
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of a novel phosphoinositide 3-kinase gamma (PI3Kγ) inhibitor against hematologic malignancies and theoretical studies on its PI3Kγ-specific binding mechanisms†
Jingyu Zhu‡
 *a,
Ke Ke‡a,
Lei Xub and
Jian Jin*a
*a,
Ke Ke‡a,
Lei Xub and
Jian Jin*a
aSchool of Pharmaceutical Sciences, Jiangnan University, Wuxi, Jiangsu 214122, China. E-mail: jingyuzhu@jiangnan.edu.cn; jianjin@jiangnan.edu.cn
bInstitute of Bioinformatics and Medical Engineering, School of Electrical and Information Engineering, Jiangsu University of Technology, Changzhou 213001, China
First published on 28th June 2019
Abstract
Class IB phosphoinositide 3-kinase gamma (PI3Kγ) is vital for regulating intracellular signaling pathways and has become an attractive drug target for the treatment of malignant tumors. In the present study, one potent PI3Kγ inhibitor (JN-PK1) with a novel scaffold against hematologic malignancies was identified based on a series of biological experiments, and then the selective mechanism of PI3Kγ inhibition was explored by a systematic computational method. JN-PK1 shows an effective antiproliferative activity on several cancer cell lines, especially blood cancer cells. Cell-free enzymatic studies demonstrated that JN-PK1 specifically inhibits PI3Kγ at low micromolar concentrations without affecting other isoforms of PI3K. In the cellular context, JN-PK1 potently inhibits PI3K/Akt/mTOR signaling pathway in a time- and concentration-dependent manner, which leads to the apoptosis of cancer cells. Further, the specific binding mode of JN-PK1 with PI3Kγ was illustrated by molecular docking, and the selective inhibition mechanism of PI3Kγ by JN-PK1 was revealed by molecular dynamics simulation. Finally, some key residues of PI3Kγ required for specificity and activity were identified. Taken together, JN-PK1 may be developed as a promising therapeutic agent for the treatment of hematologic malignancies.
1. Introduction
Phosphoinositide 3-kinases (PI3Ks) are a family of lipid kinases which play critical roles in many cellular processes, such as cell survival, proliferation, differentiation, and motility. PI3Ks are mainly grouped into three classes (Class I, II, and III) according to their structure and lipid substrate specificity. Among these classes, Class I PI3Ks are the most studied to date because of their frequent dysregulation in various cancers.1–6 Class I PI3Ks, which are further divided into Class IA (PI3Kα, PI3Kβ, PI3Kδ) and IB (PI3Kγ), have the ability to phosphorylate PIP2 (phosphatidylinositol 4,5-bisphosphate), converting it to PIP3 (phosphatidylinositol 3,4,5-trisphosphate).7 PIP3 recruits a series of downstream signaling proteins, such as Akt (protein kinase B), to the plasma membrane and activates them by directly binding to their pleckstrin homology (PH) domains.8 Extensive research conducted over the past few years highlights the association of deregulated Class I PI3K signaling with various cancers and that depicts PI3Ks as novel therapeutic targets. Therefore, a large number of PI3K inhibitors have been developed and reported over the last couple decades.9 Recently, more attention has been drawn to the development of PI3K isoform-selective inhibitors. Idelalisib (CAL-101), the first marketed selective PI3Kδ inhibitor, was successfully approved by the FDA in 2014 for the treatment of chronic lymphocytic leukemia and small lymphocytic lymphoma.10 More interestingly, the expression of PI3Kα/β is ubiquitous, while PI3Kδ/γ are restricted expressed in hematopoietic cells.11,12 Due to this, PI3Kδ and PI3Kγ may have more therapeutic potential for hematologic malignancies.12–17 Recently, Duvelisib (IPI-145), a dual PI3Kδ/γ inhibitor, entered the market in 2018,18 which will further promote the development of PI3Kγ selective inhibitors.The only member of class IB, PI3Kγ, is mainly activated by GPCRs (G-protein-coupled receptors). PI3Kγ is necessary for growth factors signaling, chemokine-stimulated chemotaxis, blocking stimulated T cell recruitment into tumors, and regulating innate immunity during inflammation and tumorigenesis.19–22 Additionally, the restriction of PI3Kγ expression to the hematopoietic system may limit the toxicity of specific inhibitors compared to pan-PI3K inhibitors. In recent years, more and more small molecule inhibitors acting on PI3Kγ have been discovered, some of which have entered clinical trials for hematologic malignancies or solid tumors, such as the specific PI3Kγ inhibitors AS604850,23 CAY10505,24 AS252424,25 CZC24832,26 and some dual PI3Kδ/γ inhibitors IPI-549 (NCT03795610),27 TG100-115,28 RP6530 (NCT02017613).29–31 However, the selective PI3Kγ inhibitors are far less developed than other PI3K isoforms inhibitors, because PI3Kγ shares high sequence similarity in the conserved ATP-kinase binding site with the Class IA isoforms, that directly leads to the great difficulty in the development of specific γ isoform inhibitors.32 Thus in this study, a high throughput virtual screening was preformed to screen the ChemBridge database, and as a result, 83 compounds were purchased and then submitted to bioassays. Finally, the novel PI3Kγ selective inhibitor JN-PK1, 1,1′-(9-methyl-9H-carbazole-3,6-diyl)diethanone, was identified for the treatment of hematologic malignancies, which was confirmed using a range of biochemical and cellular studies. Cell-free assays exhibited that JN-PK1 selectively inhibits PI3Kγ isoform. On a cellular level, JN-PK1 showed potent anti-proliferation activity against several hematologic cancer cell lines, and specifically inhibited the PI3K signaling pathway. Meanwhile, a computational investigation on this inhibitor integrating molecular docking and molecular dynamics (MD) simulation was conducted, and lastly, the selective PI3Kγ inhibition mechanism of this inhibitor was highlighted.
