
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExperimental and theoretical investigation of cyclometalated phenylpyridine iridium(III) complex based on flavonol and ibuprofen ligands as potent antioxidant†‡
Leila Tabrizi *a,
Thi Le Anh Nguyen*b and
Duy Quang Daob
*a,
Thi Le Anh Nguyen*b and
Duy Quang Daob
aSchool of Chemistry, National University of Ireland, University Road, Galway, H91 TK33, Ireland. E-mail: leila.tabrizi@nuigalway.ie
bInstitute of Research and Development, Duy Tan University, 03 Quang Trung, Danang, 550000, Vietnam. E-mail: nguyenthileanh@dtu.edu.vn
First published on 3rd June 2019
Abstract
An Ir(III) complex was synthesized using mixed ligands of biological importance, namely ibuprofen, flavonol and 2-phenylpyridine. The compound was characterized by 1H-NMR, 13C-NMR and TOF-MS spectroscopies and elemental analysis. Structures of the complex and its ligands were also calculated by density functional theory using B3LYP/Lanl2dz//6-31G(d) level of theory. Analyses of electrostatic potential, natural population, and frontier orbitals of the molecules as well as the calculation of intrinsic thermochemical properties such as bond dissociation enthalpy, ionization potential, electron affinity and proton affinity in the gas phase and in solvents (water and pentylethanoate) give the first indication that the complex is a potential antioxidant. The latter even shows better antioxidant capacity than the parent ligands. The antioxidant properties of the complex and its ligands were experimentally evaluated by studying the free radical scavenging activity towards HO˙, NO˙, DPPH˙ and ABTS˙+ radicals. Further computational work on the antioxidant processes such as the single electron transfer, the proton loss, the formal hydrogen transfer (FHT) and the radical adduct formation reactions was conducted. Results show that the FHT reaction is the mechanism responsible for the radical scavenging activity of the complex towards HO˙, HOO˙, NO˙ and DPPH˙ radicals while ABTS˙+ seems to be scavenged by an electron-donating mechanism. The FHT was further determined as a hydrogen-atom transfer but not a proton-couple electron transfer mechanism.
1. Introduction
Recent findings in biochemistry and related sciences have strengthened the connections between oxidative stress, inflammation and severe diseases in the human body.1,2 Long-time exposure to oxidative stress was proved to lead to chronic inflammation and later sickness and diseases. Both cancers and non-cancer diseases such as Parkinson's, Alzheimer's, diabetes and hypertension were reported to have a relationship with oxidative stress. For the last forty years, the use of cis-platin and its analogues for the treatment of certain kinds of cancers has helped to save and lengthen lives of many patients. Since then, there has been an exponential increasing of attention paid to the use of metallodrugs in the treatment of diseases,3 in which several late transition metal complexes, i.e. Ir, Rh, Ru and Os, have shown great potential for biological targets.4–8Meanwhile, Ir(III) complexes have found important applications in fields of research ranging from materials to catalysis based on their interesting photo-physical and photo-chemical properties.9 They are known to be efficient phosphors in OLEDs or imaging agent and sensors.10–14 In particular, there have been several attempts to use Ir-based carbene compounds or complexes as antitumor agents; a series of half-sandwich Ir(III) complexes were synthesized or modeled and reported as potential anticancer drugs.15–20 Indeed, one can expect for the new Ir(III) complexes, as well as for other metallodrugs, to (i) get stronger and more selective effects against tumors, (ii) extend the spectrum of cancer treatment and (iii) overcome the limits of Pt-based drugs such as the platin resistance and other severe side-effects.
We are interested in studying the antioxidant activity of Ir(III) complexes, which is believed as a fundamental point to search for further biological activities such as anti-inflammation or anticancer properties. One can easily imagine, in such metallodrugs, that both metal ion and ligands could play crucial roles for the deactivation and quenching of reactive oxidant species (ROS), probably through redox reaction for the metal complex and other more complicated mechanisms for ligands.3,21 In order to design such on-demand Ir complexes, the smart choice of ligands becomes obviously the key step. We mainly aim to choose ligands exhibiting strong antioxidant properties or confirmed positive biological activity in human health. The final target is to achieve metal complexes having stronger antioxidant properties than those of the parent components.
Flavonol, a member of the natural flavonoids, is acknowledged for having benefit to human health mostly as antidepressant and antioxidant. In particular, flavonol has been documented to have antibacterial, anti-inflammation and antiallergic properties or even to reduce the risk of heart diseases and to slowdown the ageing procedure.21 The presence of a hydroxyl group at the position next to the carboxyl group seems to promote stronger antioxidant properties and also makes flavonol a perfect candidate for metal ion chelation.22 Several flavonol complexes of V(IV),23 Fe(III),24 Pb(II),25 Al(III)26 and Zn(II)27 have been reported. In spite of that, there has been no in-depth study of the antioxidant or biological activities of the compounds, except in the case of the Zn(II) complex, which indeed shows strong antidiabetic activity in rats comparable to that of gliclazide, a standard drug for diabetes.27
Ibuprofen is currently one of the most popular nonsteroidal anti-inflammatory drugs to treat fever, pain and inflammation. Metal complexes of ibuprofen and its derivatives, including those of Cu(II),28 Ru(II, III)29 and Au(I)30 were documented. While Cu(II) and Ru(II, III) complexes show a similar effect on carrageenan-induced edema in rats as the parent drug (ibuprofen), they show less gastric irritation and are more protective to ulceration than ibuprofen.29 The Au(I) complex shows positive antibacterial activity in vitro against Gram-negative (E. coli and P. aeruginosa) and Gram-positive (S. aureus) microorganisms.30
The octahedral coordination of Ir(III) cation might be completed with 2-phenylpyridine. In-depth studies have been reported on the use of Ir(III) complexes of 2-phenylpyridine as well as of polypyridine as anticancer31 or imaging agents in live cells.32 For the moment, cellular uptake and the cytotoxicity of these complexes still limit their bio-applications.
In this paper, we report a combination of experimental and theoretical studies of an Ir(III) coordination compound constructed from ligands with biological activities: flavonol, ibuprofen and 2-phenylpyridine. The compound was first synthesized and characterized by 1H-NMR, 13C-NMR, and MS spectroscopies and elemental analysis. Optimized geometry and internal electronic properties of the molecule, i.e. electrostatic potential (ESP) map, natural population analysis (NPA) and frontier orbitals (HOMO, LUMO), were also computed using the density functional theory (DFT) approach. The antioxidant properties of the complex and two of the ligands (ibuprofen and flavonol) were then experimentally studied in terms of free radical scavenging activity (RSA) towards HO˙, NO˙, DDPH˙ and ABTS˙+. Theoretical investigation of thermochemical properties such as bond dissociation enthalpy (BDE), proton affinity (PA), ionization potential (IP) and electron affinity (EA) was conducted in the gas phase and in water to evaluate the antioxidant potentials of the compound through the electron- and the proton-donating/accepting capacities. Finally, DFT calculation was helpful in obtaining evidence of the free radical scavenging mechanisms by studying single electron transfer (SET), proton loss (PL), formal hydrogen transfer (FHT) and radical adduct formation (RAF) reactions between the radicals (HO˙, HOO˙, NO˙, DPPH˙ and ABTS˙+) and the Ir(III) complex at the most favorable positions. Potential energy profiles (PEPs) of reactions and natural bond orbital (NBO) analysis were used to elucidate the mechanism of the antioxidant activity through FHT and RAF reactions. Especially, further analyses of frontier orbitals of transition states (TSs) and NBO were used as key elements to distinguish the FHT mechanism as hydrogen atom transfer (HAT) or proton couple electron transfer (PCET).
2. Methods
2.1. Materials and methods
All reagents and solvents were purchased from Sigma-Aldrich and Fisher Scientific and used without further purification. Microwave-assisted reactions were carried out in an Ethos UP microwave oven using 85 mL TFM Teflon closed vessels equipped with temperature sensor, pressure controller and a magnetic stirrer bar. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded with a Bruker-400 MHz spectrometer at ambient temperature in DMSO-d6. Electrospray ionization mass spectra were recorded with a Waters LCT Premier XE spectrometer in positive- or negative-ion mode. Elemental analyses were performed with an EA 3000 CHNS. Electronic absorption spectra were obtained with a Shimadzu Lambda-1600 UV-visible spectrophotometer.Electron spin resonance (ESR) spectra were obtained with a Varian spectrometer using the following conditions: 3385.0 G field, 20.0 mV power, 100.0 kHz modulation frequency, 1.0 G amplitude and 300 s sweep time. The temperature was kept at 20 °C using a cooling water circulator.
2.2. Computational methods
All calculations were performed using the Gaussian 09, revision E.01 package.33 Geometry optimization and frequency calculations of all structures, TSs, intermediate species and products were performed using the hybrid functional B3LYP without any constraint. Two different basis sets were employed in combination, the Lanl2dz basis set for Ir and the 6-31G(d) basis set for C, H, N, and O elements, as previously recommended.34,35 The Lanl2dz set was selected as it can appropriately describe the electronic structure of transition metals while the 6-31G(d) set was chosen for balancing the accuracy of the calculation and the computed time. Moreover, calculation with the combination of these two basis sets was reported to provide good correlations with experimental results for different Ir complexes.14,35 The geometry of the complex was first studied with different spin configurations and the lowest energy one was kept for further investigations. All single-point calculations were performed with the same basis sets. Bulk effect of solvent was studied by single-point calculation using the polarizable continuum model IEF-PCM with water and pentylethanoate (PEA) as the solvents which were chosen to represent the lipid media in human bodies.36,37In order to evaluate the antioxidant potential of the Ir(III) complex and its ligands, the most conventional reactions were firstly studied as follows: HAT, PL and SET.37 The thermochemical properties of the Ir complex and ligands, such as BDE, PA, IP and EA studied in the gas phase and in solvents, were then deduced from the corresponding equations (see ESI for more details†). The second steps for the single electron transfer followed by proton transfer (SETPT) and sequential proton loss electron transfer (SPLET) reactions were only evaluated for the most probable positions on the Ir(III) complex.
Second, thermochemical properties and kinetics of the reactions between the antioxidant and free radicals, e.g. HO˙, HOO˙, NO˙, DPPH˙ and ABTS˙+, were studied in the gas phase and in solvents. Besides the SET studied for both electron-donating and -accepting pathways, the PL, the FHT and the RAF mechanisms were investigated for the most probable positions. In order to get insight into the RSA of the potent antioxidant, the PEPs of FHT (i.e. HAT or PCET) and RAF reactions between the Ir(III) complex and its free ligands with HOO˙/HO˙ radicals were selected to study in the gas phase at the same level of theory. Additional analyses at the TSs such as singly occupied molecular orbital (SOMO), Hirshfeld atomic spin density (ASD), NPA atomic charge as well as natural atomic orbital (NAO) occupancy for 1s orbital and natural electronic configuration (NEC) for the mitigating hydrogen as well as for the carbon and oxygen atoms involved in the reactions were helpful to clarify the hydrogen transfer mechanisms.
