
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the homocoupling of trialkylstannyl monomers in the synthesis of diketopyrrolopyrrole polymers and its effect on the performance of polymer-fullerene photovoltaic cells
Gaël H. L. Heintgesab and
René A. J. Janssen *ac
*ac
aMolecular Materials and Nanosystems, Institute for Complex Molecular Systems, Eindhoven University of Technology, P. O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: r.a.j.janssen@tue.nl
bInstitute for Materials Research (IMO-IMOMEC), Design & Synthesis of Organic Semiconductors (DSOS), Hasselt University, Agoralaan, 3590 Diepenbeek, Belgium
cDutch Institute for Fundamental Energy Research, De Zaale 20, 5612 AJ Eindhoven, The Netherlands
First published on 20th May 2019
Abstract
Homocoupling of monomers in a palladium-catalyzed copolymerization of donor–acceptor polymers affects the perfect alternating structure and may deteriorate the performance of such materials in solar cells. Here we investigate the effect of homocoupling bis(trialkylstannyl)-thiophene and -bithiophene monomers in two low band gap poly(diketopyrrolopyrrole-alt-oligothiophene) polymers by deliberately introducing extended oligothiophene defects in a controlled fashion. We find that extension of the oligothiophene by one or two thiophenes and creating defects up to at least 10% does not significantly affect the opto-electronic properties of the polymers or their photovoltaic performance as electron donor in solar cells in combination with [6,6]-phenyl C71 butytic acid methyl ester as acceptor. By using model reactions, we further demonstrate that for the optimized synthetic protocol and palladium-catalyst system the naturally occurring defect concentration in the polymers is expected to be less than 0.5%.
1. Introduction
The performance of conjugated polymers in organic solar cells is primarily determined by the chemical structure of the backbone and side chains that provide the material with its semiconducting and structural properties. However, other factors such as the molecular weight distribution,1–3 the presence of end groups,4,5 possible branching of the main chain,6 and impurities in the polymer can also affect the functional properties.7–9 In addition, structural defects in the polymer main chain have been found to be important. For asymmetric monomers head-to-head or tail-to-tail couplings may occur, next to the preferred head-to-tail linkages.10,11 Alternatively, in the case of crosscoupling polymerizations where two complementary comonomers are used, a coupling reaction between two identical monomers introduces a defect in the perfect alternating structure. This last phenomenon is referred to as homocoupling. Homocoupling reactions are known to occur in the Suzuki,12 Stille,13 and direct arylation14 crosscoupling polycondenstation reactions that are commonly used to synthesize donor–acceptor small-molecules and polymers that find widespread use organic solar cells. The topic of structural defects is finding increasing awareness and was recently reviewed by Maes et al.15The homocoupling of two bromide monomers in the Suzuki and Stille polymerization reaction in low band gap diketopyrrolopyrrole (DPP) polymers has previously been investigated in our group in some detail.16 The defect is caused by homocoupling of two brominated dithienyl-diketopyrrolopyrrole (T-DPP-T) units, creating T-DPP-2T-DPP-T defects in the polymer main chain. These defects generate a low-energy feature in the absorption spectrum. As a result the 2T defect can act as a trap for excitons and charges, and thereby explains the significant decrease in photovoltaic performance of solar cells when polymers with increasing 2T defect concentration were blended with [6,6]-phenyl C71 butyric acid methyl ester ([70]PCBM).16 An extensive overview of the literature showed that the same homocoupling defect is present in many DPP polymers that were reported up to 2014.16 We found it to occur when a too low ligand-to-palladium ratio is used in the reaction and that it can largely be avoided by modifying the catalyst system. The results also clearly demonstrated that this type of defect should be avoided because it is detrimental to the photovoltaic performance of these materials.
In a subsequent study Li et al. confirmed the formation of T-DPP-2T-DPP-T low-energy defects occurring in the Stille polymerization of poly(diketopyrrolopyrrole-alt-quaterthiophene) (PDPP4T).17 Li et al. also suggested that homocoupling of the trilalkylstannyl monomers would occur to a significant extent.17 They were unable to obtain direct structural or spectroscopic evidence, but argued that because of the almost quantitative reaction yield of the polymerization, the two different homocoupling reactions should occur to a similar extent. In a recent study, Costantini et al. used scanning tunneling microscopy to investigate structural defects in poly(diketopyrrolopyrrole-alt-terfurane) (PDPP3F) deposited on a Au(111) surface. They found a significant amount (ca. 12%) of defects in which an extra furan ring is present as result of homocoupling reaction between two 2,5-bis(trimethylstannyl)furan monomers.18 Coupling of the trialkylstannyl monomers in a Stille cross-coupling polymerization reaction can occur whenever palladium is existent in an oxidized form.19–22 This oxidized palladium can be present in the reaction mixture due to the age or improper storage of the catalyst, insufficient degassing of the reaction mixture, or because the catalyst system is designed as such, as is in the case of bis(triphenylphosphine)palladium(II) dichloride. In these cases a double transmetallation step occurs, followed by a reductive elimination transforming the Pd(II) into a Pd(0) species, which can then proceed to catalyze the reaction as usual, and results in a coupling between two organometallic partners.
Recently, Sommer et al. investigated this type of homocoupling in poly(N-9′-heptadecanyl-2,7-carbazole-alt-5,5-(4′,7′-di-2-thienyl-2′,1′,3′-benzothiadiazole)) (PCDTBT) polymers synthesized via a Suzuki reaction and found that the homocoupling of the boronic ester substituted carbazole monomers resulted in a remarkably lower performance in organic solar cells from defect concentrations as low as 2.4% onwards.23 Whether this decrease was due to electronic or morphological effects was, however, not very clear.
In DPP polymers, the effect of extended donor segment defects has not yet been systematically investigated, even though its occurrence, for the reasons mentioned above, can be expected. Usually, DPP donor–acceptor polymers are synthesized in Stille coupling reactions employing a dibromide monomer containing the electron-deficient DPP unit and a bis(trialkylstannyl) monomer comprising the electron-rich moiety. The reason for this choice is that oxidative addition is facilitated by electron withdrawing groups and transmetalation is facilitated by electron rich organotin compounds. Hence, homocoupling of the organotin compounds typically results in extended electron-rich segments in the polymer.24 These can have two distinct effects. First, the energy levels of the polymer might be affected, because the extended donor defect can have energy levels that deviate from those of the polymer and cause a shift of the oxidation potential. Depending on the direction of this shift of the energy levels, the defects can act as traps and contribute to an increased non-radiative recombination which lowers the open-circuit voltage (VOC) of the cells. Because DPP polymers usually display a wider optical band gap (Eg,opt) when the donor block is extended, it is not expected that these defects will form energetic traps, although optical effects could still be visible. Second, the homocoupling defect effectively reduces the number DPP units in the chain that generally carry the solubilizing side chains, such that the solubility could be affected. This could have an influence on the morphology of the active layers, resulting in differences in short-circuit current (Jsc) or fill factor (FF).3
Here we investigate the effect and extent of the homocoupling reaction of the trialkylstannyl monomers in two archetype DPP polymers by deliberate introduction of defected conjugated segments and by performing model reactions on monomer analogs. As it is a frequently used monomer, thiophene and its defect, bithiophene, are chosen initially. In a second example, the structural difference between the parent polymer and its homocoupling defect is enhanced by using bithiophene and its defect, quaterthiophene, as monomer. We demonstrate that for these polymers, this type of homocoupling does not occur to a significant extent and that if it would occur, the effects on the opto-electronic properties and photovoltaic performance are small or even absent.
