
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New ecological method for determination of different β-lactams: application to real human plasma samples
Nehal F. Farid a and
Nada S. Abdelwahab*ab
a and
Nada S. Abdelwahab*ab
aPharmaceutical Analytical Chemistry, Faculty of Pharmacy, Beni-Suef University, Beni-Suef, Egypt. E-mail: nadasayed2003@yahoo.com; nehalfayek@gmail.com; Tel: +20 1285999726 Tel: +20 1277950994
bPharmaceutical Chemistry, Faculty of Pharmacy, Nahda University (NUB), Beni-Suef, Egypt
First published on 27th June 2019
Abstract
Recently, the use of antibiotics has become widespread all over the world resulting in bacterial resistance to these antibiotics, which requires alternative medications or higher doses of antibiotics. Implementation of an easy analytical method that can analyze a wide range of β-lactam antibiotics in a single run is important to reduce the time of therapeutic drug monitoring (TDM) in hospitals and minimize the spreading of bacterial resistance. A novel environmentally harmless HPTLC method was developed and validated following FDA recommendations for analysis of four β-lactams; cefaclor, cefotaxime, cefepime, and meropenem, in human plasma. A solvent mixture of ethylacetate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol:deionized water:formic acid (60:30:15:1, by volume) was the used developing system, detection was carried out at 270 nm, and valacyclovir was used as an internal standard. A lower limit of quantitation (LLOQ) was found to be 0.1 μg per band for all the analyzed drugs. Validation parameters were calculated and found to fulfil the international requirements for bio-analytical method validation. Additionally, each of the studied antibiotics was given to a group of healthy volunteers from which blood samples were collected at tmax of each, methanol was used for precipitation of plasma protein, and the developed method was used for calculation of the concentrations in the separated plasma samples. The developed method, being a green one, and time and money saving, can be used for TDM of these drugs in clinical studies as well as for quality control analysis in pharmaceutical companies. The proposed method is the first developed HPTLC method for the simultaneous bio-analysis of the selected β-lactams.
:methanol:deionized water:formic acid (60:30:15:1, by volume) was the used developing system, detection was carried out at 270 nm, and valacyclovir was used as an internal standard. A lower limit of quantitation (LLOQ) was found to be 0.1 μg per band for all the analyzed drugs. Validation parameters were calculated and found to fulfil the international requirements for bio-analytical method validation. Additionally, each of the studied antibiotics was given to a group of healthy volunteers from which blood samples were collected at tmax of each, methanol was used for precipitation of plasma protein, and the developed method was used for calculation of the concentrations in the separated plasma samples. The developed method, being a green one, and time and money saving, can be used for TDM of these drugs in clinical studies as well as for quality control analysis in pharmaceutical companies. The proposed method is the first developed HPTLC method for the simultaneous bio-analysis of the selected β-lactams.
Introduction
β-Lactam antibiotics are the most commercially used antibiotics in the world, they work by inhibiting cell wall biosynthesis in bacteria.1 Cephalosporins and carbapenems are two classes of β-lactam antibiotics that are prescribed for serious infections that involve a wide bacterial range. The drugs under study include cephalosporins [cefaclor (2nd generation), cefotaxime (3rd generation) and cefepime (4th generation)], and meropenem which is an example of carbapenems.Cephalosporins are broad-spectrum antibacterial drugs; they are the second largest class of β-lactam antibiotics with excellent safety profiles. Third and fourth generations are able to penetrate the blood–brain barrier and reach the central nervous system with sufficient concentrations; thus they are effective in the treatment of meningitis. On the other hand, carbapenems have the broadest spectrum activity and greatest potency against Gram negative and Gram positive bacteria, hence they are used as last resort antibiotics for resistant bacteria,2 so they are considered lifesaving drugs.3
Appraising the literature, different methods were published for analysis of β-lactam antibiotics in different biological fluids (cefotaxime and cefepime reviews were reported by Consortti and Salgado4 and Omkulthom5). The published methods for β-lactam antibiotics include HPLC,6–15 LC-MS-MS,16–23 and capillary electrophoresis.24–28 Few TLC densitometric methods were found for analysis of different pharmaceutical formulations containing β-lactam antibiotics.29–34 Only one TLC densitometric method was published for in vitro determination of cefepime.35 Based on the literature survey, no HPTLC method was found in the literature for the in vivo determination of the studied β-lactam antibiotics.
Planar chromatography has wide applications ranging from simple screening tests to complicated instrumental quantitative analysis of different samples in different matrices. TLC is one of planar chromatographic methods which has the advantages of time and money saving.36,37 It has extensive applications in pharmaceutical analysis,38,39 identification of impurities,40,41 isolation and separation of biomedical metabolites or constituents from different body fluids with minimum sample pretreatment.42,43 It is also used for separation of identical compounds in a mixture.36,37 Recently, HPTLC is an improved form of TLC with better resolution and more accurate quantitative measurements. Complex mixtures can be visualized on HPTLC chromatograms at a glance.37
One of the significant problems all over the world is the environmental pollution due to the wide use of hazardous chemicals and solvents. The concept of green chemistry developed as natural evolution of pollution prevention. From the goals of green chemistry is to use alternatives to hazardous substances and to develop new analytical methods that can reduce waste and toxic solvents.44
Paying attention to environmental issues and the importance of β-lactam antibiotics, the novelty of this work is to develop and validate an ecological HPTLC method to monitor the studied β-lactam antibiotics in human plasma and to ensure a safe and effective treatment for patients, hence, decrease the extensive use of these antibiotics and minimize their bacterial resistance. Furthermore, the method was validated according to FDA guidelines45 and all results agreed with the acceptance limits. The most striking features of the proposed method are its simplicity and short analysis time. Furthermore, it is the first HPTLC method for quantitation of the cited drugs in human plasma with minimal sample pretreatment.