3. Results and discussion
3.1. Synthesis and characterization
The reaction scheme for the synthesis of the Ir(III) complex is presented in Scheme 1, which involves in a revision of the synthetic procedure previously reported by Orwat and co-workers.38 Briefly, a microwave pressure reactor was used in a one-pot synthetic method for the complex formation. Reaction time and temperature were optimized for the synthesis of the Ir(III) complex based on the starting materials including a mixture of IrCl3·3H2O and the three ligands in basic medium. Under the optimal conditions, i.e. 130 °C and 45 min, the Ir(III) complex was obtained with a yield of 62.7%. The complex was characterized by 1H-NMR, 13C-NMR and MS spectroscopies and elemental analyses.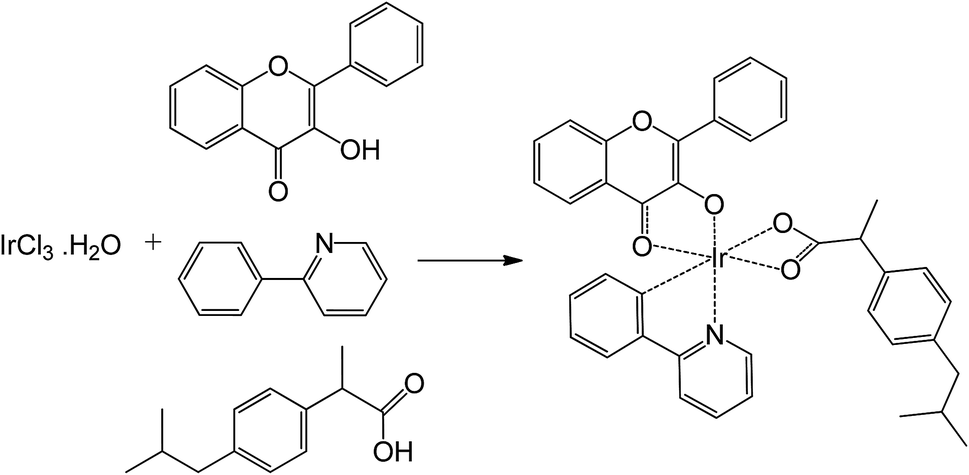 | ||
Scheme 1 Synthetic pathway of the Ir(III) complex. Reagents and conditions: 2-ethoxyethanol/water (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), NaOH (5 eq.), microwaves (5 bar, 350 W), 130 °C, 45 min, 62.7%. :1, v/v), NaOH (5 eq.), microwaves (5 bar, 350 W), 130 °C, 45 min, 62.7%. | ||
The formation of the Ir(III) complex by coordinating the metal cation (Ir) to the ligands is confirmed by the 1H-NMR spectrum (Fig. S1 of the ESI†) with the disappearance of the proton signals of OH and COOH groups of the starting flavonol and ibuprofen at 9.64 ppm and 12.21 ppm, respectively.39–41 The signal of the α proton of the carbon coordinated to iridium (HC70, Fig. 1) is significantly shifted from about 7.60 ppm in 2-phenylpyridine in free form to 6.34 ppm in the Ir(III) complex due to the formation of anionic charge of the cyclometalating phenyl ring.41 The 13C-NMR spectrum of the Ir(III) complex is in agreement with the proposed structure (Fig. S2 of the ESI†). The mass spectrum of the Ir(III) complex showed a pattern with peaks centered at m/z = 790.2144 in the positive region corresponding to [M + H]+ (Fig. S3 of the ESI†).
 | ||
| Fig. 1 Optimized geometries of ligands at the B3LYP/6-31G(d) level of theory and of Ir(III) complex at the B3LYP/Lanl2dz//6-31G(d) one. The inset shows the numbering of the 2D structure of the Ir(III) complex. | ||
3.2. Structural and electronic properties by DFT
The structure and the electronic properties of the Ir(III) complex and all ligands were studied by DFT. Optimized geometries of the Ir(III) complex at the B3LYP/Lanl2dz//6-31G(d) level of theory and all ligands at the B3LYP/6-31G(d) level of theory are shown in Fig. 1. The non-planar structure of 2-phenylpyridine is clearly observed with a dihedral angle HC80–C66–C61–N79 of about −161°. The B ring of flavonol also rotates from the plane of the AC rings to form a dihedral angle O1–C5–C7–C12 of about 144°. For ibuprofen, the carboxylate group is found to be located at one side of the phenyl ring while all alkyl chains are located at the other side.The structure of the Ir(III) complex was first optimized at various multiplicities (1, 3, 5, 7, 9) in order to study its most likely spin states. As a result, the lowest energy structure was found for the singlet spin, which was kept for all following studies. As can be seen in Fig. 1, the Ir(III) cation coordinates to the three bidentate ligands through four O sites (i.e., two O atoms of flavonol and two O atoms of ibuprofen) and directly to C80 and N79 atoms of 2-phenylpyridine. The metal center adopts a non-symmetrical hexagonal configuration with atomic distances from Ir to binding sites calculated as follows: Ir–O2 (2.07 Å), Ir–O3 (2.09 Å), Ir–O29 (2.10 Å), Ir–O30 (2.39 Å), Ir–C80 (1.98 Å) and Ir–N79 (2.03 Å). Interestingly, in the complex structure, the flavonol and 2-phenylpyridine ligands adopt planar geometry, which favors the delocalization of electrons from the ligands to the central cation for the metal–ligand coordination. Cartesian coordinates and molecular enthalpies of the Ir(III) complex are summarized in Table S1 of the ESI.†
Mapping the ESP of a molecule allows one to predict the reactivity of a particular region of atoms toward nucleophilic or electrophilic attack. The ESP maps of the three ligands and Ir(III) complex are shown in the first column of Fig. 2, where the red regions represent the most negative ESPs and the blue ones display the most positive ESPs. Accordingly, the most negative electrostatic regions are found at the N and C of 2-phenylpyridine and at oxygen atoms of flavonol and ibuprofen ligands. These atoms are obviously the favorable sites that coordinate to the metal center for complex formation. For the Ir(III) complex, the most positive electrostatic region is found on the metal center (Ir) while the most negative one is located on the phenyl rings of 2-phenylpyridine and flavonol. This observation is confirmed by quantitative calculation of NPA atomic charges (Table S2 of the ESI†).
 | ||
| Fig. 2 Electrostatic potential maps (left), HOMO (middle) and LUMO (right) of (A) 2-phenylpyridine, (B) flavonol, (C) ibuprofen and (D) Ir(III) complex (isovalue = 0.02). | ||
Understanding the frontier molecular orbitals, particularly the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), is important for predicting the electron-donating and electron-accepting capacities of a molecule. Fig. 2 shows the HOMO (middle column) and LUMO (right column) of the three ligands and Ir(III) complex. It is observed that for all three ligands, the HOMOs and LUMOs are almost equally distributed across the molecular backbone. Nevertheless, for the Ir(III) complex, the HOMOs are mainly localized at the flavonol ligand and the LUMOs are delocalized more equally at both the 2-phenylpyridine and flavonol ligands. At the neutral state, the ibuprofen ligand does not contribute to the HOMO/LUMO of the Ir(III) complex. As expected, the HOMO is mostly contributed by 2-phenylpyridine and a small d part from metal ion while the LUMO is essentially contributed by the π-conjugate system of flavonol and 2-phenylpyridine ligands. The result is in agreement with the nature of metal–ligand (e− donor).
3.3. Antioxidant properties by free radical scavenging activities
The antioxidant properties of the complex and two of the ligands (ibuprofen and flavonol) were studied by RSA towards HO˙, NO˙, DPPH˙ and ABTS˙+. UV-visible and ESR spectroscopies were employed to follow the reactions. The results are summarized in Fig. 3 and Table 1.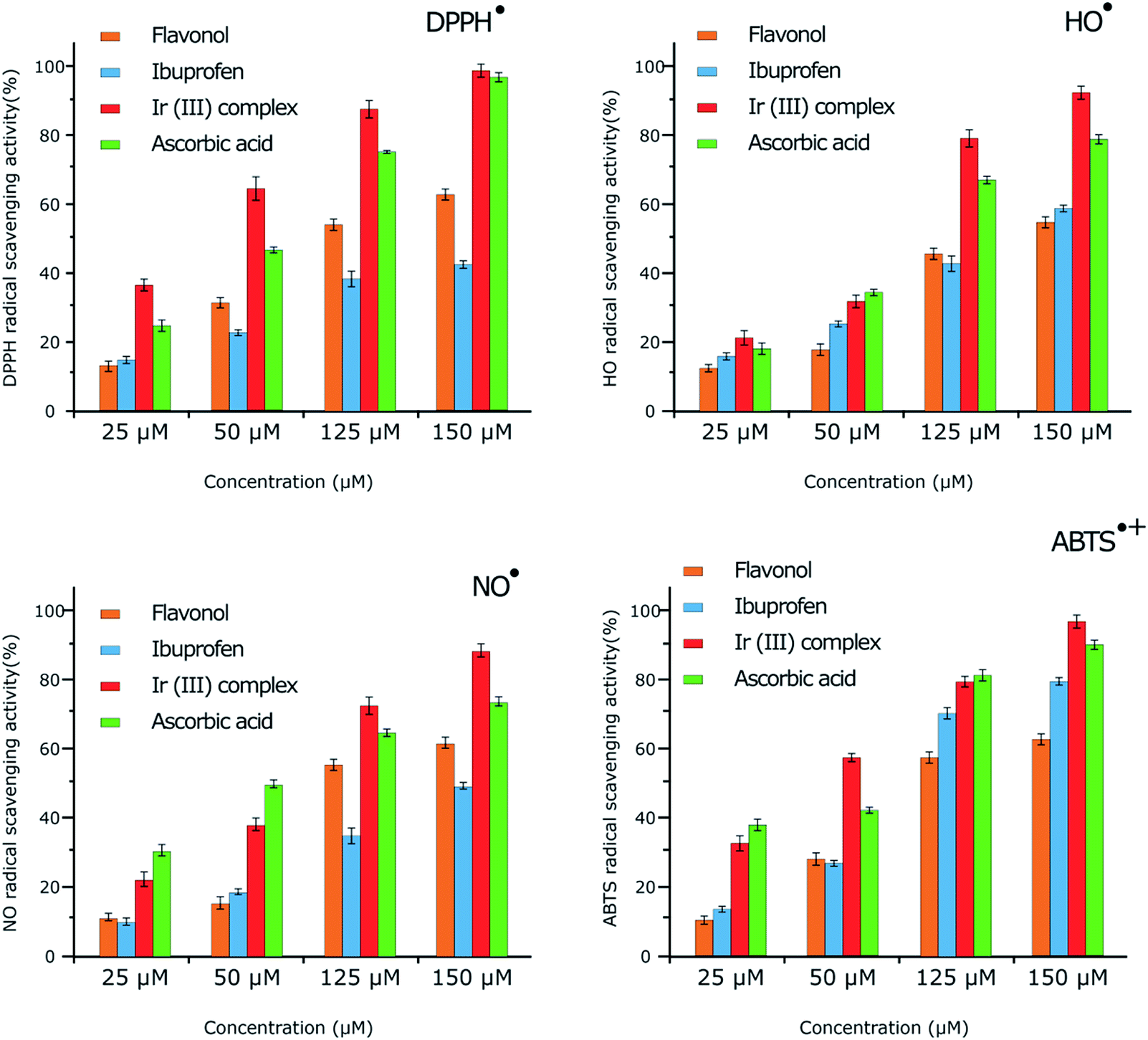 | ||
| Fig. 3 Free radical scavenging activity towards DPPH˙, HO˙, NO˙ and ABTS˙+ presented according to ESR results. The mean ± SD is shown for triplicate experiments. | ||
| Radical | IC50a (μM) | |||
|---|---|---|---|---|
| Flavonol | Ibuprofen | Ir(III) complex | Ascorbic acid | |
| a All values are the means of three measurements and given as mean ± SD. Statistically significant differences are indicated by asterisks (***p < 0.001, **p < 0.01, *p < 0.05 compared to ascorbic acid). | ||||
| DPPH˙ | 85.32 ± 0.10* | 143.55 ± 0.20** | 8.32 ± 0.05** | 14.25 ± 0.18*** |
| HO˙ | 138.24 ± 0.12** | 105.23 ± 0.05** | 19.32 ± 0.05* | 28.12 ± 0.10*** |
| NO˙ | 97.27 ± 0.12* | 127.13 ± 0.05*** | 25.42 ± 0.05* | 43.27 ± 0.14** |
| ABTS˙+ | 110.74 ± 0.12*** | 76.25 ± 0.10* | 10.14 ± 0.10*** | 65.31 ± 0.15** |
The antioxidant activity at various concentrations (25, 50, 125 and 150 μM) of flavonol, ibuprofen, and the Ir(III) complex was determined by measuring the decolorization of DPPH˙. The RSA increased with increasing concentration of the compounds. Flavonol, ibuprofen, Ir(III) complex, and ascorbic acid exhibited maximum activity of 62.45, 45.15, 98.95 and 96.10%, respectively, at 150 μM concentration (Table S3 of the ESI†). Under our reaction conditions, IC50 value of ascorbic acid was 14.25 μM, while IC50 values for flavonol, ibuprofen, and Ir(III) complex were as 85.32, 143.55 and 8.32 μM, respectively (Table 1). The RSA can be classified in the following order: Ir(III) complex > ascorbic acid > flavonol > ibuprofen.