2. Materials and methods
2.1 Antibodies and chemicals
Phospho-Akt (Thr308) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000, cat# 38449; Abacm), Phospho-Akt (Ser473) (1:1000, cat# 81283; Abacm), Akt (1:1000, cat# sc-81434; Santa Cruz Biotechnologies), Phospho-mTOR (Ser2448) (1:200, cat# sc-293133; Santa Cruz Biotechnologies), mTOR (1:200, cat# sc-517464; Santa Cruz Biotechnologies), caspase-3 (1:1000, cat# sc-56053; Santa Cruz Biotechnologies), GAPDH (1:5000, cat# 60004-1-Ig; Santa Cruz Biotechnologies), PARP (1:1000, cat# 191217; Abacm).
:1000, cat# 38449; Abacm), Phospho-Akt (Ser473) (1:1000, cat# 81283; Abacm), Akt (1:1000, cat# sc-81434; Santa Cruz Biotechnologies), Phospho-mTOR (Ser2448) (1:200, cat# sc-293133; Santa Cruz Biotechnologies), mTOR (1:200, cat# sc-517464; Santa Cruz Biotechnologies), caspase-3 (1:1000, cat# sc-56053; Santa Cruz Biotechnologies), GAPDH (1:5000, cat# 60004-1-Ig; Santa Cruz Biotechnologies), PARP (1:1000, cat# 191217; Abacm).
All compounds were purchased from ChemBridge Corporation (San Diego, CA). PI-103 was from Selleck (Shang Hai, China). The Annexin V-FITC/PI Apoptosis Detection Kit was from BD Biosciences (Becton Dickinson, UAS). MTT or 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetra-sodium bromide was from Sigma (St Louis, MO). ADP-Glo Luminescent Assay was purchased from Promega (Madison, Wisconsin, USA). BCA Protein Assay Kit was purchased from Biosharp (China).
2.2 Cell culture
Cell lines (OCI-My5, KMS11, RPMI-8226, U266, and LP1) were maintained with Iscove's modifed Dulbecco's medium (IMDM, Hyclone); cell lines (Jurkat, HL-60, u937, K562, T47D, JF-305, MKN-28, and SGC7901) were maintained in RPMI-1640 medicum (Hyclone); cell lines (Capan-1, Patu8988, SUM102, and MDA-MB-231) were maintained in DMEM medium (Gibco), and all mediums were supplemented with 10% fetal bovine serum (FBS, Gibco), 100 μg mL−1 penicillin and 100 μg mL−1 streptomycin, and all cells were cultured at 37 °C in an incubator supplied with 5% CO2.2.3 Cell growth assays
Cell growth was assessed by MTT assay. In a 96-well plate, solid tumor cells (MKN-28, SGC7901, JF-305, Capan-1, Patu8988, SUM102, T47D, and MDA-MB-231) were plated at a concentration of 3 × 103 cells in 100 μL medium, and suspension cells (OCI-My5, Jurkat, KMS11, HL-60, and u937) were plated with a concentration of 8 × 103 cells in 100 μL medium. Cells were then treated with 5 μL of 10 μM compound. After 72 h, 10 μL of 5 mg mL−1 MTT was added to each well and cells were incubated at 37 °C for 4 h. Subsequently, 100 μL MTT reagent was added to each well. On alternate days, the absorbance of each well was measured at 550 nm using a microplate reader. All data is shown as the percent viability of treated compared to non-treated cells.The anti-proliferative effect of JN-PK1 on eight hematologic cancer cell lines (HL-60, u937, K562, OCI-My5, LP1, KMS11, U266, and RPMI-8226) was assessed after cells were incubated with varying JN-PK1 concentrations (0–25 μM) at 37 °C for 72 h and viability was again measured using the MTT assay.
2.4 PI3K kinase activity in cell-free assays
JN-PK1 activity against Class I PI3K kinases was tested using ADP-Glo Luminescent Assay and PI-10333 was used as the positive control. Both compounds were tested thrice using 10 serial dilutions starting at 10 μM and subsequently diluted 2-fold for each subsequent dilution with 100% DMSO, and reacted with substrate solution (PIP2 at 50 μM, ATP at 25 μM). The two assays were done in 384-well white plates with a total reaction volume of 10 μL per well (2.5 μL of compound, 2.5 μL of PI3Kα at 0.3 μg mL−1, and 5 μL of substrate solution), or 15 μL (2.5 μL of compound, 2.5 μL of PI3Kδ at 1 μg mL−1 or PI3Kβ at 1 μg mL−1 or PI3Kγ at 1.5 μg mL−1, 5 μL of substrate solution, and 5 μL of ADP-Glo reagent). After incubating at room temperature for 1 h, the 10 μL Glo reagent assay was added to each well to stop the reaction. At the end, each plate was read for luminescence.
2.5 Immunoblotting analysis
800 rpm for 20 min at 4 °C. The supernatant was collected, and the total protein concentration was quantified using a BCA Protein Assay Kit. Loading buffer (5×) was added to all protein samples isolated for Western blot analysis and then boiled at 100 °C for 5 min.2.6 Flow cytometry assay
Using Annexin V-FITC/PI detection kits (Beyotime Company), OCI-My5 and HL-60 cells treated with JN-PK1 for 24 h were detected on a flow cytometer, and the apoptosis rates were measured according to the manufacturer's instructions.2.7 Docking studies
The 3D structure of JN-PK1 was sketched and minimized using maestro in Schrödinger. Then JN-PK1 was prepared by Ligprep module with the OPLS-2005 force field, where generated 10 low energy ring conformations per ligand. The crystal structures of four PI3K isoforms (α, β, δ, γ) with high resolution were downloaded from protein data bank (PDB code: 4JPS,34 2Y3A,35 4XEO,36 4ANW36), and prepared as the initial receptors for the docking studies using Protein Preparation Wizard in Schrödinger: removing all crystal water molecules, adding hydrogen atoms, assigning partial charges, terminating the minimization if the RMSD reached the value of 0.3 Å with the OPLS-2005 force field. The grid files of each protein were generated with the ligands as centroids using Glide Grid Generation module.Then JN-PK1 was respectively docked into each grid file of four isoforms using Glide Docking module (XP mode), with writing out 10 poses per ligand while other parameters as defaults. After completing each docking calculation, the best docked structures of JN-PK1 in each receptor–ligand system were chosen to the next analysis.