Materials and methods
Instruments
Optimization of the method was carried out using a short wavelength, 254 nm UV lamp (Vilber Lourmat, Marne La Vallee, Cedex, France).
Standards and reagents
- Cefaclor was provided by Pharco (Alexandria, Egypt), Alex for Chemical Industries & Drugs (Alexandria, Egypt) and certified to have a purity of 98.98%.- Cefotaxime sodium was supplied from Egyptian International Pharmaceutical Industries Co. (E.I.P.I.Co.), 10th of Ramadan City, Industrial Area, Egypt, with a purity of 99.02% according to the manufacturer certificate of analysis.
- Cefepime hydrochloride, with a purity of 98.95%, was given as a gift by Pharco B International, New Borg El Arab City, Third Industrial Zone, Alexandria, Egypt.
- Meropenem was purchased from (Sigma-Aldrich Chemie GmbH, Germany), with a purity of 98.89% purity.
- Valacyclovir with purity of 99.21% according to the supplier certificate of analysis and was provided by Hikma Pharma (6th of October City, Egypt).
- Ethylacetate, methanol, formic acid (EL-Nasr pharmaceutical, Chemical Co., Abu Zabaal, Cairo, Egypt).
- Deionized water (SEDICO Pharmaceuticals Co., 6th October City, Egypt).
Pharmaceutical formulations
- Bacticlor® (0.5 g cefaclor per capsule), manufactured by Pharco (Alexandria, Egypt), Alex for Chemical Industries & Drugs (Alexandria, Egypt).- Cefotax® (1 g for I.M. or I.V. injection), is labeled to contain 1048 mg cefotaxime sodium (equivalent to 1000 mg cefotaxime), and is manufactured by Egyptian International Pharmaceutical Industries Co. (E.I.P.I.Co.), 10th of Ramadan City, Industrial Area, Egypt.
- Forcetex® (1 g for I.M. or I.V. injection), containing 1213 mg cefepime hydrochloride (corresponding to 1000 mg cefepime) and is manufactured by Pharco B International, New Borg El Arab City, Third Industrial Zone, Alexandria, Egypt for Novartis Pharma. Cairo, Egypt.
- Meronem® (0.5 g for I.V. injection) is manufactured by ACS Dobfar SpA, Italy for AstraZeneca UK Limited, Macclesfield, Cheshire, SK 10 2 NA, United Kingdom.
Blank plasma samples
- Blank plasma samples were provided by Dr Khaled Nagy Laboratory, Beni-Suef, Egypt and they were collected from healthy voluntary donors to be used as a blank matrix.Chromatographic conditions
The used stationary phase was HPTLC aluminum plates (20 × 15 cm) pre-coated with silica gel 60 F254 with 200 μm thickness and 5 μm particle size (Merck, Darmstadt, Germany) and the mobile phase was consisted of a solvent mixture of ethylacetate:methanol:deionized water:formic acid (60:30:15:1, by volume). Samples was applied using Camag Linomat V applicator as bands of 4 mm width, 5 mm apart from each other, and 15 mm from the bottom edge of the plate. Chromatographic development was done in a glass jar saturated with the mobile phase mixture for 15 min. The temperature was maintained constant at 25 °C and UV scanning was carried out at 270 nm for all the studied drugs.
Solutions
Cefaclor, cefotaxime, cefepime, meropenem, and valacyclovir (IS) stock solutions (1 mg mL−1) were separately prepared in methanol using five separate 25 mL measuring flasks.Calibration curves
Administration of the studied drugs and collection of plasma samples
Blood samples were collected from healthy volunteers (informed on the experimental procedures, the nature of the study, and gave a written approval). Volunteers were divided into four groups (n = 6), group I, received a dose of 0.5 g Bacticlor® capsules, group II, took 1 g I.V. dose of Cefotax® injection, group III was injected I.V. with 1 g Forcetex® injection, and finally, group IV was injected I. V. with 0.5 g Meronem® injection. The age of the volunteers ranged from 20 to 45 years old with a weight range 50–75 kg. Blood samples of 5 mL were collected at the specified time of each drug (tmax) (tmax of cefaclor = 50 min., cefotaxime = 2–5 min., cefepime = 30 min., and meropenem = 5–6 min.46–49) in heparinized tubes and centrifuged at 4000 rpm immediately after receipt. The separated plasma samples were stored at −20 °C till the time of analysis.Preparation of the collected plasma samples
Previously collected plasma samples were thawed to room temperature just before the extraction and the preparation procedures.Chromatographic development was then done following the instructions under chromatographic conditions. The peak area ratio was then calculated and used to calculate the concentrations of each drug in the collected plasma samples using the previously computed regression equations.