2. Results and discussion
2.1 Poly(diketopyrrolopyrrole-alt-terthiophene) with quaterthiophene defects
Poly{2,2′-[(2,5-bis(2-hexyldecyl)-3,6-dioxo-2,3,5,6-tetrahydropyrrolo[3,4-c]pyrrole-1,4-diyl)dithiophene]-5,5′-diyl-alt-thiophen-2,5-diyl} (PDPP3T) is a well-known small band gap DPP polymer used for high-performing organic field-effect transistors and solar cells and a good model system to investigate the effect of homocoupling. To simulate the homocoupling of two trialkylstannyl monomers, a random terpolymer approach was used in which 3,6-bis(5-bromothiophen-2-yl)-2,5-bis(2-hexyldecyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (1) was co-polymerized with 2,5-bis(trimethylstannyl)thiophene (2) and 5,5′-bis(trimethylstannyl)-2,2′-bithiophene (3) in varying ratios (Scheme 1). This resulted in PDPP3T polymers with 5, 10, and 20% of 4T defects added in the chain.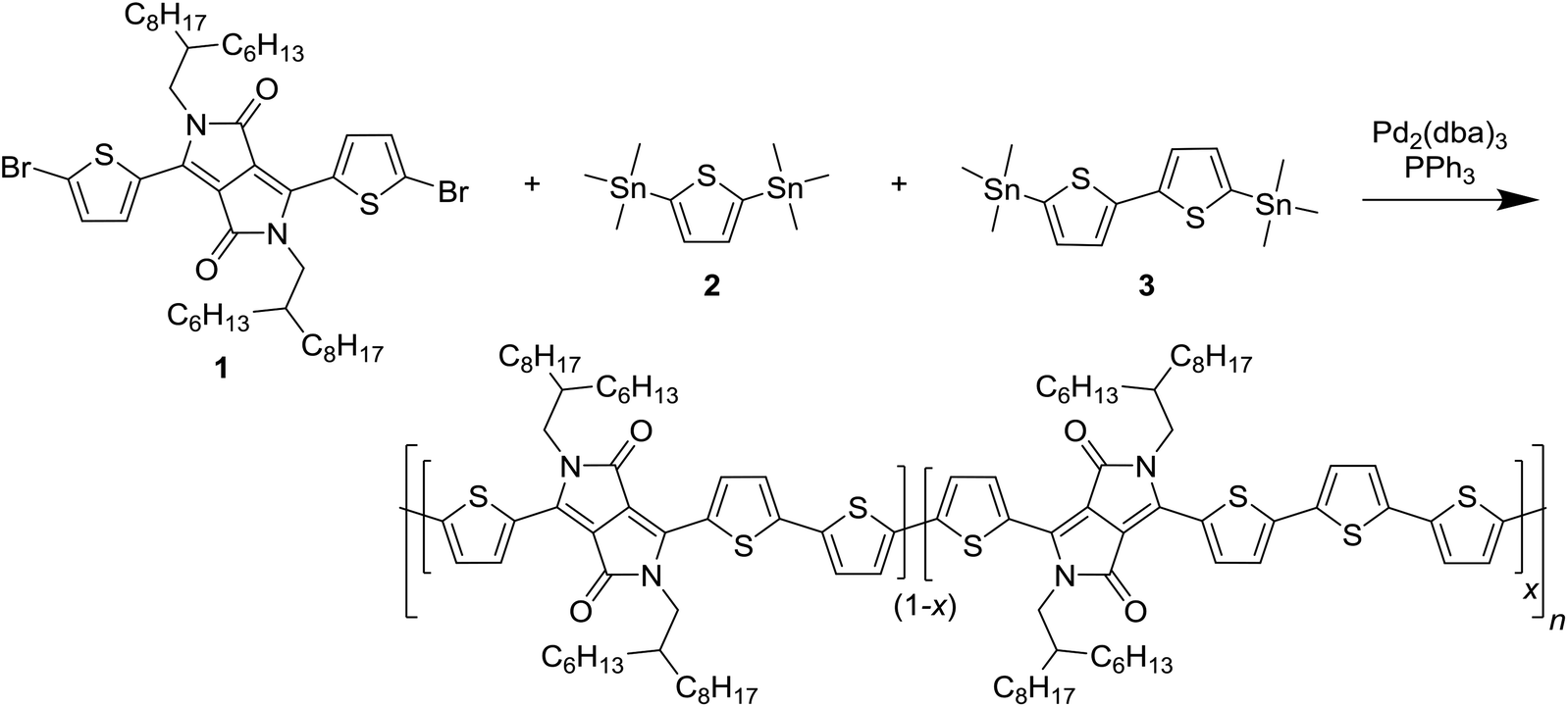 | ||
| Scheme 1 Synthesis of the PDPP3T polymers with 4T defects. The stoichiometry of monomers 2 and 3 was altered to obtain x = 5, 10, and 20% of intentional defects. | ||
Gel permeation chromatography (GPC) in 1,2-dichlorobenzene (o-DCB) at 140 °C revealed a decreasing trend in the number average molecular weight (Mn) from 148 to 88 kDa when increasing the amounts of 4T defects from 5% to 20% (Table 1). Several reasons can explain a reduced Mn. We tentatively ascribe this trend to a decrease in solubility with increasing defect content. As mentioned above, the 4T segments effectively dilute the sidechain-bearing DPP units in the main chain. This lowers the solubility and may cause the polymers above a certain length to precipitate out of solution during the polymerization. To fairly compare the defect containing polymers with defect-free PDPP3T, two batches of differing molecular weight were chosen corresponding roughly to the molecular weights obtained in defect containing polymers.
| Polymer | Mn [kDa] | Mw [kDa] | Đ | Eg,opta [eV] | HOMOb [eV] | LUMOb [eV] |
|---|---|---|---|---|---|---|
| a Determined from the absorption onset in thin films.b Determined with square wave voltammetry vs. Fc/Fc+, which was set at −4.8 eV vs. vacuum. | ||||||
| PDPP3T | 147 | 400 | 2.7 | 1.33 | −5.14 | −3.34 |
| 68 | 255 | 3.7 | ||||
| 5% 4T | 148 | 399 | 2.7 | 1.35 | −5.13 | −3.35 |
| 10% 4T | 123 | 330 | 2.7 | 1.36 | −5.03 | −3.35 |
| 20% 4T | 88 | 215 | 2.4 | 1.36 | −5.10 | −3.34 |
The UV-vis-NIR absorption spectra of the PDPP3T polymers measured in solution and thin films (Fig. 1) show no differences in shape of the absorption or optical band gap as function of the number of defects. When compared to an essentially defect-free PDPP3T, a difference in absorption maximum is visible in the solution spectra. This could be due to a different tendency towards aggregation in solution, and would suggest that the defect-free PDPP3T aggregates more than the defect-containing polymers. The shift in absorption maximum is, however, unlikely to be linked to the electronic nature of the defects as between varying levels of 4T defects no difference can be seen. We note that the signal at ∼400 nm, which is assigned to a transition to a higher-energy state, shows a gradual shift from 3.06 eV for PDPP3T, via 3.07 (5% 4T) and 3.01 (10% 4T), to 2.98 eV (20% 4T) when the defect level is increased. Such small shift with extending the donor segment is also seen in the corresponding small molecule compounds T-DPP-3T-DDP-T and T-DPP-4T-DDP-T where the corresponding peaks are found at 3.16 eV for the 3T derivative and at 3.10 eV for the 4T derivative. In thin films the differences are also small. Because the optical band gap of PDPP4T (the defect segments) is at 1.46 eV (vide infra) and larger than the band gap of PDPP3T at 1.33 eV, the presence of 4T segments will unlikely result in a clear change of the absorption spectrum and 4T defects are not expected to influence the optical band gap.
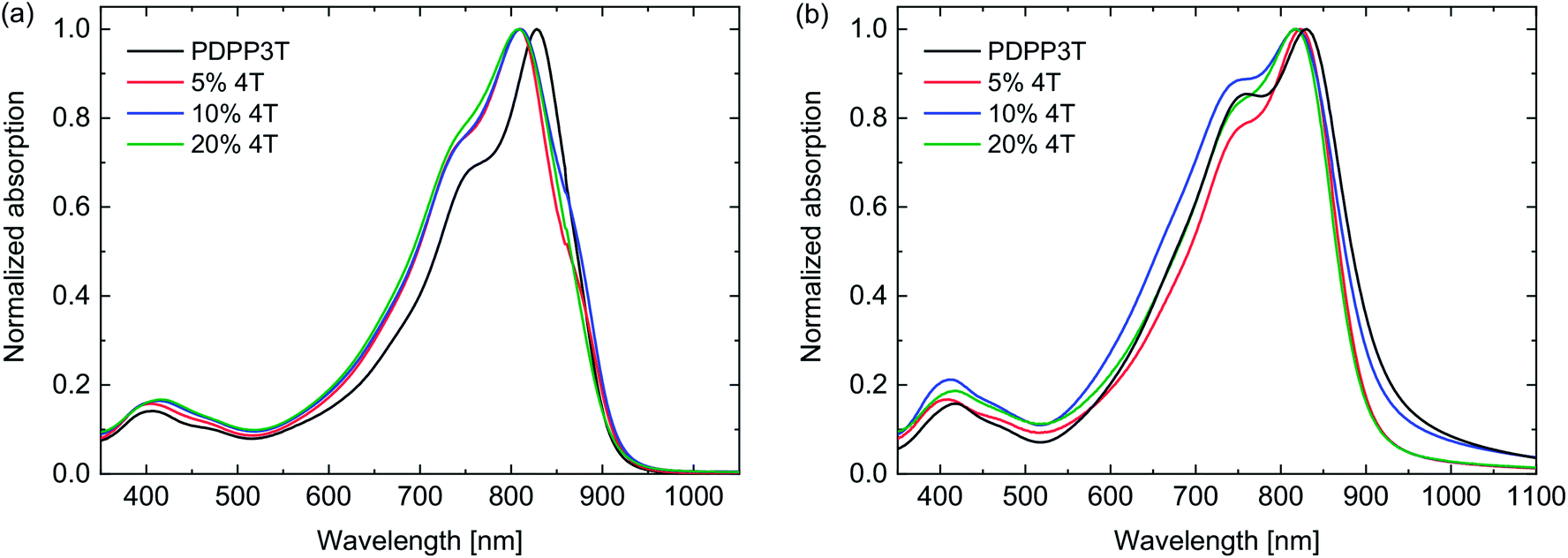 | ||
| Fig. 1 UV-vis-NIR spectra of PDPP3T polymers with 4T defects. (a) Dissolved in chloroform. (b) In thin films. | ||
Square wave voltammetry was used to determine the energy levels of the polymers. Table 1 reveals there are no significant differences in energy levels between the polymers. Again, 4T defects are also not expected to exert a strong effect because the HOMO and LUMO energy levels of PDPP4T (HOMO: −5.13 eV, LUMO: −3.26 eV) are not too dissimilar from those of PDPP3T (HOMO: −5.14 eV, LUMO: −3.34 eV).