2.8 MD simulations
The best docked JN-PK1 structures with each PI3K isoforms (α, β, δ, γ) were used as the starting structures for MD simulations with AMBER14.37 The AMBER ff0333 force filed was used for the proteins, the general AMBER force filed (GAFF)39 for the inhibitors and the semi-empirical AM1 method was employed for the optimization of ligands.40 Then the restrained electrostatic potential (RESP) technique was used to fit the electrostatic potential at the HF/6-31G* level41 and the atomic partial charges of ligands were obtained.
Each system was firstly neutralized with sodium ions and then immersed in an octagonal box of TIP3P water molecules which extended 10 Å from solute atoms. The long-range electrostatic interaction was handled by the Particle Mesh Ewald (PME) scheme,42 and the non-bonded cutoff for the van der Waals interaction was set to 10 Å. For relaxing any steric conflicts probably generated during MD simulations, each system was firstly adopted three-stage minimizations: 1000 cycles of minimizations for constraining the backbone carbons (50 kcal Å−2), 1000 cycles of minimizations with a weaker harmonic potential for constraining (10 kcal Å−2) were conducted, 5000 cycles of minimizations containing 1000 cycles of steepest descend and 4000 cycles of conjugate gradient without any constrain were finally performed. The MD simulations were run over 50 ps in the NVT ensemble with the gradually rising temperature from 0 to 300 K, which were finally performed 30000 ps. The time step was set to 2 fs, and all bonds including hydrogen atoms were constrained with the SHAKE algorithm.
2.9 MM/GBSA free energy calculation and decomposition analysis
Each system was calculated the binding free energy (ΔGbind) using the MM/GBSA method according to the following eqn (1).43,44 All components extracted from the last 10 ns stable MD trajectories were calculated with the MM_PBSA program of AMBER 14.
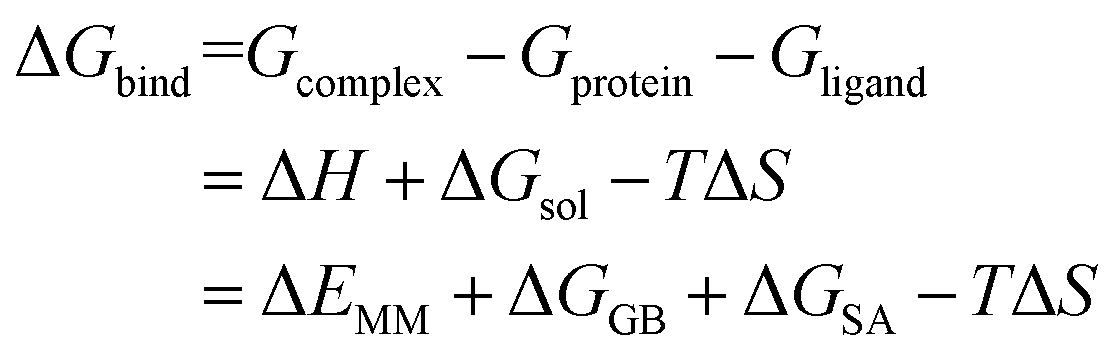 | (1) |
3. Results and discussion
3.1 JN-PK1 potently inhibits the proliferation of various cancer cells
All 83 compounds identified from virtual screening methods (data not shown) were first evaluated for their anti-proliferative activity using MTT assays in different cell lines, including hematologic cancer cell lines (OCI-My5, Jurkat, KMS11, HL-60, and u937) and different solid tumor cell lines (MKN-28, SGC7901, JF-305, Capan-1, Patu8988, SUM102, T47D, and MDA-MB-231). The known pan-PI3K inhibitor PI-103 was used as a positive control. The anti-proliferative activity of each compound is shown as viability, summarized in Fig. 1A. The results showed that some of these compounds effectively inhibited proliferation of these cell lines, such as compounds 1, 43, 65 and 79. Among these compounds, compound 1 (JN-PK1, Fig. 1B) excellently suppressed proliferation in most cancer cell lines, especially hematologic cancer cell lines such as leukemia (HL-60) and multiple myeloma (MM) (OCI-My5). This compound inhibited proliferation more potently than the positive control inhibitor PI-103 (Fig. 1C). | ||
| Fig. 1 (A) The viability values of each compound (from compound 1 to compound 83 and PI-103 as the positive control inhibitor) against 13 kinds of different malignant cell lines. (B) The structure of JN-PK1. (C) The viability values of JN-PK1 and PI-103 against 13 kinds of different malignant cell lines. (D) The viability values of JN-PK1 with gradient of concentrations against different hematologic malignant cell lines. | ||
Next, the eight hematologic tumor cell lines were treated with JN-PK1 (0–25 μM): three leukemia cell lines (HL-60, u937, and K562) and five MM cell lines (OCI-My5, LP1, KMS11, U266, and RPMI-8226). As shown in Fig. 1D, JN-PK1 treatment decreased the proliferation of all hematologic tumor cell lines in a concentration-dependent manner, with the greatest effect on the leukemia cell line HL-60 and the MM cell line RPMI-8226. Because HL-60 and OCI-My5 cells showed the highest sensitivity to JN-PK1 at the low concentration, we chose these two cell lines to verify the inhibition of PI3K by JN-PK1 in leukemia and MM.