Results and discussion
Therapeutic drug monitoring of antibiotics is essential to diminish the spreading of bacterial resistance, so, it is necessary to develop reliable analytical methods for their therapeutic monitoring and quality control analysis. β-Lactam antibiotics are still known for their excellent safety and efficacy profile, they work by interfering with bacterial cell wall synthesis which is absent in human cell.50 From the literature survey discussed above, no HPTLC method was published for the simultaneous quantitation of different β-lactam antibiotics in real plasma samples. Thin layer chromatography is solid liquid chromatographic method at which the separation depends on the difference in solubility and adsorption of different compounds between two phases (mobile phase and stationary phase) at which they are to be portioned. The developed HPTLC method has advantages of short analysis time and low solvent consumption which are economically effective. In addition, solvents used have harmless environmental impact which is essential in the field of green chemistry.Method development and optimization
The main goal during optimization of the developing system was to test green solvents and exclude harmful ones such as chloroform, methylene chloride, benzene, etc. Different mobile phase mixtures were tested starting with acetone:methanol (9:1, 7:3, and 6:3, v/v) and ethylacetate:methanol (9:1, 7:3, and 6:3, v/v). In both cases, no complete separation among the studied drugs and plasma was observed, moreover, spots of meropenem, cefepime, and plasma appeared near the baseline. However, ethylacetate was preferred than acetone as it gave compact spots and the ratio (6:3, v/v) was chosen. Looking at the structure of the studied drugs, it was found that they contained both acidic and basic groups, hence their chromatographic resolution was expected to be pH dependent. Different ratios of ammonia solution (33%), triethylamine, acetic acid, and formic acid were separately tested (0.5, 1, and 1.5%). Adjusting the medium basic with either ammonia solution or triethylamine resulted in unresolved spots between cefotaxime and cefaclor. In one hand, changing the medium with acetic acid, meropenem and cefepime co-eluted with nearly the same Rf values. On the other hand, using formic acid (1%) resulted in a reasonable separation between all spots. Water in different ratios was then added (5, 10, and 15%) in a trial to improve separation between spots of plasma and cefepime. It was noticed that addition of 15% water improved the shape of all the separated spots and produced the best separation.