Solar cells with an ITO/PEDOT:PSS/active layer/LiF/Al device configuration were produced by spin coating mixed solutions of the PDPP3T polymers and [70]PCBM (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 w/w) in chloroform containing 7.5% of o-DCB. As can be seen in Table 2 and Fig. 2 there are no clear trends in the PCEs of the defect-containing polymers. They all perform slightly worse than PDPP3T for a similar Mn. The Voc of the defect-containing polymers lies between 0.65 V and 0.67 V, which coincides with range generally observed for defect-free PDPP3T. This indicates, in accordance with the results from the square wave voltammetry, that this type of defect has little to no influence on the energy levels of the polymer and therefore does not affect the Voc. The fill factors lie between 0.64 and 0.68, and are also comparable with the FF of defect-free PDPP3T cells. This indicates that the 4T segments do not strongly increase the recombination of free charges, which is concurrent with the notion that these defects do not act as energetic traps.
:2 w/w) in chloroform containing 7.5% of o-DCB. As can be seen in Table 2 and Fig. 2 there are no clear trends in the PCEs of the defect-containing polymers. They all perform slightly worse than PDPP3T for a similar Mn. The Voc of the defect-containing polymers lies between 0.65 V and 0.67 V, which coincides with range generally observed for defect-free PDPP3T. This indicates, in accordance with the results from the square wave voltammetry, that this type of defect has little to no influence on the energy levels of the polymer and therefore does not affect the Voc. The fill factors lie between 0.64 and 0.68, and are also comparable with the FF of defect-free PDPP3T cells. This indicates that the 4T segments do not strongly increase the recombination of free charges, which is concurrent with the notion that these defects do not act as energetic traps.
| Polymer | Mn [kDa] | Jsc [mA cm−2] | Voc [V] | FF | PCE [%] |
|---|---|---|---|---|---|
| a Best cells are shown, numbers between parentheses refer to the average over 4 cells. | |||||
| PDPP3T | 147 | 15.4 (15.3) | 0.67 (0.66) | 0.69 (0.68) | 7.1 (6.9) |
| PDPP3T | 68 | 13.6 (13.5) | 0.65 (0.65) | 0.66 (0.66) | 5.9 (5.8) |
| 5% 4T | 148 | 13.4 (14.1) | 0.67 (0.66) | 0.68 (0.64) | 6.1 (6.0) |
| 10% 4T | 123 | 14.6 (14.6) | 0.66 (0.65) | 0.66 (0.65) | 6.4 (6.2) |
| 20% 4T | 88 | 12.4 (12.5) | 0.65 (0.65) | 0.67 (0.66) | 5.4 (5.3) |
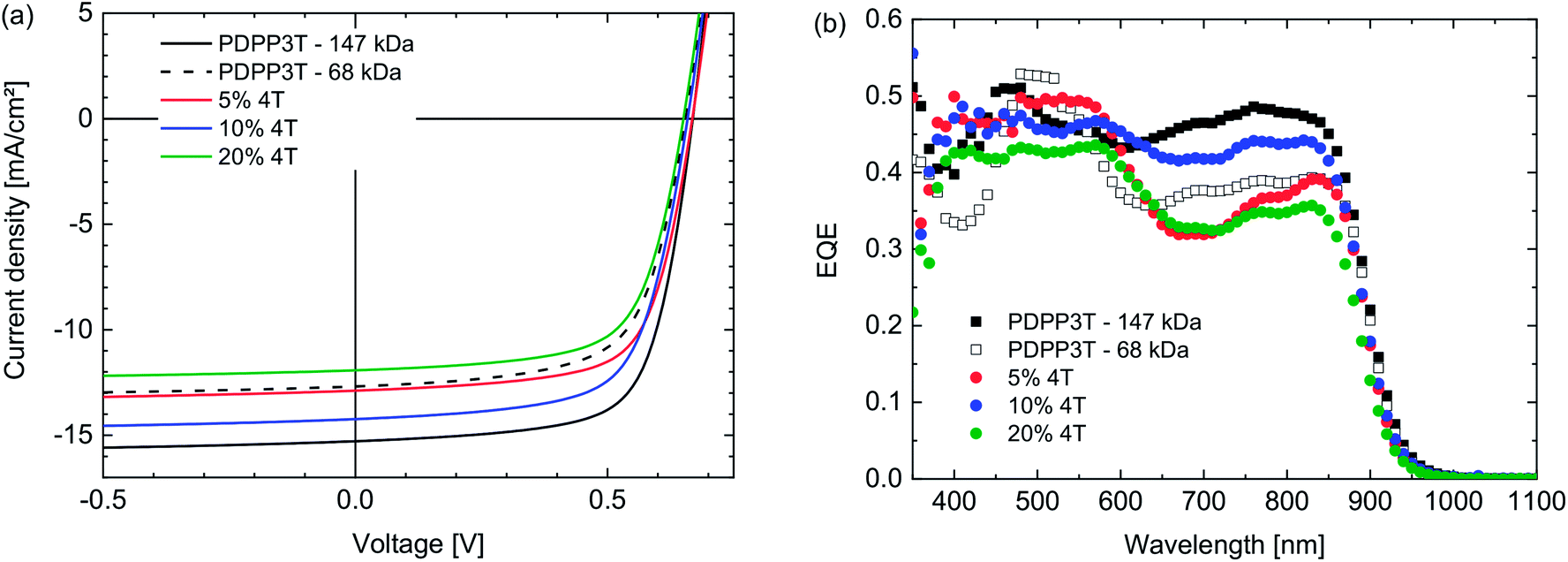 | ||
| Fig. 2 (a) J–V-characteristics of PDPP3T:[70]PCBM (1:2 w/w) polymer solar cells in which the PDPP3T polymers contain 4T defects (see legends). (b) Corresponding EQE spectra. | ||
The main difference between the cells listed in Table 2 occurs in the Jsc. When comparing the 20% defect polymer with the low Mn defect-free PDPP3T batch, a slightly lower Jsc is found, even though the molecular weights are comparable. The same is the case in the comparison between the 5% defect polymer and the high Mn defect-free PDPP3T batch. Taken separately, these observations could suggest that the homocoupling defects negatively influence the Jsc of the solar cells. However, this conclusion is weakened when considering the lack of a trend in Jsc with increasing amounts of defects. If defects negatively influence Jsc, the 10% defect polymer would be expected to have a lower Jsc than the 5% defect polymer, both on account of the higher amount of 4T segments and the lower Mn. This is, however, not the case and leads to the conclusion that these differences in Jsc are most likely caused by other factors such as molecular weight and nanomorphology and that the possible influence of the 4T defects, even in quite large concentrations (10% and 20%), is small enough to be eclipsed by other effects.
2.2 Poly(diketopyrrolopyrrole-alt-quaterthiophene) with sexithiophene defects
To introduce a larger defect, PDPP4T was chosen as the next target for modification. PDPP4T can be synthesized in polymerization of a dibrominated DPP monomer such as 1 and 5,5′-bis(trimethylstannyl)-2,2′-bithiophene (3). The associated homocoupling defect would them be a 6T segment. Introducing 6T segments in PDPP4T in a fashion analogous to Scheme 1 proved to be a synthetic challenge, owing to the difficulty of obtaining a bis(trialkylstannyl) derivative of the unsubstituted quaterthiophene. It was therefore decided to asymmetrically extend a DPP monomer. By reacting 3-(5-bromothiophen-2-yl)-2,5-bis(2-decyltetradecyl)-6-(thiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (4) with 2-([2,2′-bithiophen]-5-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (5) it was possible to obtain 6, which was converted by bromination with N-bromosuccinimide into 7 as shown in Scheme 2. We used long 2-decyltetradecyl side chains on the DPP unit in 4 to ensure sufficient solubility of the extended monomer 7 and of the resulting PDPP4T polymers.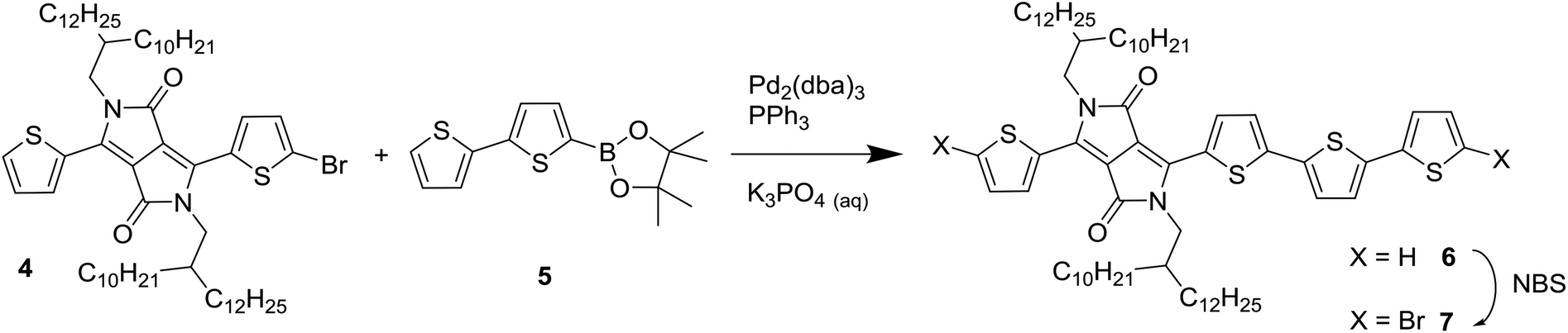 | ||
| Scheme 2 Synthesis of the asymmetrically extended monomer 7 to introduce 6T defects in a PDPP4T polymer. | ||
This asymmetrically extended DPP monomer 7 introduces the possibility of creating 8T segments, even in the absence of homocoupling reactions. To keep this possibility small and to keep the defect concentration within levels which are expected to occur during a normal polymerization reaction, polymers with 2% and 5% of defects were synthesized, together with defect-free PDPP4T as a reference, as shown in Scheme 3.