3.2 JN-PK1 selectively inhibits PI3Kγ in a cell-free system and effectively inhibits the PI3K signaling pathway
The above results indicate that JN-PK1 effectively inhibits the proliferation of hematologic tumor cells. To determine whether this effect on proliferation is caused by the inhibition of PI3Kγ, the inhibitory potency of JN-PK1 on PI3K enzymes was measured in a cell-free system. The results of the JN-PK1 treatment on four isoforms (PI3Kα, PI3Kβ, PI3Kδ and PI3Kγ) are summarized in Table 1. Compared to PI-103, JN-PK1 showed lower activity on PI3K kinases, but significantly and specifically inhibited PI3Kγ compared to the other isoforms at a lower concentration of 7.5 μM, while there was no significant inhibitory effect on Class IA PI3Ks even at high concentrations. This suggests that JN-PK1 is a selective inhibitor of PI3Kγ.| Isoform | JN-PK1 (μM) | PI-103 (nM) | Glide scores (kcal mol−1) |
|---|---|---|---|
| α | 11.30 ± 1.02 (n = 3) | 5.08 ± 1.23 (n = 3) | −8.282 |
| β | >1000 (n = 3) | 11.07 ± 2.81(n = 3) | −6.713 |
| δ | 391.90 ± 10.09 (n = 3) | 8.93 ± 1.42 (n = 3) | −6.531 |
| γ | 7.49 ± 0.09 (n = 3) | 58.17 ± 1.53 (n = 3) | −8.876 |
To further study whether JN-PK1 can be used to suppress the PI3K/Akt signaling pathway to treat hematologic malignancies, immunoblotting was used to detect the relative expression of proteins in the leukemia cell line HL-60 and MM cell line OCI-My5. It is well established in the literature that PI3K/Akt/mTOR (mammalian target of rapamycin) signaling plays significant roles in regulating survival and proliferation of hematologic cancer cells, and PI3Kγ is frequently overexpressed in different blood cancer cells.47 Therefore, the phosphorylation levels of Akt (p-AktThr308 and Ser473), the key downstream target of PI3K/Akt signaling, were detected after the treatment of JN-PK1. As illustrated in Fig. 2A, the phosphorylation of Akt at Thr308 was first downregulated by JN-PK1, and that of Ser473 site was subsequently inhibited, suggesting that the total phosphorylation of Akt was suppressed by JN-PK1 with the inhibition of PI3Kγ. And then, the phosphorylation level of mTOR (p-mTORSer2448), another key downstream protein, was measured. Fig. 2A shows that the phosphorylation of mTOR was also suppressed upon inhibition of PI3Kγ by JN-PK1 treatment. On the other hand, we found that the phosphorylation of these three proteins was notably decreased at a JN-PK1 concentration of 10 μM, but p-AktThr308 and p-mTORSer2448 decreased at a concentration as low as 5 μM. These results indicate that JN-PK1 remarkably suppresses Akt and mTOR phosphorylation in a concentration-dependent manner, and has no effect on total Akt and mTOR expression. Next, a time-course study revealed that JN-PK1 inhibits p-AktSer473 and especially p-AktThr308 within 6 h after treatment, shown in Fig. 2B. Similarly, p-mTORSer2448 was associatively suppressed after JN-PK1 treatment. Therefore, JN-PK1 inhibits the PI3K pathway in a time-dependent manner. Additionally, as shown in Fig. 2A and B, JN-PK1 suppressed p-Akt and p-mTOR more effectively in HL-60 cells than OCI-My5 cells in a time- and concentration-dependent manner, suggesting that JN-PK1 would more effectively inhibit PI3K/Akt signaling in leukemia compared to other cancer cell lines.
 | ||
| Fig. 2 JN-PK1 inhibited PI3K/Akt pathway through suppressing p-AktThr308, p-AktSer473 and p-mTORSer2448. (A) OCI-My5 and HL-60 were treated with JN-PK1 at different concentrations from 0 to 20 μM for 24 h. (B) OCI-My5 and HL-60 were treated with JN-PK1 for different time from 0 to 24 h at the indicated concentration (20 μM). | ||
3.3 JN-PK1 induces apoptosis of hematologic tumor cells
