee, Analytical Chemistry of Zirconium and Hafnium, Pergamon Press, New York, 1st edn, 1970, p. 12 Search PubMed.
- B. K. Pal and B. Chowdhury, Mikrochim. Acta, 1984, 11, 121 CrossRef.
- C. B. Ojeda, A. G. de Torres, F. S. Rojas and J. M. C. Pavon, Analyst, 1987, 112, 1499 RSC.
- M. Jamaluddin Ahmed, C. D. Stalikas, P. G. Veltsistas, S. M. Tzouwara-Karayanni and M. I. Karayannis, Analyst, 1997, 122, 221–229 RSC.
- B. K. Pal, M. Jamaluddin Ahmed and A. K. Chkraborty, Analyst, 1990, 115, 439–449 RSC.
- B. K. Pal, A. K. Chkraborty and M. Jamaluddin Ahmed, Anal. Chim. Acta, 1988, 206, 351–355 CrossRef.
- Sample Preparation Techniques in Analytical Chemistry, ed. S. Mitra, Wiley-Intersciences, New Jersey, 2003, pp. 125–133 Search PubMed.
- C. Sun, J. Y. Yang and S. R. Tzeng, Analyst, 1999, 124, 421–429 RSC.
- Standard Methods for the Examination of Water and Wastewater, ed. E. A. Greenberg, S. L. Clesceri and D. A. Eaton, American Public Health Association, Washington D. C., 18th edn, 1992, pp. 3–253 Search PubMed.
- P. Chambon, U. Lound and E. Ohanian, WHO Guidelines for Drinking Water Quality, Recommendations, WHO, Geneva, 2nd edn, 1993 Search PubMed.
- H. M. Stahr, Analytical Methods in Toxicology, John Wiley and Sons, New York, 3rd edn, 1991, p. 85 Search PubMed.
- S. Mittal and A. K. Pandey, BioMed Res. Int., 2014, 5, 126–136 CrossRef.
- M. L. Jackson, Soil Chemical Analysis, Prentice Hall, Englewood Cliffs, 1965, p. 326 Search PubMed.
- https://www.ncbi.nlm.nih.gov/pubmed/24090406, accessed October 2013.
| This journal is © The Royal Society of Chemistry 2019 |