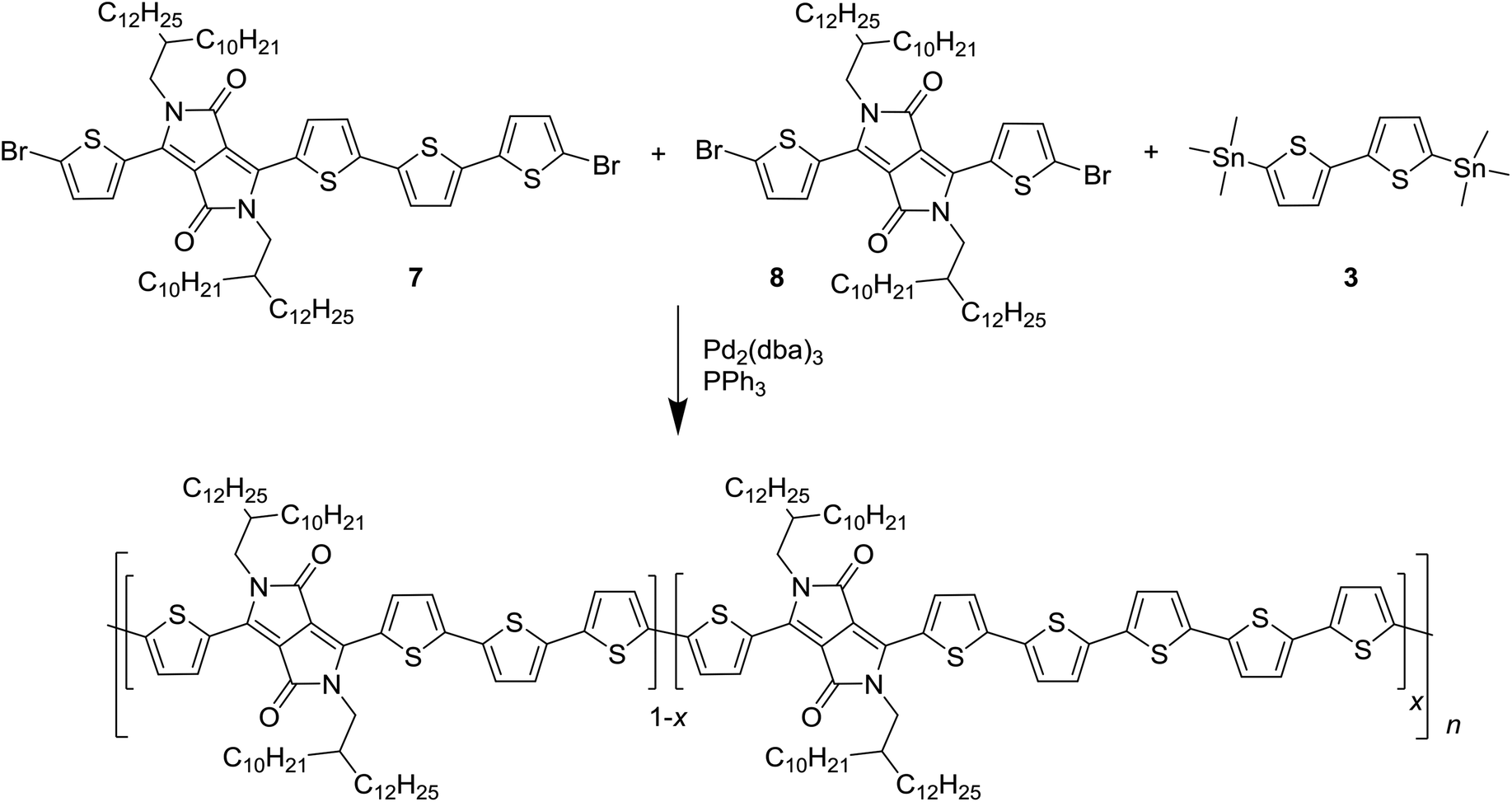 | ||
| Scheme 3 Synthesis of the PDPP4T polymers with 6T defects. The stoichiometry of the monomers 7 and 8 was altered to obtain x = 0, 2, and 5% of intentional defects. | ||
GPC analysis in o-DCB at 140 °C revealed comparable molecular weights for the three PDPP4T polymers, which should allow for a more direct comparison of the materials and enable to observe the effects of the 6T segments more clearly.
The UV-vis-NIR absorption spectra showed no significant difference between the three polymers (Fig. 3). In thin films the optical band gap is at 1.46 eV (Table 3). It is not expected that 6T segments would have a lower optical band gap than the 4T segments, because the optical band gap of a PDPP6T polymer of 1.48 eV (ref. 25) is close to that of PDPP4T (1.46 eV). Analysis of the HOMO and LUMO energy levels with square wave voltammetry also revealed no significant differences (Table 3). As in the case of PDPP3T, the defects seem to have no important influence on the electronic structure of the materials.
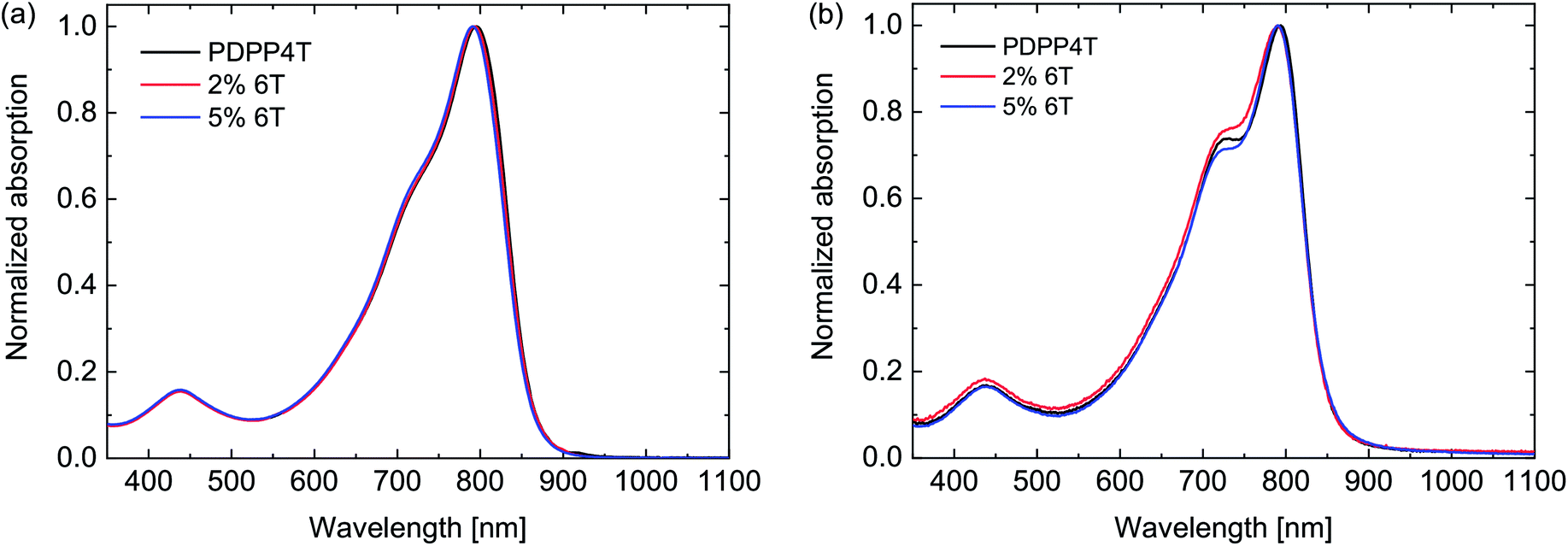 | ||
| Fig. 3 UV-vis-NIR spectra of the PDPP4T polymers with 6T defects. (a) Dissolved in chloroform. (b) In thin films. | ||
Solar cells were made from a chloroform solution containing 10% o-DCB, using the PDPP4T polymers and [70]PCBM (1:2 w/w) as active layer, employing the ITO/PEDOT:PSS/active layer/LiF/Al cell configuration. As can be seen in Fig. 4 and Table 4, the solar cells all show very similar performance. The Voc remains exactly the same, in concurrence with the results obtained from square wave voltammetry. The FF and Jsc show some variation, but differences are within the margin of error as can be seen in the averages over multiple cells. This indicates very clearly that the 6T segments, despite having a particularly low solubility, do not change the morphology of the layer significantly. Therefore it has to be concluded that this 6T defect has no influence on the optoelectronic properties or solar cell performance in concentrations up to 5%.