JN-PK1 effectively suppressed the proliferation of hematologic cancer cells. To further investigate whether JN-PK1 could induce apoptosis in hematologic cancer cells by inhibiting the PI3K/Akt pathway, Western blots were performed to detect relative levels of two apoptosis biomarkers, PARP (poly ADP-ribose polymerase) and Caspase-3, with increasing incubation time and concentration. As illustrated in Fig. 3A and B, the cleavage levels of PARP and caspase-3 were obviously increased following increased incubation time and concentration of JN-PK1 treatment, notably at a JN-PK1 concentration of 10 μM. To further observe the number of apoptotic hematologic cancer cells induced by JN-PK1, Annexin V-FITC/PI double staining was performed on OCI-My5 and HL-60 cells treated with varying concentrations of JN-PK1 for 24 h. The percentage of apoptotic cells induced by JN-PK1 was increased in a concentration-dependent manner, and over half of the cells were at a late stage of apoptosis after treatment with 10 μM (Fig. 3C and D). These results suggest that JN-PK1 efficiently inhibits the survival and proliferation of MM and leukemia cells.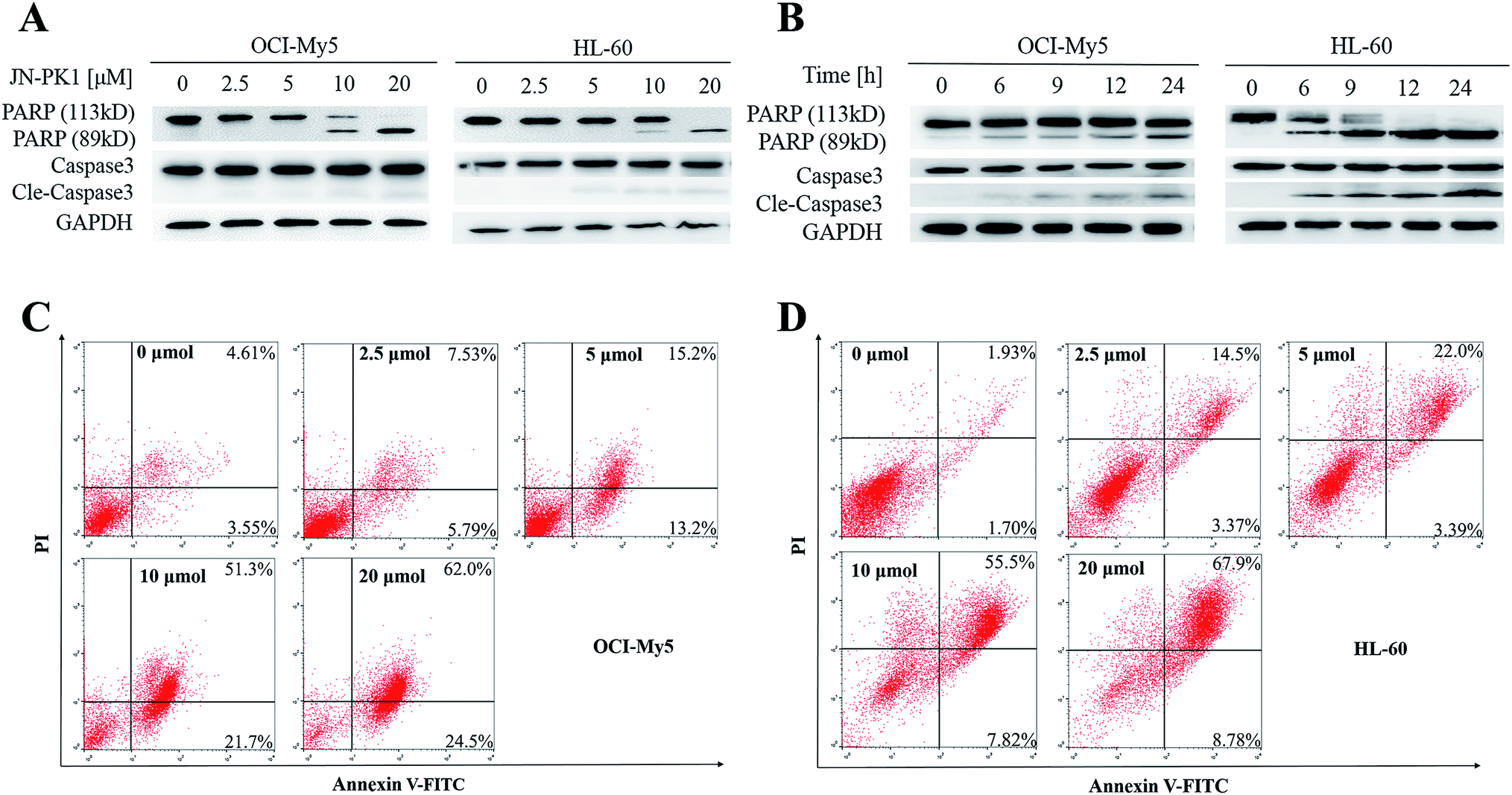 | ||
| Fig. 3 JN-PK1 induced the apoptosis of hematologic malignant cells. (A) HL-60 and OCI-My5 were treated with JN-PK1 at different concentrations from 0 to 20 μM for 24 h. (B) HL-60 and OCI-My5 were treated with JN-PK1 for different time from 0 to 24 h at the indicated concentration (20 μM). (C) OCI-My5 cells were treated with JN-PK1 at different concentrations from 0 to 20 μM for 24 h. (D) HL-60 cells were treated with JN-PK1 at different concentrations from 0 to 20 μM for 24 h. | ||
3.4 Theoretical studies on the mechanism of selective inhibition of PI3Kγ by JN-PK1
In order to elucidate the mechanism of selective inhibition of PI3Kγ by JN-PK1, a computational strategy combining molecular docking, MD simulation, free energy calculations and decomposition was employed to reveal the binding modes between JN-PK1 and the four PI3K isoform enzymes: PI3Kα, PI3Kβ, PI3Kδ and PI3Kγ. The molecular docking protocol was first performed to generate the appropriate binding poses of JN-PK1 to each PI3K isoform. The docking scores (showed in Table 1) were −8.282 for PI3Kα, −6.713 for PI3Kβ, −6.531 for PI3Kδ and −8.876 for PI3Kγ. This suggests that JN-PK1 binding is most specific to PI3Kγ, in agreement with our experimental results. And then, the interactions between JN-PK1 and the four PI3K isoforms were respectively illustrated through 2D presentations. As shown in Fig. S1B,† one of the acetyl groups of JN-PK1 formed hydrogen bonds (H-bonds) with Lys799, Ser775 and Met773 of PI3Kβ, while two acetyl groups of JN-PK1 respectively formed H-bonds with Lys779 and Ser831 of PI3Kδ (Fig. S1C†). The two carbazole phenyl-rings of JN-PK1 formed two arene–H interactions with Val850 and Ile932 of PI3Kα (Fig. S1A†), and in PI3Kγ system, JN-PK1 formed three arene–H interactions with Ile881, Ile963 and Ile879 (Fig. 4A), these interactions are essential for the binding of JN-PK1 to these proteins. Besides, as shown in Fig. 4A, an H-bond formed between one acetyl group of JN-PK1 and Lys833 of PI3Kγ, and the steric hindrance that was created by the side chain of residue Lys833 may help JN-PK1 remain in the active pocket of PI3Kγ (Fig. 4B).36,48 This may explain the stronger binding affinity of JN-PK1 to PI3Kγ despite the fewer interactions that were formed with PI3Kγ than other isoforms. Therefore, the contributions from individual residues of each isoform to JN-PK1 binding need to be further investigated. | ||
| Fig. 4 (A) The 2D presentation of the interactions between JN-PK1 and PI3Kγ. (B) The binding mode of JN-PK1 in the active site of PI3Kγ (JN-PK1 shown in green sticks and protein show in mesh model). | ||
Next, the four docked JN-PK1/PI3K complexes were used as the initial structures for the subsequent MD simulations. As shown in Fig. S2,† the RMSD (root-mean-square deviations) values of the binding pockets were quite stable in the last 10 ns, indicating that the complexes reached equilibrium within last 10 ns. Subsequently, 1000 snapshots extracted from the last 10 ns of each MD trajectory were used to calculate the binding free energies through the MM/PBSA method. As summarized in Table 2, the predicted binding free energies were −20.50 kcal mol−1 for PI3Kα/JN-PK1, −25.75 kcal mol−1 for PI3Kβ/JN-PK1, −23.43 kcal mol−1 for PI3Kδ/JN-PK1 and −27.40 kcal mol−1 for PI3Kγ/JN-PK1. This suggests that JN-PK1 specifically interacts with PI3Kγ, in agreement with our experimental data and docking results.