The use of internal standard during bio-analytical method development is important to correct for the variability in the analyte loss during sample treatment. Different internal standards were tested and the best one regarding the chromatographic behaviors and separation was valacyclovir. Sensitivity is an important factor for methods applied to biological fluids, different scanning wavelengths (230, 254, 270, and 290 nm) were tested and the highest sensitivity was obtained on scanning at 270 nm for all the proposed β-lactam antibiotics. In the same way, time of equilibration required before development is important to attain homogeneity of the atmosphere, thus diminishes the evaporation of the solvent from the HPTLC plate during the development. Effect of mobile phase saturation time on the chromatographic separation was tested (15 and 30 min.), where no considerable effect was revealed.
Finally, the optimum conditions were; mobile phase mixture of ethyl acetate:methanol:water:formic acid (6:3:1.5:0.1, by volume), the saturation time was 15 min., and UV scanning at 270 nm. The obtained Rf values were: 0.04, 0.1, 0.19, 0.38, 0.55, and 0.76 for plasma, cefepime, meropenem, IS, cefaclor, and cefotaxime, respectively, Fig. 1.
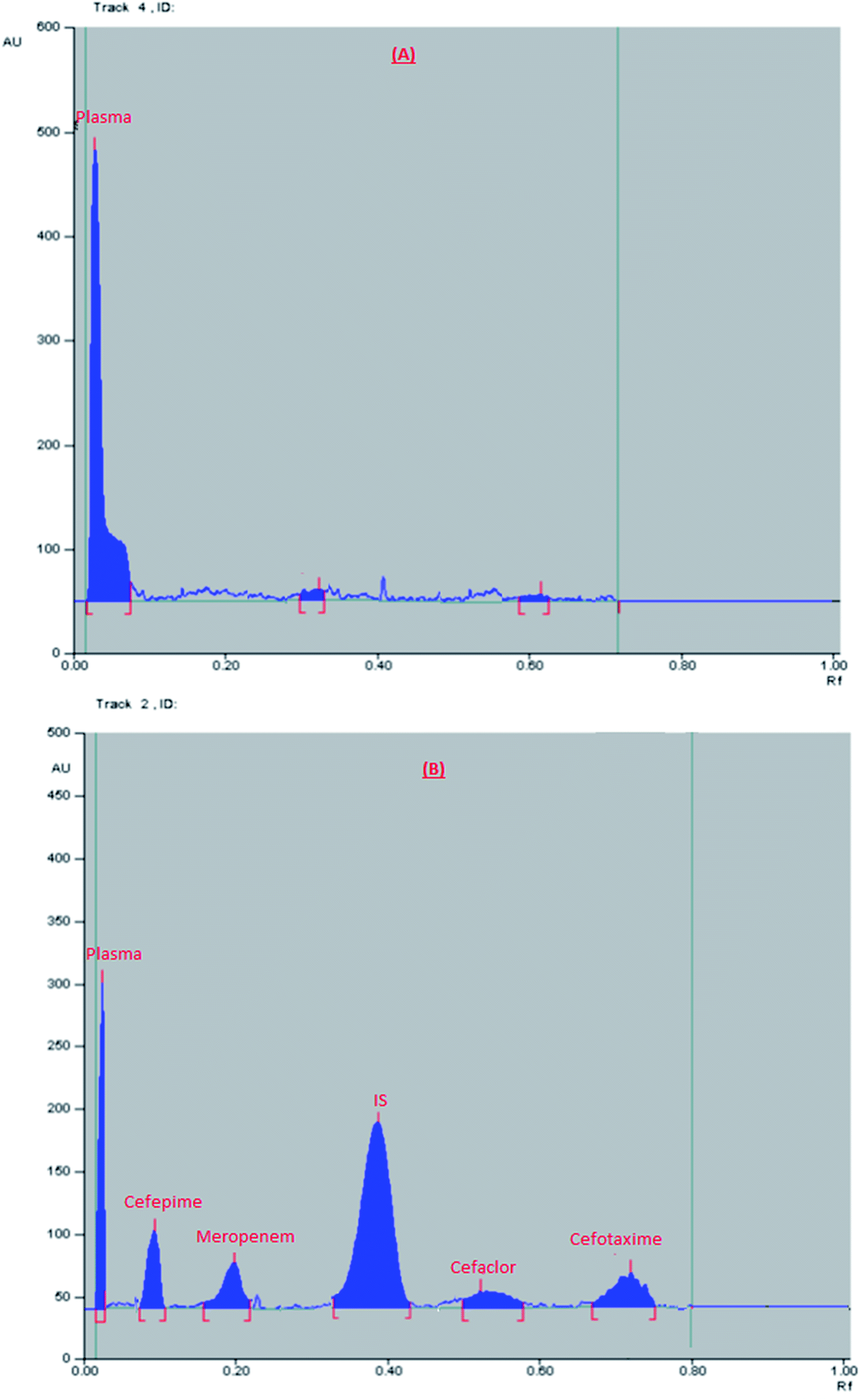 | ||
| Fig. 1 2D chromatogram of (A) blank plasma, (B) plasma sample spiked with a mixture of the studied drugs (at their LLOQ) and internal standard. | ||
After optimization of the method, linearity was first checked by construction of calibration curves using pure standards of the studied antibiotics. Calibration curves relating the peak area ratio (area of the pure standard/area of IS) to the corresponding concentrations were plotted and linearity was proven using polynomial regression in the concentration ranges of 0.06–3 μg per band for all the proposed drugs. The computed regression equations are given in Table 1.
| Parameters | Pure samples | Spiked human plasma samples | ||||||
|---|---|---|---|---|---|---|---|---|
| Cefaclor | Cefotaxime | Cefepime | Meropenem | Cefaclor | Cefotaxime | Cefepime | Meropenem | |
| a The linearity was achieved using the polynomial regression equation: A = aX2 + bX + C. a: coefficient 1. b: coefficient 2. A = peak area ratio (peak area of the analyte/peak area of IS), X= concentration μg per band. C = intercept. | ||||||||
| Range, μg per band | 0.06–3.0 | 0.1–3.0 | ||||||
| Slope | −0.0661a | −0.1419a | −0.0891a | −0.0503a | −0.0328a | −0.0103a | −0.0301a | −0.0560a |
| 0.6292b | 1.0245b | 0.4649b | 0.4006b | 0.4271b | 0.4765b | 0.4218b | 0.4423b | |
| Intercept | 0.0059 | 0.0195 | 0.0288 | 0.0151 | −0.0048 | 0.0095 | 0.0468 | 0.0421 |
| Correlation (r) | 0.9996 | 0.9998 | 0.9996 | 0.9994 | 0.9996 | 0.9998 | 0.9993 | 0.9994 |
| LLOQ | — | 0.10 | ||||||
| ULOQ | — | 3.00 | ||||||
Method validation
Validation of the proposed analytical method was carried out following FDA45 guidelines for bio-analytical method validation.Concentrations of the samples of calibration standard and QC samples were calculated using the computed regression equations given in Table 1. The results of each sample (calibration standards and quality control samples) were accepted when deviation is not more than 15% from the true concentrations (100 ± 15%) while that of LLOQ was accepted when its deviation was ≤20% (100 ± 20%) from the nominal concentration.