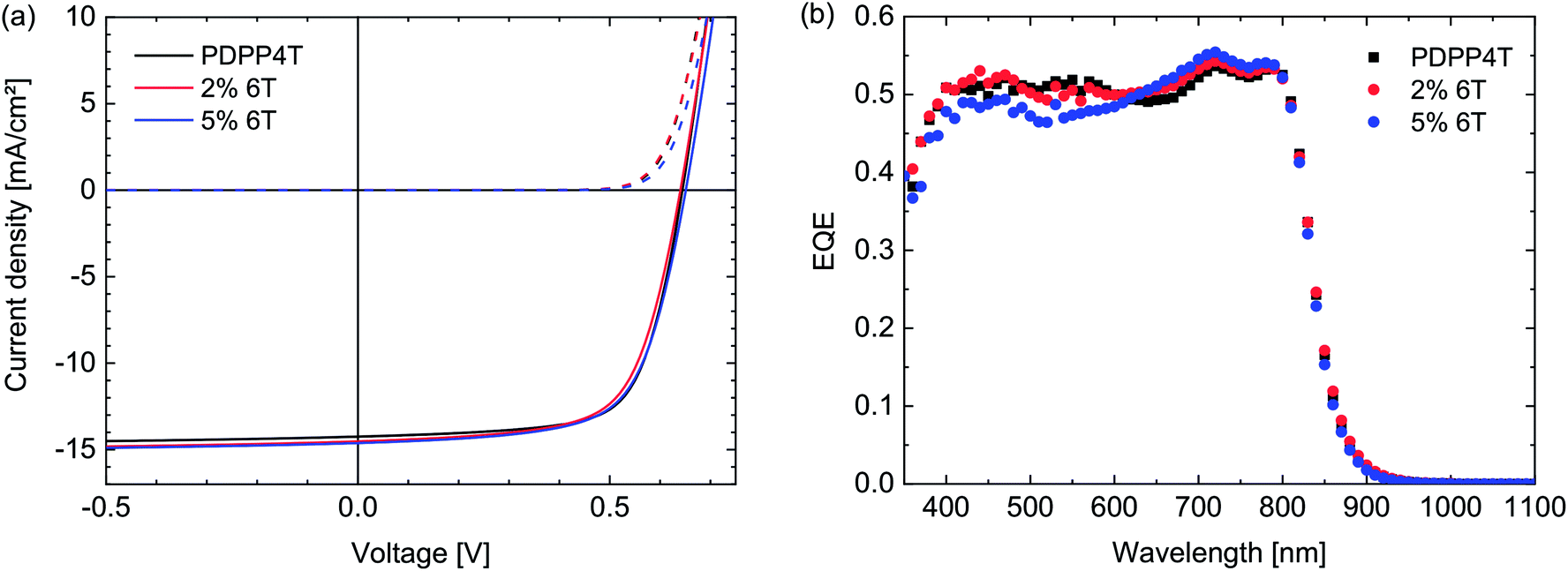 | ||
| Fig. 4 (a) J–V-characteristics of PDPP4T:[70]PCBM (1:2 w/w) polymer solar cells in which the PDPP4T polymers contain 6T defects (see legends). (b) Corresponding EQE spectra. | ||
| Polymer | Mn [kDa] | Jsc [mA cm−2] | Voc [V] | FF | PCE [%] |
|---|---|---|---|---|---|
| a Best cells are shown, numbers between parentheses refer to the average over 4 cells. | |||||
| PDPP4T | 83 | 15.3 (15.2) | 0.64 (0.64) | 0.69 (0.66) | 6.8 (6.4) |
| 2% 6T | 79 | 15.4 (15.0) | 0.64 (0.64) | 0.66 (0.66) | 6.5 (6.3) |
| 5% 6T | 79 | 15.0 (14.8) | 0.65 (0.64) | 0.66 (0.66) | 6.5 (6.3) |
2.3 Model homocoupling reactions
In the previous sections is was established that deliberately introducing homocoupling defects exerts no consistent effect on the optical and photovoltaic properties of the resulting PDPP3T and PDPP4T polymers in concentrations up to 5% or maybe even 20%. Hence, from these data it is not possible to estimate the amount of naturally occurring defect during the synthesis and the question arises how much homocoupling occurs in these Stille polymerizations. To address this question, the following experiment was conceived. A monobrominated DPP monomer, 3-(5-bromothiophen-2-yl)-2,5-bis(2-decyltetradecyl)-6-(thiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (4), was reacted with 5,5′-bis(trimethylstannyl)-2,2′-bithiophene (3) (Scheme 4). The mixture of reactants was prepared and dissolved before being separated in three flasks. One flask was treated in the same way as Stille polymerizations are usually conducted: 1.5% of tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3) and 6% of triphenylphosphine (PPh3) were added before being degassed with argon, sealed, and heated. Another flask was subjected to the same treatment except that it was not degassed, and ambient air was allowed to remain in the flask. This was considered as a simulation of an error or oversight during normal polymerization. To the last flask 3% of bis(triphenylphosphine)palladium(II) dichloride (Pd2(PPh3)2Cl2) was added as a catalyst, before the mixture was degassed with argon, sealed, and heated. This was used as a reference, as the presence of 3% of Pd(II) should cause around 3% of homocoupling defect if all Pd(II) is converted to Pd(0).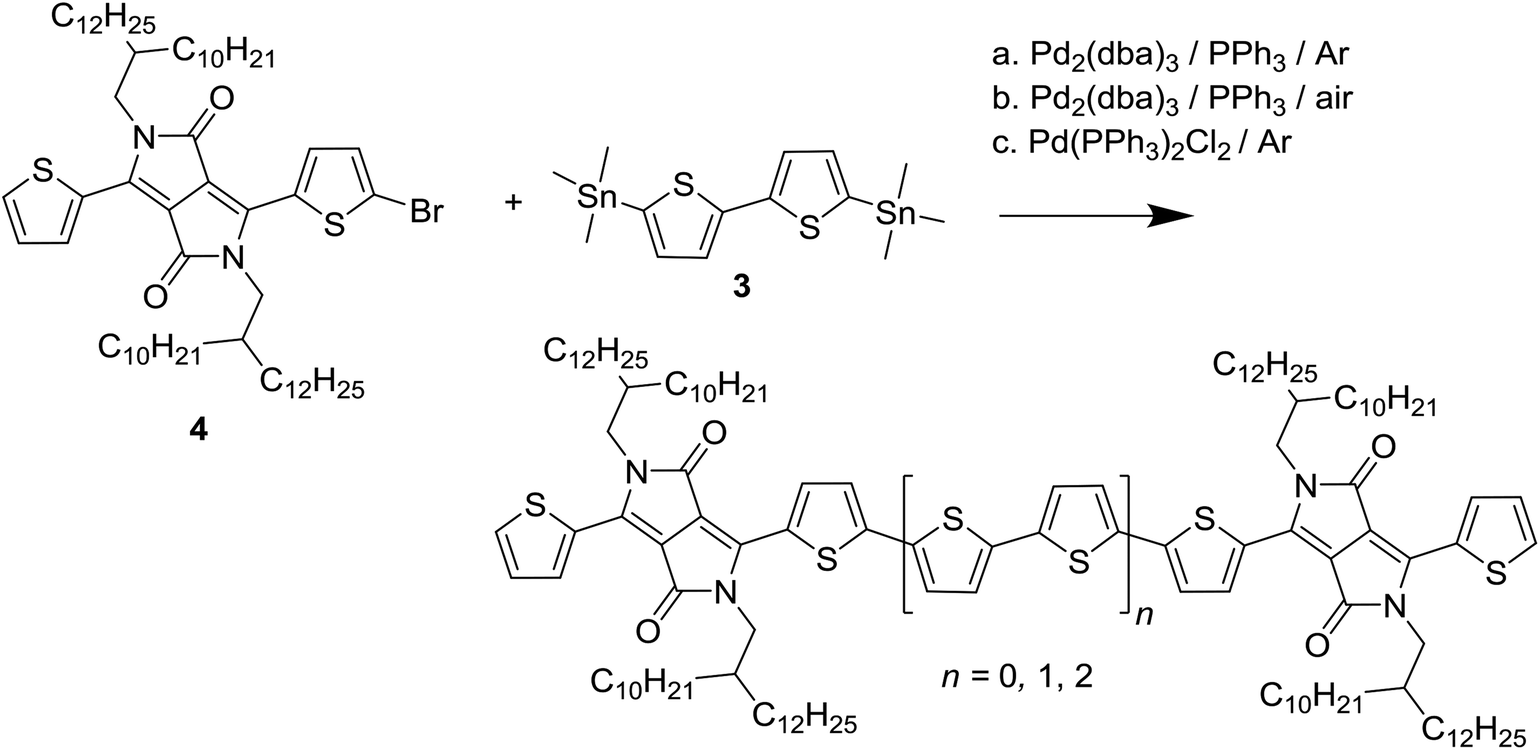 | ||
| Scheme 4 Model homocoupling Stille reaction under three different catalytic conditions. The main product is n = 1. Homocoupling of 4 will result in the product with n = 0, while homocoupling of 3 and subsequent reactions with 4 will result in the product with n = 2. | ||
All reactions were left overnight and the crude products were isolated without any purification. MALDI-TOF-MS was then used to investigate the composition of the product mixtures. Fig. 5c shows that in the case of the reaction with Pd(II) as catalyst, the main peak, belonging to the product with four thiophene rings between DPP cores (n = 1, m/z = 2107 amu), is flanked by peaks at 2271 and 1943 amu. These peaks correspond to the products with six (n = 2) and two (n = 0) thiophene rings in between the DPP moieties. Assuming that the ionization efficiency of these three species is the same, which is likely due to their similar chemical structures, the intensity of the peaks relative to the main peak can indicate the concentration of these species. The relative intensity of the 6T product is 2.8% is due to homocoupling of 3 and in good agreement with the expected value of 3% when all Pd(II) catalyst is reduced to Pd(0). The 2T product, which is the result of homocoupling of (4), is present in 3.0% relative intensity. The presence of this product is slightly unexpected but possibly due to the age of the catalyst.
