DOI:
10.1039/C9RA02823D
(Paper)
RSC Adv., 2019,
9, 17745-17753
Cell adherence and drug delivery from particle based mesoporous silica films†
Received
14th April 2019
, Accepted 27th May 2019
First published on 5th June 2019
Abstract
Spatially and temporally controlled drug delivery is important for implant and tissue engineering applications, as the efficacy and bioavailability of the drug can be enhanced, and can also allow for drugging stem cells at different stages of development. Long-term drug delivery over weeks to months is however difficult to achieve, and coating of 3D surfaces or creating patterned surfaces is a challenge using coating techniques like spin- and dip-coating. In this study, mesoporous films consisting of SBA-15 particles grown onto silicon wafers using wet processing were evaluated as a scaffold for drug delivery. Films with various particle sizes (100–900 nm) and hence thicknesses were grown onto trichloro(octadecyl)silane-functionalized silicon wafers using a direct growth method. Precise patterning of the areas for film growth could be obtained by local removal of the OTS functionalization through laser ablation. The films were incubated with the drug model 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO), and murine myoblast cells (C2C12 cells) were seeded onto films with different particle sizes. Confocal laser scanning microscopy (CLSM) was used to study the cell growth, and a vinculin-mediated adherence of C2C12 cells on all films was verified. The successful loading of DiO into the films was confirmed by UV-vis and CLSM. It was observed that the drugs did not desorb from the particles during 24 hours in cell culture. During adherent growth on the films for 4 h, small amounts of DiO and separate particles were observed inside single cells. After 24 h, a larger number of particles and a strong DiO signal were recorded in the cells, indicating a particle mediated drug uptake. The vast majority of the DiO-loaded particles remained attached to the substrate also after 24 h of incubation, making the films attractive as longer-term reservoirs for drugs on e.g. medical implants.
Introduction
The development of controlled drug delivery systems that can administer drugs locally and with a regulated release profile within the human body is of great relevance for e.g. medical implants and tissue engineering applications.1 Especially challenging is the delivery of hydrophobic drugs, which cannot be administered directly.2 An ideal scaffold material for these applications should be nontoxic, biologically active and dissolve over time. In the last 25 years mesoporous silica has gained a lot of attention in various fields of research, from catalysis to medical applications. The possibility to control the material characteristics, e.g. pore sizes and particle morphology, in combination with large surface areas up to 1000 m2 g−1 and tunable surface functionalities, are a few reasons for why this class of materials attracts extensive interest for use in drug delivery and tissue engineering applications.3–11 The high inner surface area of the particles allows the loading with a large amount of active substance molecules. The synthesis of a variety of silica-based films has been reported in literature, mainly by using evaporation-induced self-assembly in spin- or dip-coating,12,13 or e.g. electro-assisted self-assembly,14 vapor-phase deposition,15 or interfacial polymerization.16 Mesoporous films have previously been used as a drug delivery system,17,18 but a controlled release of the drugs is often limited to diffusion control or degradation of the silica matrix itself.19
Wiltschka et al. reported on coating of pre-formed mesoporous silica nanoparticles with a diameter of about 400 nm on glass slides by spin-coating.20 By a further functionalization of glass slides with hyaluronic acid it was possible to covalently link a sub-monolayer of particles to the substrate, leading to a delayed uptake of particles by cells over several days.21 This allowed a potentially delayed particle-mediated release of small hydrophobic drugs within cells. Although spin-coating is an attractive means for preparing homogeneous, particle-based coatings, coat large, or 3D substrates, is virtually not possible. Furthermore, controlled micro-patterning of surfaces remain to be demonstrated for both above described methods.
In this work, we use a direct growth (DiG) method to form films consisting of mesoporous silica particles grown onto a silicon substrate.22 Using the described method, it has previously been shown that it is possible both to grow films with controlled thickness and on 3D-substates.23 This is of great importance for fabricating porous coatings on implants without compromising the fine structures of the substrate. The particles themselves in powder form have been shown to efficiently immobilize enzymes,24 and to act as a potential drug carrier.25 By varying the particle size using various NH4F concentration in the synthesis solution, the film thickness can be altered to preserve fine structures of the substrate.23 We show in this study that it is possible to functionalize the films with ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–R–COOH after synthesis and to load the films with a hydrophobic, small molecule model drug, DiO, confirming the accessibility of the pores. Cells can adhere to all films, and the particles are taken up by the cells prior to release of their cargo. However, the particle uptake after 24 h of cultivation is still small, indicating that the films can act as long-time drug reservoir on e.g. an implant. The data show further that areas for film growth can be controlled by removal of the substrate functionalization using laser pulses prior to the film growth. This enables selective patterning of the substrate which is useful when designing the implant. Thus, the results show that silica films grown with the DiG method have an excellent potential to be used as a new material for drug delivery and tissue engineering applications, especially when a long-term drug release and a controlled surface morphology are needed.
Si–R–COOH after synthesis and to load the films with a hydrophobic, small molecule model drug, DiO, confirming the accessibility of the pores. Cells can adhere to all films, and the particles are taken up by the cells prior to release of their cargo. However, the particle uptake after 24 h of cultivation is still small, indicating that the films can act as long-time drug reservoir on e.g. an implant. The data show further that areas for film growth can be controlled by removal of the substrate functionalization using laser pulses prior to the film growth. This enables selective patterning of the substrate which is useful when designing the implant. Thus, the results show that silica films grown with the DiG method have an excellent potential to be used as a new material for drug delivery and tissue engineering applications, especially when a long-term drug release and a controlled surface morphology are needed.
Experimental
Chemicals and cells
Tetraethoxysilane (TEOS, 98%), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, ≥97.4%), N-hydroxysuccinimide (NHS, 98%), hydrochloric acid (HCl, ≥37%), nitric acid (HNO3, ≥65%), ammonia (NH3, 32% in water), heptane (99%), trichloro(octadecyl)silane (OTS), ammonium fluoride (NH4F, ≥98%), poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (P123, Mn ∼ 5800), 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI), phalloidin-tetramethylrhodamine B isothiocyanate (Phalloidin-TRITC), 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO), and C2C12 cells were purchased from Sigma-Aldrich Chemie GmbH, Schnelldorf, Germany. Hydrogen peroxide (H2O2, 30%) and (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) sodium salt (HEPES, 99.3%) was purchased from Merck KGaA, Darmstadt, Germany. Carboxyethylsilanetriol di-sodium salt (25% in water) was purchased from ABCR GmbH & Co. KG, Karlsruhe, Germany. ATTO 647N-amine was purchased from ATTO-TEC GmbH, Siegen, Germany. Dulbecco's Modified Eagle Medium (DMEM), L-glutamine, antibiotics and fetal calf serum (FCS) and Alexa488 conjugated goat anti-mouse secondary antibody (A11029) was purchased from Life Technologies – Thermo Fisher Scientific, Darmstadt, Germany. Mouse monoclonal anti vinculin antibody (ab18058/clone SPM227) was purchased at Abcam, Cambridge, UK. All chemicals were used as supplied by the manufacturer without further purification.
Synthesis
Film synthesis. The protocol for synthesizing the films followed the DiG method presented in literature.22,23 In the synthesis P123 (0.414 mmol) was dissolved in HCl (1.84 M, 80 mL) at 20 °C. Simultaneously, 0, 0.189, or 0.756 mmol of NH4F was added to the solution. Subsequently, 1 mL of heptane was mixed with 5.5 mL of TEOS and added to the P123 solution. After stirring for 4 min, the solution was kept under static conditions over night. OTS functionalized silicon wafers, prepared as reported elsewhere,22 were added to the synthesis solution under static conditions after 12, 7 or 0.5 min, depending on the NH4F concentration. Afterwards, a hydrothermal treatment step (100 °C, 24 h) was performed, followed by filtration and washing with deionized water, and finally all films were calcined at 550 °C for 5 h (heating rate: 1 °C min−1). The films are named as DiG_X, where, X corresponds to the NH4F/P123 molar ratio.
COOH-functionalization
After the synthesis, the films were washed extensively with demineralized water in an ultrasonic bath to remove free particles from the surface. Subsequently, the films were placed in a solution of carboxyethylsilanetriol di-sodium salt (25% in water) and HEPES (25 mM, pH 7.2) and stirred at ambient temperature for 2 h (1 μg mL−1). Afterwards the functionalized films were washed twice with ethanol and dried at 60 °C.
Labeling with ATTO dye
For activation of the carboxy group, the COOH-functionalized films were incubated with a mixture of NHS (69.5 μmol in HEPES) and EDC (55.8 μmol) in HEPES (25 mM, pH 7.2) for 20 min at room temperature. After washing with water, a mixture of HEPES and ATTO647N-amine (1 mg mL−1 dissolved in DMSO, 32.75 nmol) was added to the films and stirred for one hour. For purification the films were washed twice with water and dried at 60 °C over night.
DiO loading and release
Before incubation with the model drug DiO all films (either non-labelled or ATTO-647N labelled) were dried at ambient temperature for 1 h. The dried films were incubated in a mixture of cyclohexane and DiO (2.27 μM) for 4 h at RT. After the incubation all films were washed with cyclohexane and dried at 60 °C for 40 min. To determine the release of the model drug in an aqueous environment DiO loaded films were incubated with 2 mL of DMEM + 10% FCS solution for 24 h. The released amount of DiO was subsequently analyzed by UV-vis measurements as well as used for the incubation of cells in μ-Slide 8 wells (ibitreat μ-slide, IBIDI, Munich, Germany).
Substrate patterning
The patterning process was performed using a flash lamp pumped and q-switched Nd:YAG-Laser (Quanta Ray GCR-4, Spectra Physics) at a wavelength of 1064 nm, a pulse length of 10 ns and a pulse repetition rate of 10 Hz. For this purpose, an experimental setup was created. The laser beam was focused down to a spot diameter of approximately 1 mm using an antireflection coated plano-convex lens with a focal length of 500 mm. OTS functionalized silica wafers were moved with a high or low traversing speed of 2 mm s−1 or 10 mm s−1 over a length of 10 mm by use of a computer-controlled translation stage. The pulse energy was increased in steps until plasma formation was observed. This resulted in the pulse energy levels of 15 mJ, 19 mJ, and 29 mJ. Concerning a spot diameter of 1 mm this results in radiant exposure (He) values of 1.9 J cm−2, 2.4 J cm−2 and 3.7 J cm−2. The pulse energy was measured using a power meter (Nova II, OPHIR) and a corresponding measurement head (30A-P-SH, OPHIR). For each set of parameters (velocity and pulse energy) a linear array of 5 separated lines was irradiated by the laser.
Cell culture
C2C12 cells were cultivated under standard cell culture conditions (37 °C, 90% humidity, 5% CO2) in DMEM mixed with 10% FCS, 2 mM L-glutamine, and antibiotics. Films were incubated with a cell suspension (50 K cells per mL) and cultivated for 24 h under standard cell culture conditions. The cells were then fixed with paraformaldehyde (4%) in phosphate buffered saline (PBS, pH 7.4). Staining of cells was performed using DAPI (0.1 mg mL−1) for cell nuclei and Phalloidin-TRITC (50 nM) for filamentous actin. The visualization of focal adhesion contacts (FACs) was performed by using a mouse monoclonal anti vinculin antibody and Alexa488 conjugated goat anti-mouse secondary antibody as described previously.26
Material characterization
The morphology of the films was visualized by scanning electron microscopy (SEM) using a Leo 1550 Gemini Scanning Electron Microscope operated at 3 kV. The working distance was 3–5 mm. The film thickness was calculated from SEM micrographs of the film cross sections. The pore characteristics were analyzed using nitrogen sorption isotherms recorded on a Quantachrome Instruments Quadrasorb-SI operated at −196 °C. The specific surface area was calculated using the BET method at the relative pressure of 0.1–0.2. The pore size distribution was calculated using the KJS-method on the adsorption isotherm, and the total pore volume was determined at P/P0 = 0.99. Small angle X-ray diffraction (SAXRD) measurements were performed on an PANAlytical Empyrean in transmission mode using Cu Kα radiation.
Biological assessment
The growth, adhesion, and particle uptake of cells on top of films were visualized using a Leica TCS SP8 confocal laser scanning microscope and LASX software (Leica Microsystems, Wetzlar, Germany). Lasers emitting at 405 nm (used for excitation of DAPI), 488 nm (for DiO), 552 nm (for Phalloidin-TRITC) and 638 nm (for Atto647-labelled mesoporous silica nanoparticles (MSNs)) were used for the detection of integrated fluorophores. Fluorescence was detected using a HP CL APO 63×/1.40 OILCS2 oil immersion objective (Leica Microsystems) and a pinhole setting of 1 airy unit. The drug loading of the films was studied by UV-vis spectroscopy measurements of the supernatant using an Analytik Jena AG spectrophotometer SPECORD® 50.
Results and discussion
Films were synthesized using the DiG method with three concentrations of NH4F. SEM micrographs of the films are presented in Fig. 1. All films consist of mesoporous SBA-15 particles directly grown onto the substrate with the pores oriented parallel to the surface. No free particles are observed on the substrate after ultrasonication treatment. The particle width and film thickness decreases with an increasing salt concentration (Table 1), which is consistent with the data previously shown by Wu et al.23 The micrographs clearly shows that a vast majority of the particles are well attached to the substrate. It has been suggested that this type of films is formed through nucleation of silica coated micelles on the hydrophobic substrate, and that the particles grow from these sites.22 However, defects in the interface between the particles and substrate can appear, as is indicated by the black arrow in Fig. 1(a). This defect yields a particle that is only partly bound to the substrate and can thus influence the particle detachment rate. The time for adding the substrates to the synthesis solution increased with decreasing salt content to gain the desired film morphology since the material formation rate is significantly increased by the addition of NH4F.23,27 The salt affects the condensation rate of the silica precursor, and also the structure of the silica network, and therefore it is of great importance that the substrates are added during a time window in the formation of the siliceous network where micelles can nucleate onto the substrate, but prior to the micelle aggregation. In order to demonstrate that DiG films can also be grown on rough, 3D surfaces, a DiG_0.00 was synthesized onto an alumina sand blasted silicon wafer. The SEM micrograph in Fig. 1(d) reveal that the film follows the 3D structure of the substrate in all directions. Hence, the films are not limited to flat substrates, and coating of e.g. an implant should be possible as long as the surface is pre-hydrophobized.
 |
| | Fig. 1 SEM micrographs of top view and cross sections of (a) DiG_0.00, (b) DiG_0.45, (c) DiG_1.83 showing the difference in particle size when the NH4F concentration is altered, and (d) DiG_0.00 grown on rough substrate, where the film follow the inclines of the substrate. The arrow is showing a defect in the interface between particle and substrate. | |
Table 1 Physiochemical characteristics of the films and corresponding powders
| Material |
SSABET [m2 g−1] |
Pore size [nm] |
Total pore volume [cm3 g−1] |
Unit cell parameter [nm] |
Wall thickness [nm] |
Particle length [nm] |
Particle width [nm] |
Film thickness [nm] |
| DiG_0.00 |
724 |
10.4 |
0.93 |
13.0 |
2.6 |
550 ± 60 |
920 ± 220 |
400 ± 40 |
| DiG_0.45 |
549 |
10.5 |
0.98 |
12.5 |
2.0 |
350 ± 40 |
510 ± 60 |
225 ± 20 |
| DiG_1.83 |
563 |
11.0 |
0.96 |
13.3 |
2.3 |
280 ± 30 |
110 ± 20 |
115 ± 15 |
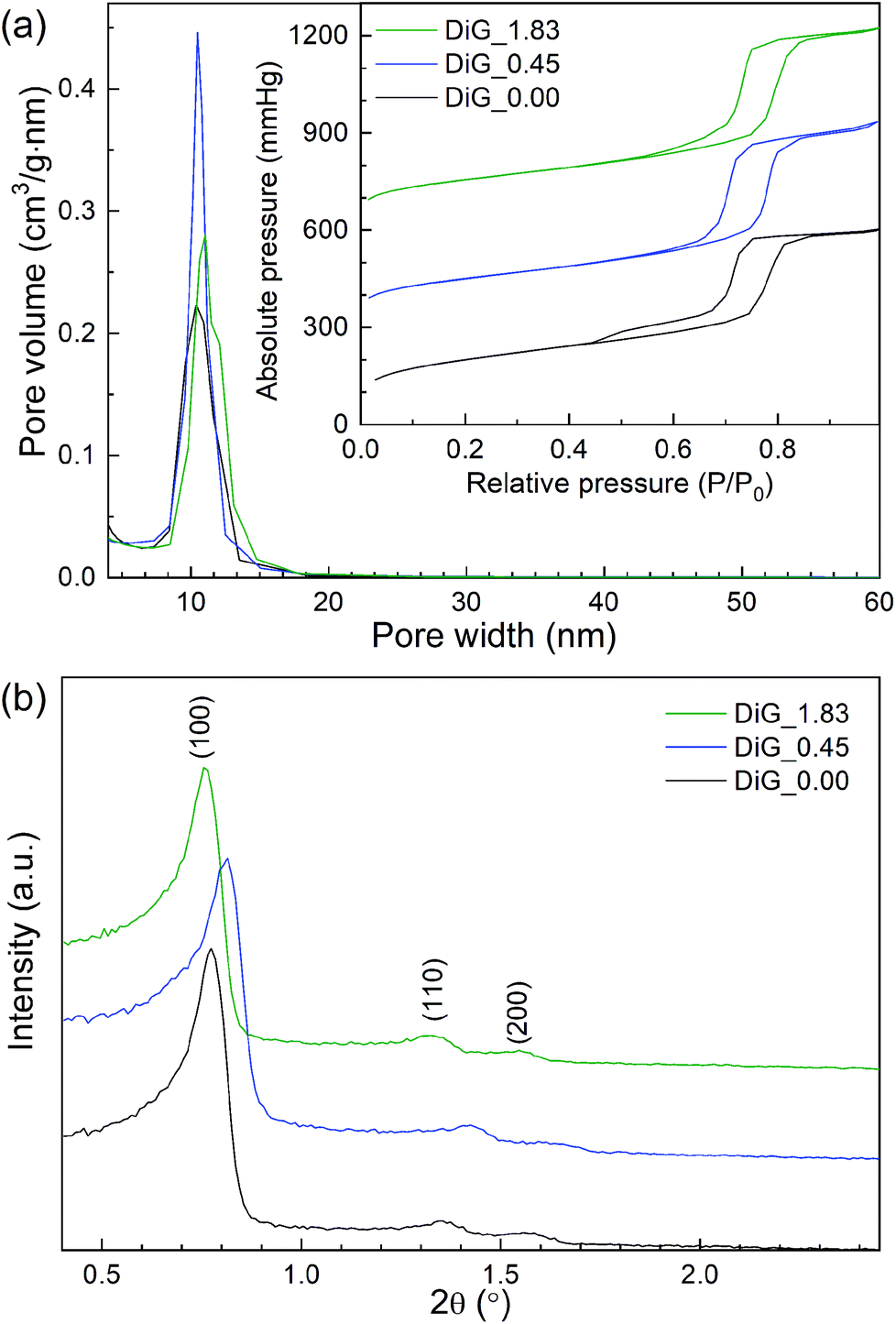
Nitrogen physisorption and SAXRD analysis of the corresponding SBA-15 powders from the film syntheses were performed. The results are presented in Fig. 2 and Table 1. As can be observed in Fig. 2(a), all materials give type IV nitrogen isotherms with type 1 hysteresis loops, typical for mesoporous materials with cylindrical pores. DiG_0.00 shows a small tail in the desorption branch of the isotherm, indicating plugs or constrictions in the mesopores.28 The pore size distributions show that the pore size varies from 10.4 nm to 11.0 nm with decreasing NH4F concentration. Fig. 2(b) illustrates the SAXRD diffractograms of the powders. All materials show three peaks corresponding to the hexagonal p6mm structure of SBA-15. The silica wall thickness is calculated as unit cell parameter – pore size and shows that the silica walls are decreased for DiG_0.45. A previous study has shown that the wall thickness of SBA-15 is decreased when the NH4F concentration is increased.29 This was attributed to the change in solubility of the PEO chains of P123 when the salt concentration is altered. A TEM micrograph of DiG_0.00 powder showing the hexagonal pore ordering is provided in the ESI, Fig. S1.†
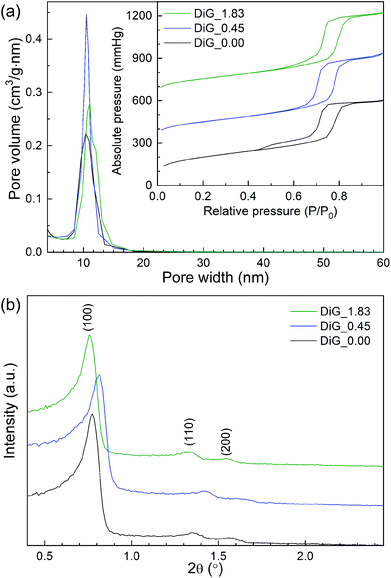 |
| | Fig. 2 (a) Nitrogen isotherms and pore size distributions, and (b) SAXRD diffractograms of the synthesized materials. | |
Cell culture and adherence
To study the potential of utilizing films synthesized with the DiG method in medical applications, and to visualize the particles, fluorescence labeling of the DiG film particles was performed using an ATTO647N dye. The successful binding of the fluorescent dye was corroborated by CLSM, and a homogeneous distribution of MSNs on the surface of the films, comparable to the previously shown SEM images in Fig. 1, could be confirmed (Fig. S2, ESI†).
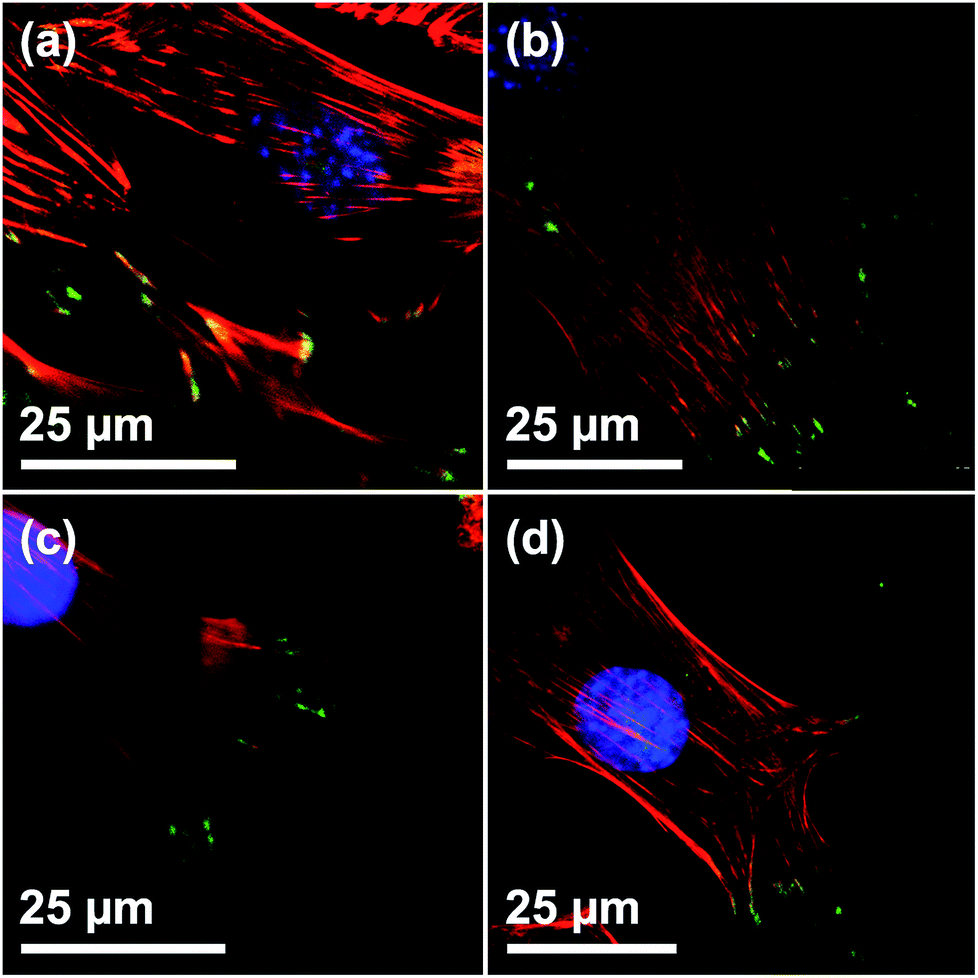
To determine the biocompatibility of the films, C2C12 cells were seeded on the film surfaces. Cells grown on blank silicon wafers were used as reference. The number of focal adhesion contacts (FACs), characterized by vinculin staining at the end of actin filaments, indicate adhesion strength on a given surface.30 CLSM verified a regular formation of the actin cytoskeleton and vinculin-mediated FACs on all of the investigated surfaces, indicating that growth and adhesion was not compromised in presence of the films (Fig. 3). Furthermore, the presence of a multitude of FACs suggests a good adherence of cells on the surfaces of the films, independent of the film morphology (Fig. 3(a)–(c)). Also, the shape and appearance of the adhesion points of C2C12 cells on all surfaces are in good agreement with the work of others.31
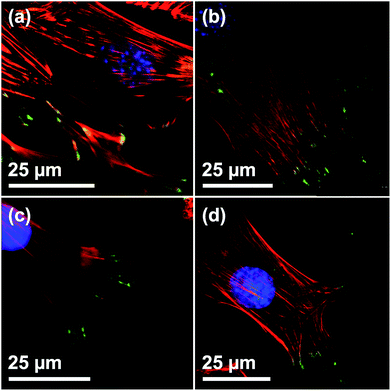 |
| | Fig. 3 Visualization of FACs of C2C12 on Films with different particle sizes by vinculin staining. CLSM micrographs of C2C12 on (a) DiG_1.83, (b) DiG_0.45, (c) DiG_0.00, and (d) a silicon wafer without film. Staining: blue = nucleus, red = microfilaments, green = vinculin stained FACs. The specificity of secondary antibody binding was demonstrated in a negative control omitting the primary anti vinculin antibody (ESI S3†). | |
Model drug loading and release
Due to their high inner surface area, MSNs have been extensively used as drug delivery vehicles especially for drugs showing a low solubility in aqueous environments.32,33 Moreover, with such a system a premature uptake of active substances by cells could be excluded as these drugs are only released from the porous system after the particles have been taken up by cells. To show the general accessibility of the film porosity for drug loading, DiG_0.00 was loaded with DiO, a hydrophobic model drug. The DiG_0.00 was chosen for these studies since it consists of the largest particles and hence more available effective surface area and pore volume per cm2. However, as the conclusions drawn are naturally also fully relevant also for the thinner films.
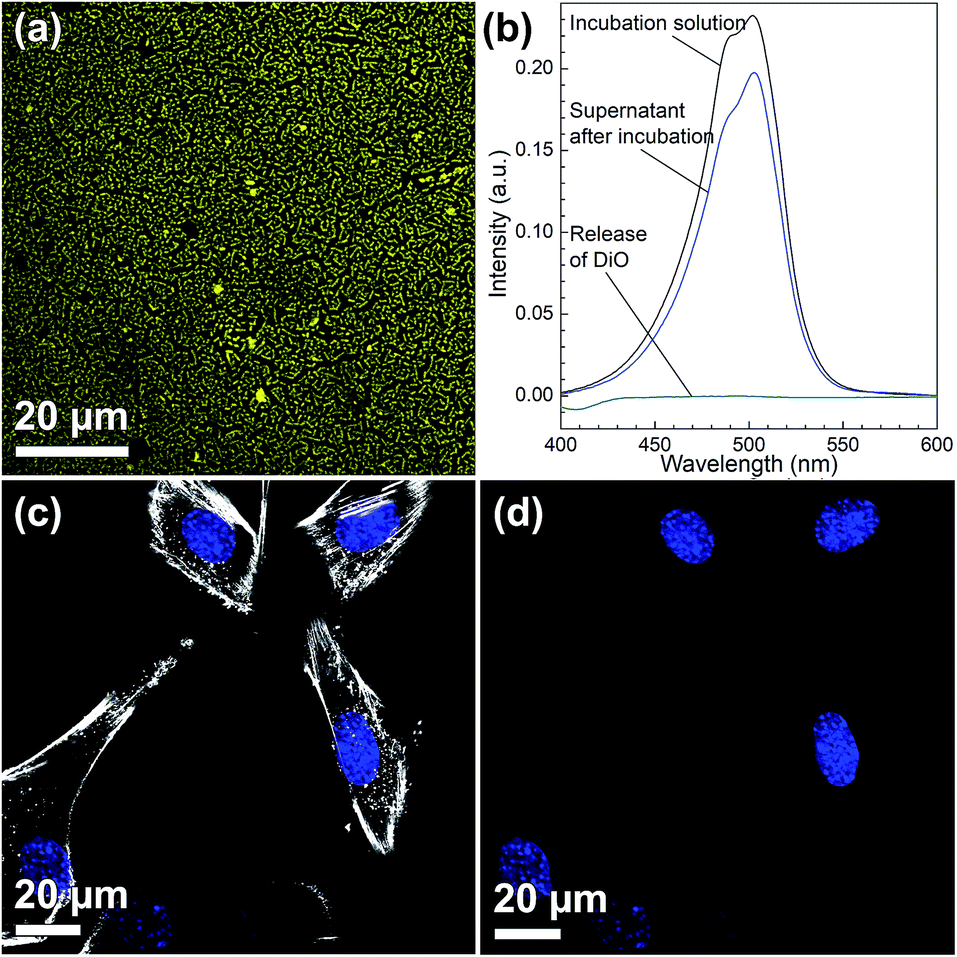
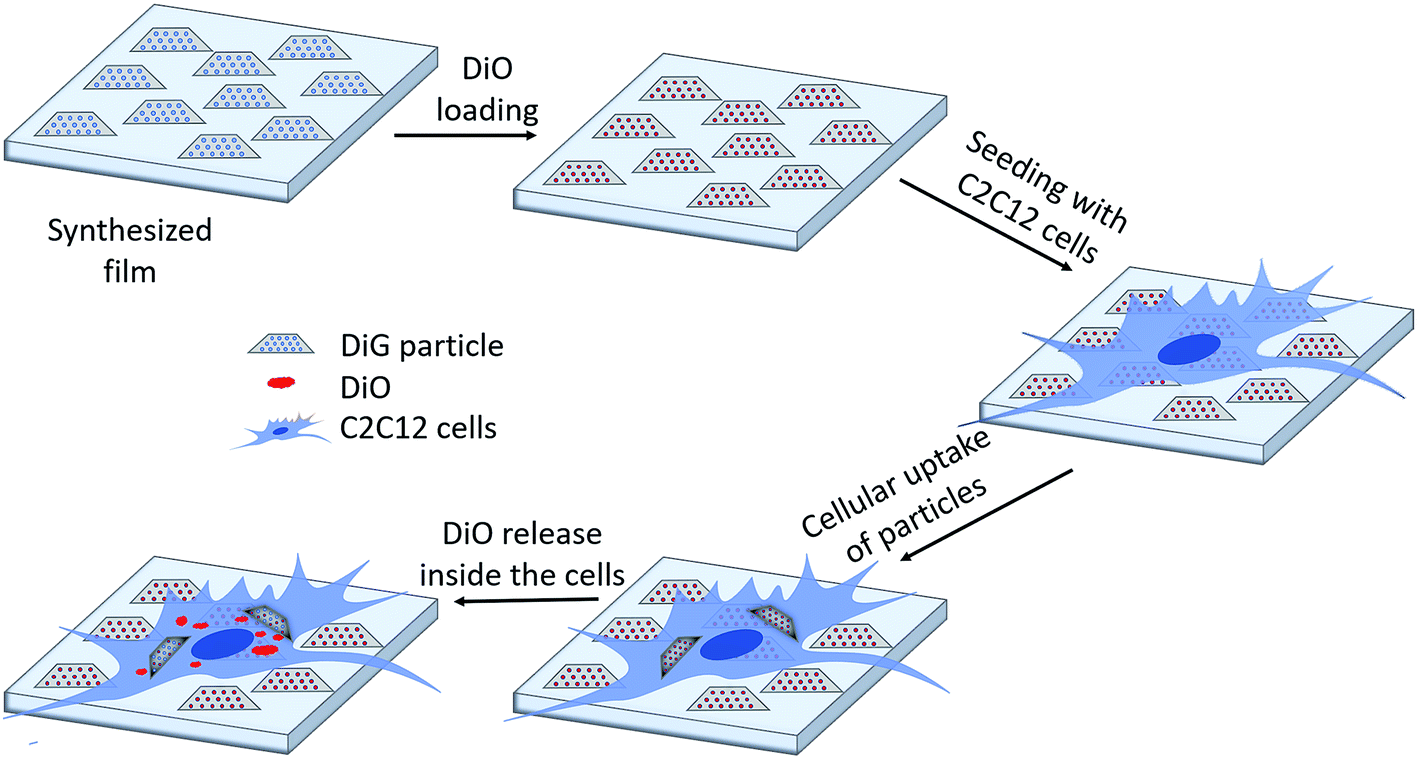
Fig. 4(a) shows a CLSM micrograph of the DiO loaded film. It is clear that all particles on the film surface are filled with DiO. The drug loading into the films was also verified by UV-vis measurements on the supernatant before and after DiO adsorption (Fig. 4(b)). The intensity of the DiO signal (503 nm) decreased by 15% as a result of the incubation of DiG_0.00 with a substrate area of about 5 cm2. The stability of the DiO loading was observed by submerging DiO loaded films in an FCS containing DMEM solution for 24 h. No DiO signal could be detected in the supernatant, see Fig. 4(b), indicating that no DiO is leaching out from the films. Further, no DiO-mediated intracellular fluorescence could be detected subsequent to incubation of C2C12 cells with the supernatant of the release experiment (Fig. 4(c) and (d)), which additionally confirms the stability of the model drug loading under cell culture conditions.34 The lack of free model drug in the supernatant indicates that the drug must be delivered to the cells according to a different mechanism. Previous studies have reported that a cellular uptake of hydrophobic drugs is favored by intracellular release of drugs from loaded particles taken up from the film surface.20 The possibility of particle-mediated transport of drugs was investigated by further cell experiments. Therefore, cells were seeded on the surface of DiG_0.00 films loaded with the model drug DiO and the uptake of drug and particles was investigated by CLSM. The corresponding micrographs are presented in Fig. 5. After an incubation of 4 hours only a weak signal of DiO could be detected inside C2C12 cells (Fig. 5(a)), whereas after an incubation time of 24 hours a strong signal of the model drug could be visualized within cells (Fig. 5(b)). To investigate the drug uptake in more detail, the particles forming films were labeled with ATTO647N dye prior to the loading with DiO. The DiO concentration in this experiment was reduced with a factor of 5 to enable clear visualization of the ATTO647N dye signal. Hence, a co-localization of DiO (yellow) and the fluorescently labeled MSNs (red) inside cells could be observed after 8 and 24 h of cell culture (Fig. 5(c)). This suggests a particle mediated uptake of drugs from the films. The particle uptake and particle-mediated transport of active agents shown here is in accordance with former work of us and others where the uptake of MSNs as potential drug carriers occurred from cell culture media in both monolayer and hydrogel cultures.6,7,35 Fig. 5(d) demonstrates comparatively weak staining subsequent to incubation of free DiO on film free silicon wafers prior to the cell cultivation, illustrating the need of a particle-mediated uptake for lipophilic active substances by cells. The particle mediated drug uptake mechanism is illustrated in Fig. 6. It has though been observed that lipophilic drugs can be taken up by cells through a kiss-and-run mechanism when drug loaded nanoparticles come in contact with the hydrophobic membrane surface of the cells.36 This mechanism cannot fully be excluded based on the present results. However, the amounts of drugs available on the films surface is limited to the molecules close to the pore openings, and the CLSM micrographs only show DiO signal when particles are internalized in the cells. After a cultivation of 24 h it was additionally possible to observe that only a small fraction of the film particles had been taken up by cells while the majority of the particles remained on the substrates surface (Fig. S2, ESI†). No particle-free areas around adhered cells could be detected. As discussed above, a gradual detachment of particles from the substrate can be due to differences in the particle growth. A previous study, employing DiG films as a catalyst host in esterification, revealed minor particle detachment from the substrate after 1 h of reaction.23 After washing and reuse of the film, no additional detachment was observed, showing the stability of the films after the initial particle detachment. The film stability has also been shown for different surfactant removal techniques, where the films did not show morphological changes upon ethanol extraction at 78 °C for 24 h, or sonication in methanol.23 In the presented study, the films were treated in an ultrasonic bath three times prior to the cell cultivation in order to remove any free particles from the surface. Fig. 1(a) shows a defect in the particle/substrate interface formed during the film growth, e.g. only one side of the half hexagonal prism is bound to the substrate. In addition, smaller particles can be observed in the cross section. These defects are leading to a faster loosening of the particles, and hence yield the initial drug release. One can hypothesize that a continued drug release will depend on dissolution of the silica framework in the body fluid,37 which can lead to a continued particle loosening from the substrate or exposure of the drugs through disintegration of the pore walls at the film surface. This can be compared to the spin-coated particulate system developed by Wiltschka et al. that showed a significantly increased uptake of particles by the cells and particle-free areas were easily observed around the adhered cells.20 A consequence of an increased particle uptake is a non-desired increased cell toxicity as shown by Böcking et al., who observed an enhanced toxicity when more than 5 layers of particles were spin-coated onto the substrate.38 In addition, a long-lasting uptake of particles from spin-coated films by cells, as it is advantageous in potential later applications, can only be achieved by further time consuming surface modifications.21 For these reasons, the presented films have the potential to be used as an implant coating material without further surface modification, providing a reservoir for a long-time uptake of drug loaded particles by surrounding cells.
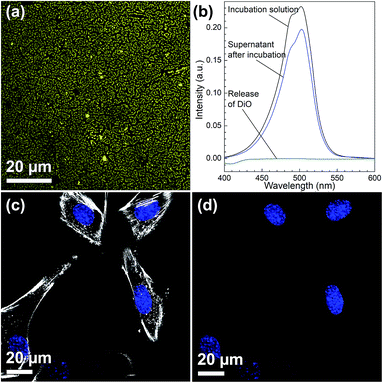 |
| | Fig. 4 (a) CLSM micrograph of DiO (yellow) loaded film (DiG_0.00), (b) UV-vis measurement of supernatants before (black) and after loading DiO into DiG_0.00 film (4 h, blue). Release of DiO during a 24 h incubation period into the surrounding cell culture media (turquoise). CLSM micrographs after incubation of cells (blue = nucleus) with the supernatant of the DiO release experiment for 24 h with (c) and without (d) staining the cytoskeleton of cells (white). | |
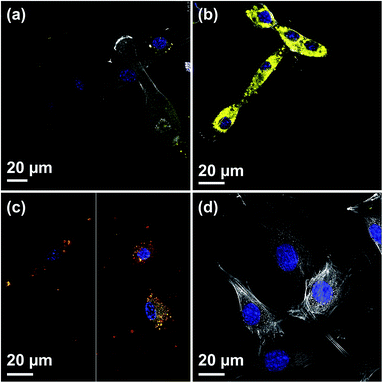 |
| | Fig. 5 CLSM micrographs showing the DiO (yellow) uptake from DiG_0.00 in cells after a cultivation time of (a) 4 h and (b) and 24 h, (c) co-localization of MSN (red) and DiO (yellow) signal within cells after different time of incubation (left = 8 h, right = 24 h), and (d) C2C12 cells grown on film free silicon wafers incubated with free DiO-solution in cell culture media for 24 h (blue = nucleus, white = microfilaments, yellow = DiO). | |
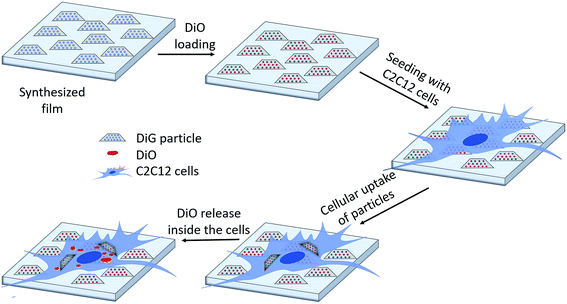 |
| | Fig. 6 Schematic illustration of the drug loading and release mechanism from DiG films to C2C12 cells. | |
Patterning
The microstructure of surfaces has in many studies been shown to be important for how cells respond to the substrate, including cellular alignment and stem cell differentiation. (See for example ref. 39–41.) Thus, as a further development, micropattern DiG films were synthesized by local removal of the hydrophobic surface film on the substrate, as it has been shown that functionalization of the substrate with hydrophobic molecules, like OTS or TMCS, is required for a densely packed film to form.23 Here, OTS functionalized substrates were partially irradiated with a Nd:YAG-Laser using tuneable pulse energy. An irradiation with a pulse energy of 29 mJ using a traversing speed of 2 mm s−1 and 10 mm s−1 resulted in a selective removal of the hydrophobic groups on Si-wafer. Both lower pulse energies (15 mJ and 19 mJ) showed almost no removal effect. This indicates a threshold between 19 mJ and 29 mJ at a spot diameter of 1 mm or 2.4 J cm−2 and 3.7 J cm−2 in terms of radiant exposure. Also, the plasma formation on the sample surface during the laser irradiation starts between 19 mJ and 29 mJ. Film synthesis using the irradiated substrates revealed that the particles only grow at the non-irradiated areas on the substrates, i.e. where the functionalization was intact (Fig. 7). The various sample velocities resulted in different shapes of the irradiated area, where a fast movement resulted in separate, circular domains and a slow movement gave linear structures, shown by the photographs in Fig. 7(a) and (b). The irradiated and film containing areas are indicated by red and green arrows, respectively. The film growth was confirmed using CLSM, where the films were functionalized with COOH-groups and subsequently marked with ATTO647N (Fig. 7(c) and (d)). These micrographs show film growth solely on non-irradiated areas of the substrate. The green lines in the micrographs are artefacts due to interference.
 |
| | Fig. 7 Photographs of films grown on substrates treated with pulsed laser at a traversing speed of (a) 10 mm s−1 and (b) 2 mm s−1 and a pulse energy of 29 mJ, where the blue arrows indicating areas with particles, red arrows indicating irradiated areas. CLSM micrographs of the patterned areas showing that particles only grow outside of the irradiated areas (c) and (d), where the green signal refers to the ATTO647N fluorescently labeled films. | |
The different appearance of the two sample velocities was expected and can be explained by the different overlapping areas. In case of 10 mm s−1 at 10 Hz two adjacent laser pulses with a diameter of approximately 1 mm shows no overlapping at all and can be clearly separated. Using a sample velocity of 2 mm s−1 the distance between two adjacent pulses is smaller by a factor of 5 which makes it impossible to distinguish the single pulses. With both sample velocities the irradiated areas showed a significant irregular boundary structure (Fig. 7(c) and (d)). This irregularity and the observation of a threshold between 2.4 J cm−2 and 3.7 J cm−2 that coincides with the beginning of plasma formation indicates a removal mechanism based on a photo-mechanical effect. Also, a formation of small dots near the center of the irradiated areas (both velocities) was observed and can be seen in Fig. 7(c) and (d). The arrangement of the dots is very similar for each irradiated area in case of 10 mm s−1 and shows a repeating appearance at 2 mm s−1. This indicates that at these dots the Si-substrate shows ablation induced by local maxima of the radiant exposure within the laser beam profile. It is hence clear that laser irradiation can be used for pattering a substrate and control the film growth on a substrate, but there are several points for further investigations, like the hot spot formation, the influence of the substrate ablation threshold or the behavior at higher levels of radiation exposure. Also, to support the hypothesis of photo-mechanical interaction some further experiments should be performed.
Conclusions
We have shown that films synthesized with the DiG method can be used as a drug delivery system. C2C12 cells adhere well on films comprising of particles with various sizes. The accessible pores make it possible to load the films with potential drugs, DiO, and functionalization of the film surfaces with e.g. COOH-groups is possible. The films are biocompatible with good growth and adherence of C2C12 cells. The DiO is distributed to the cells mainly through particle uptake where the particles release their cargo inside cells. No release of DiO from the films could be detected in the supernatant after 24 h. The particles are bound to the substrate, resulting in a slow uptake of the drugs and hence, the films are suitable as a drug-reservoir. In combination with the 3D growth of particles23 and the possibility of a local control of the growth areas of particles on substrates by pre-treatment with lasers enables new potential applications, especially in the field of implant coatings where necessary areas can be provided with film while other areas can be omitted to enable an optimal healing process. As an outlook, one can imagine DiG film growth on other substrates than Si-wafers, e.g. titanium or flexible polymers to come closer to a medical application.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The Swedish Research Council (VR) (2015-00624) (E. B.), and the German Research Foundation (WI3868/4-1) (R. W.) are acknowledged for financial support.
References
- E. Gultepe, D. Nagesha, S. Sridhar and M. Amiji, Nanoporous inorganic membranes or coatings for sustained drug delivery in implantable devices, Adv. Drug Delivery Rev., 2010, 62(3), 305–315 CrossRef CAS PubMed.
- J. M. Rosenholm, C. Sahlgren and M. Lindén, Towards multifunctional, targeted drug delivery systems using mesoporous silica nanoparticles – opportunities & challenges, Nanoscale, 2010, 2(10), 1870–1883 RSC.
- M. Vallet-Regí, M. Colilla, I. Izquierdo-Barba and M. Manzano, Mesoporous Silica Nanoparticles for Drug Delivery: Current Insights, Molecules, 2018, 23(1), 47 CrossRef PubMed.
- C. Argyo, V. Weiss, C. Bräuchle and T. Bein, Multifunctional Mesoporous Silica Nanoparticles as a Universal Platform for Drug Delivery, Chem. Mater, 2014, 26(1), 435–451 CrossRef CAS.
- J. M. Rosenholm, J. Zhang, M. Linden and C. Sahlgren, Mesoporous silica nanoparticles in tissue engineering--a perspective, Nanomedicine, 2016, 11(4), 391–402 CrossRef CAS.
- B. Baumann, R. Wittig and M. Lindén, Mesoporous silica nanoparticles in injectable hydrogels: factors influencing cellular uptake and viability, Nanoscale, 2017, 9(34), 12379–12390 RSC.
- B. Baumann, T. Jungst, S. Stichler, S. Feineis, O. Wiltschka, M. Kuhlmann, M. Lindén and J. Groll, Control of Nanoparticle Release Kinetics from 3D Printed Hydrogel Scaffolds, Angew. Chem., Int. Ed., 2017, 56(16), 4623–4628 CrossRef CAS.
- J. M. Rosenholm, C. Sahlgren and M. Lindén, Multifunctional Mesoporous Silica Nanoparticles for Combined Therapeutic, Diagnostic and Targeted Action in Cancer Treatment, Curr. Drug Targets, 2011, 12(8), 1166–1186 CrossRef CAS.
- J. P. Jahnke, M. N. Idso, S. Hussain, M. J. N. Junk, J. M. Fisher, D. D. Phan, S. Han and B. F. Chmelka, Functionally Active Membrane Proteins Incorporated in Mesostructured Silica Films, J. Am. Chem. Soc., 2018, 140(11), 3892–3906 CrossRef CAS.
- M. Bardhan, A. Majumdar, S. Jana, T. Ghosh, U. Pal, S. Swarnakar and D. Senapati, Mesoporous silica for drug delivery: Interactions with model fluorescent lipid vesicles and live cells, J. Photochem. Photobiol., B, 2018, 178, 19–26 CrossRef CAS.
- M. Manzano and M. Vallet-Regí, Ultrasound responsive mesoporous silica nanoparticles for biomedical applications, Chem. Commun., 2019, 55(19), 2731–2740 RSC.
- C. J. Brinker, Y. Lu, A. Sellinger and H. Fan, Evaporation-Induced Self-Assembly: Nanostructures Made Easy, Adv. Mater., 1999, 11(7), 579–585 CrossRef CAS.
- D. Grosso, F. Cagnol, G. J. d. A. A. Soler-Illia, E. L. Crepaldi, H. Amenitsch, A. Brunet-Bruneau, A. Bourgeois and C. Sanchez, Fundamentals of Mesostructuring Through Evaporation-Induced Self-Assembly, Adv. Funct. Mater., 2004, 14(4), 309–322 CrossRef CAS.
- A. Goux, M. Etienne, E. Aubert, C. Lecomte, J. Ghanbaja and A. Walcarius, Oriented Mesoporous Silica Films Obtained by Electro-Assisted Self-Assembly (EASA), Chem. Mater, 2009, 21(4), 731–741 CrossRef CAS.
- N. Nishiyama, S. Tanaka, Y. Egashira, Y. Oku and K. Ueyama, Vapor-Phase Synthesis of Mesoporous Silica Thin Films, Chem. Mater, 2003, 15(4), 1006–1011 CrossRef CAS.
- A. M. A. Abdelsamad, A. S. G. Khalil and M. Ulbricht, Influence of controlled functionalization of mesoporous silica nanoparticles as tailored fillers for thin-film nanocomposite membranes on desalination performance, J. Membr. Sci., 2018, 563, 149–161 CrossRef CAS.
- N. Ehlert, M. Badar, A. Christel, S. J. Lohmeier, T. Luessenhop, M. Stieve, T. Lenarz, P. P. Mueller and P. Behrens, Mesoporous silica coatings for controlled release of the antibiotic ciprofloxacin from implants, J. Mater. Chem., 2011, 21(3), 752–760 RSC.
- R. Lensing, A. Bleich, A. Smoczek, S. Glage, N. Ehlert, T. Luessenhop, P. Behrens, P. P. Müller, M. Kietzmann and M. Stieve, Efficacy of nanoporous silica coatings on middle ear prostheses as a delivery system for antibiotics: An animal study in rabbits, Acta Biomater., 2013, 9(1), 4815–4825 CrossRef CAS.
- H. Qu, S. Bhattacharyya and P. Ducheyne, Silicon oxide based materials for controlled release in orthopedic procedures, Adv. Drug Delivery Rev., 2015, 94, 96–115 CrossRef CAS.
- O. Wiltschka, D. Böcking, L. Miller, R. E. Brenner, C. Sahlgren and M. Lindén, Preparation, characterization, and preliminary biocompatibility evaluation of particulate spin-coated mesoporous silica films, Microporous Mesoporous Mater., 2014, 188, 203–209 CrossRef CAS.
- O. Wiltschka, P. Scheitenberger and M. Lindén, Control of particle uptake kinetics from particulate mesoporous silica films by cells through covalent linking of particles to the substrate – towards sequential drug delivery for tissue engineering applications, J. Mater. Chem. B, 2016, 4(47), 7669–7675 RSC.
- E. M. Björk, F. Söderlind and M. Odén, Single-pot synthesis of ordered mesoporous silica films with unique controllable morphology, J. Colloid Interface Sci., 2014, 413, 1–7 CrossRef PubMed.
- P.-H. Wu, P. Mäkie, M. Odén and M. E. Björk, Growth and Functionalization of Particle-Based Mesoporous Silica Films and Their Usage in Catalysis, Nanomaterials, 2019, 9(4), 562 CrossRef PubMed.
- H. Gustafsson, E. M. Johansson, A. Barrabino, M. Odén and K. Holmberg, Immobilization of lipase from Mucor miehei and Rhizopus oryzae into mesoporous silica - The effect of varied particle size and morphology, Colloids Surf., B, 2012, 100, 22–30 CrossRef CAS PubMed.
- R. Senthilkumar, D. Sen Karaman, P. Paul, E. M. Björk, M. Odén, J. E. Eriksson and J. M. Rosenholm, Targeted delivery of a novel anticancer compound anisomelic acid using chitosan-coated porous silica nanorods for enhancing the apoptotic effect, Biomater. Sci., 2015, 3(1), 103–111 RSC.
- R. Wittig, E. Waller, G. von Freymann and R. Steiner, Direct laser writing-mediated generation of standardized topographies for dental implant surface optimization, J. Laser Appl., 2012, 24(4), 042011 CrossRef.
- E. M. Björk, P. Mäkie, L. Rogström, A. Atakan, N. Schell and M. Odén, Formation of block-copolymer-templated mesoporous silica, J. Colloid Interface Sci., 2018, 521, 183–189 CrossRef PubMed.
- E. B. Celer, M. Kruk, Y. Zuzek and M. Jaroniec, Hydrothermal stability of SBA-15 and related ordered mesoporous silicas with plugged pores, J. Mater. Chem., 2006, 16(27), 2824–2833 RSC.
- E. M. Björk, F. Söderlind and M. Odén, Tuning the Shape of Mesoporous Silica Particles by Alterations in Parameter Space: From Rods to Platelets, Langmuir, 2013, 29(44), 13551–13561 CrossRef PubMed.
- U. Horzum, B. Ozdil and D. Pesen-Okvur, Step-by-step quantitative analysis of focal adhesions, MethodsX, 2014, 1, 56–59 CrossRef.
- P. D'Andrea, D. Civita, M. Cok, L. Ulloa Severino, F. Vita, D. Scaini, L. Casalis, P. Lorenzon, I. Donati and A. Bandiera, Myoblast Adhesion, Proliferation and Differentiation on Human Elastin-Like Polypeptide (HELP) Hydrogels, J. Appl. Biomater. Funct. Mater., 2017, 15(1), 43–53 Search PubMed.
- V. Mamaeva, C. Sahlgren and M. Lindén, Mesoporous silica nanoparticles in medicine—Recent advances, Adv. Drug Delivery Rev., 2013, 65(5), 689–702 CrossRef CAS PubMed.
- C. Bharti, U. Nagaich, A. K. Pal and N. Gulati, Mesoporous silica nanoparticles in target drug delivery system: A review, Int. J. Pharm. Invest., 2015, 5(3), 124–133 CrossRef CAS PubMed.
- J. Huang, C. Zhu, P. Zhang, Q. Zhu, Y. Liu, Z. Zhu, M. Wang, W. Li, G. Yang, N. Dong, J. Liu, L. Chen, Y. Zhang, R. Yang, L. Deng, J. Fan, X. Wang, J. Liu, B. Ma, Q. Fu and K. Wu, S100+ cells: A new neuro-immune cross-talkers in lymph organs, Sci. Rep., 2013, 3, 1114 CrossRef PubMed.
- J. M. Rosenholm, A. Meinander, E. Peuhu, R. Niemi, J. E. Eriksson, C. Sahlgren and M. Lindén, Targeting of Porous Hybrid Silica Nanoparticles to Cancer Cells, ACS Nano, 2009, 3(1), 197–206 CrossRef CAS PubMed.
- D. Hofmann, C. Messerschmidt, M. B. Bannwarth, K. Landfester and V. Mailänder, Drug delivery without nanoparticle uptake: delivery by a kiss-and-run mechanism on the cell membrane, Chem. Commun., 2014, 50(11), 1369–1371 RSC.
- K. Braun, A. Pochert, M. Beck, R. Fiedler, J. Gruber and M. Lindén, Dissolution kinetics of mesoporous silica nanoparticles in different simulated body fluids, J. Sol-Gel Sci. Technol., 2016, 79(2), 319–327 CrossRef CAS.
- D. Böcking, O. Wiltschka, J. Niinimäki, H. Shokry, R. Brenner, M. Lindén and C. Sahlgren, Mesoporous silica nanoparticle-based substrates for cell directed delivery of Notch signalling modulators to control myoblast differentiation, Nanoscale, 2014, 6(3), 1490–1498 RSC.
- J. Y. Lim and H. J. Donahue, Cell Sensing and Response to Micro- and Nanostructured Surfaces Produced by Chemical and Topographic Patterning, Tissue Eng., 2007, 13(8), 1879–1891 CrossRef CAS PubMed.
- J. L. Charest, M. T. Eliason, A. J. García and W. P. King, Combined microscale mechanical topography and chemical patterns on polymer cell culture substrates, Biomaterials, 2006, 27(11), 2487–2494 CrossRef CAS PubMed.
- J. B. Recknor, J. C. Recknor, D. S. Sakaguchi and S. K. Mallapragada, Oriented astroglial cell growth on micropatterned polystyrene substrates, Biomaterials, 2004, 25(14), 2753–2767 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details on substrate preparation, TEM micrographs of the materials, a CLSM micrograph of a film after 24 h of cell cultivation, and a CLSM micrograph of C2C12 cells on a film. See DOI: 10.1039/c9ra02823d |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Bernhard Baumanna,
Florian Hausladenc,
Rainer Wittig
*ab,
Bernhard Baumanna,
Florian Hausladenc,
Rainer Wittig