 | ||
| Fig. 2 2D chromatogram of real plasma samples obtained from healthy volunteers at their tmax. | ||
| Component | Concentrationa (μg per band) | Intraday | Interday | ||||
|---|---|---|---|---|---|---|---|
| Recovery% | RSD% | Bias%b | Recovery% | RSD% | Bias%b | ||
| a Average of 3 experiments.b Bias = [(measured concentration − nominal concentration)/nominal concentration] × 100. | |||||||
| Cefaclor | 0.10 (LLOQ) | 102.56 | 7.29 | 2.56 | 91.98 | 8.13 | −8.02 |
| 0.60 (LQC) | 108.02 | 2.83 | 8.02 | 99.79 | 8.60 | −0.21 | |
| 1.50 (MQC) | 99.57 | 0.04 | −0.43 | 109.34 | 9.40 | 9.34 | |
| 2.50 (HQC) | 104.64 | 7.49 | 4.64 | 104.86 | 9.72 | 4.86 | |
| Cefotaxime | 0.10 (LLOQ) | 105.5 | 8.47 | 5.75 | 93.07 | 9.63 | −6.93 |
| 0.60 (LQC) | 93.97 | 5.09 | −6.03 | 107.56 | 13.42 | 7.56 | |
| 1.50 (MQC) | 94.32 | 7.92 | −5.68 | 91.48 | 10.09 | −8.52 | |
| 2.50 (HQC) | 99.13 | 6.76 | −0.87 | 89.66 | 7.47 | −10.30 | |
| Cefepime | 0.10 (LLOQ) | 116.77 | 3.91 | 16.77 | 118.46 | 1.84 | 18.46 |
| 0.60 (LQC) | 105.52 | 4.42 | 5.52 | 103.65 | 6.24 | 3.65 | |
| 1.50 (MQC) | 106.34 | 5.77 | 6.34 | 108.67 | 9.55 | 3.65 | |
| 2.50 (HQC) | 96.21 | 6.53 | −3.79 | 95.29 | 9.15 | −4.71 | |
| Meropenem | 0.10 (LLOQ) | 102.56 | 4.57 | 2.56 | 107.22 | 10.53 | 7.22 |
| 0.60 (LQC) | 98.74 | 6.19 | −1.26 | 109.01 | 3.31 | 8.99 | |
| 1.50 (MQC) | 90.00 | 4.20 | −10.00 | 97.67 | 9.17 | −2.33 | |
| 2.50 (HQC) | 106.35 | 0.31 | 6.35 | 99.21 | 8.84 | −0.79 | |
| Analyte | Concentration of the analyte (μg per band) | % recoverya |
|---|---|---|
| a Average of 3 determinations. | ||
| Cefaclor | 0.60 | 88.56 |
| 1.50 | 93.25 | |
| 2.50 | 95.57 | |
| Mean ± % RSD | 92.46 ± 3.862 | |
|
||
| Cefotaxime | 0.60 | 76.86 |
| 1.50 | 73.19 | |
| 2.50 | 80.80 | |
| Mean ± % RSD | 76.75 ± 4.946 | |
|
||
| Cefepime | 0.60 | 91.24 |
| 1.50 | 86.96 | |
| 2.50 | 96.94 | |
| Mean ± % RSD | 91.71 ± 5.459 | |
|
||
| Meropenem | 0.60 | 94.95 |
| 1.50 | 95.68 | |
| 2.50 | 97.41 | |
| Mean ± % RSD | 90.01 ± 1.316 | |
|
||
| IS | 1.00 | 95.07 ± 2.987 |
| Analyte | % recoverya | ||
|---|---|---|---|
| Concentration of the analyte (μg per band) | Three freeze–thaw cycles | Bench top stability | |
| a Average of 3 determinations. | |||
| Cefaclor | 0.60 | 103.79 | 96.72 |
| 1.50 | 99.30 | 111.39 | |
| 2.50 | 89.90 | 109.67 | |
| Mean ± % RSD | 97.66 ± 7.256 | 105.93 ± 7.57 | |
|
|||
| Cefotaxime | 0.60 | 106.89 | 99.63 |
| 1.50 | 97.54 | 107.50 | |
| 2.50 | 98.78 | 96.8 | |
| Mean ± % RSD | 101.07 ± 5.024 | 101.31 ± 5.473 | |
|
|||
| Cefepime | 0.60 | 108.37 | 89.87 |
| 1.50 | 107.18 | 92.37 | |
| 2.50 | 110.63 | 89.78 | |
| Mean ± % RSD | 108.73 ± 1.612 | 90.67 ± 1.621 | |
|
|||
| Meropenem | 0.60 | 99.08 | 110.13 |
| 1.50 | 96.24 | 111.33 | |
| 2.50 | 104.79 | 100.00 | |
| Mean ± % RSD | 100.04 ± 4.353 | 107.15 ± 5.808 | |
Results of method application to real human plasma samples
The developed HPTLC method was successfully used to determine the concentration of a single dose of the studied antibiotics in real human plasma of healthy volunteers. From previous studies, it was reported that Cmax (μg mL−1) for the given doses was 18.16, 102, 78.7, and 70 μg mL−1 for cefaclor, cefotaxime, cefepime, and meropenem, respectively.46–49 Chromatograms obtained from selected plasma samples are shown in Fig. 2. Additionally, the concentrations of the given doses were calculated from the previously computed regression equations and summarized in Table 5. All the plasma concentrations measured in volunteers samples were within the calibration range of the proposed HPTLC method.| Volunteers | Expected concentration (μg per band)46 | The found concentration (μg per band) | Expected concentration (μg per band) | The found concentration (μg per band)47 |
|---|---|---|---|---|
| Cefaclor | Cefotaxime | |||
| 1 | 0.454 | 0.480 | 1.020 | 1.120 |
| 2 | 0.476 | 0.800 | ||
| 3 | 0.570 | 1.187 | ||
| 4 | 0.510 | 1.053 | ||
| 5 | 0.560 | 1.144 | ||
| 6 | 0.420 | 1.013 | ||
| Mean ± % RSD | 0.519 ± 10.873 | 1.053 ± 13.184 | ||
| Patients | Expected concentration (μg per band)48 | The found concentration (μg per band) | Expected concentration (μg per band)49 | The found concentration (μg per band) |
|---|---|---|---|---|
| Cefepime | Meropenem | |||
| 1 | 0.787 | 0.890 | 0.700 | 0.575 |
| 2 | 0.720 | 0.712 | ||
| 3 | 0.707 | 0.637 | ||
| 4 | 0.827 | 0.688 | ||
| 5 | 0.613 | 0.588 | ||
| 6 | 0.693 | 0.737 | ||
| Mean ± % RSD | 0.742 ± 13.467 | 0.656 ± 10.170 | ||
Conclusion
An innovative HPTLC method was established for in vivo analysis of four β-lactam antibiotics. The method had the advantages of simple sample preparation and short analysis time. Moreover, the proposed method adopted the use of green solvents with harmless environmental impact. All validation parameters agreed with FDA acceptance criteria. This method permitted the accurate determination of the studied antibiotics thus, it can be used during therapeutic drug monitoring in daily clinical practice, hence minimize the antibiotics microbial resistance.Ethics
The study was approved by the Animal Care and Use Committee of the Faculty of Medicine, Beni-Suef University (FM-BSU REC) according to guidelines of the declaration of Helsinki, International Conference on Harmonization (ICH) and United States Codes of Federal Regulation and registered in under the Federal Wide Assurance number (REC-A-PHBSU18005) for protection of animals (appendix 1).Conflicts of interest
Both authors declare that they have no conflicts of interest.Acknowledgements
All authors wish to thank the staff of Dr Khaled Nagy Laboratory, Beni-Suef, Egypt for their efforts in collecting the blank plasma samples.References
- R. P. Elander, Industrial production of β-lactam antibiotics, Appl. Microbiol. Biotechnol., 2003, 61, 385–392 CrossRef CAS.
- J. A. Torres, M. V. Pillegas and J. P. Quinn, Current concepts in antibiotic resistant gram negative bacteria, Expert Rev. Anti-Infect. Ther., 2007, 5, 833–843 CrossRef CAS.
- A. M. Queenan and K. Bush, Carbapenemases: the versatile β-lactamases, Clin. Microbiol. Rev., 2007, 20, 440–485 CrossRef CAS.
- L. P. Consortti and H. R. N. Salgado, A critical review of analytical methods for quantification of cefotaxime, Crit. Rev. Anal. Chem., 2017, 47, 359–371 CrossRef CAS.
- M. A. Omkulthom, Review on determination of cefepime in biological fluids by different analytical methods, J. Innovations Pharm. Biol. Sci., 2016, 3, 141–146 CAS.
- V. Granados-Soto, M. E. Aguilar-Cota, G. Reyes-Garcia, R. Medina-Santillán and F. J. Flores-Murrieta, Simple method for the determination of cefaclor in human plasma samples by HPLC, J. Liq. Chromatogr. Relat. Technol., 2003, 26, 3315–3323 CrossRef CAS.
- B. J. Z. Ruan, H. Lou, D. Xu and H. Yuan, Determination of cefaclor in human plasma by reversed-phase high-performance liquid chromatography with UV detection and its application to the bioequivalence studies, Anal. Lett., 2009, 42, 210–2179 Search PubMed.
- S. Naz, M. H. Shoaib, L. Bashir, R. I. Yousuf, F. Anjum, F. Siddiqui and S. Yaseen, HPLC method development and validation for the determination of cefaclor in human plasma, Pak. J. Pharm. Sci., 2017, 30, 1645–1649 CAS.
- S. E. Briscoe, B. C. McWhinney, J. Lipman, J. A. Roberts and J. P. J. Ungerer, A method for determining the free (unbound) concentration of ten betalactam antibiotics in human plasma using high performance liquid chromatography with ultraviolet detection, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 907, 178–184 CrossRef CAS.
- T. Legrand, D. Vodovar, N. Tournier, N. Khoudour and A. Hulina, Simultaneous determination of eight β-Lactam antibiotics, amoxicillin, cefazolin, cefepime, cefotaxime, ceftazidime, cloxacillin, oxacillin, and piperacillin, in human plasma by using ultra-high- performance liquid chromatography with ultraviolet detection, Antimicrob. Agents Chemother., 2016, 60, 4734–4742 CrossRef CAS PubMed.
- T. Legrand, S. Chhun, E. Rey, B. Blanchet, J. Zahar, F. Lanternierd, G. Ponsa and V. Jullien, Simultaneous determination of three carbapenem antibiotics in plasma by HPLC with ultraviolet detection, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 875, 551–556 CrossRef CAS PubMed.
- B. C. McWhinney, S. C. Wallis, T. Hillister, J. a. Roberts, J. Lipman and J. P. J. Ungerer, Analysis of 12 beta-lactam antibiotics in human plasma by HPLC with ultraviolet detection, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878, 2039–2043 CrossRef CAS PubMed.
- K. Kipper, K. Anier, I. Leito, J. Karjagin, K. Oselin and K. Herodes, Rapid determination of meropenem in biological fluids by LC: comparison of various methods for sample preparation and investigation of meropenem stability, Chromatographia, 2009, 70, 1423–1427 CrossRef CAS.
- T. Roth, S. Fiedler, S. Mihai and H. Parsch, Determination of meropenem levels in human serum by high-performance liquid chromatography with ultraviolet detection, Biomed. Chromatogr., 2017, 31, 1–7 CrossRef PubMed.
- E. Dailly, R. Bouquié, G. Deslandes, P. Jolliet and R. Le Floch, A liquid chromatography assay for a quantification of doripenem, ertapenem, imipenem, meropenem concentrations in human plasma: application to a clinical pharmacokinetic study, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 1137–1142 CrossRef CAS PubMed.
- X. Chen, D. Zhong, B. Huang and J. Cui, Determination of cefaclor in human plasma by a sensitive and specific liquid chromatographic-tandem mass spectrometric method, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2003, 784, 17–24 CrossRef CAS.
- C. H. W. Wang and L. Miao, Determination of cefaclor by UPLC–MS-MS for a Chinese pharmacokinetic study, J. Chromatogr. Sci., 2014, 52, 636–640 Search PubMed.
- M. Carlier, V. Stove, J. A. Roberts, E. Van de Velde, J. J. De Waele and A. G. Verstraete, Quantification of seven β-lactam antibiotics and two β-lactamase inhibitors in human plasma using a validated UPLC-MS/MS method, Int. J. Antimicrob. Agents, 2012, 40, 416–422 CrossRef CAS PubMed.
- F. B. Sime, M. S. Roberts, J. A. Roberts and T. A. Robertson, Simultaneous determination of seven β-lactam antibiotics in human plasma for therapeutic drug monitoring and pharmacokinetic studies, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 960, 134–144 CrossRef CAS PubMed.
- R. Cazorla-Reyes, R. Romero-Gonzalez, A. Garrido Frenich, M. A. R. Maresca and J. L. M. Vidal, Simultaneous analysis of antibiotics in biological samples by ultra high performance liquid chromatography-tandem mass spectrometry, J. Pharm. Biomed. Anal., 2014, 89, 203–212 CrossRef CAS.
- P. Colin, L. De Bock, H. T'Jollyn, K. Boussery and J. Van Bocxlaer, Development and validation of a fast and uniform approach to quantify beta-lactam antibiotics in human plasma by solid phase extraction-liquid chromatography-electrospray-tandem mass spectrometry, Talanta, 2013, 103, 285–293 CrossRef CAS.
- T. Ohmori, A. Suzuki, T. Niwa, H. Ushikoshi, K. Shirai and S. Yoshida, et al., Simultaneous determination of eight β-lactam antibiotics in human serum by liquid chromatography-tandem mass spectrometry, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 1038–1042 CrossRef CAS PubMed.
- M. Carlier, V. Stove, J. J. de Waele and A. G. Verstraete, Ultrafast quantification of β-lactams antibiotics in human plasma using UPLC-MS/MS, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 978–979, 89–94 CrossRef CAS PubMed.
- M. Kummer, N. Šestáková, R. Theurillat, U. Huynh-Do, A. Endimiani, P. Sendi and W. Thormann, Monitoring of cefepime in urine by micellar electrokinetic capillary chromatography with UV detection and liquid chromatography coupled to mass spectrometry, J. Sep. Sci., 2018, 41, 4067–4074 CrossRef CAS.
- R. Theurillat, J. Joneli, U. Wanzenried, J. Schiess, M. Hurni, T. Weber, P. Sendi and W. Thormann, Therapeutic drug monitoring of cefepime with micellar electrokinetic capillary chromatography: assay improvement, quality assurance, and impact on patient drug levels, J. Sep. Sci., 2016, 39, 2626–2632 CrossRef CAS.
- N. Šestáková, R. Theurillat, P. Sendi and W. Thormann, Monitoring of cefepime in human serum and plasma by micellar electrokinetic capillary chromatography. Improvement of sample preparation and validation by liquid chromatography coupled to mass spectrometry, J. Sep. Sci., 2017, 40, 1805–1814 CrossRef.
- A. Al-Attas, J. J. Nasr, N. El-Enany and F. Belal, A green capillary zone electrophoresis method for the simultaneous determination of piperacillin, tazobactam and cefepime in pharmaceutical formulations and human plasma, Biomed. Chromatogr., 2015, 29, 1811–1818 CrossRef CAS PubMed.
- T. Kitahashi and I. Furuta, Determination of meropenem by capillary electrophoresis using direct injection of serum, J. Chromatogr. Sci., 2005, 43, 430–433 CAS.
- D. Agbaba, S. Eric, D. Zivanov Stakic and S. Vladimirov, HPTLC assay of cephalexin and cefaclor in pharmaceuticals, Biomed. Chromatogr., 1998, 12, 133–135 CrossRef CAS.
- S. Eric-Jovanovic, D. Agbaba, D. Zivanov-Stakic and S. Vladimirov, HPTLC determination of ceftriaxone, cefixime and cefotaxime in dosage forms, J. Pharm. Biomed. Anal., 1998, 18, 893–898 CrossRef CAS.
- R. K. Nanda, S. E. Bhagwat, S. E. Potawale and S. C. Hamane, Development and validation of HPTLC method for simultaneous densitometric analysis of cefotaxime sodium and sulbactam sodium as the bulk drugs and in the pharmaceutical dosage form, J. Pharm. Res., 2010, 3, 1667–1669 CAS.
- M. Dąbrowska, M. Starek and S. Pikulska, Simultaneous identification and quantitative analysis of eight cephalosporins in pharmaceutical formulations by TLC–densitometry, J. Planar Chromatogr.--Mod. TLC, 2011, 24, 23–29 CrossRef.
- M. Dąbrowska, M. Starek, J. Krzek, E. Papp and P. Król, A degradation study of cefepime hydrochloride in solutions under various stress conditions by TLC–densitometry, Biomed. Chromatogr., 2015, 29, 388–395 CrossRef PubMed.
- N. A. Elragehy, E. M. Abdel-Moety, N. Y. Hassan and M. R. Rezk, Stability-indicating determination of meropenem in presence of its degradation product, Talanta, 2008, 77, 28–36 CrossRef CAS PubMed.
- M. Dabrowska, W. Opoka and M. Starek, Determination of cefuroxime axetil and cefepime in biological materials by thin-layer chromatography–densitometry, J. Planar Chromatogr.--Mod. TLC, 2017, 30, 291–298 CrossRef CAS.
- M. Tomczyka, A. Bazylkob and J. Bonarewicz, Method development and validation for optimized separation of quercetin derivatives in selected Potentilla species using high-performance thin-layer chromatography photodensitometry method, J. Pharm. Biomed. Anal., 2012, 61, 265–270 CrossRef PubMed.
- B. Renger, Z. Végh and K. Ferenczi-Fodor, Validation of thin layer and high performance thin layer chromatographic methods, J. Chromatogr. A, 2011, 1218, 2712–2721 CrossRef CAS PubMed.
- N. S. Abdelwahab, Determination of atenolol, chlorthalidone and their degradation products by TLC-densitometric and chemometric methods with application of model updating, Anal. Methods, 2010, 2, 1994–2001 RSC.
- N. F. Farid and N. S. Abdelwahab, Stability-indicating HPTLC method for studying stress degradation behavior of sulbutiamine HCl, J. Chromatogr. Sci., 2016, 4, 609–617 Search PubMed.
- E. A. Abdelaleem and N. S. Abdelwahab, Green chromatographic method for analysis of some anti-cough drugs and their toxic impurities with comparison to conventional methods, Saudi Pharm. J., 2018, 26, 1185–1191 CrossRef PubMed.
- N. S. Abdelwahab, N. F. Fared, M. Elagawany and E. H. Abdelmomen, Different spectrophotometric and chromatographic methods for determination of mepivacaine and its toxic impurity, J. AOAC Int., 2017, 100, 1392–1399 CrossRef CAS PubMed.
- N. S. Abdelwahab, H. A. H. Elshemy and N. F. Farid, determination of flutamide and two major metabolites using HPLC–DAD and HPTLC methods, Chem. Cent. J., 2018, 12, 1–15 CrossRef PubMed.
- M. Y. Fares, N. S. Abdelwahab, M. M. Abdelrahman and H. M. Abdel-Rahman, Determination of sofosbuvir with two co-administered drugs; paracetamol and DL-methionine by two chromatographic methods, Bioanalysis, 2019, 11(5), 349–364 CrossRef CAS PubMed.
- T. Rojanarata, Green pharmaceutical chemistry for the sustainability, Silpakorn Univ. Sci. Technol. J., 2012, 6(1), 7–13 Search PubMed.
- FDA, Guidance for Industry Bio-analytical Method Validation, 2013 Search PubMed.
- M. Tutunji, O. Jarrar, M. Musameh, S. M. Alam, Quamruzaman and R. Dham, Bioequivalence evaluation of two brands of cefaclor 500 mg capsules: quantification of cefaclor using solid phase extraction technique, J. Clin. Pharm. Ther., 2001, 26, 149–153 CrossRef CAS PubMed.
- Through web site: https://webcache.googleusercontent.com/search?q=cache:9Avp6yAV6mUJ: https://www.medsafe.govt.nz/profs/Datasheet/c/Cefotaximeinjaft.pdf+%26cd=8%26hl=en%26ct=clnk%26gl=eg, accessed February 2019, 10.0 PM.
- Through web site: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/050679s036lbl.pdf, accessed February 2019, 10.0 PM.
- H. C. Kelly, M. Hutchison and S. J. Haworth, A comparison of the pharmacokinetics of meropenem after administration by intravenous injection over 5 min and intravenous infusion over 30 min, J. Antimicrob. Chemother., 1995, 36, 35–41 CrossRef CAS PubMed.
- Through website: https://pharmafactz.com-medicinal-chemistry-of-beta-lactam-antibiotics, accessed March 2019, 6.00 PM.
| This journal is © The Royal Society of Chemistry 2019 |