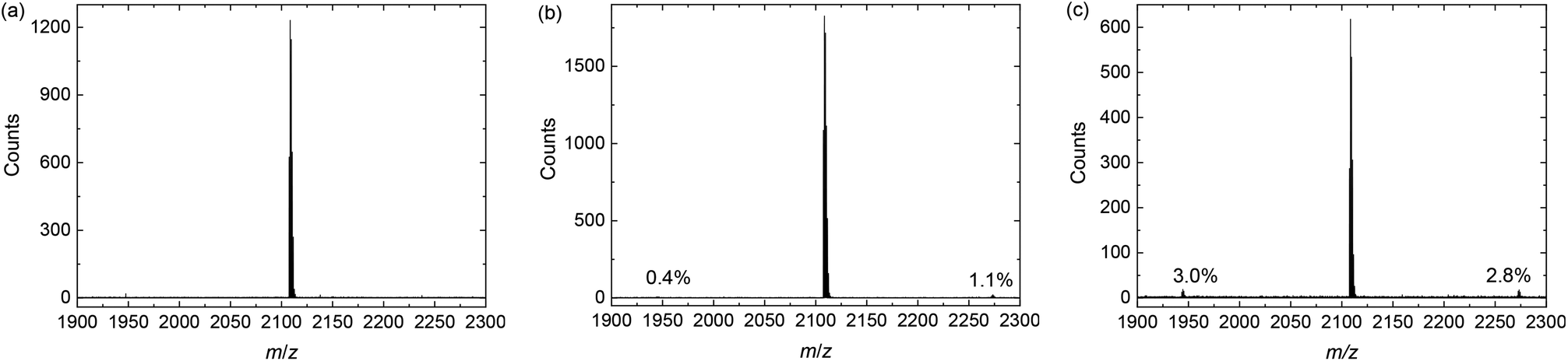 | ||
| Fig. 5 MALDI-TOF-MS of the homocoupling model reaction mixtures (Scheme 4). (a) With standard conditions. (b) With air. (c) With Pd(II). | ||
Repeating this analysis for the degassed sample and Pd(0) catalyst (Fig. 5a), it is clear that no homocoupling of any kind can be detected above the noise level. This indicates that if any homocoupling is present in this sample, its relative concentration is under 0.5%. For the Pd(0) catalyzed reaction exposed to ambient air (Fig. 5b), both kinds of homocoupling can be detected: 1.1% of the trialkylstannyl-based homocoupling product (n = 2 in Scheme 4) is present, which suggests that oxidation of palladium can occur when the reaction mixture is exposed to air. Also, 0.4% of the bromide-based homocoupling (n = 0 in Scheme 1) can be observed, indicating that exposure to air can also influence this coupling mechanism. This is possibly due to oxidation of the triphenylphosphine ligand, leading to an imbalance in the ligand to palladium ratio, which was proven before to have an effect on this type of homocoupling.16 Fig. 6 shows details of the MALDI-TOF mass spectra in the region where homocoupling products n = 0 and n = 2 are expected.
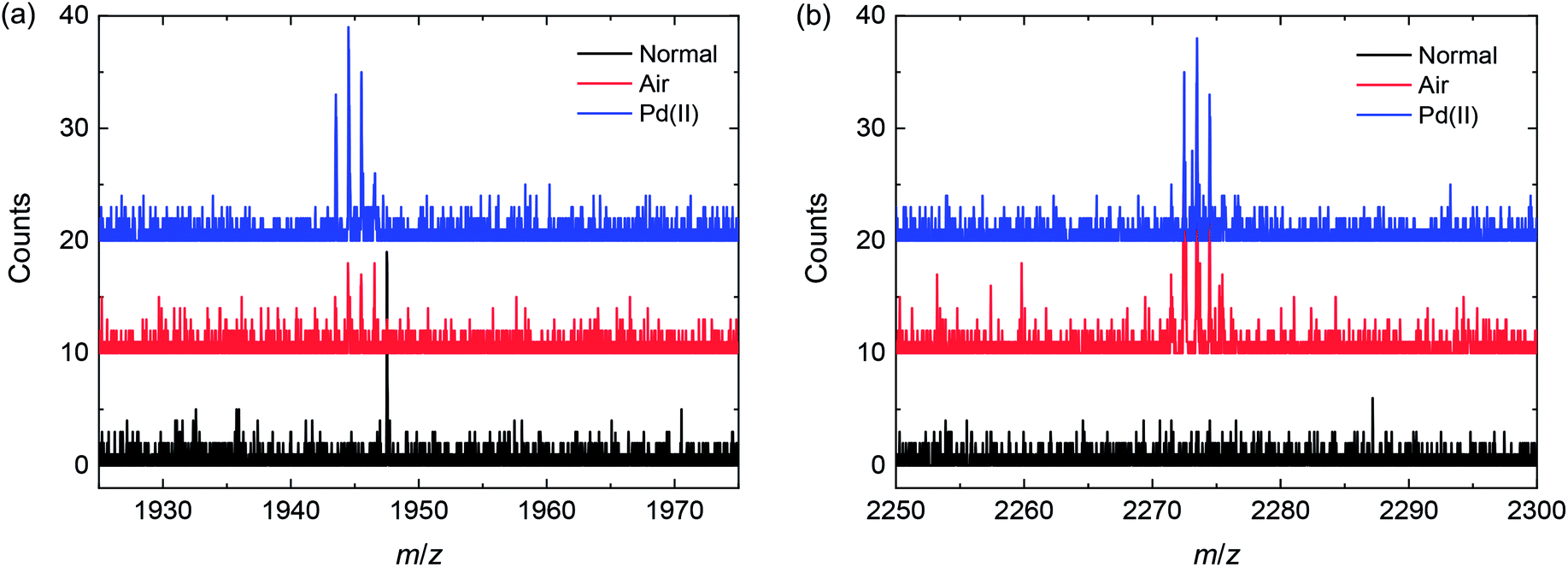 | ||
| Fig. 6 Detail of the MALDI-TOF mass spectra of the three mixtures, showing the peaks corresponding to the product with two central thiophene rings (n = 0 in Scheme 4) (left) and the product with six central thiophene rings (n = 2 in Scheme 4) (right). | ||
3. Conclusion
By deliberately creating defects in the monomers of donor–acceptor DPP polymers, we investigated the effect of homocoupling reactions between two organometallic trialkylstannyl monomers in palladium-catalyzed cross-coupling polymerization reactions. In the case of PDPP3T, the 4T defects that were introduced seemed to have little effect on the optoelectronic properties of the resulting materials. Also in solar cells no consistent effect of the homocoupling defects on the photovoltaic performance was observed, even though the defect-containing polymers generally gave a reduced performance as a result of a lower short-circuit current density than the defect-free polymers of comparable molecular weight. In all cases open-circuit voltage and fill factor were identical. The variation in short-circuit current density is not uncommon for different batches of the same polymer and can result from variations in molecular weight and polydispersity, which often affect the nanomorphology, or from other factors such as the presence of impurities. The results suggests that even for large concentrations (at least up to 10%), the effects of these defects are smaller than common batch-to-batch variations. In the case of PDPP4T, the 6T defects also did not have any measurable effect on the energy levels of the polymer. The solar cells all showed very similar performance, thus excluding that the 6T segments had any large effect on the morphology of the layer.With a model reaction, the naturally occurring amount of homocoupling during a Stille reaction was investigated. It was found that under optimum reaction conditions no homocoupling of trialkylstannyl monomers could be detected and that the level of defects is therefore below 0.5%. This validates the experiments with the intentionally introduced defects, by confirming that the naturally occurring defect level is far below the amounts that were deliberately added to the polymers. It was also found that exposure to air could induce 1.1% of homocoupling defects, thus confirming that this side reaction could easily take place if mistakes or inaccuracies occur during the polymerization reaction. Interestingly, also 0.4% homocoupling of the bromide monomer was detected, indicating that air exposure could also influence this side-reaction.