By ESR spectroscopy, the Ir(III) complex displayed significant DPPH˙ RSA in a dose-dependent manner (Fig. 3). The radical scavenging activity of the Ir(III) complex increased with increasing concentration, which presented as 37.86% and 98.27% (calculated from the ESR signal intensity, Fig. S5 of the ESI†) at 25 μM and 150 μM in comparison with 25.73% and 96.46% for ascorbic acid at the same concentrations, respectively. The scavenging activities of the DPPH˙ radicals obtained by ESR spectroscopy are in the same order as that of UV-visible spectroscopic study: Ir(III) complex > ascorbic acid > flavonol > ibuprofen.
In ESR monitoring, HO˙ radicals generated in Fe2+/H2O2 system are trapped by DMPO forming spin adduct and detected by an ESR spectrometer. Ascorbic acid was used as positive control. The Ir(III) complex displayed significant HO˙ RSA with increasing concentration (Fig. S6 of the ESI†). The ESR results show that the Ir(III) complex and ascorbic acid suppress about 92.12% and 78.64% (calculated from the ESR signal intensity, Fig. 3) of the hydroxyl radicals at 150 μM, respectively. The order of potent antioxidant activity was found to be identical to that obtained from UV-visible spectroscopy.
Furthermore, antioxidant activities of flavonol, ibuprofen, Ir(III) complex and ascorbic acid were also investigated for NO˙ radical scavenging by ESR spectroscopy. NO˙ radical was released from a nitric oxide donor, S-nitroso-N-acetylpenicillamine. NO˙ was trapped with iron-dithiocarbamate to produce the [(MGD)2–Fe2+–NO] adduct which gave a triplet-line ESR signal (Fig. S7 of the ESI†). Hemoglobin, as positive control, inhibited the ESR signal that indicated the production of this spin adduct from NO˙. The Ir(III) complex displayed significant scavenging activity towards NO˙ when increasing the concentration. The ESR results show that the Ir(III) complex and ascorbic acid suppress about 88.87% and 72.35% (calculated from the ESR signal intensity, Fig. 3) of the NO˙ radicals at 150 μM, respectively. Antioxidant activity decreases in the following order: Ir(III) complex > ascorbic acid > flavonol > ibuprofen, similar to the results of UV-visible spectroscopic study.
In addition, antioxidant activities of flavonol, ibuprofen, Ir(III) complex and ascorbic acid were also considered for ABTS˙+ radical scavenging by ESR. The Ir(III) complex presents considerable ABTS˙+ RSA with increasing concentration (Fig. S8 of the ESI†). The ESR results confirmed that Ir(III) complex and ascorbic acid suppress about 95.94% and 89.72% (calculated from the ESR signal intensity, Fig. 3) of ABTS˙+ at 150 μM, respectively. Antioxidant activity decreases in the following order: Ir(III) complex > ascorbic acid > flavonol > ibuprofen, similar to the results of UV-visible spectroscopic study.
Firstly, the scavenging activities of DPPH˙, OH˙, NO˙ and ABTS˙+ radicals obtained by ESR spectroscopy are in excellent agreement with the results obtained from UV-visible spectroscopy. Secondly, the antioxidant activity results of the free ligands and Ir(III) complex against the free radicals DPPH˙, OH˙, NO˙ and ABTS˙+ showed that the Ir(III) complex displays greater scavenging activity than the free ligands and also the standard antioxidant ascorbic acid, with decreasing order as follows: Ir(III) complex > ascorbic acid > flavonol > ibuprofen. Thirdly, the antioxidant activity of the Ir(III) complex against the DPPH˙ radical is better than that against the other radicals and the trend is decreasing in the following order: DPPH˙ > ABTS˙+ > OH˙ > NO˙. In the following part, we choose to discuss in more detail the mechanism of DPPH˙ scavenging activity by the Ir(III) complex.
:PBS (1:9) solutions (pH 7.4) (Fig. 4). On increasing the concentration of the Ir(III) complex, we simultaneously observed the decreasing of the absorption band of DPPH˙ form (518 nm) and the increasing of the absorption band of DPPH− anion (around 425 nm). This observation shows the consumption of the DPPH˙ present in the reaction to produce the DPPH− anion form.48 The 425 nm absorption bands were maximized at 40 μM then decreased upon increasing the concentration of the Ir(III) complex to 50 or 60 μM due to the exchange of the anion form of DPPH− to DPPH-H. The DPPH-H molecule was formed as a result of accepting one proton from the Ir(III) complex. The result is totally in agreement with our recent findings for a Cu(II) complex and others.49–51
 | ||
| Fig. 4 UV-visible spectra monitoring the reaction between DPPH˙ (50 μM) and Ir(III) complex (10–60 μM) in methanol:PBS (1:9) solutions (pH 7.4). | ||
Furthermore, in the preliminary phase of another study, we also investigated the in vitro antitumor activity of the Ir(III) complex and two of the ligands towards A549, A2780 and A2780cis cells in comparison with cis-platin as reference. From the results, the studied complex shows considerably higher activity than free ligands and cis-platin (Table S7 and Fig. S9 of the ESI†). This excellent cytotoxicity of the Ir(III) complex seems to relate to its ability of ROS production (Fig. S10 of the ESI†).
3.4. Intrinsic reactivity by DFT
The principal intrinsic reactivities characteristic for the antioxidant properties of a molecule such as the H-atom donor (lowest BDEs only), the proton donor (lowest PAs only), the electron donor (IP) and electron acceptor (EA) capacities of all compounds are presented in Table 2. Calculations were performed in the gas phase and in water and PEA solvent (IEF-PCM) at the B3LYP/6-31G(d) and the B3LYP/Lanl2dz//6-31G(d) levels of theory for ligands and Ir(III) complex, respectively.| Properties (kcal mol−1) | Bond position | 2-Phenylpyridinea | Flavonola | Ibuprofena | Ir(III) complexb | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gas | Water | PEA | Gas | Water | PEA | Gas | Water | PEA | Gas | Water | PEA | ||
| a Calculated at the B3LYP/6-31G(d) level of theory.b Calculated at the B3LYP/Lanl2dz//6-31G(d) level of theory. | |||||||||||||
| BDE | C8–O–H | 69.2 | 69.6 | 69.4 | — | — | |||||||
| C13–H | 105.9 | 106.4 | — | — | |||||||||
| C17–H | 111.0 | — | — | ||||||||||
| C31–H | 92.2 | 97.0 | — | ||||||||||
| C32–H | 87.1 | 86.9 | 87.2 | 86.9 | |||||||||
| C35–H | 78.9 | 79.4 | 78.6 | 78.7 | 79.3 | 78.3 | |||||||
| C62–H | 108.8 | — | — | ||||||||||
| C65–H | 104.7 | 104.9 | 102.0 | — | — | ||||||||
| C67–H | 108.8 | — | — | ||||||||||
| PA (ETE) | C8–O–H | 335.1 | — | ||||||||||
| C13–H | 335.1 | 105.2 | 276.7 | 401.7 | 110.6 | ||||||||
| C31–H | 348.9 | 125.8 | 406.1 | 126.1 | |||||||||
| C35–H | 360.8 | 73.5 | 243.4 | 355.5 (37.9) | 72.1 (78.4) | 242.3 | |||||||
| C62–H | 398.7 | 107.1 | 283.8 | 384.8 | 99.5 | ||||||||
| C63–H | 401.7 | 108.2 | 388.3 | 102.4 | |||||||||
| C64–H | 402.7 | 108.3 | 386.7 | 101.2 | |||||||||
| IP (PDE) | 179.5 | 156.2 | 151.5 | 170.1 | 149.1 | 142.3 | 183.4 | 160.2 | 153.8 | 136.4 (291.5) | 130.5 (35.2) | 119.8 | |
| EA | −5.9 | 54.1 | 24.9 | 12.7 | 68.3 | 41.7 | −22.5 | 35.8 | 8.4 | 26.8 | 79.3 | 52.1 | |
As seen in Table 2, in the gas phase, the lowest BDE value found for free ligands is at the C8–O–H position on flavonol (69.2 kcal mol−1). Ibuprofen shows smallest BDE value for the C–H bond located between the carboxylic groups and the phenyl ring, i.e. C35–H (78.9 kcal mol−1), followed by the para C–H position next to the phenyl rings, i.e. C32–H (87.1 kcal mol−1). The H-donating capacity of these positions results from the electron-withdrawing effect of the carboxylic groups and the conjugated π-system of the phenyl ring. In contrast, almost all of the H atoms of 2-phenylpyridine and flavonol (except for C8–O–H) are directly linked to the C atoms of the conjugated π-system and show much higher BDEs; the lowest values stand at around 105–110 kcal mol−1. For the Ir(III) complex, the lowest BDE values are also found on the same C–H positions of the easy-to-break ibuprofen ligand. We observe nearly identical BDEs for C35–H and C32–H of the complex as well as of the ibuprofen ligand (78.7 and 87.2 kcal mol−1 in the gas phase for C35 and C32 positions, respectively). The H-atom donating capacity decreases as follows: flavonol > Ir(III) complex ≈ ibuprofen > 2-phenylpyridine. Both the Ir(III) complex and two ligands demonstrate strong antioxidant capacity through the H-donating mechanism which is higher than that of phenol (BDE of O–H: 88.3 ± 0.8 kcal mol−1).52
There is almost no effect on the BDE values of all compounds in water and in PEA. For example, we observed for C35–H of ibuprofen BDE values of 79.4 kcal mol−1 and 78.6 kcal mol−1, and for C8–O–H of flavonol BDE values of 69.6 and 69.4 kcal mol−1 in water and in PEA, respectively. Similarly, a BDE value of 79.3 kcal mol−1 in water vs. 78.3 kcal mol−1 in PEA was found for the Ir(III) complex.