| Isoform | ΔEvdw | ΔEele | ΔGSA | ΔGGB | ΔGtotal | PDB |
|---|---|---|---|---|---|---|
| α | −30.20 ± 0.66 | −3.16 ± 1.36 | −2.72 ± 0.55 | 15.58 ± 0.39 | −20.50 ± 0.17 | 4JPS |
| β | −36.30 ± 1.31 | −19.23 ± 1.68 | −3.04 ± 0.13 | 32.82 ± 1.75 | −25.75 ± 1.97 | 2Y3A |
| δ | −33.23 ± 1.11 | −7.72 ± 1.57 | −2.94 ± 0.20 | 20.46 ± 0.14 | −23.43 ± 1.02 | 4XE0 |
| γ | −32.17 ± 0.65 | −14.71 ± 0.89 | −2.95 ± 0.20 | 22.42 ± 0.35 | −27.40 ± 0.40 | 4ANW |
In order to understand the γ-specific binding modes of JN-PK1, the binding free energies of all systems were decomposed into the contributions from individual residues and the JN-PK1-residues interaction spectrums are illustrated in Fig. S3.† Furthermore, we calculated the free energy changes, ΔΔGtotal = ΔGtotal (PI3Kγ) − ΔGtotal (PI3Kα/PI3Kβ/PI3Kδ), and the residues located in the ATP-binding pockets were selected to discussed in detail.49 As shown in Fig. 5E, the negative values of ΔΔGtotal represent a favorable contribution for PI3Kγ. In general, as shown in Fig. 5D, JN-PK1 binding with PI3Kγ was sandwiched between residues Ile831 and Ile963, and encompassed by residues Ile881, Val882 and Trp812 above and residues Tyr867 and Ile879 below, which formed strong hydrophobic interactions and highly improved the binding affinity of JN-PK1. Compared to the binding mode of PI3Kα (Fig. 5A), JN-PK1 was packed more tightly with PI3Kγ, especially residues Phe961, Ile831, Ile881 and Val882, shown in Fig. 5E. This tight binding prevented JN-PK1 from exiting the active pocket of PI3Kγ. Compared to PI3Kδ (Fig. 5C), JN-PK1 remained in closer proximity to the active pocket of PI3Kγ through tighter binding with residues Ile831, Tyr867, Ile879, Ile881, Val882 and Phe961. Comparing the free energy change ΔΔGtotal of Ile800 of PI3Kα and Ile777 of PI3Kδ (Fig. 5E), the free energy ΔGtotal of Ile797 of PI3Kβ was similar to that of Ile831 of PI3Kγ combined with JN-PK1, which may explain the reason why JN-PK1 bound more tightly to PI3Kβ (Fig. 5B) with better predicted binding free energies. Therefore, residue Ile831 of PI3Kγ is important for the binding selectivity of JN-PK1 to PI3Kγ. Moreover, comparing the free energy changes ΔΔGtotal as showed in Fig. 5E, the backbone residue Val882 uniquely formed an H-bond between one acetyl group of JN-PK1 and PI3Kγ, common for selective PI3Kγ inhibitors.48 Additionally, hydrophobic interactions formed by the hinge residue Ile881 and Trp812 may restrain the movement of Trp812 and further promote JN-PK1 stabilization in the active pocket of PI3Kγ.50 As a potent PI3Kγ-selective inhibitor, JN-PK1 exhibited stronger affinity for PI3Kγ (Tables 1 and 2), and more tightly connected with important residues Ile831, Ile881 and Val882 in the active pockets of PI3Kγ compared to the other three PI3K isoforms (Fig. 5E).
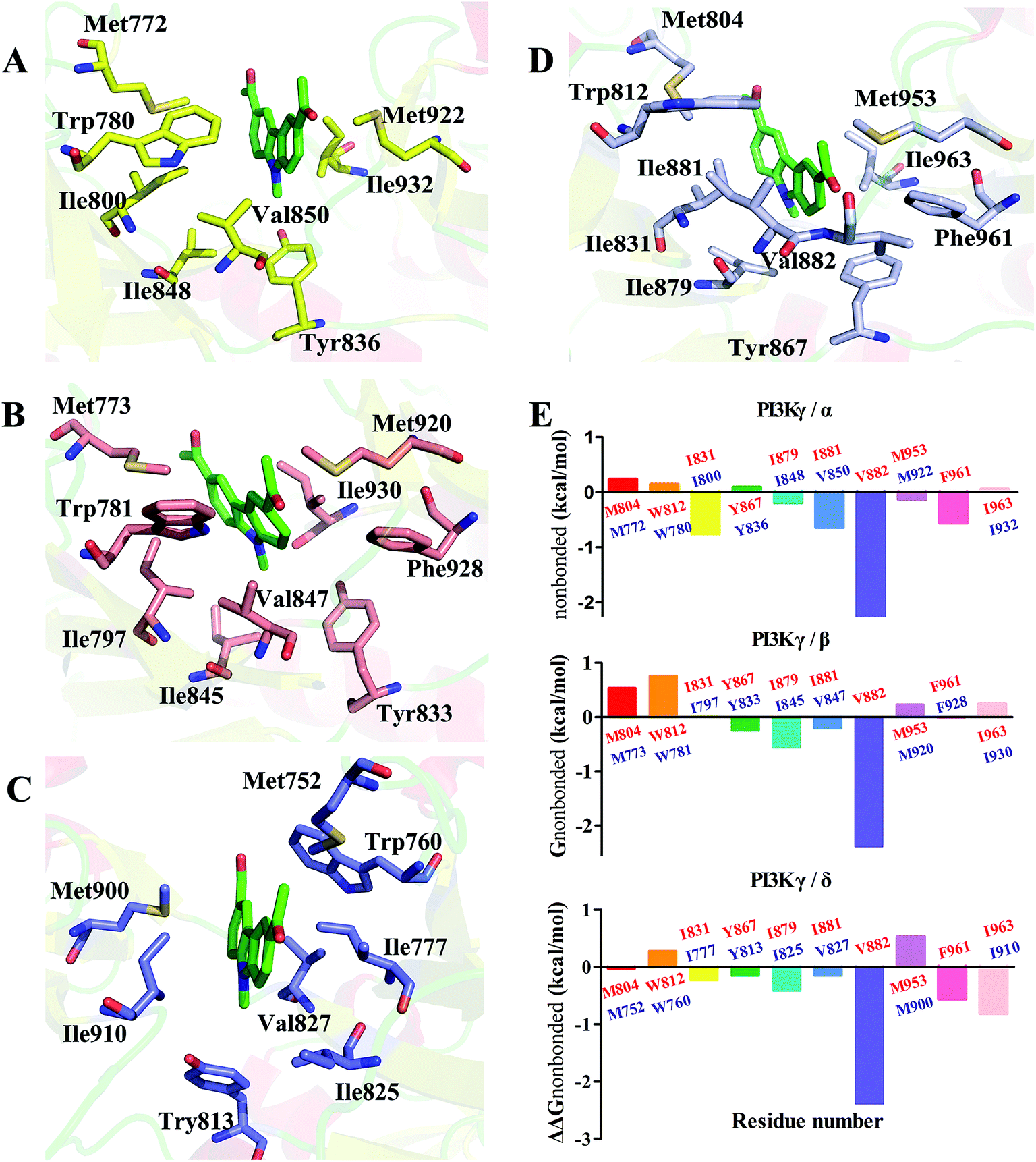 | ||
| Fig. 5 The 3D presentations of the binding modes in the active sites of PI3Kα (A)/PI3Kβ (B)/PI3Kδ (C)/PI3Kγ (D) with JN-PK1 shown in green. (E) The free energy changes ΔΔGtotal which were defined as ΔGtotal (PI3Kγ) − ΔGtotal (PI3Kα/PI3Kβ/PI3Kδ), with PI3Kγ residues colored in red and PI3Kα/PI3Kβ/PI3Kδ residues colored in purple. | ||
4. Conclusions
In summary, using a series of studies at both cellular and computational models, the highly potent and selective PI3Kγ inhibitor JN-PK1 was identified for treating hematologic malignancies. JN-PK1 exhibited high anti-proliferative activity on most cancer cell lines, especially hematologic cell lines. The selective inhibition on PI3Kγ of JN-PK1 was confirmed by a cell-free enzymatic system, and then the PI3K/Akt signaling pathway was specifically inhibited in a time- and concentration-dependent manner after JN-PK1 treatment. Immunoblotting and flow cytometry analysis visually presented that JN-PK1 induced apoptosis of cancer cells in association with its potent PI3K inhibition. Furthermore, the molecular docking and MD simulation were used to deeply study the binding modes between JN-PK1 and the four PI3K isoforms, and the selective inhibition mechanism of JN-PK1 on PI3Kγ was revealed with the highlight of some key residues of PI3Kγ relating to selectivity. In general, JN-PK1 may be an effective treatment of hematologic malignancies as a promising PI3Kγ inhibitor with a novel scaffold, and the theoretical studies on its γ-specific inhibition mechanisms may provide some important guidelines for further study.Conflict of interest
The authors declare no conflict of interest.Acknowledgements
The study was supported by the National Natural Science Foundation of China (No. 21807049) and the Fundamental Research Funds for the Central Universities (JUSRP11892).References
- K. D. Courtney, R. B. Corcoran and J. A. Engelman, J. Clin. Oncol., 2010, 28, 1075–1083 CrossRef CAS PubMed.
- D. A. Fruman and C. Rommel, Nat. Rev. Drug Discovery, 2014, 13, 140–156 CrossRef CAS PubMed.
- P. T. Hawkins and L. R. Stephens, Biochim. Biophys. Acta, 2015, 1851, 882–897 CrossRef CAS PubMed.
- B. Hennessy, D. Smith, P. Ram and Y. Lu, Nat. Rev. Drug Discovery, 2005, 4, 988–1004 CrossRef CAS PubMed.
- P. Liu, H. Cheng, T. M. Roberts and J. J. Zhao, Nat. Rev. Drug Discovery, 2009, 8, 627–644 CrossRef CAS PubMed.
- B. Jiang and L. Liu, Adv. Cancer Res., 2009, 102, 19–65 CrossRef CAS PubMed.
- F. Dituri, A. Mazzocca, L. Lupo, C. E. Edling, A. Azzariti and S. Antonaci, Int. J. Cancer, 2012, 130, 2505–2513 CrossRef CAS PubMed.
- E. Lien, C. Dibble and A. Toker, Curr. Opin. Cell Biol., 2017, 45, 62–71 CrossRef CAS PubMed.
- J. Zhu, M. Wang, B. Cao, T. Hou and X. Mao, Curr. Med. Chem., 2014, 21, 3173–3187 CrossRef CAS PubMed.
- B. Miller, D. Przepiorka, R. de Claro, K. Lee, N. Lei, S. Natalie and G. Ramadevi, Clin. Cancer Res., 2015, 21, 1525–1529 CrossRef CAS PubMed.
- A. Setti, M. Kumar, K. Babu and A. Rasagna, J. Recept. Signal Transduction Res., 2015, 36, 26–36 CrossRef PubMed.
- E. Jabbour, O. G. Ottmann, M. Deininger and A. Hochhaus, Haematologica, 2014, 99, 7–18 CrossRef CAS PubMed.
- J. Zhu, T. Hou and X. Mao, Drug Discovery Today, 2015, 20, 988–994 CrossRef CAS PubMed.
- M. Li and V. Sala, Circulation, 2018, 138, 696–711 CrossRef CAS PubMed.
- K. Kawauchi, T. Ogasawara, M. Yasuyama, K. Otsuka and O. Yamada, Anti-Cancer Agents Med. Chem., 2009, 9, 1024–1038 CrossRef CAS PubMed.
- D. Barrett, V. I. Brown, S. A. Grupp and D. T. Teachey, Paediatr. Drugs, 2012, 14, 299–316 Search PubMed.
- K. Balakrishnan, M. Peluso, M. Fu, N. Y. Rosin, J. A. Burger, W. G. Wierda, M. J. Keating and K. Faia, Leukemia, 2015, 29, 1811–1822 CrossRef CAS PubMed.
- H. A. Blair, Drugs, 2018, 78, 1847–1853 CrossRef PubMed.
- M. C. Schmid, C. J. Avraamides, H. C. Dippold, I. Franco, P. Foubert, L. G. Ellies and L. M. Acevedo, Cancer Cell, 2011, 19, 715–727 CrossRef CAS PubMed.
- O. De Henau, M. Rausch, D. Winkler, L. F. Campesato, C. Liu and D. H. Cymerman, Nature, 2016, 539, 443–447 CrossRef CAS PubMed.
- M. M. Kaneda, P. Cappello, A. V. Nguyen, N. Ralainirina, C. R. Hardamon and P. Foubert, Cancer Discovery, 2016, 6, 870–885 CrossRef CAS PubMed.
- M. M. Kaneda, K. S. Messer, N. Ralainirina, H. Li, C. J. Leem and S. Gorjestani, Nature, 2016, 539, 437–442 CrossRef CAS PubMed.
- C. Billottet, L. Banerjee, B. Vanhaesebroeck and A. Khwaja, Cancer Res., 2009, 69, 1027–1036 CrossRef CAS PubMed.
- S. Tyagi, S. Sharma and R. D. Budhiraja, Can. J. Physiol. Pharmacol., 2012, 90, 881–885 CrossRef CAS PubMed.
- M. Jin, Q. Zhou, E. Lee, S. Dan, H. Q. Duan and D. Kong, Inflammation, 2014, 37, 1254–1260 CrossRef CAS PubMed.
- K. Bell, M. Sunose, K. Ellard, A. Cansfield, J. Taylor, W. Miller and N. Ramsden, Bioorg. Med. Chem. Lett., 2012, 22, 5257–5263 CrossRef CAS PubMed.
- A. Tolcher, D. Hong, R. Sullivan, J. Mier, G. Shapiro, B. Chmielowski and A. Ribas, Cancer Res., 2017, 77, CT089 Search PubMed.
- J. Doukas, W. Wrasidlo, G. Noronha, E. Dneprovskaia, R. Fine, S. Weis, J. Hood, A. DeMaria, R. Soll and D. Cheresh, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19866–19871 CrossRef CAS PubMed.
- C. Carlo-Stella, P. Barde, R. Delarue, L. Scarfò, S. Viswanadha, S. Locatelli and S. Gandolfi, Hematol. Oncol., 2017, 35, 263 CrossRef.
- M. Girardi, S. Vakkalanka, S. Viswanadha and F. Bertoni, Blood, 2013, 122, 4418 Search PubMed.
- S. Vakkalanka, S. Nyayapathy and S. Viswanadha, Blood, 2014, 124, 4497 Search PubMed.
- R. Thomas, M. Schwarz and C. Rommel, Nat. Rev. Drug Discovery, 2006, 5, 903–918 CrossRef PubMed.
- S. Park, N. Chapuis, V. Bardet, J. Tamburini, N. Gallay, L. Willems and Z. A. Knight, Leukemia, 2008, 22, 1698–1706 CrossRef CAS PubMed.
- P. Furet, V. Guagnano, R. A. Fairhurst, P. Imbach-Weese, I. Bruce, M. Knapp, C. Fritsch, F. Blasco, J. Blanz, R. Aichholz, J. Hamon, D. Fabbro and G. Caravatti, Bioorg. Med. Chem. Lett., 2013, 23, 3741–3748 CrossRef CAS PubMed.
- X. Zhang, O. Vadas, O. Perisic, K. Anderson, J. Clark, P. Hawkins and L. Stephens, Mol. Cell, 2011, 41, 567–578 CrossRef CAS PubMed.
- J. Somoza and K. David, J. Biol. Chem., 2015, 290, 8439–8446 CrossRef CAS PubMed.
- D. Case, T. E. Cheatham, T. Darden and H. Gohlke, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- Y. Duan, C. Wu, S. Chowdhury, M. C. Lee and G. M. Xiong, J. Comput. Chem., 2003, 24, 1999–2012 CrossRef CAS PubMed.
- J. Wang, R. Wolf, J. Caldwell, P. A. Kollman and D. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- L. Xu, Y. Li, L. Li, S. Zhou and T. Hou, Mol. BioSyst., 2012, 8, 2260–2273 RSC.
- C. Bayly, P. Cieplak, W. Cornell and P. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Phys. Chem., 1993, 98, 10089–10092 CrossRef CAS.
- D. A. Sabbah, J. L. Vennerstrom and H. A. Zhong, J. Chem. Inf. Model., 2012, 52, 3213–3224 CrossRef CAS PubMed.
- P. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong and M. Lee, Acc. Chem. Res., 2000, 32, 889–897 CrossRef.
- J. Weiser, P. Shenkin and W. Still, J. Comput. Chem., 1999, 20, 217–230 CrossRef CAS.
- S. Zhao, J. Zhu, L. Xu and J. Jin, Chem. Biol. Drug Des., 2017, 89, 846–855 CrossRef CAS PubMed.
- I. Hiroshi, H. Teru, F. Mariateresa, P. Giulia, M. Naoya, Y. Hiroshi and O. Yutaka, Blood, 2010, 116, 1460–1468 CrossRef PubMed.
- E. H. Walker, M. E. Pacold, O. Perisic, L. Stephens, P. T. Hawkins, M. P. Wymann and R. L. Williams, Mol. Cell, 2000, 6, 909–919 CrossRef CAS PubMed.
- J. Zhu, K. Li, L. Xu and J. Jin, Chem. Biol. Drug Des., 2019, 93, 818–831 CrossRef CAS PubMed.
- A. Berndt, S. Miller, O. Williams and D. Le, Nat. Chem. Biol., 2010, 6, 117–124 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02649e |
| ‡ Equivalent authors. |
| This journal is © The Royal Society of Chemistry 2019 |