In previous work we have shown that in the case of homocoupling of dibromide monomers (Br-T-DPP-T-Br) in the synthesis of DPP polymers, the resulting T-DPP-2T-DPP-T defects have a lower band gap and constitute traps for excitons and charges. In contrast, in organometallic-based homocoupling of bis(trialkylstannyl) monomers (Sn-nT-Sn), the DPP units in the resulting DPP-T-2nT-T-DPP chain defects will be further apart. Hence their interaction is weakened, such that these defects will not give rise to traps in the same way. This was confirmed by the defect-containing polymers because even with 20% of defects no change in the optoelectronic properties or the Voc could be observed. As it is clear from the PDPP4T polymers that homocoupling defects, even 6T segments with significantly lower solubility, up to 5% are unlikely to have a significant impact on morphology, the conclusion must be that these homocoupling defects can be ignored when assessing the photovoltaic performance of these DPP polymers. We emphasize that this is not a general conclusion, but depends on the actual chemical structure of the polymer main chain and the precise nature of the defect. As an example, if the functional groups on the reaction components would be inverted, i.e. a DPP unit incorporated in the bis(trialkylstannyl) monomer in a polymerization reaction with dibromodithiophene, the defect resulting from organometallic-based homocoupling will have a large impact on the performance of the resulting PDPP4T. Also, Sommer et al. established a large impact of carbazole homocoupling on the photovoltaic performance in PCDTBT polymers with [70]PCBM.23 Sommer et al. showed that mainly the JSC and FF are affected, while the VOC remained essentially unchanged. The exact reason could not be identified. In PCDTBT, the carbazole units bear 1-octylnonyl sidechains. Hence homocoupling of the carbazoles could increase the solubility of the polymers and give rise to an unfavorable more mixed morphology.3
Summarizing, defects that can be associated with homocoupled trialkylstannyl thiophene monomers do not give rise to significant change of the opto-electronic properties of DPP based polymers PDPP3T and PDPP4T. Moreover, using the optimized synthesis protocol for DPP polymers, we find no evidence of significant homocoupling of any kind. We stress that this conclusion holds for the specific cases investigated and that for other donor–acceptor polymers the result could be different.
4. Experimental section
4.1 Materials and methods
Unless indicated otherwise, all synthetic procedures were performed under a protective argon atmosphere. Commercial (dry) solvents and reactants were used without further purification, unless stated otherwise. N-bromosuccinimide (NBS) was recrystallized from deionized water prior to use. 2,5-Bis(trimethylstannyl)thiophene (2), 5,5′-bis(trimethylstannyl)-2,2′-bithiophene (3), and triphenylphosphine (PPh3) were recrystallized from methanol prior to polymerization. Tris(dibenzylideneacetone)dipalladium (Pd2(dba)3) and bis(triphenylphosphine)palladium(II) dichloride (Pd(PPh3)2Cl2) were purchased from Strem Chemicals, Inc., [70]PCBM (purity 90–95%) was purchased from Solenne BV. All other chemicals and solvents were obtained from Sigma-Aldrich Co. 3,6-Bis(5-bromothiophen-2-yl)-2,5-bis(2-hexyldecyl)-2,5-pyrrolo[3,4-c]pyrrole-1,4-dione (1),26 3-(5-bromothiophen-2-yl)-2,5-bis(2-decyltetradecyl)-6-(thiophen-2-yl)-2,5-pyrrolo[3,4-c]pyrrole-1,4-dione (4),27 and 3,6-bis(5-bromothiophen-2-yl)-2,5-bis(2-decyltetradecyl)-2,5-pyrrolo[3,4-c]pyrrole-1,4-dione (8)28 were synthesized according to previously published procedures.1H-NMR (400 MHz) spectra were recorded on a Varian Mercury spectrometer. Chemical shifts are given in ppm with respect to tetramethylsilane as internal standard. Matrix assisted laser desorption ionization time of flight (MALDI-TOF) mass spectrometry was measured on a Bruker Autoflex Speed spectrometer. For the analysis of the model homocoupling reaction, trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) was used as matrix for all samples. Polymer molecular-weight distributions were estimated by GPC at 140 °C on a PL-GPC 120 system using a PL-GEL 10 mm MIXED-C column with 1,2-dichlorobenzene (o-DCB) as the eluent and using polystyrene internal standards. Samples were dissolved at a concentration of 0.1 mg ml−1 in o-DCB at 140 °C for 1 h before being measured.
UV-vis-NIR absorption spectroscopy was conducted on a PerkinElmer Lambda 1050 spectrophotometer with a 3D WB PMT/InGaAs/PbS detector module. Solid films were obtained by spin coating (2000 rpm) solutions of the materials (4 mg ml−1) in chloroform onto glass substrates which were pre-cleaned with acetone and isopropanol, before being treated with UV–ozone for 30 min. Square wave voltammetry was measured on the polymers in solid state which were deposited onto a platinum wire by dipping the wire in the hot solutions (chloroform). A silver rod was employed as counter electrode and a silver chloride coated silver rod (Ag/AgCl) was used as a quasi-reference electrode. 0.1 M tetrabutylammonium hexafluorophosphate in acetonitrile was used as electrolyte solution. The measurement was carried out under inert atmosphere with an AutoLab PGSTAT 12 at a scan speed of 0.125 V s−1, a modulation amplitude of 20 mV, and at a frequency of 25 Hz. Ferrocene/ferrocenium (Fc/Fc+) was employed as a standard with E = − 4.8 eV.
Photovoltaic devices with an active area of 0.09 and 0.16 cm2 were fabricated in air on patterned indium tin oxide (ITO) glass substrates (Naranjo Substrates). The substrates were cleaned by sonication in acetone for 15 min, followed by scrubbing with a sodium dodecyl sulfate solution (99%, Acros), rinsing with deionized water, and a final sonication step in 2-propanol. Before deposition of the device layers the substrates underwent a 30 min. UV–ozone treatment. Clevios PEDOT:PSS Al 4083 (Heraeus), was deposited by spin coating at 3000 rpm. The active layers were deposited from a solution of polymer (4 mg ml−1) and [70]PCBM (8 mg ml−1) in chloroform containing 7.5% of o-DCB in the case of the PDPP3T polymers, and 10% of o-DCB in the case of the PDPP4T polymers. The solutions were heated to 90 °C for one hour to ensure that the polymers were completely dissolved, then kept at 60 °C, and were finally cooled to room temperature under vigorous stirring during 2 min. Before spin coating at 2000 or 3000 rpm. LiF (1 nm) and Al (100 nm) were deposited by thermal evaporation under high vacuum (∼3 × 10−7 mbar) as a back contact.
Current density–voltage (J–V) characteristics were measured with a Keithley 2400 source meter under ∼100 mW cm−2 white light illumination from a tungsten–halogen lamp filtered by a Schott GG385 UV filter and a Hoya LB120 daylight filter. The short-circuit current density (Jsc) was determined more accurately from external quantum efficiency (EQE) measurements by integration of the EQE with the AM1.5 G solar spectrum. EQE measurements were carried out under 1 sun equivalent operating conditions in a setup consisting of a modulated monochromatic light, a preamplifier (Stanford Research Systems SR570), and a lock-in amplifier (Stanford Research Systems, SR830). The modulated monochromatic light was generated by using an optical chopper (Stanford Research Systems, SR540), a monochromator (Oriel Cornerstone 130) and a 50 W (Osram 64610) tungsten–halogen lamp. The 1 sun conditions were provided by the use of a 730 nm light-emitting diode (Thorlabs) at different intensities for appropriate bias illumination. The devices were kept in a nitrogen-filled box with a quartz window. A calibrated silicon cell was used as reference for J–V and EQE measurements.