In the gas phase, flavonol exhibits the best proton-donating capacity among the three ligands; the lowest PA values were found equal at 335.1 kcal mol−1 for C13–H and C8–O–H. Ibuprofen shows the lowest PA values for the C31–H and C35–H positions, at 348.9 and 360.8 kcal mol−1, respectively. 2-Phenylpyridine displays lowest PA values for C62–H, C63–H, and C64–H, at an average of about 50 kcal mol−1 higher than that of the other ligands. For the Ir(III) complex, the lowest PA is found at C35–H, being 355.5 kcal mol−1. The proton-donating capacity in the gas phase decreases as follows: flavonol > ibuprofen > Ir(III) complex > 2-phenylpyridine.
Interestingly, in water, we observed much lower PA values for all compounds than in the gas phase, by a factor of from 3 to 5 times. For example, in water, the lowest PA found for C35–H of the Ir(III) complex (72.1 kcal mol−1) and for the same bond of ibuprofen (73.5 kcal mol−1) are all about five times lower than in the gas phase. In the non-polar solvent (PEA), the PA value of the Ir(III) complex rises to 242.3 kcal mol−1 for the same bond (C35–H).
This result indicates that the solvent, especially a polar solvent, has an important impact on promoting solvation of molecules and the deprotonation process. In water, the proton-donating capacity decreases in the order: Ir(III) complex ≈ ibuprofen > flavonol ≈ 2-phenylpyridine. Proton loss occurs even more conveniently than the hydrogen abstraction mechanism in polar solvent (lowest PA values < lowest BDE values). Similar phenomena of decreasing of PA values in polar solvents have been previously reported.52 However, if we take into account the ETE value (78.4 kcal mol−1) of the second step of the SPLET mechanism, i.e. electron transfer from the radical anion resulting from deprotonation at the C35–H position of the Ir(III) complex, the total energy for transferring the whole package of proton and electron in water is considerably higher than the BDE value, 150.5 kcal mol−1 compared to 79.3 kcal mol−1. Thus, for the Ir(III) complex the SPLET mechanism is less favorable than the FHT one in terms of the intrinsic properties.
One may notice that the IP values for all ligands in the gas phase are quite similar (about 170–180 kcal mol−1) and considerably higher than the IP of the Ir(III) complex (136.4 kcal mol−1). In comparison with the IP value of the standard phenol (about 185 kcal mol−1),53 the result indicates that the Ir(III) complex can act as a good electron donor molecule and better than all the ligands. In water, these values were reduced to 130.5 kcal mol−1 for the Ir(III) complex, 149.1 kcal mol−1 for flavonol and 160.2 kcal mol−1 for ibuprofen. In PEA, the IP value of the Ir(III) complex even decreases to 119.8 kcal mol−1. The electron donor capacity of molecules was clearly favored in solvent compared to the gas phase, especially in non-polar solvent, and it decreased in the following order: Ir(III) complex > flavonol > 2-phenylpyridine > ibuprofen.
In the gas phase, the electron acceptor capacity of all compounds could be classified as being in the order Ir(III) complex (26.8 kcal mol−1) > flavonol (12.7 kcal mol−1) > 2-phenylpyridine (−5.9 kcal mol−1) > ibuprofen (−22.5 kcal mol−1). This order, though unchanged in water, but with higher values than in the gas phase, e.g. 79.3 vs. 26.8 kcal mol−1 for the Ir(III) complex in water vs. in the gas phase, indicates that the electron-accepting mechanism is more favored in polar solvent than in vacuo. The Ir(III) complex is evidently the best electron donor and electron acceptor compared to its ligands.
Furthermore, the PDE value characterizing the second step of the SETPT mechanism ((R5), eqn (5) in the ESI†) for the radical cation of the Ir(III) complex was also calculated. The obtained PDEs in the gas phase and in water are 291.5 and 35.2 kcal mol−1, respectively. Thus, it is observed that the SETPT mechanism is less favored than the FHT one in terms of the intrinsic properties.
Overall, we note that the Ir(III) complex is a good antioxidant and its capacity of giving a hydrogen atom or donating or accepting one electron are all better than those of the ligands. The results in the next part obtained by RSA confirm the superior antioxidant properties of Ir(III) complex compared to its ligands.
3.5. Thermochemical properties and kinetics of the reaction by DFT
The consideration of the influence of reactive media as well as chemical nature of the free radicals provides a more complete picture of the scavenging reaction. For that reason, we evaluate in this section the thermochemical properties (enthalpy and Gibbs free energy) of reactions of the Ir(III) complex and free ligands with HO˙, HOO˙, NO˙, DPPH˙ and ABTS˙+ radicals in the gas phase, in water and in PEA. The HO˙ and HOO˙ radicals are the most common radicals found in living bodies, whereas NO˙, DPPH˙ and ABTS˙+ are the ones used in the experimental antioxidant essays. Five antioxidant mechanisms including SET, PL, FHT (HAT/PCET) and RAF are evaluated in detail.The thermochemical property results for the complex are all presented in Table 3. The data obtained for the ligands can be found in Tables S8 and S9 of the ESI.† As can be seen in Table 3, the RAF reaction with DPPH˙ radical was not performed because of the steric effect of the radical, whereas only the SET mechanism was taken into account for the ABTS˙+ radical due to the electron transfer process of the antioxidant assays.
| Radical | Gas | Water | PEA | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HO˙ | HOO˙ | NO˙ | DPPH˙ | ABTS˙+ | HO˙ | HOO˙ | NO˙ | DPPH˙ | ABTS˙+ | HO˙ | HOO˙ | NO˙ | DPPH˙ | ABTS˙+ | |
| a The PL reaction is considered at C35 as: AntioxH + R˙ → (Antiox)− + RH˙+.b The FHT reaction is considered at C35 as: AntioxH + R˙ → (Antiox)˙ + RH.c The RAF reaction is considered at C13 as: Antiox + R˙ → (Antiox-R)˙. | |||||||||||||||
| ΔH0 (kcal mol−1) | |||||||||||||||
| SET-ed | 137.0 | 142.4 | 183.9 | 71.2 | −11.3 | 33.7 | 46.9 | 89.7 | 14.0 | 1.5 | 53.8 | 65.5 | 107.8 | 28.1 | −1.8 |
| SET-ea | 341.6 | 247.8 | 199.5 | 128.2 | 204.2 | 223.1 | 137.8 | 86.9 | 55.0 | 70.1 | 248.7 | 161.7 | 110.9 | 71.8 | 100.0 |
| PL(C35)a | 213.0 | 199.3 | 245.1 | 129.3 | — | 95.0 | 90.7 | 130.2 | 51.7 | — | 120.3 | 114.1 | 155.4 | 69.9 | — |
| FHT(C35)b | −30.6 | 1.2 | 75.0 | 5.2 | — | −31.7 | 5.6 | 71.8 | 5.8 | — | −31.7 | 0.8 | 72.2 | 5.7 | — |
| RAF(C13)c | −21.6 | 6.6 | — | — | — | −20.4 | 7.9 | — | — | — | −20.5 | 7.5 | — | — | — |
|
|||||||||||||||
| ΔG0 (kcal mol−1) | |||||||||||||||
| SET-ed | 137.0 | 142.2 | 183.7 | 70.6 | −12.1 | 33.9 | 46.9 | 89.8 | 13.9 | 0.2 | 53.8 | 65.2 | 107.7 | 27.5 | −2.6 |
| SET-ea | 341.1 | 247.5 | 199.2 | 127.7 | 203.9 | 221.8 | 136.7 | 85.8 | 54.0 | 69.7 | 248.2 | 161.4 | 111.2 | 71.3 | 99.7 |
| PL(C35)a | 213.1 | 200.7 | 245.0 | 130.4 | — | 94.9 | 91.9 | 130.0 | 52.8 | — | 120.4 | 115.6 | 155.3 | 71.0 | — |
| FHT(C35)b | −31.4 | 1.3 | 73.9 | 4.9 | — | −32.7 | 5.3 | 70.4 | 5.1 | — | −32.4 | 0.9 | 71.1 | 5.3 | — |
| RAF(C13)c | −11.2 | 18.5 | — | — | — | −9.9 | 20.2 | — | — | — | −10.1 | 19.4 | — | — | — |
In addition, the PEPs of FHT (HAT or PCET) and RAF of the studied compounds with HO˙ and HOO˙ radicals are also established in order to provide more insight into the free radical scavenging mechanism.
| Anti-ox + (R˙) → (Anti-ox˙+) + (R−) | (R1) |
| ΔH0 = H(Anti-ox˙+) + H(R−) − H(Anti-ox) − H(R˙) | (1) |
| ΔG0 = G(Anti-ox˙+) + G(R−) − G(Anti-ox) − G(R˙) | (2) |
| Anti-ox + (R˙) → (Anti-ox˙−) + (R+) | (R2) |
| ΔH0 = H(Anti-ox˙−) + H(R+) − H(Anti-ox) − H(R˙) | (3) |
| ΔG0 = G(Anti-ox˙−) + G(R+) − G(Anti-ox) − G(R˙) | (4) |
The reaction enthalpies (ΔH0) and Gibbs free energies (ΔG0) of reactions (R1) and (R2) for the Ir(III) complex are calculated in the gas phase and in solvents at 298.15 K as indicated by eqn (1)–(4). Values are presented in Table 3. The corresponding results for the ligands are shown in Tables S8 and S9 of the ESI.†
It is clearly observed that for each of the investigated radicals, the electron-donating and electron-accepting capacities are all in the same order as Ir(III) complex > flavonol > ibuprofen, indicating the stronger radical scavenging capacity of the Ir(III) complex compared to the ligands. For example, in the gas phase, the Ir(III) complex quenches ABTS˙+ by −11.3 kcal mol−1, while flavonol and ibuprofen scavenge the same radical by 21.6 and 35.7 kcal mol−1. In water, to scavenge ABTS˙+, 1.5, 18.7 and 31.2 kcal mol−1 are required for the Ir(III) complex, flavonol and ibuprofen, respectively. In addition, through the SET mechanism, we evaluate the scavenging activity of the Ir(III) complex towards different free radicals both in the gas phase and in solvents (water and PEA). From the results, the antioxidant capacity of the Ir(III) complex towards radicals in water is better than that in PEA and in the gas phase, except for ABTS˙+. The scavenging capacities of the Ir(III) complex towards different radicals are in the order ABTS˙+ > DPPH˙ > HO˙ > HOO˙ > NO˙, with the ΔH0 value for ABTS˙+ and NO˙ varying from 1.5 to 89.7 kcal mol−1, from −1.8 to 107.8 kcal mol−1 and from −11.3 to 183.9 kcal mol−1 in water, in PEA and in the gas phase, respectively. This order is somehow shared with the result reported in the experimental part as DPPH˙ ≈ ABTS˙+ > HO˙ > NO˙, despite different solvents used in the experiment. For the electron-accepting reaction, all the SET-ea reactions of the Ir(III) complex with the studied radicals are endergonic and unfavorable in water with both ΔH0 and ΔG0 values being positive (Table 3). The scavenging activity of free radicals by the Ir(III) complex is in the same order both in the gas phase and in solvents: DPPH˙ > ABTS˙+ ≈ NO˙ > HOO˙ > HO˙.
Two typical ROS, the hydroperoxyl radical HOO˙ and hydroxyl radical HO˙, for which ΔH0 and ΔG0 were found to be the lowest, were subjects of an in-depth study of the free radical scavenging activity of the Ir(III) complex through the formal H-abstraction. Reactions could be described as (R3) and (R4) for HOO˙ and HO˙, respectively:
| RH + HOO˙ → R˙ + HOOH | (R3) |
| RH + HO˙ → R˙ + HOH | (R4) |
Based on the BDE values, one can determine the weakest C–H bonds of the Ir(III) complex at C32 and C35 positions; these latter correspond to the most favorable sites for the FHT reaction. The free radical scavenging reactions of HOO˙ and HO˙ radicals therefore were studied at these two positions of the Ir(III) complex. Similar reactions were performed with ibuprofen for comparison and results are reported in Table S8 of the ESI.† C35 happened to be the most reactive position for both complex and ligand with lower energies compared to C32 (about 8 kcal mol−1, Table S10 of the ESI†).
A similar trend for the scavenging activity of the complex and ligand was observed with a very small difference in term of energy. For example, the HOO˙ scavenging by the Ir(III) complex occurring at C35 is endergonic and non-spontaneous with positive Gibbs free energies (ΔG0) being 1.3 kcal mol−1 in the gas phase and 5.3 kcal mol−1 in water (Table 3), while the ones of the ligand are 1.2 kcal mol−1 (gas phase) and 5.3 kcal mol−1 (water) (Table S8†). Contrarily, the HO˙ scavenging through hydrogen transfer mechanism by the Ir(III) complex is exergonic and spontaneous, with negative ΔG0 of −31.4 kcal mol−1 in the gas phase and −32.7 kcal mol−1 in water for the same position (Table 3). Values of ΔG0 of the reaction with corresponding positions on ibuprofen ligand were −31.4 and −32.8 kcal mol−1 in the gas phase and in water, respectively (Table S8†).
PEPs of the reaction between the Ir(III) complex and the ligand ibuprofen were also established in order to (i) provide more insight into the chemical kinetics of hydrogen transfer process and (ii) distinguish the nature of FHT reaction: HAT or PCET. For these purposes, geometry optimizations of TSs were firstly performed in the gas phase at the B3LYP/Lanl2dz//6-31G(d) level of theory. Imaginary frequencies and relative vibrations along the reaction pathway are carefully verified following by intrinsic reaction coordination (IRC) calculation. The imaginary frequencies of all studied reactions are summarized in Table S11 in the ESI.† Reactant intermediates (INT-1) and product intermediates (INT-2) at the lowest energies obtained from IRC for both directions are structurally optimized and enthalpy values are calculated for each molecule, all at the same level of theory.
The obtained results allow one to establish the PEPs of the reaction with HOO˙ and HO˙ radicals (Fig. 5). Analysis of optimized structures and frontier orbitals of TSs which are responsible for the hydrogen transfer are presented in Fig. 6.
 | ||
| Fig. 5 Potential energy profiles of FHT reaction of Ir(III) complex and ibuprofen with HOO˙ (A) and HO˙ (B) radicals calculated in the gas phase at the B3LYP/Lanl2dz/6-31G(d) level of theory for C32 and C35 positions. | ||
 | ||
| Fig. 6 Optimized transition structures (left) and frontier orbitals (right) responsible for the HAT reaction between HOO˙/HO˙ and Ir(III) complex occurring at C32–H and C35–H positions (isovalue = 0.02 for the contour). | ||
As has been described previously by many authors for the FHT mechanism, the initial step consists of the approach of the oxygen atom of the HOO˙ radical to the H atom of the weak C–H bond to form a reactant intermediate complex (INT-1). In the following step, the mitigating H-atom moves away from the carbon atom of Ir(III) complex and tends to form a covalent bond with the oxygen of HOO˙ radical, consequently forming the TS. As seen in Fig. 6, at TSs, the O–H distances are somewhat shorter than the C–H distances (1.23 Å vs. 1.33 Å for C32 position and 1.25 Å vs. 1.31 Å for C35 position). In the next step, another intermediate (INT-2) forms between H2O2 and the radicalized Ir(III) complex. Finally, donated H-atom leaves the complex to form H2O2 and the corresponding radical products. In terms of energy, the reaction barriers (Ea) rise up to 15.0 and 10.3 kcal mol−1, respectively, while final products were stabilized at 9.7 and 1.2 kcal mol−1 above the reactants for C32 and C35, respectively (Fig. 5A). We observed very similar results for ibuprofen ligand, with Ea found at 15.1 and 13.6 kcal mol−1 while the final products are stabilized at 9.6 and 1.4 kcal mol−1 for C32 and C35, respectively. Shorter O–H distances compared to C–H distances at TSs were also found for the reaction of the ligand, i.e. 1.21 vs. 1.34 Å for C32 position and 1.23 Å vs. 1.32 Å for C35 position (Fig. S11 of the ESI†). C35 is confirmed as the more thermodynamically favored position, consistent with the fact that –C35 is a tertiary C-center radical, which is more stable than a secondary radical such as –C32.54 It is also found that the FHT reaction of the Ir(III) complex at the C35 position presents a lower Ea value, i.e. 3.3 kcal mol−1, than the one of ibuprofen ligand. The lower barrier of the Ir(III) complex may explain the higher RSA of the complex as experimentally observed (Fig. 3).
The FHT reaction with HO˙ radical was studied for the same positions, i.e. C32 and C35, on the Ir(III) complex and also compared with the corresponding positions on ibuprofen ligand. PEP results are shown in Fig. 5B. It is observed that the HO˙ scavenging through the FHT mechanism occurs as a barrier-less reaction with Ea lying from −6.0 to −3.2 kcal mol−1 lower than the reactants. The H-abstraction is exothermic and spontaneous for all positions with final products as the sum of Ir(III) complex radical and H2O of −30.6 to −22.1 kcal mol−1 lower than the reactants. At C32 position, the scavenging process by the Ir(III) complex and by ibuprofen takes place almost in the same manner in terms of energy. For example, the reactant intermediates (INT-1) were found at −1.5 kcal mol−1 for the Ir(III) complex and −2.0 kcal mol−1 for ibuprofen while the TSs at −3.2 and −3.6 kcal mol−1 and the products at −22.1 and −22.2 kcal mol−1 were determined for complex and ligand, respectively. At C35 position, bigger gaps of energy between the Ir(III) complex and ibuprofen were observed. Indeed, the reactant intermediates (INT-1) were found at −7.7 and −5.6 kcal mol−1, the TSs at −6.0 and −5.1 kcal mol−1, and the product intermediate (INT-2) at −37.5 and −33.2 kcal mol−1 for Ir(III) complex and ibuprofen, respectively. The C35 position on the Ir(III) complex is also the most thermodynamically favored position for the FHT reaction with HO˙, which is in agreement with the HOO˙ scavenging result.
Regarding the H-abstraction steps, hydrogen bonding intermediates formed between the oxygen atom of HO˙ radical and the H-mitigating atom, similarly as for the HOO˙ scavenging. At TSs, elongated C–H bonds were observed, while the H–O distances are shortened, e.g. 1.18 Å vs. 1.43 Å for C–H vs. O–H of C32 and 1.21 Å vs. 1.36 Å for C–H vs. O–H of C35 position (Fig. 6). Similar results were obtained for corresponding positions on ibuprofen ligand (Fig. S8 of the ESI†). Finally, the Ea value of the reaction between the Ir(III) complex and HO˙ radical at the C35 position is also lower than that of ibuprofen ligand, i.e. −6.0 and −5.1 kcal mol−1, respectively. This relative tendency is identical to the results for HOO˙ radical scavenging and probably corresponds to higher activity of complex compared to ligands as observed in the experimental part.
Concerning the H mitigation, it is essential to understand the precise mechanism as HAT or PCET. Although reactants and products of the two mechanisms are identical, the electron and the proton are transferred differently in two distinct ways. In the HAT process, the electron and the proton are transferred together as a single entity (H˙) in a single step, while in the PCET process, the proton (H+) and the electron (e−) are separately transferred as two subatomic entities. In order to distinguish the HAT/PCET mechanism, the frontier orbitals as well as the NBO of TSs are carefully analyzed. NPA charges, Hirshfeld ASD, NAO occupancy and NEC analyses provide more information for the H transfer mechanism.
Indeed, when analyzing the frontier orbitals of TSs, the SOMOs, as expected, are found to be located along the H transfer pathway (donor–H–acceptor) in the case of ibuprofen ligand, for both C32 and C35 positions (Fig. S11 of ESI†). However, for the Ir(III) complex, all SOMOs are found to be always on 2-phenylpyridine moiety, similarly to the HOMO of the original complex, but not on ibuprofen and certainly not at the reactive positions. This unexpected result drives us to examine other frontier orbitals of the complex to search for the H-transfer-responsible orbital. Consequently, for HOO˙ scavenging, the SOMO-3 orbitals are found to be located on the ibuprofen moiety as well as the H-transition vector while the SOMO-5 orbitals are responsible in the case of HO˙ scavenging (Fig. 6). As can be seen on the SOMO-3 orbitals, 2p orbitals of H-donor (C atom on the complex) and H-acceptor (O atom of HOO˙/HO˙ radical) are similarly distributed along the H-transferring vector with a node plan located at the transferred H atom. This observation corresponds to H-atom transfer process from the ibuprofen moiety to HOO˙/HO˙ free radical via an interaction of the SOMO-3 or SOMO-5 orbitals.55,56
NBO analysis (Table 4) shows that at TSs of the HAT reaction with HOO˙, the electron densities are transferred from the third lone pair of the reactive oxygen on HOO˙, LP(3) O81, to the first unoccupied anti-bonding orbitals of the donated H atom, i.e. LP*(1) H45 and LP*(1) H47, with stabilization energies corresponding to 87.1 and 119.7 kcal mol−1. Another interaction was found between the σ-bonding orbitals of the newly formed bond H45–O81, σ(1) H45–O81, and the first unoccupied anti-bonding orbitals of C32, LP*(1) C32, with stabilization energy of 53.6 kcal mol−1. The unoccupied anti-bonding orbital of the newly formed bond H45–O81, σ*(1) H45–O81, also interacts with the first lone pair anti-bonding orbital of C32, LP*(1) C32, with stabilization energy of 47.9 kcal mol−1. In the case of C35, the third lone pair of O81, LP(3) O81, is transferred to the first unoccupied anti-bonding orbital of the C35–H47 bond, σ*(1) C35–H47, with a stabilization energy equal to 63.2 kcal mol−1 while the second lone pair of O81, LP(2) O81, is transferred to the first lone pair unoccupied of H atom, LP*(1) H47, with a stabilization energy of 26.2 kcal mol−1. The orbital interactions are in agreement with previous reports on the HAT reaction of HOO˙.56
| Reaction | Bond | Donor NBO (i) | Acceptor NBO (j) | E2 (Ei − Ej) (kcal mol−1) |
|---|---|---|---|---|
| HAT-HOO˙ | C32⋯H45⋯O81 | LP(3) O81 | LP*(1) H45 | 87.1 |
| LP*(1) C32 | σ*(1) H45–O81 | 47.9 | ||
| σ(1) H45–O81 | LP*(1) C32 | 53.6 | ||
| C35⋯H47⋯O81 | LP(3) O81 | LP*(1) H47 | 119.7 | |
| LP(2) O81 | LP*(1) H47 | 26.2 | ||
| LP(3) O81 | σ*(1) C35–H47 | 63.2 | ||
| HAT-HO˙ | C32⋯H45⋯O81 | LP(3) O81 | σ*(1) C32–H45 | 31.5 |
| LP(2) O81 | LP*(1) H45 | 14.1 | ||
| LP*(3) O81 | LP*(1) H45 | 66.5 | ||
| C35⋯H47⋯O81 | LP(3) O81 | σ*(1) C35–H47 | 40.8 | |
| LP(2) O81 | LP*(1) H47 | 18.4 | ||
| LP*(3) O81 | LP*(1) H47 | 80.3 | ||
| RAF-HOO˙ | C12⋯O83 | LP(3) O83 | LP*(1) C12 | 64.5 |
| LP(1) O83 | LP*(1) C12 | 6.8 | ||
| LP(2) O81 | σ*(1) C12–O83 | 9.7 | ||
| C13⋯O83 | LP(3) O83 | LP*(1) C13 | 63.6 | |
| LP(1) O83 | LP*(1) C13 | 6.9 | ||
| LP(2) O81 | σ*(1) C13–O83 | 9.7 | ||
| RAF-HO˙ | C12⋯O81 | LP*(3) O81 | LP(1) C12 | 319.4 |
| C13⋯O81 | σ(2) C13–C17 | LP*(4) O81 | 29.6 | |
| LP(1) O81 | LP*(1) H82 | 15.0 | ||
| LP(3) O81 | LP*(1) H82 | 362.4 |
For the HAT reaction with HO˙, very similar donor–acceptor orbital couples were observed for the C32 and C35 positions. For example, the third lone pair of the reactive oxygen atom, LP(3) O81, is transferred to the σ anti-bonding orbital of the C–H bond, σ*(1) C32–H45 or σ*(1) C35–H47, with 31.5 or 40.8 kcal mol; the second lone pair of the reactive oxygen atom, LP(2) O81, is transferred to the first lone pair unoccupied anti-bonding orbital of mitigating hydrogen atom, LP*(1) H45 or LP*(1) H47, with 14.1 or 18.4 kcal mol; the third unoccupied lone pair of reactive oxygen atom, LP*(3) O81, transfers to the first unoccupied lone pair of mitigating hydrogen atom, LP*(1) H45 or LP*(1) H47, with 66.5 or 80.3 kcal mol−1.
As can be seen in Table 5, the NPA charges of the mitigating H are found as positive at about 0.31–0.4 e−, similar to that reported in the literature for the HAT mechanism (0.3 e−).57 Very small Hirshfeld ASDs varying from 0.0171 to 0.0297 were found for the mitigating H, while significantly higher values were obtained on the donor and acceptor atoms (0.176–0.306 for C atoms and 0.351–0.636 for O atoms). The NPA charges and Hirshfeld ASD values of carbon and oxygen atoms involved in the reaction are also presented in Table 5 for comparison. In addition, the NEC also shows that the mitigating H is characterized by 1s0.60−0.69 orbital configurations which seem similar to 1s1 orbital. The mitigating H atom therefore has a hydrogen-atom-like character rather than being a proton-like species. Overall, the NBO analysis is in agreement with the frontier orbital analysis and strongly supports the H-transfer mechanism as HAT process.
| Radical | TS | Atoms | NPA charge | Hirshfeld ASD | 1s occupancy | NEC |
|---|---|---|---|---|---|---|
| HOO˙ | Ir–C32 | H | 0.38441 | 0.026972 | 0.61255 | 1s0.61 |
| C | −0.36909 | 0.279536 | ||||
| O | −0.35820 | 0.359035 | ||||
| Ir–C35 | H | 0.39686 | 0.020084 | 0.59962 | 1s0.60 | |
| C | −0.27871 | 0.256208 | ||||
| O | −0.32228 | 0.381153 | ||||
| Ibu-C32 | H | 0.39130 | 0.027329 | 0.60584 | 1s0.61 | |
| C | −0.35440 | 0.306068 | ||||
| O | −0.35308 | 0.351018 | ||||
| Ibu-C35 | H | 0.39688 | 0.017137 | 0.59982 | 1s0.60 | |
| C | −0.28856 | 0.257628 | ||||
| O | −0.33374 | 0.372410 | ||||
| HO˙ | Ir–C32 | H | 0.30869 | 0.029091 | 0.68880 | 1s0.69 |
| C | −0.24770 | 0.208034 | ||||
| O | −0.64785 | 0.635898 | ||||
| Ir–C35 | H | 0.34248 | 0.021048 | 0.65441 | 1s0.65 | |
| C | −0.27487 | 0.176278 | ||||
| O | −0.67564 | 0.482783 | ||||
| Ibu-C32 | H | 0.31026 | 0.029696 | 0.68733 | 1s0.69 | |
| C | −0.39111 | 0.205974 | ||||
| O | −0.64913 | 0.626864 | ||||
| Ibu-C35 | H | 0.33735 | 0.027386 | 0.65967 | 1s0.66 | |
| C | −0.27383 | 0.235012 | ||||
| O | −0.65995 | 0.607433 |
| R + HOO˙ → (ROOH)˙ | (R5) |
| R + HO˙ → (ROH)˙ | (R6) |
Results from semi-empirical calculations show that four positions, i.e. C12 and C13 on flavonol ligand and C38 and C40 on ibuprofen ligand, seem to be the most probable sites for the RAF reaction with HOO˙ (Table S12 of the ESI†). Based on the reaction enthalpy and Gibbs free energy calculated at higher level of DFT calculation for all positions of the Ir(III) complex with HOO˙ (Table S13 of the ESI†), we selected to illustrate the RAF reaction on C12 and C13 (Table S14 of ESI†). The RAF reaction with HO˙ radicals was also performed on the same positions for Ir(III) complex and flavonol ligand.
As can be seen in Table 3, the RAF seems to be the second-most-favored scavenging mechanism for all studied radicals with very low reaction enthalpy (ΔH0) and Gibbs free energy (ΔG0). The ΔH0 value of the RAF towards HO˙ radical in water is equal to −21.6 kcal mol−1, only about 10 kcal mol−1 higher than ΔH0 of the HAT reaction but much lower than ΔH0 of SET (137.0 kcal mol−1) and PL (213.0 kcal mol−1) reactions. The same observations can be found for the HOO˙ radical. In addition, almost identical enthalpies were found for the same radical both in the gas phase and in solvent, e.g. for HO˙ we found an enthalpy of −21.6 in the gas phase, −20.4 in water and −20.5 kcal mol−1 in PEA. The addition process for HOO˙ is endothermic and non-spontaneous while the same process for HO˙ is exothermic and spontaneous for both Ir(III) complex and flavonol; the trend is very similar to that for the HAT mechanism. We can however observe that the reaction seems to be favored at C13 rather than C12 with lower enthalpy and Gibbs free energy (of about 2–10 kcal mol−1, Table S14 of the ESI†). In particular, the flavonol ligand seems to be better than the Ir(III) complex in scavenging HOO˙ and HO˙ radicals with the Gibbs free energy almost equal to or lower than that of the Ir(III) complex, e.g. 14.3 vs. 18.5 for HOO˙ and −21.4 vs. −11.2 for HO˙ radicals at the C13 position (Table S14 of the ESI†).
The PEPs of the RAF reaction with HOO˙ and with HO˙ radicals are displayed in Fig. 7. As can be seen in Fig. 7, the HOO˙ radical adduct formation is an endothermic and non-spontaneous process. The reactant intermediate complexes (INT) formed with an energy lying several kcal mol−1 below the separated reactants, e.g. −1.4 kcal mol−1 to −5.5 kcal mol−1 in the case of Ir–C13 to Flav-C12. Reaction barriers (Ea) of about 14 kcal mol−1 were observed in most cases, except for Flav-C13, which is characterized by a much lower TS energy (6.6 kcal mol−1). The radical products obtained by forming a chemical bond between the reactive O and C sites have stabilized energies of 9.5, 8.7, 6.6 and 2.0 kcal mol−1 for Flav-C12, Ir–C12, Ir–C13 and Flav-C13, respectively. The C13 position is more favored than C12 in the HOO˙ adduct formation for both Ir(III) complex and flavonol ligand.
 | ||
| Fig. 7 Potential energy profiles of RAF reaction between Ir(III) complex and HOO˙ (A) and OH˙ (B) radicals calculated in the gas phase at B3LYP/Lanl2dz//6-31G(d) level of theory for C12 and C13 positions and for the corresponding positions on flavonol ligand. | ||
Regarding the HO˙ adduct formation at the same C12 and C13 positions on the Ir(III) complex and the corresponding positions on flavonol, the reaction is exothermic and exergonic in all cases. We observed a similar behavior for Flav-C12 and Flav-C13, while almost identical energies were observed for Ir–C12 and Ir–C13. The reaction is barrier-less with energies of all TS (Ea) complexes lying at −2.2, −3.2, −11.1 and −12.5 kcal mol−1 for Ir–C13, Ir–C12, Flav-C12 and Flav-C13, respectively. Final radical products were stabilized at −21.6, −22.2, −30.2 and −32.3 kcal mol−1 below the reactants for Ir–C13, Ir–C12, Flav-C12 and Flav-C13, respectively. Again in HO˙ scavenging through the RAF mechanism, the C13 position is more favored than the C12 position, for both Ir(III) complex and ligand. It is seen that for the RAF mechanism, the flavonol ligand shows better RSA than the Ir(III) complex, towards both HO˙ and HOO˙, which is not surprising considering the very good antioxidant properties known for flavonol.
Optimized structures and frontier orbital analysis of TSs of the Ir(III) complex are presented in Fig. 8. At TSs, the reactive oxygen atoms is 1.85 Å distant from the reactive carbon atom in the case of HOO˙ radical, while this distance is about 2.20–2.23 Å in the case of HO˙ radical. Similar to the HAT reaction, frontier orbital analysis revealed that SOMO-3 orbitals are responsible for the HOO˙ scavenging while the SOMO-5 orbitals are responsible in case of HO˙ scavenging, in which we can clearly observe an overlap between the 2p orbitals on O atoms with the π orbitals of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds (Fig. 8). NBO analysis (Table 4) revealed that for the HOO˙ scavenging reaction by the Ir(III) complex, the third lone pair of the reactive oxygen atom (O83), LP(3) O83, donates electrons to the unoccupied anti-bonding orbital of the reactive carbon atoms (C12 or C13), LP*(1) C12 or LP*(1) C13, with stabilization energies of 64.5 or 63.6 kcal mol−1. Meanwhile, the interaction between the first lone pair of the reactive oxygen atom (O83), LP(1) O83, and the unoccupied anti-bonding orbital of the reactive carbon atoms (C12 or C13), LP*(1) C12 or LP*(1) C13, is determined as only 6.8 or 6.9 kcal mol−1. This result is in good agreement with the SOMO analysis which demonstrated that SOMO-3 are the responsible orbitals for the RAF reaction with HOO˙ radical. Results obtained for HO˙ scavenging through the RAF mechanism show that the reaction at the C12 position involved the third unoccupied lone pair of the oxygen atom, LP* (3) O81, and the first lone pair of the carbon atom, LP(1) C12, with stabilization energy of 319.4 kcal mol−1. For the C13 position, the σ-bonding orbitals of the C13–C17 bonds, σ(2) C13–C17, transfer to the fourth lone pair anti-bonding orbital of O81, LP*(4) O81, with stabilization energy of 29.6 kcal mol−1.
C double bonds (Fig. 8). NBO analysis (Table 4) revealed that for the HOO˙ scavenging reaction by the Ir(III) complex, the third lone pair of the reactive oxygen atom (O83), LP(3) O83, donates electrons to the unoccupied anti-bonding orbital of the reactive carbon atoms (C12 or C13), LP*(1) C12 or LP*(1) C13, with stabilization energies of 64.5 or 63.6 kcal mol−1. Meanwhile, the interaction between the first lone pair of the reactive oxygen atom (O83), LP(1) O83, and the unoccupied anti-bonding orbital of the reactive carbon atoms (C12 or C13), LP*(1) C12 or LP*(1) C13, is determined as only 6.8 or 6.9 kcal mol−1. This result is in good agreement with the SOMO analysis which demonstrated that SOMO-3 are the responsible orbitals for the RAF reaction with HOO˙ radical. Results obtained for HO˙ scavenging through the RAF mechanism show that the reaction at the C12 position involved the third unoccupied lone pair of the oxygen atom, LP* (3) O81, and the first lone pair of the carbon atom, LP(1) C12, with stabilization energy of 319.4 kcal mol−1. For the C13 position, the σ-bonding orbitals of the C13–C17 bonds, σ(2) C13–C17, transfer to the fourth lone pair anti-bonding orbital of O81, LP*(4) O81, with stabilization energy of 29.6 kcal mol−1.
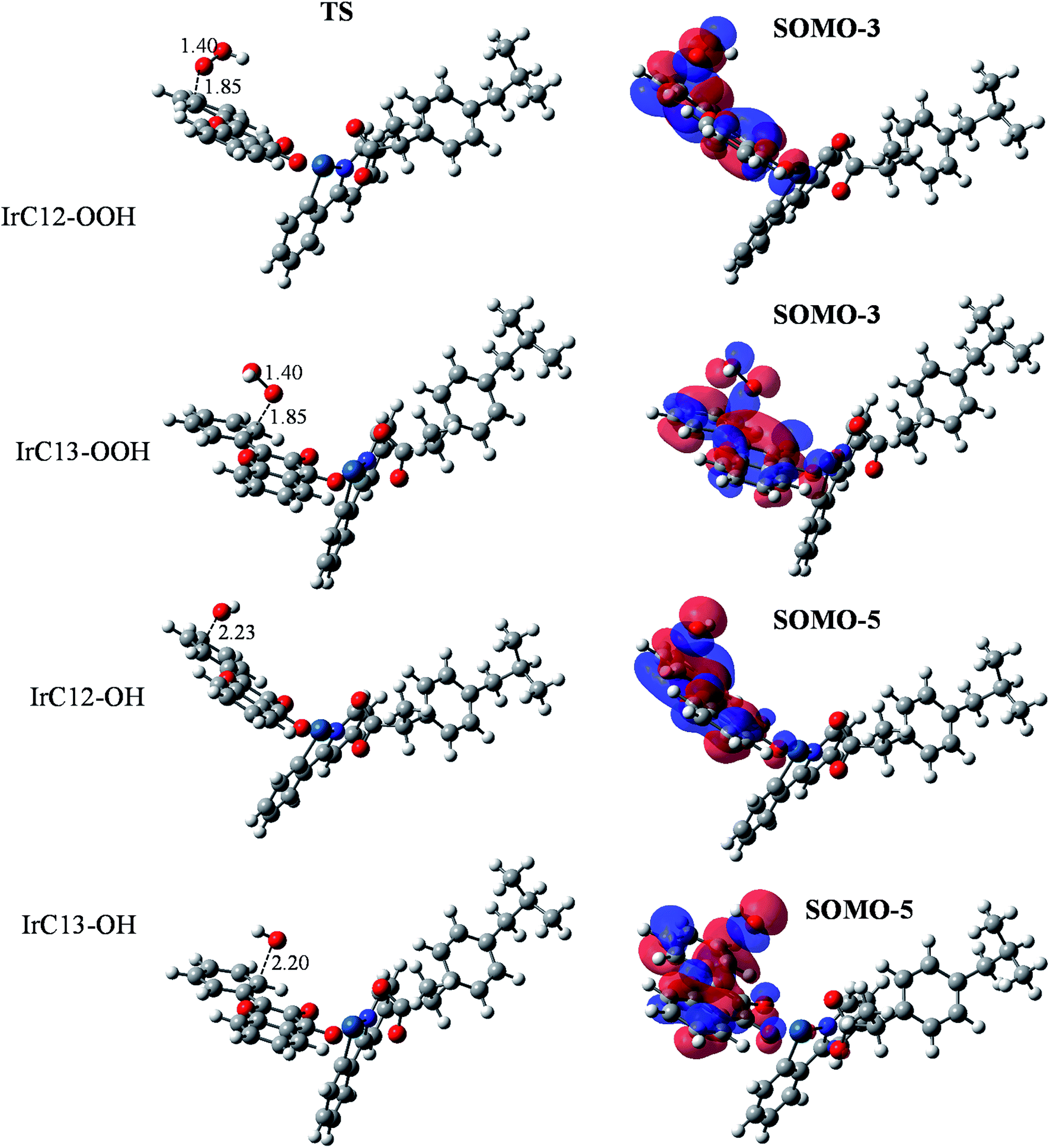 | ||
| Fig. 8 Optimized transition structures (TS) and frontier orbitals of the RAF reaction between HOO˙/HO˙ and Ir(III) complex, occurring at C12 and C13 positions (isovalue = 0.02). | ||
4. Concluding remarks
In summary, we have reported the synthesis and characterization of an Ir(III) complex based on biologically important ligands, ibuprofen and flavonol. The complex demonstrated better free radical scavenging activity than the ligands for most common radicals (ABTS˙+, DPPH˙, NO˙, HO˙). The complex preliminarily shows good in vitro antitumor activity towards A549, A2780 and A2780cis cells and its cytotoxicity is significantly greater than that of cis-platin and the ligands in the same conditions.In parallel, theoretical investigation by DFT was employed to study the geometrical and electronic structures of the complex as well as of the ligands. Intrinsic thermochemical properties of all compounds calculated in both the gas phase and in two solvents (water and PEA) revealed also that the Ir(III) complex is a potent antioxidant and its antioxidant capacity is greater than that of the ligands, in agreement with the experimental investigation. Principal reactions of free radical scavenging of the Ir(III) complex were chosen to study in detail, including the HAT, the RAF and the SET mechanisms. Findings are summarized as follows:
- for the SET reaction, electron-donating process is preferred over electron-accepting one both in the gas phase and in solvents, for all studied radicals.
- The PL mechanism is among the less favored pathways for free radical scavenging. Nevertheless, it is noted that the radicals could be scavenged in the order DPPH˙ > HOO˙ > HO˙ > NO˙ in both phases.
- The FHT mechanism was proved to be the most active pathway for free radical scavenging towards HO˙, HOO˙, NO˙ and DPPH˙. The FHT reaction towards HOO˙ radical is endothermic and non-spontaneous while the same reaction towards HO˙ radical is exothermic and spontaneous. C35 is the most favorable position for the FHT mechanism in both cases. The H-abstraction was confirmed to be directed by the HAT mechanism but not PCET by analyzing the frontier orbitals and from NBO analyses of TSs. In our computational conditions (gas phase), the Ir(III) complex shows similar enthalpy of reactions but lower barrier for the same position as ibuprofen, which might be an indication for the higher radical scavenging activity of the complex compared to ligands.
- Similar results were obtained for the RAF reaction with HOO˙/HO˙ radicals. The radical adduct of HOO˙ is endothermic and non-spontaneous while the RAF reaction towards HO˙ radical is exothermic and spontaneous. C13 and C12 are the most favored positions for HOO˙ and HO˙, respectively. Flavonol, in our computational condition (gas phase), shows better antioxidant property via RAF mechanism than the Ir(III) complex.
The study also demonstrated that theoretical and experimental investigation could be used effectively as complementary to evaluate structures and properties, especially antioxidant properties of molecules. Finally, we hope that the present work will mark a further step in the design and synthesis of new anticancer metallodrugs based on bioactive ligands.
Conflicts of interest
The authors declare no competing financial interest.Abbreviations
| ROS | Reactive oxygen species |
| ABTS | 2,2′-Azinobis(3-ethylbenzthiazoline-6-sulphonic acid) |
| DPPH | 2,2-Diphenyl-1-picrylhydrazyl |
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest unoccupied molecular orbital |
| SOMO | Singly occupied molecular orbital |
| FHT | Formal hydrogen transfer |
| RAF | Radical adduct formation |
| BDE | Bond dissociation enthalpy |
| IP | Ionization potential |
| EA | Electron affinity |
| ΔH0 | Adiabatic reaction enthalpy |
| ΔG0 | Gibbs free energy |
| NBO | Natural bond orbital |
| SET | Single electron transfer |
| PL | Proton loss |
| PA | Proton affinity |
| ZPE | Zero-point vibrational energy |
| ESR | Electron spin resonance |
Acknowledgements
The authors acknowledge general support from the School of Chemistry, National University of Ireland (NUI), Galway. This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant no. 103.03-2018.366.References
- C. C. Benz and C. Yau, Nat. Rev. Cancer, 2008, 8, 875–879 CrossRef CAS PubMed.
- S. Reuter, S. C. Gupta, M. M. Chaturvedi and B. B. Aggarwal, Free Radical Biol. Med., 2010, 49, 1603–1616 CrossRef CAS PubMed.
- E. R. Milaeva, Curr. Top. Med. Chem., 2011, 11, 2703–2713 CrossRef CAS PubMed.
- B. Englinger, C. Pirker, P. Heffeter, A. Terenzi, C. R. Kowol, B. K. Keppler and W. Berger, Chem. Rev., 2019, 119, 1519–1624 CrossRef CAS PubMed.
- C. G. Hartinger, A. D. Phillips and A. A. Nazarov, Curr. Top. Med. Chem., 2011, 11, 2688–2702 CrossRef CAS PubMed.
- F. Ponte, I. Ritacco, G. Mazzone, N. Russo and E. Sicilia, Inorg. Chim. Acta, 2018, 470, 325–330 CrossRef CAS.
- Z. Liu, I. Romero-Canelón, B. Qamar, J. M. Hearn, A. Habtemariam, N. P. E. Barry, A. M. G. Pizarro, J. Clarkson and P. J. Sadler, Angew. Chem., Int. Ed., 2014, 53, 3941–3946 CrossRef CAS PubMed.
- G. S. Yellol, A. J. Donaire, G. Yellol, V. Vasylyeva, C. Janiak and J. Ruiz, Chem. Commun., 2013, 49, 11533–11535 RSC.
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143–203 CrossRef CAS.
- S. Ladouceur, K. N. Swanick, S. Gallagher-Duval, Z. Ding and E. Zysman-Colman, Eur. J. Inorg. Chem., 2013, 30, 5329–5343 CrossRef.
- Y. You and S. Y. Park, J. Am. Chem. Soc., 2005, 127, 12438–12439 CrossRef CAS PubMed.
- E. Orselli, R. Q. Albuquerque, P. M. Fransen, R. Fröhlich, H. M. Janssen and L. D. Cola, J. Mater. Chem., 2008, 18, 4579–4590 RSC.
- X.-N. Li, Z.-J. Wu, Z.-J. Si, H.-J. Zhang, L. Zhou and X.-J. Liu, Inorg. Chem., 2009, 48, 7740–7749 CrossRef CAS PubMed.
- P. Brulatti, R. J. Gildea, J. A. K. Howard, V. Fattori, M. Cocchi and J. A. G. Williams, Inorg. Chem., 2012, 51, 3813–3826 CrossRef CAS PubMed.
- Y. Yang, L. Guo, Z. Tian, Y. Gong, H. Zheng, S. Zhang, Z. Xu, X. Ge and Z. Liu, Inorg. Chem., 2018, 57, 11087–11098 CrossRef CAS PubMed.
- A. Petrini, R. Pettinari, F. Marchetti, C. Pettinari, B. Therrien, A. Galindo, R. Scopelliti, T. Riedel and P. J. Dyson, Inorg. Chem., 2017, 56, 13600–13612 CrossRef CAS PubMed.
- V. Novohradsky, Z. Liu, M. Vojtiskova, P. J. Sadler, V. Brabec and J. Kasparkova, Metallomics, 2014, 6, 682–690 RSC.
- C. Wang, J. Liu, Z. Tian, M. Tian, L. Tian, W. Zhao and Z. Liu, Dalton Trans., 2017, 46, 6870–6883 RSC.
- J. P. Cerón-Carrasco, J. Ruiz, C. Vicente, C. de Haro, D. Bautista, J. Zuniga and A. Requena, J. Chem. Theory Comput., 2017, 13, 3898–3910 CrossRef PubMed.
- J. Li, L. Guo, Z. Tian, M. Tian, S. Zhang, K. Xu, Y. Qian and Z. Liu, Dalton Trans., 2017, 46, 15520–15534 RSC.
- D. Malešev and V. Kuntić, J. Serb. Chem. Soc., 2007, 72, 921–939 CrossRef.
- M. M. Kasprzak, A. Erxleben and J. Ochocki, RSC Adv., 2015, 5, 45853–45877 RSC.
- S. I. Pillai, S. P. Subramanian and M. Kandaswamy, Eur. J. Med. Chem., 2013, 63, 109–117 CrossRef CAS PubMed.
- M. D. Engelmann, R. Hutcheson and F. Cheng, J. Agric. Food Chem., 2005, 53, 2953–2960 CrossRef CAS PubMed.
- L. Dangleterre and J.-P. Cornard, Polyhedron, 2005, 24, 1593–1598 CrossRef CAS.
- Y. A. Davila, M. I. Sancho, M. C. Almandoz and S. E. Blanco, Spectrochim. Acta, Part A, 2012, 95, 1–7 CrossRef CAS PubMed.
- K. Vijayaraghavan, S. I. Pillai and S. P. Subramanian, Eur. J. Pharmacol., 2012, 680, 122–129 CrossRef CAS PubMed.
- N. A. E. Ragehy, M. Abdelkawy and A. E. Bayoumy, Anal. Lett., 1994, 27, 2127–2139 CrossRef.
- A. Andrade, S. F. Namora, R. G. Woisky, G. Wiezel, R. Najjar, J. A. A. Sertié and D. de O. Silva, J. Inorg. Biochem., 2000, 81, 23–27 CrossRef CAS PubMed.
- A. T. M. Fiori, W. R. Lustri, A. Magalhães and P. P. Corbi, Inorg. Chem. Commun., 2011, 14, 738–740 CrossRef CAS.
- C.-H. Leung, H.-J. Zhong, H. Yang, Z. Cheng, D. S.-H. Chan, V. P.-Y. Ma, R. Abagyan, C.-Y. Wong and D.-L. Ma, Angew. Chem., Int. Ed., 2012, 51, 9010–9014 CrossRef CAS PubMed.
- K. K.-W. Lo and K. Y. Zhang, RSC Adv., 2012, 2, 12069–12083 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision E.01Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- I. Avilov, P. Minoofar, J. Cornil and L. D. Cola, J. Am. Chem. Soc., 2007, 129, 8247–8258 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- A. Galano, G. Mazzone, R. Alvarez-Diduk, T. Marino, J. R. Alvarez-Idaboy and N. Russo, Annu. Rev. Food Sci. Technol., 2016, 7, 335–352 CrossRef CAS PubMed.
- B. Orwat, E. Witkowska, I. Kownacki, M.-J. Oh, M. Hoffmann, M. Kubicki, I. Grzelak, B. Marciniec, I. Glowacki, B. Luszczynska, G. Wiosna-Salyga, J. Ulanski, P. Ledwon and M. Lapkowski, Dalton Trans., 2017, 46, 9210–9226 RSC.
- A. Kurzwernhart, W. Kandioller, C. Bartel, S. Bachler, R. Trondl, G. Muhlgassner, M. A. Jakupec, V. B. Arion, D. Marko, B. K. Kepplerab and C. G. Hartinger, Chem. Commun., 2012, 48, 4839–4841 RSC.
- H. Chiniforoshan, L. Tabrizi, M. Hadizade, M. R. Sabzalian, A. N. Chermahini and M. Rezapour, Spectrochim. Acta, Part A, 2014, 128, 183–190 CrossRef CAS PubMed.
- M. Y. Wong, G. Xie, C. Tourbillon, M. Sandroni, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel and E. Zysman-Colman, Dalton Trans., 2015, 44, 8419–8432 RSC.
- Y. Zhong and F. Shahidi, Woodhead Publ. Ser. Food Sci., Technol. Nutr., 2015, 287–333 CAS.
- S. Sampath, B. Kalimuthu, V. Veeramani, S. Janardhanam, M. A. Baran and R. Chellan, RSC Adv., 2016, 6, 12382–12390 RSC.
- N. Söylemez, E. Yabas, S. Sahin-Bolukbasi and M. Sülü, J. Porphyrins Phthalocyanines, 2018, 22, 233–242 CrossRef.
- S. Gligorovski, R. Strekowski, S. Barbati and D. Vione, Chem. Rev., 2015, 115, 13051–13092 CrossRef CAS PubMed.
- N. Sreejayan and M. N. A. Rao, J. Pharm. Pharmacol., 1997, 49, 105–107 CrossRef PubMed.
- I. Ejidike and P. Ajibade, Molecules, 2015, 20, 9788–9802 CrossRef CAS PubMed.
- O. Friaa and D. Brault, Org. Biomol. Chem., 2006, 4, 2417–2423 RSC.
- Q. U. Ain, U. Ashiq, R. A. Jamal and M. Mahrooof-Tahir, Spectrochim. Acta, Part A, 2013, 115, 683–689 CrossRef CAS PubMed.
- I. Nakanishi, T. Kawashima, K. Ohkubo, H. Kanazawa, K. Inami, M. Mochizuki, K. Fukuhara, H. Okuda, T. Ozawa, S. Itoh, S. Fukuzumi and N. Ikota, Org. Biomol. Chem., 2005, 3, 626–629 RSC.
- L. Tabrizi, D. Q. Dao and T. A. Vu, RSC Adv., 2019, 9, 3320–3335 RSC.
- J. S. Wright, E. R. Johnson and G. A. DiLabio, J. Am. Chem. Soc., 2001, 123, 1173–1183 CrossRef CAS PubMed.
- IP value phenol calculated at the same B3LYP/6-31G(d) level of theory is 185.56 kcal/mol in the gas phase..
- A. Galano and A. Martínez, J. Phys. Chem. B, 2012, 116, 1200–1208 CrossRef CAS PubMed.
- J. R. Leon-Carmona and A. Galano, J. Phys. Chem. B, 2011, 115, 4538–4546 CrossRef CAS PubMed.
- A. Galano, Phys. Chem. Chem. Phys., 2011, 13, 7178–7188 RSC.
- A. Galano, J. Mex. Chem. Soc., 2015, 59, 231–262 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H-NMR, 13C-NMR, TOF MS spectra of Ir(III) complex; HPLC chromatogram; experimental methods for DPPH˙, HO˙, NO˙, and ABTS˙+ radical scavenging assays and cytotoxicity, cell culture and ROS production experiments; results for percentage of DPPH˙, HO˙, NO˙, and ABTS˙+ radicals scavenged and ESR spectra of four studied radicals as a function of concentration; in vitro antitumor activity of Ir(III) complex and two ligands towards A549, A2780 and A2780cis cancer cells; IC50 concentrations of Ir(III) complex and cis-platin against three studied cancer cell lines; ROS levels in A549 cells as determined by NBT assays; NBO charges of Ir(III) complex; ΔH0 and ΔG0 for HAT reactions with HOO˙ and HO˙ radicals; energy of the optimized structures of TSs for the RAF reaction with HOO˙ radical; ΔH0 and ΔG0 for RAF reactions with HOO˙ and HO˙ radicals; ΔH0 and ΔG0 for RAF and HAT reactions of HOO˙ and HO˙ with flavonol and ibuprofen; TS and SOMO orbitals of HAT and RAF reaction between ibuprofen and flavonol with HO˙/HOO˙ radicals; Cartesian coordinates of molecules and TSs of HAT and RAF reactions. See DOI: 10.1039/c9ra02726b |
| ‡ The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. |
| This journal is © The Royal Society of Chemistry 2019 |