4.2 Synthesis
:1). The mixture was degassed with argon for 15 min. And then sealed in the tube for reaction at 115 °C overnight. 1,1,2,2-Tetrachloroethane (TCE) was then added to dissolve and dilute the mixture before it was precipitated in methanol. The solids were then filtered, dissolved in TCE at 110 °C and stirred with ethylenediaminetetraacetic acid (EDTA) for one hour after which water was added and the mixture was stirred for another hour. The organic layer was then separated, washed twice with water and partially reduced in volume under reduced pressure. The polymer was then precipitated in methanol and the solids were subjected to Soxhlet extraction with subsequently acetone, hexane, and dichloromethane. The remaining solids were then dissolved in TCE, the solution was filtered and precipitated in acetone. The polymers were then filtered and dried at 40 °C in vacuum overnight.:1) (1.5 ml). GPC (o-DCB, 140 °C): Mn = 148 kDa, Mw = 399 kDa, Đ = 2.7.:1) (1.5 ml). GPC (o-DCB, 140 °C): Mn = 123 kDa, Mw = 330 kDa, Đ = 2.7.:1) (1.5 ml). GPC (o-DCB, 140 °C): Mn = 148 kDa, Mw = 399 kDa, Đ = 2.7.:3 heptane:dichloromethane, gradient to 5:5) and the obtained solid was recrystallized in methanol to yield the product as a blue solid (78 mg, yield 48%). 1H NMR (400 MHz, CDCl3) δ: 8.92 (d, J = 4.2 Hz, 1H), 8.87 (dd, J = 3.9, 1.2 Hz, 1H), 7.61 (dd, J = 5.0, 1.2 Hz, 1H), 7.30 (d, J = 4.2 Hz, 1H), 7.26 (m, 2H), 7.22 (m, 2H), 7.13 (d, J = 3.9 Hz, 1H), 7.05 (dd, J = 5.1, 3.6 Hz, 1H), 4.03 (d, J = 7.6 Hz, 4H), 1.94 (m, 2H), 1.40–1.11 (m, 80H), 0.87 (td, J = 6.3, 3.3 Hz, 12H).:6 heptane:dichloromethane, gradient to 5:5) and recrystallized three times from methanol and once from acetone to achieve maximum purity. The product was isolated as a blue solid (30 mg, yield 40%). 1H NMR (400 MHz, CDCl3) δ: 8.92 (d, J = 4.2 Hz, 1H), 8.62 (d, J = 4.2 Hz, 1H), 7.30 (d, J = 4.1 Hz, 1H), 7.22 (d, J = 4.0 Hz, 2H), 7.06 (d, J = 3.8 Hz, 1H), 7.00 (d, J = 3.9 Hz, 1H), 6.96 (d, J = 3.8 Hz, 1H), 4.02 (d, J = 7.6 Hz, 2H), 3.95 (d, J = 7.7 Hz, 2H), 1.94 (m, 2H), 1.40–1.15 (m, 80H), 0.91–0.82 (m, 12H). MALDI-TOF-MS: predicted: 1292.55, found: 1292.58.:1). The mixture was degassed with argon for 15 min. And then sealed in the tube for reaction at 115 °C overnight. TCE was then added to dissolve and dilute the mixture before being precipitated in methanol. The solids were then filtered, dissolved in TCE at 110 °C, and stirred with EDTA for one hour after which water was added and the mixture was stirred for another hour. The organic layer was then separated, washed twice with water, and partially reduced in volume under reduced pressure. The polymer was then precipitated in methanol and the solids were subjected to Soxhlet extraction with subsequently acetone, hexane, and dichloromethane. The remaining solids were then dissolved in TCE, the solution was filtered and precipitated in acetone. The polymers were then filtered and dried at 40 °C in vacuum overnight.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Koen Hendriks for valuable discussions. G. H. L. Heintges acknowledges the Agency for Innovation by Science and Technology in Flanders (IWT). This project has further received funding from the European Research Council under the European Union's Seventh Framework Programme (FP/2007–2013)/ERC Grant Agreement No. 339031 and from the Ministry of Education, Culture and Science (Gravity program 024.001.035).References
- A. Katsouras, N. Gasparini, C. Koulogiannis, M. Spanos, T. Ameri, C. J. Brabec, C. L. Chocos and A. Avgeropoulos, Macromol. Rapid Commun., 2015, 36, 1778–1797 CrossRef CAS PubMed.
- K. H. Hendriks, G. H. L. Heintges, V. S. Gevaerts, M. M. Wienk and R. A. J. Janssen, Angew. Chem., Int. Ed., 2013, 52, 8341–8344 CrossRef CAS PubMed.
- J. J. van Franeker, G. H. L. Heintges, C. Schaefer, G. Portale, W. Li, M. M. Wienk, P. van der Schoot and R. A. J. Janssen, J. Am. Chem. Soc., 2015, 137, 11783–11794 CrossRef CAS PubMed.
- Z. B. Henson, K. Müllen and G. C. Bazan, Nat. Chem., 2012, 4, 699–704 CrossRef CAS PubMed.
- J. K. Park, J. Jo, J. H. Seo, J. S. Moon, Y. D. Park, K. Lee, A. J. Heeger and G. C. Bazan, Adv. Mater., 2011, 23, 2430–2435 CrossRef CAS PubMed.
- G. H. L. Heintges, J. J. van Franeker, M. M. Wienk and R. A. J. Janssen, Chem. Commun., 2016, 52, 92–95 RSC.
- C. Bracher, H. Yi, N. W. Scarratt, R. Masters, A. J. Pearson, C. Rodenburg, A. Iraqi and D. G. Lidzey, Org. Electron., 2015, 27, 266–273 CrossRef CAS.
- M. P. Nikiforov, B. Lai, W. Chen, S. Chen, R. D. Schaller, J. Strzalka, J. Maser and S. B. Darling, Energy Environ. Sci., 2013, 6, 1513–1520 RSC.
- W. R. Mateker, J. D. Douglas, C. Cabaneto, I. T. Sachs-Quintana, J. A. Bartelt, E. T. Hoke, A. El Labban, P. Beaujuge, J. M. J. Fréchet and M. D. McGehee, Energy Environ. Sci., 2013, 6, 2529–2537 RSC.
- I. Osaka and R. D. McCullough, Acc. Chem. Res., 2008, 41, 1202–1214 CrossRef CAS PubMed.
- L. Ying, F. Huang and G. C. Bazan, Nat. Commun., 2017, 8, 14047 CrossRef CAS PubMed.
- F. Koch and W. Heitz, Macromol. Chem. Phys., 10097, 198, 1531–1544 CrossRef.
- V. Farina, B. Krishnan, D. R. Marshall and G. P. Roth, J. Org. Chem., 1993, 58, 5434–5444 CrossRef CAS.
- F. Lombeck, H. Komber, S. I. Gorelsky and M. Sommer, ACS Macro Lett., 2014, 3, 819–823 CrossRef CAS.
- G. Pirotte, P. Verstappen, D. Vanderzande and W. Maes, Adv. Electron. Mater., 2018, 4, 1700481 CrossRef.
- K. H. Hendriks, W. Li, G. H. L. Heintges, G. W. P. van Pruissen, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2014, 136, 11128–11133 CrossRef CAS PubMed.
- W. Hong, S. Chen, B. Sun, M. A. Arnould, Y. Meng and Y. Li, Chem. Sci., 2015, 6, 3225–3235 RSC.
- D. A. Warr, L. M. A. Perdigão, H. Pinfold, J. Blohm, D. Stringer, A. Leventis, H. Bronstein, A. Troisi and G. Costantini, Sci. Adv., 2018, 4, eaas9543 CrossRef PubMed.
- S. Ostrowska, S. Rogalski, J. Lorkowski, J. Walkowiak and C. Pietraszuk, Synlett, 2018, 29, 1735–1740 CrossRef CAS.
- C. A. Contreras-Celedón, J. A. Rincón-Medina, D. Mendoza-Rayo and L. Chacón-Garcia, Appl. Organomet. Chem., 2015, 29, 439–442 CrossRef.
- H. Bohra and M. Wang, J. Mater. Chem. A, 2017, 5, 11550–11571 RSC.
- M. Moreno-Mañas, M. Perez and R. Pleixats, J. Org. Chem., 1996, 61, 2346–2351 CrossRef.
- F. Lombeck, H. Komber, D. Fazzi, D. Nava, J. Kuhlmann, D. Stegerer, K. Strassel, J. Brandt, A. Diaz, Z. Mendaza, C. Müller, W. Thiel, M. Caironi, R. Friend and M. Sommer, Adv. Energy Mater., 2016, 6, 1601232 CrossRef.
- B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493–1528 CrossRef CAS PubMed.
- W. Li, R. S. C. Roelofs, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2012, 134, 13787–13795 CrossRef CAS PubMed.
- L. Bürgi, M. Turbiez, R. Pfeiffer, F. Bienewald, H. J. Kirner and C. Winnewisser, Adv. Mater., 2008, 20, 2217–2224 CrossRef.
- E. Y. Ko, G. E. Park, D. H. Lee, H. A. Um, J. Shin, M. J. Cho and D. H. Choi, ACS Appl. Mater. Interfaces, 2015, 7, 28303–28310 CrossRef CAS PubMed.
- S. Cho, J. Lee, M. H. Tong, J. H. Seo and C. Yang, Adv. Funct. Mater., 2011, 21, 1910–1916 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |