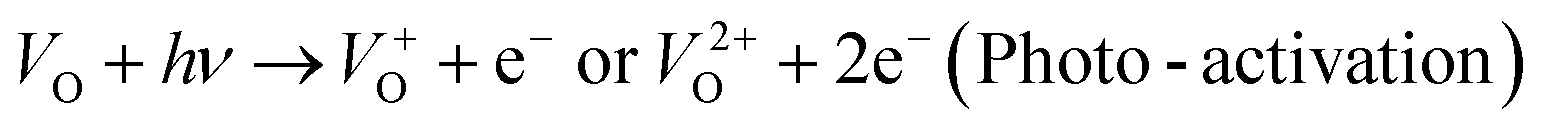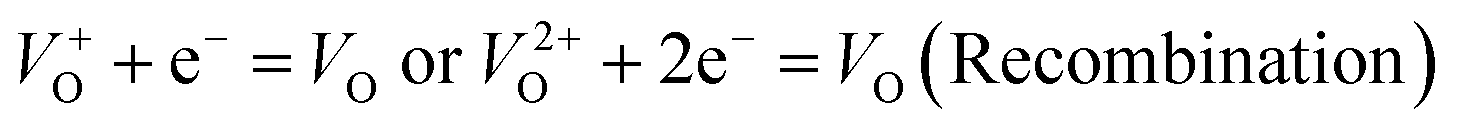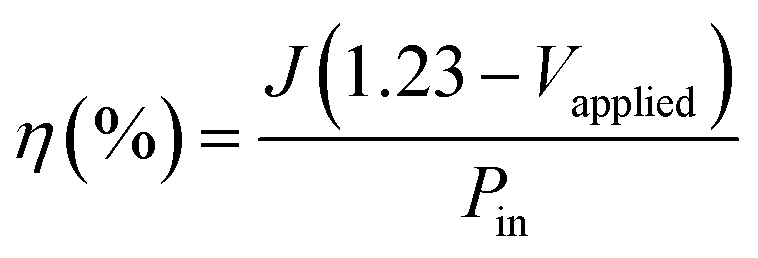DOI:
10.1039/C9RA02553G
(Paper)
RSC Adv., 2019,
9, 17165-17178
Surfactant-assisted microwave processing of ZnO particles: a simple way for designing the surface-to-bulk defect ratio and improving photo(electro)catalytic properties
Received
4th April 2019
, Accepted 21st May 2019
First published on 3rd June 2019
Abstract
ZnO nanopowders were produced using microwave processing of a precipitate and applied as a photoanode for photoelectrochemical water splitting. Two different surfactants, cetyltrimethylammonium bromide (CTAB) as the cationic and Pluronic F127 as the non-ionic one, were employed to in situ adjust the surface-to-bulk defect ratio in the ZnO crystal structure and further to modify the photo(electro)catalytic activity of the ZnO photoanode. The crystal structure, morphological, textural, optical and photo(electro)catalytic properties of ZnO particles were studied in detail to explain the profound effects of the surfactants on the photoanode activity. The ZnO/CTAB photoanode displayed the highest photocurrent density of 27 mA g−1, compared to ZnO (10.4 mA g−1) and ZnO/F127 photoanodes (20 mA g−1) at 1.5 V vs. SCE in 0.1 M Na2SO4 under visible illumination of 90 mW cm−2. A significant shift of the overpotential toward lower values was also observed when photoanodes were illuminated. The highest shift of the overpotential, from 1.296 to 0.248 V vs. SCE, was recorded when the ZnO/CTAB photanode was illuminated. The ZnO/CTAB photoanode provides efficient charge transfer across the electrode/electrolyte interface, with a longer lifetime of photogenerated electron–hole pairs and reduced possibility of charge recombination. The photoconversion efficiency was improved from 1.4% for ZnO and 0.9% for ZnO/F127 to 4.2% for ZnO/CTAB at 0.510 mV. A simple procedure for the synthesis of ZnO particles with improved photo(electro)catalytic properties was established and it was found that even a small amount of CTAB used during processing of ZnO increases the surface-to-bulk defect ratio. Optimization of the surface-to-bulk defect ratio in ZnO materials enables increase of the absorption capacity for visible light, rendering of the recombination rate of the photogenerated pair, as well as increase of both the photocurrent density and photoconversion efficiency.
1. Introduction
Since 1972 when Fujishima and Honda published the first paper on the photoelectrolysis of water using TiO2 as a photoanode,1 numerous efforts have been made by materials scientists to develop photocatalysts that would be more efficient under sunlight irradiation.2–4 In the 21st century, when we are facing an energy crisis and environmental pollution as one of the major global problems, the idea developed by Fujishima and Honda is more popular than ever since solar-driven photoelectrochemical (PEC) water splitting of semiconductor catalysis can be used for efficient conversion of solar energy to chemical energy or electricity.5–7 An efficient photoactive material should meet both kinetic and thermodynamic criteria for the photoconversion of solar energy to chemical energy or electricity.7 In order to be suited for this purpose, photoactive materials need to: (1) absorb both ultraviolet and visible light irradiation producing electron–hole pairs, (2) separate electrons and holes in space to prevent their recombination, (3) provide rapid charge transfer across the electrode/electrolyte interface, (4) have suitable redox potentials to drive photo-oxidative reactions, (5) have a high photo-corrosion resistivity, and (6) to be recyclable, either for the sake of reuse or to prevent further water contamination.3,7–12 Over the years, metal oxides such as TiO2, ZnO, SnO2, Fe2O3, CoO, V2O5, WO3, etc., have been studied as the photoanodes for solar-driven PEC water splitting due to their high photoactivity and photostability.4,8,13–19 Noble metal nanocomposites, especially Ag-based,20 as well as metal chalcogenides such as CuSe, CdS, MoS2, Ag2S, etc., have also been studied as promising visible light photocatalysts and electrocatalysts due to their suitable band gap (<2.5 eV) and excellent absorption of visible light.21–23 Newly developed sophisticated materials such as: metal–organic frameworks (MOFs), covalent–organic frameworks (COFs), zeolite–imidazole frameworks (ZIFs), porous coordination polymers (PCPs) have shown high photocatalytic or electrocatalytic activity.24 Over the past few years, since the pioneering study in 2009 by Wang and coworkers,25 semiconductor polymeric graphitic carbon nitride (g-C3N4) photocatalysts have attracted intensely growing interests in the research area of visible-light-induced hydrogen evolution reaction because of their facile synthesis, easy functionalization, attractive electronic band structure, high physicochemical stability and photocatalytic activity.26
Although their application as a photoanode under direct sunlight illumination is restricted by a wide band gap (>3 eV), allowing the absorption of UV light only, ZnO-based materials, together with TiO2, are the most intensively investigated photo(electro)catalytic materials. ZnO is a significantly better conductor of both electrons and holes than many photocatalysts, even TiO2,8 and due to this it is worth trying to overcome the disadvantages of ZnO-based materials related to optical absorption and sunlight photonic activity. Different approaches have been developed for enhancing the optical properties of ZnO materials: incorporation of transition metal ions into the crystal structure, sensitization of particle surface, hydrogenation, incorporation of crystalline defects in metal oxide semiconductors in the form of vacancies and interstitials, etc.9,27–30
Crystalline defects can strongly influence electrical and optical properties of a semiconductor. Defects often introduce levels in the band gap of semiconductors;31 these levels involve transitions between different charge states of the same defect. Surface defects can improve absorption capacity while bulk defects, commonly oxygen vacancies, can act as recombination centers leading to the loss of photocatalytic efficiency.32 It has been reported that tuning the relative concentration ratio of surface to bulk defects in semiconductor nanocrystals can improve the separation of photogenerated electron–hole and therefore can enhance photocatalytic efficiency.33 Accordingly, understanding the relationship between point defects, especially the surface-to-bulk defect ratio, and photo(electro)catalytic efficiency is important for successful application of ZnO as a photoanode materials.
In our previous work, we studied the influence of point defects on the crystal structure, optical and photocatalytic properties of microwave processed ZnO particles. To vary the concentration and the type of point defects, we used polyethylene oxide (PEO) with different molecular weights (200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 600000 and 900000 g mol−1).30 We demonstrated that oxygen interstitials at the surface of ZnO particles had profound effects on the direct sunlight photodegradation of methylene blue. The aim of this research is to in situ modify the surface-to-bulk defects ratio in ZnO particles by surfactant-directed microwave processing. Surfactants have so far been used mostly as an aid in hydrothermal, solvothermal or sol–gel processes, as they may help prepare monodisperse particles with narrow size distributions, low tendency towards agglomeration, controlled crystal size and shape, and good redispersibility.34,35 According to the authors' best knowledge, there is no data about surfactant-directed microwave processing of ZnO particles. In this study, we employed two types of surfactants, CTAB as the cationic and Pluronic F127 as the non-ionic one. A small amount of surfactants was used, as it has been shown that an excess of surfactants in a solution could led to high viscosities, potentially reducing the mobility of nanoparticles and hindering the self-assembly process.34,36 We examined in detail the crystal structure, morphological, textural, optical and photo(electro)catalytic properties of microwave (MW) processed ZnO particles. We also discussed the influence of surfactants on the surface-to-bulk defect ratio, and correlated the surface-to-bulk defect ratio with charge separation and radiative recombination of photogenerated electron–hole pairs, as well as their impact on photo(electro)catalytic properties of MW processed ZnO particles.
000, 600000 and 900000 g mol−1).30 We demonstrated that oxygen interstitials at the surface of ZnO particles had profound effects on the direct sunlight photodegradation of methylene blue. The aim of this research is to in situ modify the surface-to-bulk defects ratio in ZnO particles by surfactant-directed microwave processing. Surfactants have so far been used mostly as an aid in hydrothermal, solvothermal or sol–gel processes, as they may help prepare monodisperse particles with narrow size distributions, low tendency towards agglomeration, controlled crystal size and shape, and good redispersibility.34,35 According to the authors' best knowledge, there is no data about surfactant-directed microwave processing of ZnO particles. In this study, we employed two types of surfactants, CTAB as the cationic and Pluronic F127 as the non-ionic one. A small amount of surfactants was used, as it has been shown that an excess of surfactants in a solution could led to high viscosities, potentially reducing the mobility of nanoparticles and hindering the self-assembly process.34,36 We examined in detail the crystal structure, morphological, textural, optical and photo(electro)catalytic properties of microwave (MW) processed ZnO particles. We also discussed the influence of surfactants on the surface-to-bulk defect ratio, and correlated the surface-to-bulk defect ratio with charge separation and radiative recombination of photogenerated electron–hole pairs, as well as their impact on photo(electro)catalytic properties of MW processed ZnO particles.
2. Experimental
2.1. Materials
All chemicals were of analytical grade and used directly as received from the manufacturers without additional purification. Zinc chloride (ZnCl2, purity >99.5, Lach-Ner, Neratovice, Czech Republic) was used as a zinc source, sodium hydroxide (NaOH, purity >98%, Carlo Erba Reagents) as a precipitating agent, cetyltrimethylammonium bromide (CTAB, C19H42BrN, Alfa Aesar, Ward Hill, MA, USA) as a cationic surfactant and triblock copolymer Pluronic F127 (EO106PO70EO106, Sigma-Aldrich) was employed as a nonionic surfactant. Distilled water was used as the solvent and for powder rinsing, while absolute ethanol (C2H5OH, 96%, Zorka Pharma, Serbia) was used for final rinsing.
2.2. Synthesis of catalysts
The powders were prepared by microwave processing of a precipitate. In the first case, 20 mL of 1.75 M NaOH was added dropwise to 100 mL of 0.066 M ZnCl2 solution with continuous stirring for about 20 min. After being stirred at 50 °C for 90 min in total, the as-prepared white precipitate was microwave processed in a domestic oven (2.45 GHz, 130 W) for 5 min. After cooling to room temperature, the suspension was centrifuged at 5000 rpm for 10 min, rinsed at least five times with distilled water and consecutively with absolute ethanol to remove the surface residual of the starting chemical solutions, and air dried in an oven at 60 °C for 24 h.
The same procedure was applied for synthesis of CTAB and Pluronic F127 surface modified ZnO particles except that 0.0472 g of surfactant (CTAB or Pluronic F127) was dissolved in 100 mL of 0.066 M ZnCl2 solution; the surfactant amount was tuned to be 5 wt% with respect to ZnCl2.
Throughout this paper, the powders synthesized without and with the assistance of CTAB and Pluronic F127 surfactants have been designated as ZnO, ZnO/CTAB and ZnO/F127, respectively.
2.3. Characterization
X-ray diffraction (XRD) data were recorded on a Rigaku Ultima IV diffractometer in Bragg–Brentano geometry, with Ni-filtered CuKα radiation (λ = 1.54178 Å). The data were collected over a 2θ range 20–80° with a step of 0.02° and acquisition rate of 2° min−1. The crystal phases were identified by comparing the collected data with those reported in the International Centre for Diffraction Data (ICDD) database. The unit cell parameters were calculated using the least-squares method by the LSUCRI computing program.37 The crystallite sizes (D) were calculated from XRD line-broadening using the Scherrer equation D = 0.9λ/βmcosθ, where λ is the X-ray wavelength (1.54178 Å); βm the full width at half maximum (FWHM) for the XRD reflection, and θ the diffraction angle (°).38,39 Fourier transform infrared (FT-IR) spectra were recorded on a Thermo Scientific™ Nicolet™ iS™10 FT-IR Spectrometer equipped with an attenuated total reflectance (ATR) accessory. ATR/FT-IR measurements were done in the wavenumber region of 400–2000 cm−1, with a resolution of 4 cm−1. Room-temperature μ-Raman spectra were recorded with a DXR Raman microscope (Thermo Scientific, Waltham MA, USA), equipped with an optical microscope and a CCD detector. The spectra were recorded in the frequency interval of 50–3500 cm−1 with a resolution of 4 cm−1. The 532 nm line of a diode-pumped solid-state high-brightness laser was used as the excitation source; the laser power was tuned on 10 mW. The morphology of zinc oxide particles was observed by field emission scanning electron microscopy (FE-SEM, Ultra plus, Carl Zeiss, Germany). Samples for FE-SEM analysis were dispersed in water, in an ultrasonic bath, for 5 min; after dispersion a few drops were filtered through a polycarbonate membrane. The membrane was put on carbon tape on an aluminum stub and carbon-coated in order to prevent electron charging. Before analysis, the sample was vacuumed for 15 min. The specific surface area (SSA) and porous properties of photocatalysts were determined based on a N2 adsorption–desorption isotherm at −195.8 °C using an ASAP 2020 (Micromeritics Instrument Corporation, Norcross, GA, USA). Prior to analysis samples were degassed under reduced pressure at 120 °C for 10 h. The SSA was calculated according to the Brunauer–Emmett–Teller (BET) method from the linear part of the N2 adsorption isotherm.40 The total pore volume (Vtotal) was estimated from the adsorbed amount at a relative pressure p/p0 = 0.998. The volume of mesopores (Vmeso) and pore size distribution were analysed according to the Barrett–Joyner–Halenda (BJH) method from the desorption branch of the isotherm.41 The volume of micropores (Vmicro) was calculated from the alpha-S plot. The particle size distribution in water suspensions was determined by a laser light-scattering particle size analyzer (PSA) (Mastersizer 2000; Malvern Instruments Ltd., Malvern, Worchestershire, U.K.). Prior to measurement, the powders were dispersed in distilled water, using an ultrasonic bath, for 5 min. The measurements were done in a dynamic condition with pump rotation of 1100 rpm. Optical properties were studied by UV-Vis diffuse reflectance (DRS) and photoluminescence (PL) spectroscopy. The UV-Vis DRS were collected using an Agilent Cary 5000 spectrophotometer equipped with a diffuse reflectance accessory. The measurements were performed in the 800–200 nm region, with a 1 nm data interval and 600 nm min−1 scan rate, using a commercial PTFE standard for baseline correction. The PL spectra were recorded on a Horiba Jobin Yvon Fluorolog FL3-22 spectrofluorometer using Xe lamp excitation.
2.4. Photo(electro)catalytic tests
The photocatalytic activity of prepared powders was studied by decolorization of methylene blue (MB) dye under direct sunlight illumination. A stock solution with the concentration of 1000 ppm was prepared by dissolving 1.0 g of methylene blue (Methylen blay B extra, E. Merck, Darmstadt, Germany) in 1 L of distilled water. The MB solutions for photocatalytic activity experiments were prepared by diluting the stock solutions to the appropriate concentration. In each experiment, 0.1 g of the prepared powder was mixed with 100 mL of the MB aqueous solution with a concentration of 10 ppm in a beaker. To distinguish the efficiency of degradation from adsorption, prior to illumination the suspension was magnetically stirred for 1 h in dark to establish the adsorption–desorption equilibrium. After equilibrium was established, the concentration of MB was measured and taken as the initial concentration Co. During illumination stirring was maintained to keep the mixture in suspension. At specific time intervals, 3 mL aliquots were withdrawn and centrifuged (7000 rpm, 5 min) to remove particles from the solution before absorbance measurements. Solution concentrations were monitored by a GBC Cintra UV-Vis spectrophotometer in the wavelength range of 450–750 nm; the concentration of MB was calculated according to the absorbance value at 665 nm. The experiments were done under direct sunlight illumination between 12 a.m. and 15 p.m. during the month of May 2018 and at ambient temperature (25 to 28 °C). The intensity of light was measured by a PeakTech 5165 Digital-Lux-Meter and it varied between 900 and 1100 lux.
Photoelectrocatalytic (PEC) efficiency was investigated using linear sweep voltammetry (LSV), chronoamperometric (CA) and electrochemical impedance spectroscopy (EIS) measurements. The PEC measurements were performed on a Gamry PCI4/750 using a three-electrode quartz cell consisting of a working electrode, platinum foil as the counter electrode and a saturated calomel electrode (SCE) as the reference electrode. The working electrode paste was prepared by mixing 90 wt% of zinc oxide powder as an active material with 10 wt% polyvinylidene fluoride (PVDF) binder in N-methyl-2-pyrrolidone (NMP) solvent. This slurry was homogenized for 1 h in an ultrasonic bath, and subsequently coated on FTO glass (Sigma-Aldrich 20 Ω cm−2) as a thin film. To evaporate the solvent, the electrode was dried under vacuum at 170 °C for 4 h. The surface area of the working electrode was 3 cm2. In all electrochemical measurements, an aqueous solution of 0.1 M Na2SO4 was used as the electrolyte. Dark conditions during measurements were provided by protecting the cell from light. An Osram Ultra-Vitalux 300 W lamp was used as the light source. The distance between the light source and the cell was adjusted to 20 cm, yielding about 90 mW cm−2 illumination intensity. LSV was measured with a scan rate of 20 mV s−1 in the voltage range between 0.2 V and 1.5 V vs. SCE. The impedance diagrams of ZnO samples were measured in the frequency interval from 105 to 0.01 Hz, using an AC voltage amplitude of 5 mV at a fixed potential of 1.5 V vs. SCE. The impedance data were fitted to the electrical analogue under study using Z-View2 (version 3.5) software. The chronoamperometric study was performed under chopped light illumination in a constant time interval of 60 s at 1.5 V vs. SCE.
3. Results and discussion
3.1. The influence of CTAB and F127 on the crystal structure, morphology and textural properties of ZnO particles
It has been demonstrated that the photocatalytic activity of metal-oxide photocatalysts for both water purification and overall water splitting, comprehensively depends on the crystallinity, crystallite size and polarity, as well as the particle size and textural properties.13,34,42,43 In particular, for ZnO-based photocatalytic particles, it has been established that hexagonal platelike crystals exhibit 5-fold better photocatalytic activity than rodlike ones.34,42,43 An improved activity of platelike crystals can be explained by ZnO crystalline structure. ZnO has a wurtzite crystalline structure with layer contains all positive Zn2+ ions or all negative O2− ions stack alternatively along the [0001] direction, resulting in the Zn-terminated (0001) and O-terminated (000![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) polar planes;43 an illustrative stacking model of ZnO with a wurtzite crystalline structure is presented in Fig. 1(a). The terminal polar planes (0001) and (000) were reported to be much more active in photocatalysis as compared to the nonpolar facets perpendicular to them (for example 100, or 010), mainly due to a higher density of defects such as oxygen vacancies.34 Surfactants can significantly affect crystallite polarity, morphology and specific surface area of ZnO particles prepared by the hydrothermal method.34 Up to now, there has been no data about the influence of surfactants on the crystal structure, morphology and textural properties of ZnO particles synthesized by fast, energy-effective microwave processing. The XRD patterns of microwave processed ZnO, ZnO/CTAB and ZnO/F127 particles are shown in Fig. 1(b). The patterns indicate highly crystalline particles with a wurtzite-type hexagonal symmetry, without other crystal phases or impurities (ICDD no. 89-0510) (the existence of solely Zn and O atoms in the ZnO samples is confirmed by EDX analysis). The unit cell parameters calculated for ZnO particles, synthesized without the aid of surfactants, were found to be a = b = 3.2509(6) Å, c = 5.2103(2) Å, V = 47.69(2) Å3 which is in good agreement with the parameters for ZnO crystals with a wurtzite-type hexagonal structure (ICDD no. 89-0510). To determine the orientation of crystal growth, crystallite size was calculated for three specific crystallographic directions [100], [002] and [101] and was found to be 19.9, 18.1 and 18.0 nm, respectively, indicating an almost isotropic nano-crystallites growth. The calculated polarity value, I(002)/I(100), was 1.27. The unit cell parameters, crystallite sizes and polarity were also calculated for ZnO/CTAB and ZnO/F127; the calculated values are listed in Table 1. As can be noticed, neither the presence of surfactants nor their type, cationic or non-ionic, influenced the lattice parameters. However, the crystallite size and isotropy were affected by the presence of surfactants. A more profound effect on crystallite growth and polarity was observed when CTAB was used; I(002)/I(100) = 1.11 indicating a well-developed polar (0001) plane (the plane normal to the [0001]-axis, Fig. 1(c)) with a hexagonal platelike morphology, implicating that ZnO/CTAB could be very effective towards the decolorization of methylene blue.34 We demonstrated that a relatively small amount of surfactants influenced the crystallite size and polarity despite the short time period (5 min) of microwave irradiation, leading to a fast crystallization of the Zn(OH)2 precursor into the ZnO phase.30
) polar planes;43 an illustrative stacking model of ZnO with a wurtzite crystalline structure is presented in Fig. 1(a). The terminal polar planes (0001) and (000) were reported to be much more active in photocatalysis as compared to the nonpolar facets perpendicular to them (for example 100, or 010), mainly due to a higher density of defects such as oxygen vacancies.34 Surfactants can significantly affect crystallite polarity, morphology and specific surface area of ZnO particles prepared by the hydrothermal method.34 Up to now, there has been no data about the influence of surfactants on the crystal structure, morphology and textural properties of ZnO particles synthesized by fast, energy-effective microwave processing. The XRD patterns of microwave processed ZnO, ZnO/CTAB and ZnO/F127 particles are shown in Fig. 1(b). The patterns indicate highly crystalline particles with a wurtzite-type hexagonal symmetry, without other crystal phases or impurities (ICDD no. 89-0510) (the existence of solely Zn and O atoms in the ZnO samples is confirmed by EDX analysis). The unit cell parameters calculated for ZnO particles, synthesized without the aid of surfactants, were found to be a = b = 3.2509(6) Å, c = 5.2103(2) Å, V = 47.69(2) Å3 which is in good agreement with the parameters for ZnO crystals with a wurtzite-type hexagonal structure (ICDD no. 89-0510). To determine the orientation of crystal growth, crystallite size was calculated for three specific crystallographic directions [100], [002] and [101] and was found to be 19.9, 18.1 and 18.0 nm, respectively, indicating an almost isotropic nano-crystallites growth. The calculated polarity value, I(002)/I(100), was 1.27. The unit cell parameters, crystallite sizes and polarity were also calculated for ZnO/CTAB and ZnO/F127; the calculated values are listed in Table 1. As can be noticed, neither the presence of surfactants nor their type, cationic or non-ionic, influenced the lattice parameters. However, the crystallite size and isotropy were affected by the presence of surfactants. A more profound effect on crystallite growth and polarity was observed when CTAB was used; I(002)/I(100) = 1.11 indicating a well-developed polar (0001) plane (the plane normal to the [0001]-axis, Fig. 1(c)) with a hexagonal platelike morphology, implicating that ZnO/CTAB could be very effective towards the decolorization of methylene blue.34 We demonstrated that a relatively small amount of surfactants influenced the crystallite size and polarity despite the short time period (5 min) of microwave irradiation, leading to a fast crystallization of the Zn(OH)2 precursor into the ZnO phase.30
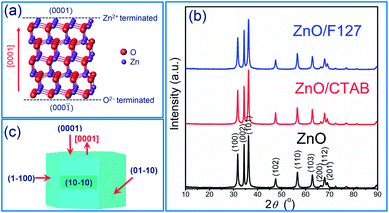 |
| | Fig. 1 (a) Illustration of ZnO wurtzite crystalline structure, (b) XRD patterns of microwave processed ZnO particles, and (c) scheme of ZnO crystallite. | |
Table 1 Unit cell parameters with standard deviations, crystallite sizes in specific crystallographic directions, polarity values and Raman intensity ratio with specific band position
| Sample |
Unit cell parameters |
Crystallites size, D (nm) |
I(002)/I(100) |
Raman |
| a = b (Å) |
c (Å) |
V (Å3) |
D100 |
D002 |
D101 |
I440/I570 |
E2H (cm−1) |
| ZnO |
3.2509(6) |
5.2103(2) |
47.69(2) |
19.9 |
18.1 |
18.0 |
1.27 |
1.165 |
434.6 |
| ZnO/CTAB |
3.2519(7) |
5.2093(2) |
47.71(2) |
21.0 |
19.8 |
18.6 |
1.11 |
1.295 |
436.5 |
| ZnO/F127 |
3.2513(7) |
5.2114(3) |
47.71(2) |
19.6 |
18.0 |
17.1 |
1.22 |
1.195 |
436.5 |
The representative FE-SEM micrographs of MW processed zinc oxide particles are shown in Fig. 2. As can be seen, in the ZnO powder processed without surfactants nanocrystallites are organized in cone-shaped particles with a uniform size and morphology. The average length of particles, deduced from the image by measuring more than 250 particles employing the SemAfore digital slow scan image recording system, is 93 nm. The uniformity of zinc oxide particles deteriorated when CTAB was used. The ZnO/CTAB powder consisted of spheroidal particles with an estimated average diameter of 58 nm; the particles were scatteredly organized in thin platelike aggregates with lengths between 220 and 670 nm and an average thickness of cc. 30 nm. The ZnO/F127 powder consisted of cone-shaped particles, similar to those in ZnO but with a reduced average length, estimated to about 80 nm. The particle size distributions determined from the micrographs using the computing program are included in Fig. 2 as insets. The particle size distributions with characteristic parameters (d(0.1), d(0.5), d(0.9) and span) determined by laser diffraction scattering are presented in the bottom part of Fig. 2. The particles size distributions are presented over a number since it gives insight into the statistically predominant particles. The characteristic parameters measured for ZnO, ZnO/CTAB and ZnO/F127 implicate that surfactants slightly influenced the particle size distributions; for example, d(0.5) varied from 285 nm for ZnO, to 336 nm for ZnO/CTAB and 270 nm for ZnO/F127, revealing the same trend as the values for the average crystallite size and the particle size distributions determined from the micrographs, ZnO/F127 < ZnO < ZnO/CTAB. As expected, the average particle sizes measured in water suspensions were about threefold larger than those deduced from FE-SEM images, which is due to both the hydrodynamic diameter and particle sticking in dynamic fluid conditions.9
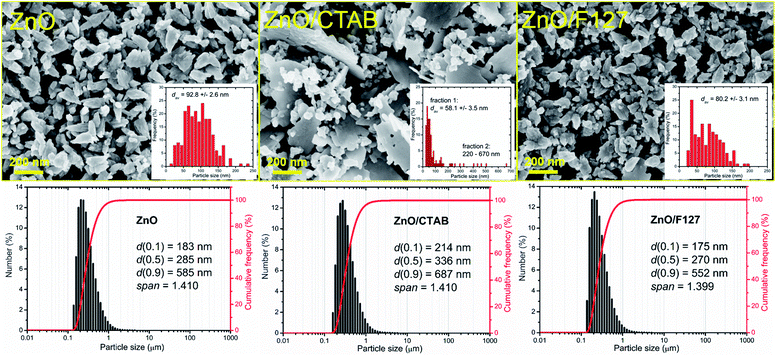 |
| | Fig. 2 FE-SEM images of microwave processed ZnO particles, with particles size distribution deduced from the images as insets, and particle size distribution determined by laser diffraction from water suspensions (bottom). | |
It is known that the presence of CTAB in different concentrations (from 0.1 to 0.8 M) can greatly change the shape of ZnO crystals obtained by hydrothermal or solvothermal synthesis.44 In this study we established that even small amounts of surfactant, 0.0013 mol L−1 CTAB or 0.0029 mol L−1 Pluronic F127, affected the shape and size of microwave processed zinc oxide particles. We also showed that platelike sub-micrometer sized agglomerates could be prepared even with energy-efficient MW processing.
The textural properties of metal-oxide particles influence their catalytic properties; more accurately, a larger surface area and a larger pore volume of the samples provided a larger number of active sites for photocatalytic reactions.45 Different surfactants are often used to control the morphology and texture of hydrothermally processed ZnO particles.46–49 The influence of surfactants on the textural properties of microwave processed zinc oxide particles are listed in Table 2. The specific surface area of ZnO particles processed without the aid of surfactants was 19.1 m2 g−1, the total pore volume was found to be 0.0710 cm3 g−1 with about 97% of mesopores and average pore diameter 14.2 nm. The BET results show that even a small amount of CTAB as a surfactant enlarged SSA for more than 60%, i.e. to 24.8 m2 g−1; the total pore volume was 0.1147 cm3 g−1, mesopores were also predominant while the average pore diameter was increased to 20.5 nm. The BET surface area of ZnO/F127 was 15.5 m2 g−1, the total pore volume was 0.0723 cm3 g−1 with more than 95% of mesopores and an average pore diameter of 25.5 nm. These values indicate profound effects of CTAB on all textural properties: SSA, the total pore volume, and the average pore diameter, while the same amount of Pluronic F127 slightly decreased SSA, had no influence on the total pore volume, but played an important role in the development of larger mesopores. Accordingly, both samples synthesized with the aid of surfactants had good catalytic potential.
Table 2 Textural (SSA, volume and size of pores), and morphological properties of ZnO particles
| Sample |
SBETa (m2 g−1) |
Vtotalb (cm3 g−1) |
Vmesoc (cm3 g−1) |
Vmicrod (cm3 g−1) |
rave (nm) |
dav,FE-SEMf (nm) |
dN(0.5)PSAg (nm) |
Particles shape |
| SBET – BET specific surface area. Vtotal – total pore volume. Vmeso – volume of mesopores (2–50 nm). Vmicro – volume of micropores (<2 nm). rav – BJH adsorption average pore diameter. dav – average particle size determined from FE-SEM images. dN(0.5)PSA – average particle size (based on number) determined by PSA. |
| ZnO |
19.1 |
0.0710 |
0.0687 |
0.0054 |
14.2 |
92.7 |
285 |
Conelike |
| ZnO/CTAB |
24.8 |
0.1147 |
0.1121 |
0.0088 |
20.5 |
355.0 |
336 |
Platelike |
| ZnO/F127 |
15.5 |
0.0732 |
0.0698 |
0.0062 |
25.7 |
80.2 |
270 |
Conelike |
ATR/FT-IR spectra were employed to inspect functional groups in ZnO particles and identify possible residues of the reagents used for MW processing. As can be seen from Fig. 3, all the spectra are practically identical. The most prominent band is in the 400–600 cm−1 region and it is attributed to Zn–O vibrations in the ZnO lattice.50 The weak peak centered at 890 cm−1 can be assigned to –C–H bending vibrations; it is most probably due to the residue of ethanol used to rinse the sample after microwave processing. The broad bands in the regions 1320–1575 and 3200–3550 cm−1 are attributed to bending and stretching vibrations, respectively, of O–H groups from the adsorbed water molecules hydrogen bonded to the surface of zinc oxide particles.51,52 Very weak intensities and relatively small peak areas indicate that a negligible amount of hydroxyl groups is adsorbed on the surface of ZnO particles and that we are dealing with ZnO rather than Zn(OH)2.51 No residues of either the processing reagents or surfactants can be observed.
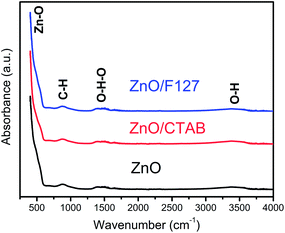 |
| | Fig. 3 FTIR spectra of microwave processed ZnO particles. | |
The room-temperature μ-Raman spectra, used to study the influence of surfactants on lattice defects in microwave processed ZnO particles, are shown in Fig. 4(a). The most intensive band at 98 cm−1, assigned as E2L, is characteristic of the wurtzite crystal structure and can be attributed to vibrations of the zinc sublattice in ZnO.30,53,54 The low intensity peak centered near 200 cm−1 and a sharp peak at 327 cm−1 are attributed to the second-order phonon mode 2E2L and the multi phonon mode E2H–E2L, respectively.55 The weak shoulder near 380 cm−1 (marked with a dark cyan triangle in Fig. 4(a)) is attributed to A1 (TO). The second weak shoulder at 410 cm−1 (marked with a pink asterisk in Fig. 4(a)) corresponds to the E1 (TO) mode; it indicates that ZnO crystallites have propagation in a direction other than the c-axis typical of ZnO particles.56 This is in accordance with the (almost) isotropic crystallite geometry calculated from the XRD data for the most characteristic crystallographic directions (Table 1). The peak centered at 435 cm−1 is due to the E2H mode and it is assigned to oxygen vibrations.55 According to literature data, the asymmetry of the E2H peak points to lattice disorder while its intensity indicates crystallinity.57,58 In the Raman spectrum of MW processed ZnO nanocrystals, a relatively high intensity of the E2H peak indicates a good crystallinity which is in accordance with the crystallinity of about 80% calculated from the XRD data. A wide optical phonon band in the 510–740 cm−1 interval holds two peaks; the one centered near 570 cm−1 represents A1 (LO) and E1 (LO) modes, whereas the other, at 635 cm−1 represents a combination of acoustic and optical modes (TA + LO). The A1 (LO) mode is associated with the bulk defects: oxygen vacancies, zinc interstitials or defect complexes containing both.55 It has been shown that the presence of impurities and/or defects strongly influences these modes, especially E1 (LO).58,59 The relatively high-intensity of the A1 (LO) + E1 (LO) modes in the Raman spectrum of the ZnO sample (Fig. 4(a)) points to a relatively large density of intrinsic bulk defects (oxygen vacancies and zinc interstities); these bulk defects were incited by rapid crystallization driven by microwave irradiation.30 The A1 (LO) mode is accompanied with a weak band at 483 cm−1 (marked with an orange circle), which is assigned as the interfacial surface phonon mode 2LA.55 The wide band in the wavenumber region 1090−1150 cm−1 is attributed to the optical overtone 2LO, namely to 2A1 (LO) and 2E1 (LO).
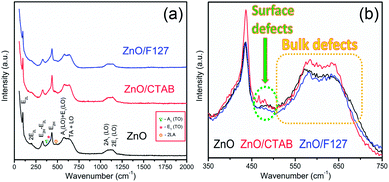 |
| | Fig. 4 (a) Raman spectra of microwave processed ZnO particles, and (b) expanded 350–750 cm−1 region of E2H and A1 (LO) + E1 (LO) mode. | |
As Fig. 4(a) shows, all vibrational modes appearing in the Raman spectra of ZnO particles processed with the aid of surfactants correspond well to those for the bare one. However, we found that surfactants caused a red shift of the E2H mode (Fig. 4(b) & Table 1). Since the E2H mode is mainly due to the motion of oxygen atoms perpendicular to the c-axis of ZnO crystals, any change on the bond bending caused by the tensile strain along the c-axis results in a red shift of the E2H mode.60 The slight red shift of the E2H mode, from 434.6 to 436.5 cm−1, indicates an increase of the tensile strain caused by a decrease of oxygen vacancies compared to bare ZnO. As expected, since oxygen atoms are less massive than zinc atoms, redistribution of electron density due to defects has a stronger influence on the oxygen sublattice, represented by the E2H mode, than on the vibrations of the zinc sublattice assigned as E2L. Another impact of the surfactants on the Raman spectra of MW processed ZnO particles is proved by the relationship between intensities of the E2H mode to the A1 (LO) + E1 (LO) mode, Fig. 4(b) and Table 1. An increase of the I440/I570 intensity ratio confirms the decrease of the concentration of intrinsic lattice defects. The decreased amount of oxygen vacancies in the crystal lattices of ZnO/CTAB and ZnO/F127, compared to bare ZnO, can be attributed to the in situ action of polymers as passivating agents.61 The bromide anions of CTAB or oxygen ions of Pluronic can fill up the singly 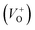 or doubly
or doubly 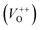 charged states of oxygen vacancies in the ZnO lattice.62
charged states of oxygen vacancies in the ZnO lattice.62
3.2. The influence of CTAB and F127 on the optical properties of ZnO particles
To investigate the effect of the cationic and non-ionic surfactants on the optical properties of ZnO particles synthesized by MW processing, UV-Vis DR and PL spectra were recorded. UV-Vis DRS plots show characteristic reflectance curves with the adsorption edge near 370 nm, Fig. 5(a). While all samples absorbed more than 95% of UV light, they showed a slightly different absorption capacity of visible light. Actually, the ZnO sample revealed the highest reflectance; compared to ZnO/CTAB and ZnO/F127, ΔR increased from 5 to 10% in the 400–800 nm spectral region. The absorption capacity varied in the following manner: ZnO < ZnO/F127 < ZnO/CTAB. The different absorption capacity in the visible spectral region could be attributed to both different morphologies of ZnO particles and the density of surface defects. As previously shown, particles with a larger diameter or thickness have a longer optical path for light transport, resulting in a greater absorption capacity; particle size can influenced the capacity of visible-light absorption even without affecting the band gap energy.48,63 Surface defects can additionally enhance visible light absorption.30 To determine both, direct and indirect band gap energies (Ebg) of ZnO particles the Kubelka–Munk method was employed.30 The diffuse reflectance R is related to the Kubelka–Munk function F(R) by eqn (1):| |
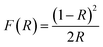 | (1) |
where R is the percentage reflectance. After finding the F(R) value from eqn (1) graphs of [F(R) × E]2 and [F(R) × E]1/2 vs. energy E (eV) were plotted. The band gap energies were estimated by extrapolation of the linear part of the curves to [F(R) × E]2 i.e. [F(R) × E]1/2 to 0, Fig. 5(b) and (c). The estimated values for the direct band gaps were 3.36 eV for ZnO and ZnO/F127, and 3.34 eV for ZnO/CTAB, which is in accordance with the one for bulk ZnO (3.37 eV).64 The estimated values for the indirect bandgaps were 3.23, 3.22 and 3.24 eV for ZnO, ZnO/CTAB and ZnO/F127, respectively. Thus, CTAB caused a slight shift of the band gap energy toward the visible light region while the same weight amount of Pluronic had no influence on the band gap of zinc oxide particles.
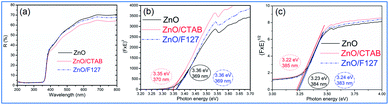 |
| | Fig. 5 (a) UV-Vis DR spectra of microwave processed ZnO particles, (b) Kubelka–Munk plots for direct, and (c) indirect bandgap semiconductors. | |
The photoluminescence spectra of ZnO-based materials commonly exhibit UV emission due to exciton recombination which is accompanied with defect related visible emission.65 Among a variety of point defects, the main types in ZnO are zinc interstitials (Zni) and oxygen vacancies (VO); the former are surface defects, also known as shallow donors, while the latter make deep-level i.e. bulk defects.66 To ascertain the influence of surfactants used in microwave processing on the surface-to-bulk defect ratio in the synthesized ZnO particles, room temperature PL spectra were recorded with the excitation wavelength of 290 nm, Fig. 6(a). The most peculiar observation in the PL spectra is the absence of the near band edge (NBE) emission of ZnO, which is usually a sharp band near 380 nm, originating from the recombination of photo-induced charge carriers through an exciton–exciton collision process.67 The absence of the NBE emission in PL spectra indicates that recombination of photo-induced electrons and holes in the ZnO particles can be efficiently inhibited by microwave processing.30 All PL spectra show a similar pattern; two emission bands appear: one in the 370–470 nm region and the other in the wide spectral region, from 470 to 700 nm, and they are attributed to surface and deep-level defects, respectively. As can be seen from Fig. 6(a), the type of surfactant used in MW processing affected the PL intensity, as well as the intensity of the surface-to-bulk defect ratio, attributed to the I620/I410 ratio. To reveal the precise position, intensity and area (%) of the emission bands, the PL spectra were mathematically deconvoluted. Before fitting, for the sake of comparison, the areas of the PL spectra were normalized to 1. Deconvolution of the PL spectra was successfully done by three Gaussian components centered near 420, 570, and 630 nm, and attributed to the violet-blue, green-yellow and orange-red emissions, respectively, Fig. 6(b)–(d). Fig. 6(e) is an illustrative representation of the PL emission spectra in the visible spectral range which is distinguished by deconvolution. The data obtained by deconvolution are listed in Table 3. There is still controversy about the origin of the violet(-blue) emission, as it has been reported to originate from Zni but also from VZn.68 The main assumption is that the probability of forming VZn is slight since the enthalpy of VZn defects is higher than the enthalpy of Zni defects. In this paper, the violet-blue emission band centered near 420 nm is attributed to the existence of interstitial zinc; it is caused by the electron transition from the interstitial and extended interstitial levels of zinc to the valence band.66–68 To be more precise, transition from the interstitial to the valence band is typical of violet emission, while transition from the extended Zni states, which are slightly below the simple Zni state, to the valence band is typical of blue emission. The band centered at 560 nm corresponds to the green-yellow emission correlated to the existence of both surface and deep-level defects. Actually, the green emission is due to the transition from the conduction band to the deep levels of the doubly charged oxygen vacancy states 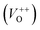 , while the yellow emission is induced by the transition from the Zni energy level to the deep-levels of
, while the yellow emission is induced by the transition from the Zni energy level to the deep-levels of 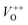 .66 The most intensive and the broadest band in the orange-red spectral region centered near 630 nm is correlated to the existence of zinc interstitials and oxygen interstitials; it is probably due to the transition from the Zni level to the Oi level in ZnO.69,70 The energy levels of intrinsic defects in ZnO have been calculated using the full-potential linear muffin-tin orbital method;71,72 the position of the Zni level is theoretically located at 0.22 eV below the conduction band while the position of the Oi level is located approximately at 2.28 eV below the conduction band. It is expected that the transition from the Zni energy level to the Oi energy level is approximately 2.06 eV, which is in good agreement with the orange-red peaks centered approximately at 1.97 eV. The energy band diagram (Fig. 7), based on the theoretically calculated energy values for different point defects and emission energies deduced after deconvolution of the PL spectra, indicates the most probable defects in the structure of microwave processed zinc oxide particles.
.66 The most intensive and the broadest band in the orange-red spectral region centered near 630 nm is correlated to the existence of zinc interstitials and oxygen interstitials; it is probably due to the transition from the Zni level to the Oi level in ZnO.69,70 The energy levels of intrinsic defects in ZnO have been calculated using the full-potential linear muffin-tin orbital method;71,72 the position of the Zni level is theoretically located at 0.22 eV below the conduction band while the position of the Oi level is located approximately at 2.28 eV below the conduction band. It is expected that the transition from the Zni energy level to the Oi energy level is approximately 2.06 eV, which is in good agreement with the orange-red peaks centered approximately at 1.97 eV. The energy band diagram (Fig. 7), based on the theoretically calculated energy values for different point defects and emission energies deduced after deconvolution of the PL spectra, indicates the most probable defects in the structure of microwave processed zinc oxide particles.
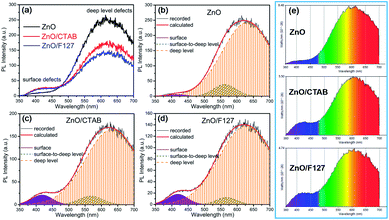 |
| | Fig. 6 (a) Photoluminescence spectra of microwave processed ZnO particles under excitation at 290 nm, (b–d) PL spectra deconvoluted with a Gaussian function, (e) PL emission spectra in the visible spectral range which is distinguished by a deconvolution. | |
Table 3 Position, intensity and area of emission bands in PL spectra obtained after deconvolution by a Gaussian function
| Sample |
Peak |
Emission |
| Violet-blue |
Green-yellow |
Orange-red |
| ZnO |
Position (nm) |
417 |
562 |
632 |
| Intensity |
5.72 |
38.4 |
243.0 |
| Area (%) |
0.81 |
7.88 |
91.31 |
| ZnO/CTAB |
Position (nm) |
418 |
567 |
633 |
| Intensity |
21.1 |
18.7 |
165.3 |
| Area (%) |
5.36 |
5.33 |
89.10 |
| ZnO/F127 |
Position (nm) |
418 |
567 |
628 |
| Intensity |
17.9 |
12.2 |
138.5 |
| Area (%) |
5.22 |
3.63 |
91.14 |
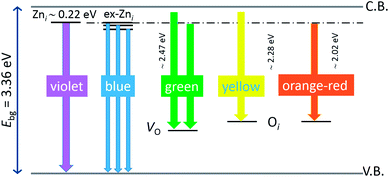 |
| | Fig. 7 Schematic band energy diagram based on the full potential linear muffin-tin orbital method,71,72 and the recorded data with denoted possible point defects in the microwave processed ZnO particles. | |
The low-intensity violet-blue emission and the strong orange-red emission, including the green-yellow emission, in the PL spectrum of ZnO indicate a negligible concentration of Zn interstitial defects and a high concentration of both oxygen vacancies and oxygen interstitials. As explained in our previous paper, a large amount of intrinsic bulk defects is due to rapid crystallization caused by the high energy provided to the precipitated precursor via microwave irradiation.30 The intensity and the surface area of the emission bands in the PL spectrum of ZnO/CTAB are somewhat different from those in the PL spectrum of ZnO. The violet-blue emission indicates a larger concentration of surface defects while the sum of green-yellow and orange-red emissions indicates a reduced concentration of oxygen vacancies and interstitials. These results are in agreement with the Raman spectroscopy data (Fig. 4, Table 1), which reveal an increase in crystallinity simultaneously with a reduced concentration of oxygen vacancies as a result of bonding between bromide anions in CTAB with 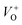 or
or 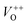 in the ZnO lattice. The PL spectrum of ZnO/F127 is almost the same as the corresponding spectrum of ZnO/CTAB, though the surface-to-bulk area ratio is somewhat different. In this case, oxygen ions of Pluronic F127 were involved in the neutralization of
in the ZnO lattice. The PL spectrum of ZnO/F127 is almost the same as the corresponding spectrum of ZnO/CTAB, though the surface-to-bulk area ratio is somewhat different. In this case, oxygen ions of Pluronic F127 were involved in the neutralization of 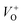 or
or 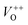 in the ZnO lattice. The recorded PL spectra with additional deconvolution confirm the profound effects of surfactants (used in MW processing) on the relative surface-to-bulk defect ratio. An illustrative representation of the impact of surfactants on the defect chemistry and radiative recombination in the microwave processed zinc oxide particles is given by the chromaticity (CIE 1931) plot in the (x, y) coordinate system, Fig. 8. The chromaticity coordinates are (0.4724, 0.4559), (0.4474, 0.4213), and (0.4393, 0.4202) with correlated color temperatures 2860, 2967, and 3088 K for ZnO, ZnO/CTAB, and ZnO/F127, respectively. This CIE plot displaying ZnO particles with color tunability, indicates great possibilities of the microwave processing of ZnO by merely varying surfactants or polymers. ZnO particles synthesized in this way may be applied as white light emitting devices (WLEDs).73
in the ZnO lattice. The recorded PL spectra with additional deconvolution confirm the profound effects of surfactants (used in MW processing) on the relative surface-to-bulk defect ratio. An illustrative representation of the impact of surfactants on the defect chemistry and radiative recombination in the microwave processed zinc oxide particles is given by the chromaticity (CIE 1931) plot in the (x, y) coordinate system, Fig. 8. The chromaticity coordinates are (0.4724, 0.4559), (0.4474, 0.4213), and (0.4393, 0.4202) with correlated color temperatures 2860, 2967, and 3088 K for ZnO, ZnO/CTAB, and ZnO/F127, respectively. This CIE plot displaying ZnO particles with color tunability, indicates great possibilities of the microwave processing of ZnO by merely varying surfactants or polymers. ZnO particles synthesized in this way may be applied as white light emitting devices (WLEDs).73
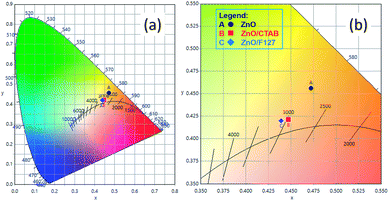 |
| | Fig. 8 (a) The CIE 1931 x, y chromaticity space of microwave processed ZnO particles and (b) magnified image with CRI values. | |
It is noteworthy that apart from the nature of point defects, PL also reflects band structures: in direct bandgap materials, the PL spectrum is narrow with a high intensity, while in indirect bandgap materials the PL spectrum can be broad with small intensity peaks. Therefore, the PL spectra imply that microwave processed ZnO materials have an indirect bandgap, which is more probably due to a large amount of point defects.
3.3. The influence of CTAB and F127 on the photocatalytic properties of ZnO particles
As we deduced from the PL spectra, a high efficiency of charge separation is achieved in the microwave processed ZnO particles, resulting in a great potential for photocatalytic activity. As a simple check of photocatalytic activity, MW processed ZnO powders were tested for decolorization of methylene blue (as a model system) under direct sunlight illumination. Fig. 9(a) reveals the efficiency of the photocatalytic degradation of MB dye in the presence of the microwave processed ZnO powders. ZnO/CTAB and ZnO/F127 showed a 100% efficiency after 2 h of direct sunlight illumination, while the efficiency of ZnO was just 67% after 5 hours of illumination. The time necessary to decolorize 50% of the dye (t1/2) was calculated from the kinetic plots, Fig. 9(b); the calculated t1/2 values for ZnO, ZnO/CTAB and ZnO/F127 are equal to 135, 26.42 and 32.92 min, respectively. The photocatalytic activities of different ZnO-based materials were compared in terms of the rate constant of degradation of some typical dyes, and are listed in Table 4.
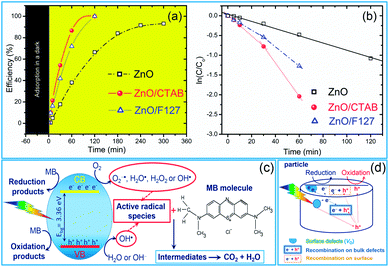 |
| | Fig. 9 (a) Photocatalytic efficiency, (b) appropriate first-order reaction kinetic plots for de-colorization of [MB]i = 10 ppm in the presence of microwave processed ZnO particles under natural sunlight illumination, (c) illustration of the reaction scheme for MB photodegradation on ideal ZnO crystal, and (d) illustration of exciton recombination pathways on ZnO with surface and bulk defects. | |
Table 4 Reaction rate constants for different ZnO-based photocatalysts
| Sample |
Rate constant (min−1) |
Degradation parameters |
Ref. |
| ZnO-600 |
0.0203 |
MO under UV light |
74 |
| Ag-ZnO-600 |
0.06838 |
MO under UV light |
74 |
| Ce-ZnO/CNT |
0.00579 |
MB under UV light |
75 |
| Ce-ZnO |
0.00211 |
MB under UV light |
75 |
| ZnO |
0.00072 |
MB under UV light |
75 |
| ZnO |
0.027 |
MB under UV light |
76 |
| ZnO rodlike particles |
0.06329 |
RhB under UV light |
77 |
| ZnO ricelike particles |
0.0431 |
RhB under UV light |
77 |
| ZnO disclike particles |
0.02448 |
RhB under UV light |
77 |
| ZnO |
0.22 |
MB under UV light |
78 |
| ZnO, 180 min milled |
0.0097 |
MB under UV light |
78 |
| ZnO nanospheres |
0.017 |
MO under Vis light |
79 |
| ZnO hollow nanospheres |
0.0288 |
MO under Vis light |
79 |
| ZnO yolk–shell nanospheres |
0.0321 |
MO under Vis light |
79 |
| ZnO |
0.0023 |
MB under UV light |
80 |
| ZnO/PEO-2 |
0.0042 |
MB under sunlight |
30 |
| ZnO/PEO-6 |
0.0100 |
MB under sunlight |
30 |
| ZnO/PEO-9 |
0.0057 |
MB under sunlight |
30 |
| ZnO |
0.0356 |
MB under UV light |
9 |
| ma-ZnO/SnO2 |
0.0736 |
MB under UV light |
9 |
| ZnO/SnO2-400 |
0.0638 |
MB under UV light |
9 |
| ZnO/SnO2-700 |
0.00342 |
MB under UV light |
9 |
| ZnO |
0.136 |
MB under sunlight |
9 |
| ma-ZnO/SnO2 |
0.180 |
MB under sunlight |
9 |
| ZnO/SnO2-400 |
0.0440 |
MB under sunlight |
9 |
| ZnO/SnO2-700 |
0.0120 |
MB under sunlight |
9 |
| ZnO |
0.0051 |
MB under sunlight |
This work |
| ZnO/CTAB |
0.0262 |
MB under sunlight |
This work |
| ZnO/F127 |
0.0210 |
MB under sunlight |
This work |
Generally, photocatalytic activity is driven by crystallinity, optical properties, specific surface area, particle size and morphology. In particular a higher surface-to-bulk defect ratio and larger particle sizes are expected to increase the photocatalytic activity of ZnO materials. A higher surface-to-bulk defect ratio diminishes the probability of recombination of photoexcited electrons and holes, simultaneously enhancing photocatalytic activity; while larger particles have a better absorption capacity. In the case of microwave processed ZnO particles, an excellent photocatalytic activity of ZnO/CTAB can be attributed to: (a) the larger surface area and larger pore volume (Table 2), providing a larger number of active sites for photocatalytic reactions,45 (b) a specific platelike particle morphology with an average size of about 550 nm enhanced the absorption capacity of visible light, ZnO/CTAB particles exhibited ∼5% higher absorbance capacity than ZnO particles (Fig. 5), (c) the increased surface-to-bulk defect ratio (Fig. 4, Table 2) hindered recombination of photogenerated electrons and holes. Both surfactants decreased the concentration of bulk oxygen vacancies, while sensitizing the surfaces.
It has been shown that mechanism of MB photodegradation depends on point defects in photocatalysts.81,82 Generally, a reaction pathway for MB degradation on a photocatalyst is very complex, it assume series of reactions starting with photoexcitation eqn (2). Actually, when ZnO absorb light with energy equal or greater than the band gap of the semiconductor, electrons are excited from the valence band to the conduction band. In following, the photogenerated holes (h+) and electrons (e−) migrate from bulk to surface. The photogenerated holes at the VB react with water molecules adsorbed at the particle surface to produce hydroxyl radical OH˙ eqn (3), while electrons in CB react with oxygen molecules generating anionic superoxide radical O2−˙ eqn (4). In further steps radicals can be transformed in highly reactive OH˙ eqn (5)–(8).81,82
| | |
ZnO + hν → ZnO (e− (CB) + h+ (VB))
| (2) |
| | |
h+ (VB) + H2O or OH− → OH˙ + H+
| (3) |
| | |
2HO2˙ + 2H+ → H2O2 + 2OH˙
| (7) |
Degradation of MB molecule obey via reaction with radicals (9), especially with O2−˙, HO2˙, and OH˙ as highly reactive ones, and may be accompanied with oxidation (10) and reduction (11) processes on the surface of the photoexcited catalysts.81 After several steps and intermediates, the final products of MB photodegradation are CO2, H2O and inorganic molecules (9).82
| | |
MB dye + reactive radicals → MB dye intermediates + reactive radicals → … → CO2 + H2O + inorganic molecules
| (9) |
| | |
MB dye + h+ (VB) → oxidation products
| (10) |
| | |
MB dye + e− (CB) → reduction products
| (11) |
The proposed photodegradation mechanism of MB dye on ZnO catalysts under direct sunlight irradiation is illustratively represented in Fig. 9(c). Mechanism of radical formation and further dye degradation depends on the surface-to-bulk defect ratio. In the case of a ZnO crystal free of any defects electron–hole recombination is in competition with charge transfer to adsorbed species. Mechanistic studies demonstrate that the majority of photogenerated electrons and holes recombine, resulting in a moderate photoactivation efficiency. The recombination can occurs in both the bulk and the surface.83 Theoretical calculations and experimental studies have shown that the surface defects can trap the photogenerated holes, helping the separation of photogenerated electron–hole pairs and hindering the recombination. What's more, the photogenerated holes trapped by surface defects are ready to react with electron donors (e.g. OH−) and the photocatalytic reaction can be greatly promoted.83 Although, the defects in the bulk can act as the recombination centers leading to the deterioration of photocatalytic efficiency.84–86 With all the other influencing factors, surface defects play a significant role in the photodegradation mechanism. Fig. 9(d) illustrate an general exciton recombination pathways on ZnO crystal with surface and bulk defects. Fig. 10 illustrate crystal structures of the ZnO, ZnO/CTAB, and ZnO/F127, pointing to different surface-to-bulk defect ratio.
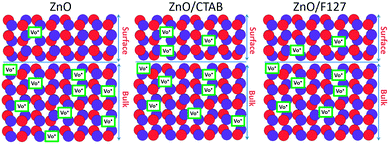 |
| | Fig. 10 Illustration of ZnO, ZnO/CTAB and ZnO/F127 crystal structures. | |
An electrochemical impedance spectroscopy analysis was carried out to investigate the charge transfer and charge recombination processes when microwave processed ZnO were used as photoanodes. The LSV of ZnO photoanodes, recorded in dark conditions and under illumination, are shown in Fig. 11(a). The current density values measured in the dark conditions at 1.5 V vs. SCE were 1.5, 13.6 and 7.6 mA g−1 for ZnO, ZnO/CTAB and ZnO/F127, respectively. When the photoanodes were illuminated with 90 mW cm−2 the current density increased and at 1.5 V vs. SCE the measured values were 10.4, 27.0 and 20.0 mA g−1 for ZnO, ZnO/CTAB and ZnO/F127, respectively. Illumination of the photoanodes also induced a significant shift of the overpotential toward lower values; the most significant decrease in potential, from 1.296 to 0.248 V vs. SCE, was measured when ZnO/CTAB was used as the photoanode. Fig. 11(b) shows the typical chronoamperometric response of the ZnO photoanodes recorded in the on/off regime under illumination of 90 mW cm−2 at the potential of 1.5 V vs. SCE. As can be seen from Fig. 11(b), all microwave processed ZnO materials are slightly active in dark conditions. However, these materials become active when they absorb visible light; light energy knocks electrons from the valence band to the conduction band, generating photoexcitons.87 The on/off ratio for the ZnO, ZnO/CTAB and ZnO/F127 photoanodes were found to be 5.9, 14 and 6.9, respectively. The ZnO/CTAB photoanode showed a higher photoresponse than ZnO and ZnO/F127 ones, which is 1.9 and 1.5-fold higher. The higher on/off ratio confirmed the profound effects of the optimal surface-to-bulk defect ratio in ZnO/CTAB by prolonging the electron–hole pair recombination time and faster carrier transport to the electrode.88 These results are in accordance with LSV measurements. The stability of photocurrent signals is verified under repeated on/off regimes and under continual illumination confirming the resistivity of the of the point defects to photocorrosion and prolonged catalysts lifetime of the microwave processed ZnO materials. This confirmation of the point defects stability is more important knowing that VO sites can be easily passivated by the adsorbed H2O and CO2 during photocatalytic reactions.85,89 Besides, the activation energies needed for the photo-ionization of VO to 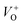 and
and 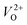 are low, ∼2.0 eV and ∼2.3 eV, respectively.89 The photo-ionized oxygen vacancies can be neutralized to VO by a recombination process with the photo-generated electron or intrinsic electrons, eqn (12) and (13):85
are low, ∼2.0 eV and ∼2.3 eV, respectively.89 The photo-ionized oxygen vacancies can be neutralized to VO by a recombination process with the photo-generated electron or intrinsic electrons, eqn (12) and (13):85
| |
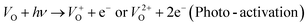 | (12) |
| |
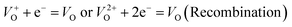 | (13) |
 |
| | Fig. 11 (a) Linear sweep voltammograms of ZnO photoanodes in 0.1 M Na2SO4 solution at scan rate 20 mV s−1 in dark condition and under visible light illumination, and (b) chronoamperometric current density vs. time (J–t) response of the ZnO photoanodes measured at 1.5 V vs. SCE in dark condition and under visible light illumination. | |
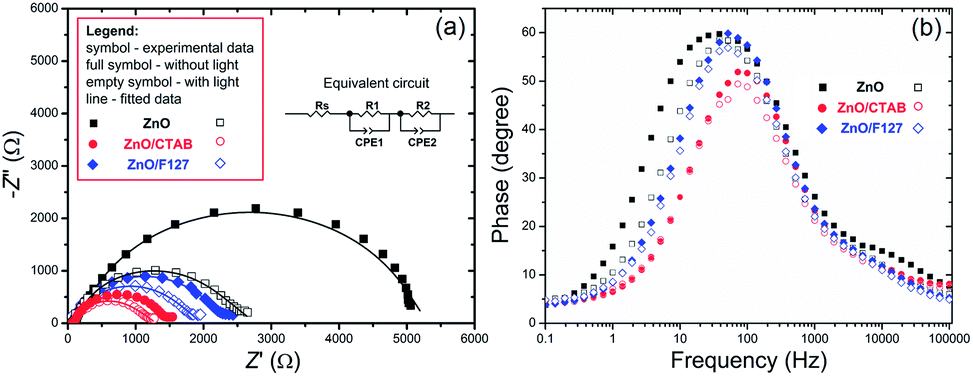
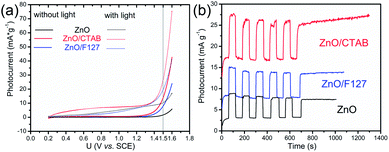
To explain the effects of the surfactants, i.e. surface-to-bulk defects ratio on the photoresponse and charge transport phenomena,90,91 Nyquist plots for the three ZnO photoanodes were recorded in dark and under illumination, at the potential of 1.5 V vs. SCE. The Nyquist plots display similar semicircles with different radii, Fig. 12(a); the radii were significantly reduced during illumination. The smallest semicircle was recorded for the ZnO/CTAB photoanode, which indicates that its resistance to the carrier transfer from the photoanode bulk to the surface was the smallest, i.e. the separation efficiency was the largest.90 Even though one semicircle can be clearly observed in the Nyquist plots, two semicircles can be distinguished by enlarging the high frequency range: smaller one in the higher frequency region (1–100 kHz) and predominant one in the medium frequency region (0.1 Hz to 1 kHz). According to literature data, the higher frequency region semicircle is attributed to the charge transfers and recombination processes occurring at the electrode/electrolyte interface, while the medium frequency region semicircle is attributed to electrochemical processes occurring at the photoanode/redox electrolyte interface.91 The diameters of the semicircles in the Nyquist plots signify the charge transfer kinetics at the electrode interface.17 Therefore, to determine the diameter of the semicircles, it was necessary to fit the Nyquist plots by an equivalent circuit. For the given data-set (Nyquists plots), it is always possible to apply more than one equivalent circuit model that can fit the data and it is necessary to find an appropriate physics-based mathematical model for fitting. Among several tested models we choose to fit it with a circuit consisting of two parallel resistor (R) and constant phase elements (CPE) connected in a series plus one more R connected in a series, inset in Fig. 12(a); the parameters obtained by fitting of the Nyquists plots are listed in Table 5. The Rs is the series resistance of the electrochemical device the value of which represents the sum of the FTO substrate resistance, the resistance related to the ionic conductivity in the electrolyte and the external contact resistance. The Rs values obtained by fitting varied between 55 and 60 Ω; however, since Rs should be a constant independent of the photoanode, the values are not presented in Table 5. The CPE1 and R1 are attributed to the high-frequency arc describing the charge transfer in the bulk, while the CPE2 and R2 are attributed to the predominant arc, describing the charge transfer processes on the photoanode/electrolyte interface.92,93 Generally, the charge transfer processes in the bulk are faster than the charge transfer processes on the interface of the photoanode/electrolyte.94 The fitting of the Nyquist plots recorded in the dark conditions gives R2 values of 5271, 1361 and 2204 Ω cm−2 for ZnO, ZnO/CTAB and ZnO/F127, respectively. For the Nyquist plots recorded under illumination, the fitting shows reduced R2 values of 2454, 1119 and 1754 Ω cm−2. Therefore, ZnO/CTAB shows the smallest resistance to the charge transfer processes on the photoanode/electrolyte interfaces; this result is in accordance with the highest photocurrent obtained by the LSV measurement, as well as with the photoresponse. The EIS results indicate that the ZnO/CTAB photoanode provides the most efficient charge transfer across the electrode/electrolyte interface, with a longer lifetime of photogenerated electron–hole pairs and reduced possibility of charge recombination.95,96
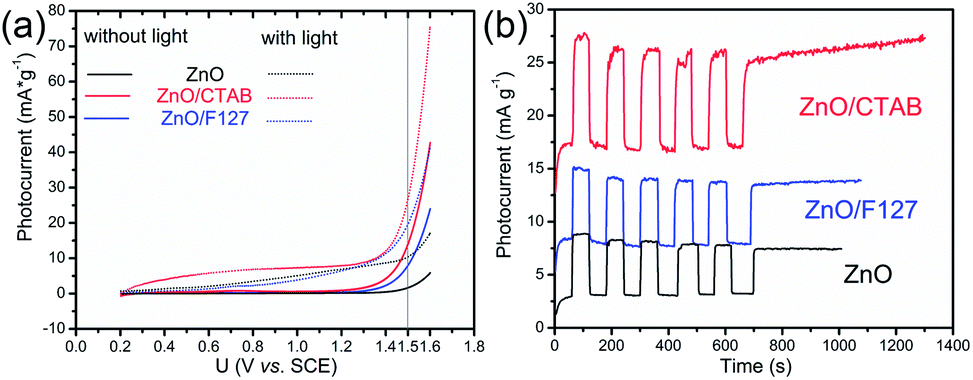
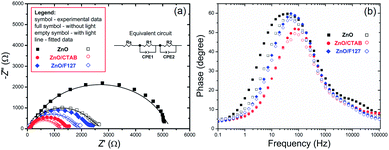 |
| | Fig. 12 (a) EIS of the ZnO photoanodes recorded in dark condition and under visible light illumination with the equivalent circuit model as an inset, and (b) Bode phase plots. | |
Table 5 Parameters obtained by fitting of EIS plots with the equivalent circuit presented in Fig. 12(a)
| Sample |
Dark conditions |
Light illumination |
| R1 (Ω) |
CPE1 (F) |
R2 (Ω) |
CPE2 (F) |
R1 (Ω) |
CPE1 (F) |
R2 (Ω) |
CPE2 (F) |
| ZnO |
57.1 |
2.67 × 10−5 |
5271 |
1.32 × 10−5 |
40.9 |
2.24 × 10−5 |
2454 |
1.49 × 10−5 |
| ZnO/CTAB |
37.4 |
2.08 × 10−5 |
1361 |
1.34 × 10−5 |
33.7 |
2.34 × 10−5 |
1119 |
1.94 × 10−5 |
| ZnO/F127 |
31.9 |
1.41 × 10−5 |
2204 |
1.27 × 10−5 |
30.4 |
1.75 × 10−5 |
1754 |
1.52 × 10−5 |
The charge separation dynamics of ZnO photoanodes is estimated according to the electron lifetime (τe) which can be calculated from the maximum peak frequency (fmax) in the mid-frequency region of the Bode plots (Fig. 12b) using eqn (14):19
The calculated electron lifetimes of ZnO, ZnO/CTAB and ZnO/F127 are 4.1, 2.2 and 3.1 ms, respectively. The shortest electron lifetime found for ZnO/CTAB suggests the fastest charge transfer of electrons across electrode/electrolyte interface in ZnO/CTAB as the photoanode.
The photoconversion efficiency values are calculated based on the J–V curves using eqn (15):97
| |
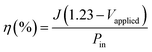 | (15) |
where:
J is the photocurrent (A g
−1),
Vapplied is the applied potential
vs. SCE, and
Pin is the power of the incident light which is in this case 90 mW cm
−2. The photoconversion efficiency of the ZnO photoanodes as a function of applied potential is shown in
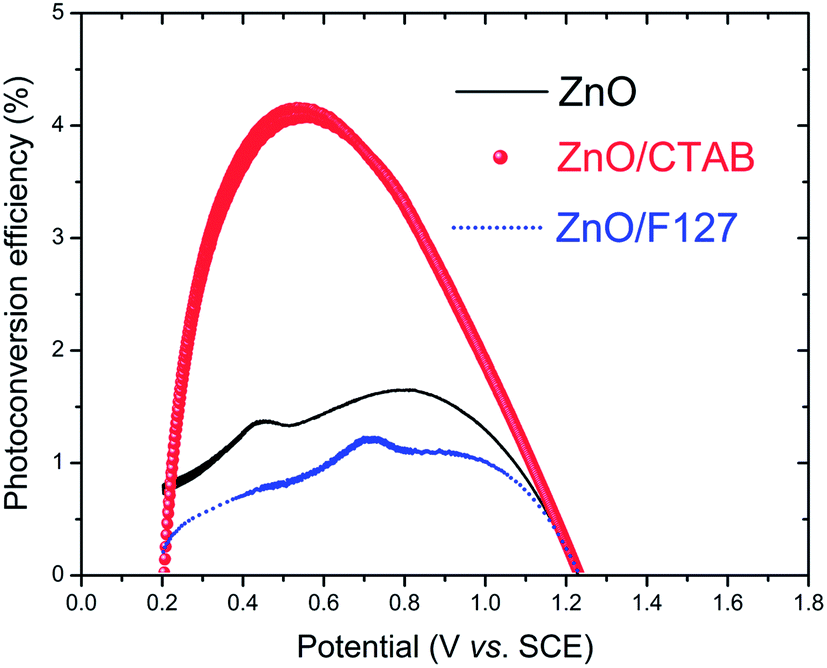
Fig. 13; the ZnO/CTAB photoanode shows the largest photoconversion efficiency attaining 4.2% at 0.520 V
vs. SCE which is, at the same potential, about 3.0 and 4.7-fold larger than that of ZnO and ZnO/F127, respectively. The ZnO photoanode reaches the maximal efficiency of 1.65% at 0.791 V
vs. SCE while the ZnO/F127 photoanode achieves the maximal efficiency of 1.23% at 0.713 V
vs. SCE.
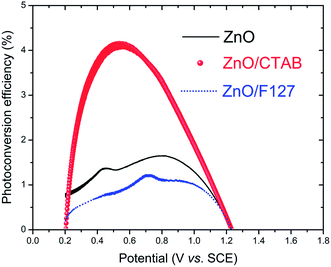 |
| | Fig. 13 Photoconversion efficiency of ZnO photoanodes as a function of applied potential. | |
4. Conclusions
We have established an eco-friendly, rapid and low-cost processing method for obtaining highly photo(electro) catalytically active ZnO particles. The proposed method, which uses a small amount of surfactants (CTAB and Pluronic F127), allows tuning of nano-crystallite polarity, particle texture, morphology, and surface-to-bulk defect ratio. In our study, the synergy of these properties resulted in enhanced optical and photo(electro)catalytic properties of ZnO particles. The best photo(electro)catalytic properties were exhibited by the ZnO/CTAB sample. After 2 hours under direct sunlight illumination, ZnO/CTAB photocatalysts efficiently (100%) decolorized 10 ppm MB dye water solution, with a decolorization halftime, t1/2, of 26.42 min. ZnO/CTAB exhibited excellent properties as a photoanode. When the ZnO/CTAB photoanode was illuminated with 90 mW cm−2 the current density, measured at 1.5 V vs. SCE, reached 27.0 mA g−1. Illumination of the ZnO/CTAB photoanode induced a significant shift of the overpotential toward lower values, namely from 1.296 to 0.248 V vs. SCE. Compared to ZnO and ZnO/F127, the ZnO/CTAB photoanode showed a higher photoresponse which was 1.9 and 1.5-fold higher. The ZnO/CTAB photoanode provides an efficient charge transfer across the electrode/electrolyte interface, with a longer lifetime of photogenerated electron–hole pairs and reduced possibility of charge recombination. The fastest charge transfer of electrons across the electrode/electrolyte interface acts with ZnO/CTAB as the photoanode. The photoconversion efficiency of ZnO/CTAB was 4.2%. XRD, Raman, PL, and EIS analyses reveal that the optimal surface-to-bulk defect ratio is favorable for improvement of photo(electro)catalytic properties.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported by the Ministry of Education, Science and Technological Development of the Republic of Serbia, Grant no III45004, and the bilateral cooperation program between the Republic of Serbia and the Republic of Slovenia “Nanostructured and mesoporous functional materials with enhanced solar light driven photocatalytic activity” for 2018–2019.
References
- A. Fujishima and K. Honda, Nature, 1972, 238(5358), 37–38 CrossRef CAS PubMed.
- S. Girish Kumar and K. S. R. Koteswara Rao, RSC Adv., 2015, 5, 3306–3351 RSC.
- X. Chen, Y. Li, X. Pan, D. Cortie, X. Huang and Z. Yi, Nat. Commun., 2016, 7, 12273 CrossRef CAS PubMed.
- P. Kumar, R. Boukherroub and K. Shankar, J. Mater. Chem. A, 2018, 6, 12876–12931 RSC.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS.
- S. Guo, X. Zhao, W. Zhang and W. Wang, Mater. Sci. Eng., B, 2018, 227, 129–135 CrossRef CAS.
- K. Afroz, M. Moniruddin, N. Bakranov, S. Kudaibergenov and N. Nuraje, J. Mater. Chem. A, 2018, 6, 21696–21718 RSC.
- Y. Li, X. Zhang, S. Jiang, H. Dai, X. Sun and Y. Li, Sol. Energy Mater. Sol. Cells, 2015, 132, 40–46 CrossRef CAS.
- S. Marković, A. Stanković, J. Dostanić, Lj. Veselinović, L. Mančić, S. D. Škapin, G. Dražić, I. Janković-Častvan and D. Uskoković, RSC Adv., 2017, 7, 42725–42737 RSC.
- G. Liao, J. Chen, W. Zeng, C. Yu, C. Yi and Z. Xu, J. Phys. Chem. C, 2016, 120, 25935–25944 CrossRef CAS.
- G. Liao, W. Zhao, Q. Li, Q. Pang and Z. Xu, Chem. Lett., 2017, 46, 1631–1634 CrossRef CAS.
- G. Liao, Q. Li, W. Zhao, Q. Pang, H. Gao and Z. Xu, Appl. Catal., A, 2018, 549, 102–111 CrossRef CAS.
- A. A. Ismail and D. W. Bahnemann, Sol. Energy Mater. Sol. Cells, 2014, 128, 85–101 CrossRef CAS.
- Z. Liu, C. Ma, Q. Cai, T. Hong, K. Guo and L. Yan, Sol. Energy Mater. Sol. Cells, 2017, 161, 46–51 CrossRef CAS.
- C. Y. Toe, H. L. Tan, C. Boyer, A. Rawal, S. C. Thickett, J. Scott, R. Amala and Y. H. Ng, J. Mater. Chem. A, 2017, 5, 4568–4575 RSC.
- J. Abed, M. AlMheiri, F. Alexander, N. S. Rajput, J. Viegas and M. Jouiad, Sol. Energy Mater. Sol. Cells, 2018, 180, 228–235 CrossRef CAS.
- R. Kant, S. Pathak and V. Dutta, Sol. Energy Mater. Sol. Cells, 2018, 178, 38–45 CrossRef CAS.
- W. Zhang, W. Wang, H. Shi, Y. Liang, J. Fu and M. Zhu, Sol. Energy Mater. Sol. Cells, 2018, 180, 25–33 CrossRef CAS.
- X. Zhang, Y.-Z. Zhou, D.-Y. Wu, X.-H. Liu, R. Zhang, H. Liu, C.-K. Dong, J. Yang, S. A. Kulinich and X.-W. Du, J. Mater. Chem. A, 2018, 6, 9057–9063 RSC.
- G. Liao, J. Fang, Q. Li, S. Li, Z. Xu and B. Fang, Nanoscale, 2019, 11, 7062–7096 RSC.
- W. Zhong, S. Shen, S. Feng, Z. Lin, Z. Wang and B. Fang, CrystEngComm, 2018, 20, 7851–7856 RSC.
- W. Zhong, W. Tu, S. Feng and A. Xu, J. Alloys Compd., 2019, 772, 669–674 CrossRef CAS.
- Y. Liu, S. Shen, J. Zhang, W. Zhong and X. Huang, Appl. Surf. Sci., 2019, 478, 762–769 CrossRef CAS.
- H. Zhu, D. Liu, D. Zou and J. Zhang, J. Mater. Chem. A, 2018, 6, 6130–6154 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- G. Liao, Y. Gong, L. Zhang, H. Gao, G. Yang and B. Fang, Energy Environ. Sci., 2019 10.1039/c9ee00717b.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- W. Li, R. Liang, A. Hu, Z. Huanga and Y. N. Zhou, RSC Adv., 2014, 4, 36959–36966 RSC.
- T. Xia, P. Wallenmeyer, A. Anderson, J. Murowchick, L. Liu and X. Chen, RSC Adv., 2014, 4, 41654–41658 RSC.
- S. Marković, V. Rajić, A. Stanković, Lj. Veselinović, J. Belošević-Čavor, K. Batalović, N. Abazović, S. D. Škapin and D. Uskoković, Sol. Energy, 2016, 127, 124–135 CrossRef.
- A. Janotti and C. G. Van de Walle, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 165202–165222 CrossRef.
- J. Highfield, Molecules, 2015, 20, 6739–6793 CrossRef CAS PubMed.
- M. Kong, Y. Li, X. Chen, T. Tian, P. Fang, F. Zheng and X. Zhao, J. Am. Chem. Soc., 2011, 133, 16414–16417 CrossRef CAS PubMed.
- H. Silva, C. Mateos-Pedrero, C. Magen, D. A. Pacheco Tanaka and A. Mendes, RSC Adv., 2014, 4, 31166–31176 RSC.
- Q. Liu, Z. Sun, Y. Dou, J. H. Kim and S. X. Dou, J. Mater. Chem. A, 2015, 3, 11688–11699 RSC.
- P. Bai, P. Wu, Z. Yan, J. Zhou and X. S. Zhao, J. Phys. Chem. C, 2007, 111, 9729–9733 CrossRef CAS.
- R. Garvey, LSUCRIPC, Least squares unit-cell refinement with indexing on the personal computer, Powder Diffr., 1986, 1, 114–118 CrossRef.
- H. P. Klug and L. E. Alexander, X-ray diffraction procedures for polycrystalline and amorphous materials, Wiley, New York, 4th edn, 1954 Search PubMed.
- S. K. Pardeshi and A. B. Patil, J. Mol. Catal. A: Chem., 2009, 308, 32–40 CrossRef CAS.
- F. Rouquerol, J. Rouquerol and K. Sing, Adsorption by powders and porous solids, Academic Press, London, 1999 Search PubMed.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- G. R. Li, T. Hu, G. L. Pan, T. Y. Yan, X. P. Gao and H. Y. Zhu, J. Phys. Chem. C, 2008, 112, 11859–11864 CrossRef CAS.
- A. Mclaren, T. Valdes-Solis, G. Li and S. C. Tsang, J. Am. Chem. Soc., 2009, 131, 12540–12541 CrossRef CAS.
- Y.-X. Wang, J. Sun, X. Y. Fan and X. Yu, Ceram. Int., 2011, 37, 3431–3436 CrossRef CAS.
- I. Stambolova, V. Blaskov, D. Stoyanova, I. Avramova, L. Dimitrov, K. Milenova, K. Balashev, S. Simeonova, A. Tzonev, L. Aleksandrov and A. Eliyas, Bull. Mater. Sci., 2017, 40, 483–492 CrossRef CAS.
- Y. Wang, X. Li, G. Lu, G. Chen and Y. Chen, Mater. Lett., 2008, 62, 2359–2362 CrossRef CAS.
- R. Yi, N. Zhang, H. Zhou, R. Shi, G. Qiu and X. Liu, Mater. Sci. Eng., B, 2008, 153, 25–30 CrossRef CAS.
- A. Stanković, Z. Stojanović, Lj. Veselinović, S. D. Škapin, I. Bračko, S. Marković and D. Uskoković, Mater. Sci. Eng., B, 2012, 177, 1038–1045 CrossRef.
- A. Stanković, S. Dimitrijević and D. Uskoković, Colloids Surf., B, 2013, 102, 21–28 CrossRef PubMed.
- A. Sadollahkhani, I. Kazeminezhad, J. Lu, O. Nur, L. Hultman and M. Willander, RSC Adv., 2014, 4, 36940–36950 RSC.
- R. N. Moussawi and D. Patra, RSC Adv., 2016, 6, 17256–17268 RSC.
- I. M. El-Nahhal, J. K. Salem, N. S. Tabasi, R. Hempelmann and F. S. Kodeh, Chem. Phys. Lett., 2018, 691, 211–218 CrossRef CAS.
- R. Cuscó, E. Alarcón-Lladó, J. Ibáñez, L. Artús, J. Jiménez, B. Wang and M. J. Callahan, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 165202 CrossRef.
- K. F. Lin, H. M. Cheng, H. C. Hsu and W. F. Hsieh, Appl. Phys. Lett., 2006, 88, 263117 CrossRef.
- R. Sánchez Zeferino, M. Barboza Flovers and U. Pal, J. Appl. Phys., 2011, 109, 014308 CrossRef.
- Y. Li, G. Dai, C. Zhou, Q. Zhang, Q. Wan, L. Fu, J. Zhang, R. C. Cao, A. Pan, Y. Zhang and B. Zou, Nano Res., 2010, 3, 326–338 CrossRef CAS.
- R. Jothilakshmi, V. Ramakrishnan, R. Thangavel, J. Kumar, A. Saruac and M. Kuball, J. Raman Spectrosc., 2009, 40, 556–561 CrossRef CAS.
- M. Šćepanović, M. Grujić-Brojčin, K. Vojisavljević, S. Bernik and T. Srećković, J. Raman Spectrosc., 2010, 41, 914–921 CrossRef.
- H. J. Fan, R. Scholz, F. M. Kolb, M. Zacharias, U. Gösele, F. Heyroth, C. Eisenschmidt, T. Hempel and J. Christen, Appl. Phys. A: Mater. Sci. Process., 2004, 79, 1895–1900 CrossRef CAS.
- S. Sahoo, G. L. Sharma and R. S. Katiyar, J. Raman Spectrosc., 2012, 43, 72–75 CrossRef CAS.
- X. Sui, C. Shao and Y. Liu, Polymer, 2007, 48, 1459–1463 CrossRef CAS.
- C.-K. Wu, K. Sivashanmugan, T.-F. Guo and T.-C. Wen, Materials, 2018, 11, 378 CrossRef PubMed.
- S. Liu, C. Li, J. Yu and Q. Xiang, CrystEngComm, 2011, 13, 2533–2541 RSC.
- N. S. Han, H. S. Shim, J. H. Seo, S. Y. Kim, S. M. Park and J. K. Song, J. Appl. Phys., 2010, 107, 084306 CrossRef.
- S. B. Rana, J. Mater. Sci.: Mater. Electron., 2017, 28, 13787–13796 CrossRef CAS.
- H. Zeng, G. Duan, Y. Li, S. Yang, X. Xu and W. Cai, Adv. Funct. Mater., 2010, 20, 561–572 CrossRef CAS.
- J.-H. Lin, R. A. Patil, R. S. Devan, Z.-A. Liu, Y.-P. Wang, C.-H. Ho, Y. Liou and Y.-R. Ma, Sci. Rep., 2014, 4, 6967 CrossRef CAS PubMed.
- B. Jin and D. Wang, J. Lumin., 2012, 132, 1879–1884 CrossRef CAS.
- C. H. Ahn, Y. Y. Kim, D. C. Kim, S. K. Mohanta and H. K. Cho, J. Appl. Phys., 2009, 105, 013502 CrossRef.
- C. H. Ahn, Y. Y. Kim, D. C. Kim, S. K. Mohanta and H. K. Cho, J. Appl. Phys., 2009, 105, 089902 CrossRef.
- B. Lin, Z. Fu and Y. Jia, Appl. Phys. Lett., 2001, 79, 943–945 CrossRef CAS.
- H. Zeng, Z. Li, W. Cai and P. Liu, J. Appl. Phys., 2007, 102, 104307 CrossRef.
- J. R. Sadaf, M. Q. Israr, O. Nur, M. Willander, Y. Ding and Z. L. Wang, Nanoscale Res. Lett., 2011, 6, 513 CrossRef PubMed.
- Y. Hong, C. Tian, B. Jiang, A. Wu, Q. Zhang, G. Tian and H. Fu, J. Mater. Chem. A, 2013, 1, 5700–5708 RSC.
- Md. Elias, Md. Khairul Amin, S. H. Firoz, Md. Asjad Hossain, S. Akter, Md. Awlad Hossain, Md. Nizam Uddin and I. Ahmed Siddiquey, Ceram. Int., 2017, 43, 84–91 CrossRef CAS.
- M. Irani, T. Mohammadi and S. Mohebbi, J. Mex. Chem. Soc., 2016, 60, 218–225 CAS.
- S.-Y. Pung, W.-P. Lee and A. Aziz, Int. J. Inorg. Chem., 2012, 608183 Search PubMed.
- C. A. Aggelopoulos, M. Dimitropoulos, A. Govatsi, L. Sygellou, C. D. Tsakiroglou and S. N. Yannopoulos, Appl. Catal., B, 2017, 205, 292–301 CrossRef CAS.
- Q. Xie, J. Li, Q. Tian and R. Shi, J. Mater. Chem., 2012, 22, 13541–13547 RSC.
- H. Vahdat Vasei, S. M. Masoudpanah, M. Adeli and M. R. Aboutalebi, Ceram. Int., 2018, 44, 7741–7745 CrossRef.
- A. Ajmal, I. Majeed, R. Naseem Malik, H. Idriss and M. Amtiaz Nadeem, RSC Adv., 2014, 4, 37003–37026 RSC.
- R. S. Dariani, A. Esmaeili, A. Mortezaali and S. Dehghanpour, Optik, 2016, 127, 7143–7154 CrossRef CAS.
- X. Zhang, J. Qin, Y. Xue, P. Yu, B. Zhang, L. Wang and R. Liu, Sci. Rep., 2014, 4, 4596 CrossRef PubMed.
- H. Zhao, F. Pan and Y. Li, J. Materiomics, 2017, 3, 17–32 CrossRef.
- L. Tsui and G. Zangari, J. Electrochem. Soc., 2014, 161, 3066–3077 CrossRef.
- J. Yan, G. Wu, N. Guan, L. Li, Z. Li and X. Cao, Phys. Chem. Chem. Phys., 2013, 15, 10978–10988 RSC.
- I. Khan, A. A. M. Ibrahim, M. Sohail and A. Qurashi, Ultrason. Sonochem., 2017, 37, 669–675 CrossRef CAS PubMed.
- T. Majumder, J. J. L. Hmar, S. Dhar and S. P. Mondal, Chem. Phys., 2017, 490, 1–6 CrossRef CAS.
- Z. Yang, T. Meng, Q. Zhang and H.-P. D. Shieh, IEEE Electron Device Lett., 2016, 37, 437–440 CAS.
- Z. Zhou, S. Wu, L. Qin, L. Li, L. Li and X. Li, J. Mater. Chem. A, 2018, 6, 15593–15602 RSC.
- M. S. Ansari, A. Banik, A. Kalita, P. K. Iyer and M. Qureshi, J. Mater. Chem. A, 2018, 6, 15868–15887 RSC.
- T. Majumder and S. P. Mondal, J. Electroanal. Chem., 2016, 769, 48–52 CrossRef CAS.
- S. Guo, S. Wang, N. Wu, J. Liu, Y. Ni and W. Liu, RSC Adv., 2015, 5, 103767–103775 RSC.
- T. Lopes, L. Andrade, F. L. Formal, M. Gratzel, K. Sivula and A. Mendes, Phys. Chem. Chem. Phys., 2014, 16, 16515–16523 RSC.
- F. Rasouli, A. Rouhollahi and F. Ghahramanifard, Mater. Sci. Semicond. Process., 2019, 93, 371–378 CrossRef CAS.
- Y. C. Weng and K. T. Hsiao, Int. J. Hydrogen Energy, 2015, 40, 3238–3248 CrossRef CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Ivana Stojković Simatović
*a,
Ivana Stojković Simatović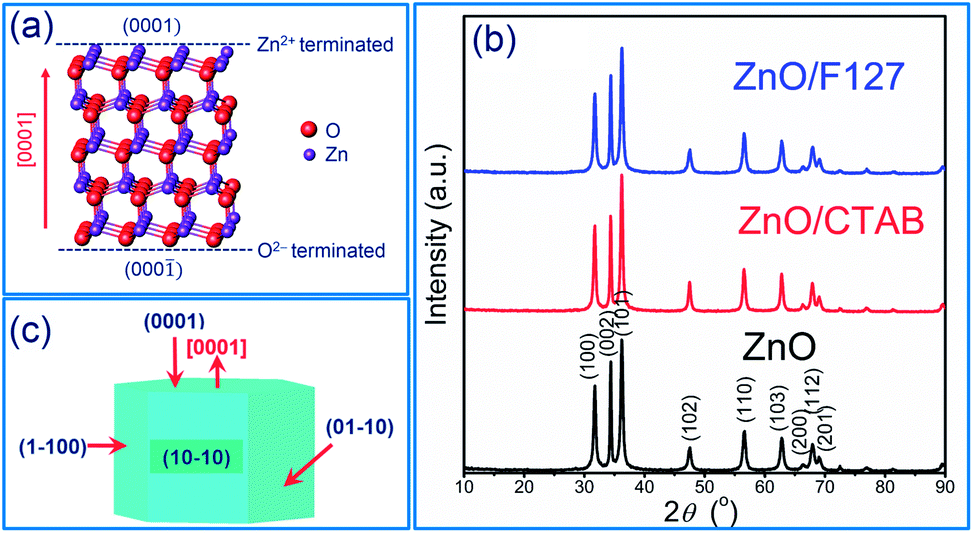
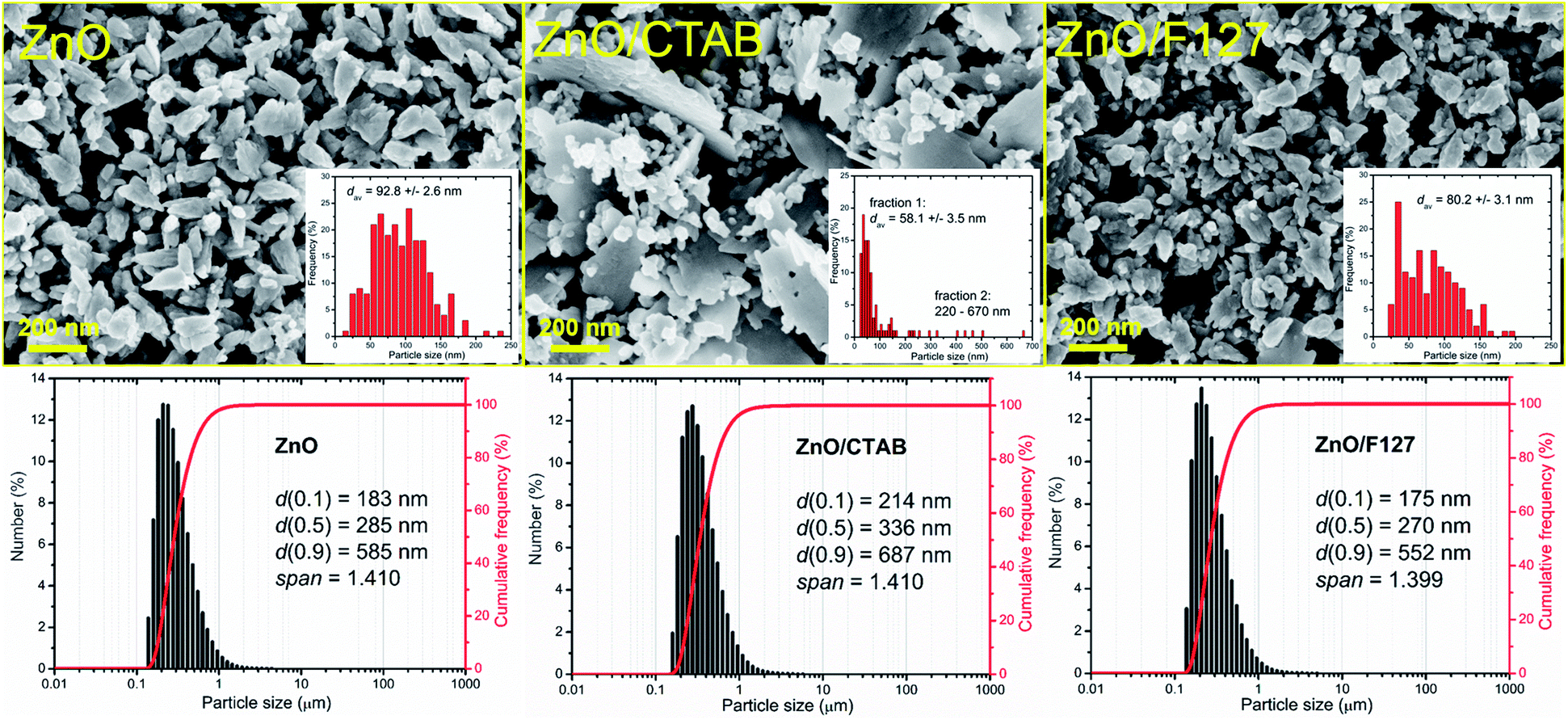
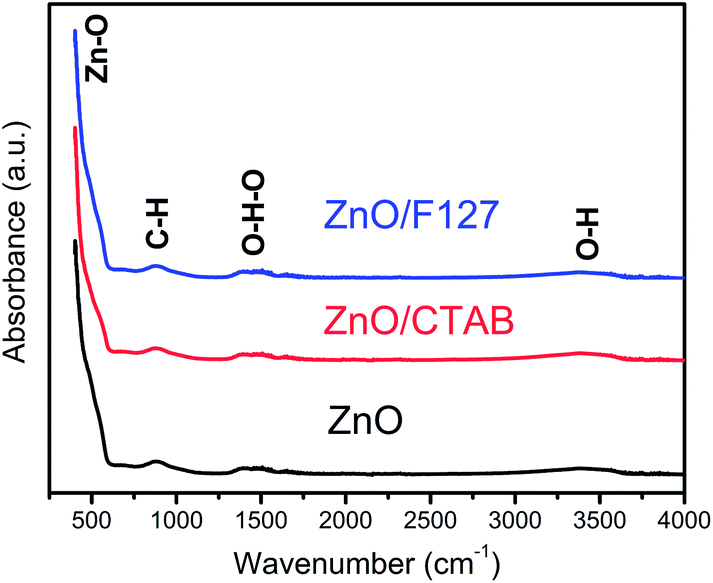
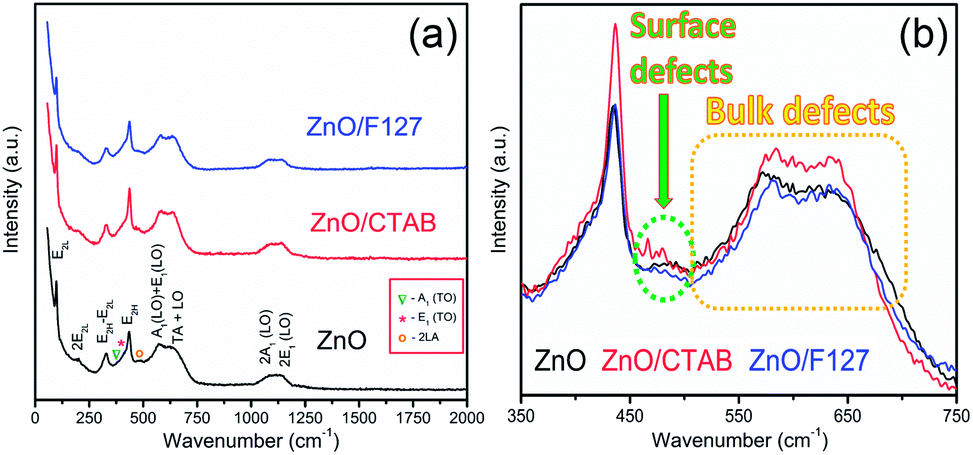
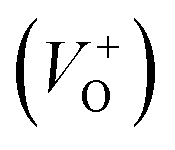 or doubly
or doubly 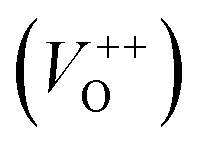 charged states of oxygen vacancies in the ZnO lattice.62
charged states of oxygen vacancies in the ZnO lattice.62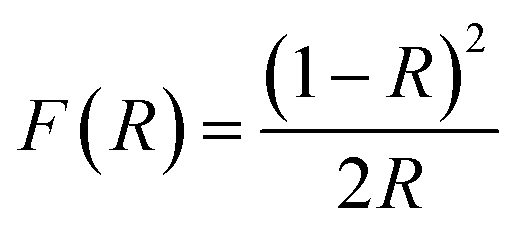
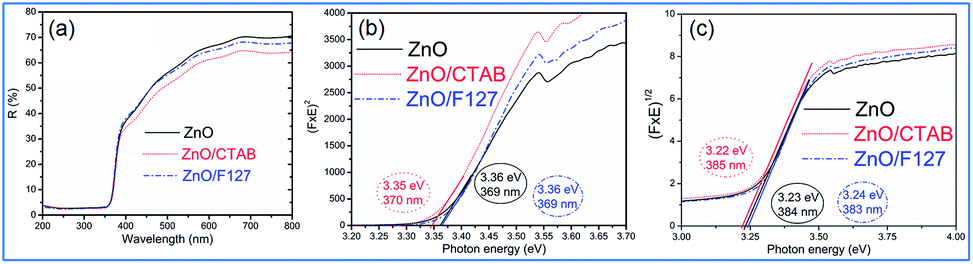
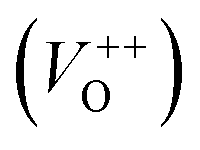 , while the yellow emission is induced by the transition from the Zni energy level to the deep-levels of
, while the yellow emission is induced by the transition from the Zni energy level to the deep-levels of 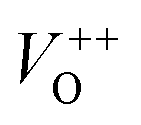 .66 The most intensive and the broadest band in the orange-red spectral region centered near 630 nm is correlated to the existence of zinc interstitials and oxygen interstitials; it is probably due to the transition from the Zni level to the Oi level in ZnO.69,70 The energy levels of intrinsic defects in ZnO have been calculated using the full-potential linear muffin-tin orbital method;71,72 the position of the Zni level is theoretically located at 0.22 eV below the conduction band while the position of the Oi level is located approximately at 2.28 eV below the conduction band. It is expected that the transition from the Zni energy level to the Oi energy level is approximately 2.06 eV, which is in good agreement with the orange-red peaks centered approximately at 1.97 eV. The energy band diagram (Fig. 7), based on the theoretically calculated energy values for different point defects and emission energies deduced after deconvolution of the PL spectra, indicates the most probable defects in the structure of microwave processed zinc oxide particles.
.66 The most intensive and the broadest band in the orange-red spectral region centered near 630 nm is correlated to the existence of zinc interstitials and oxygen interstitials; it is probably due to the transition from the Zni level to the Oi level in ZnO.69,70 The energy levels of intrinsic defects in ZnO have been calculated using the full-potential linear muffin-tin orbital method;71,72 the position of the Zni level is theoretically located at 0.22 eV below the conduction band while the position of the Oi level is located approximately at 2.28 eV below the conduction band. It is expected that the transition from the Zni energy level to the Oi energy level is approximately 2.06 eV, which is in good agreement with the orange-red peaks centered approximately at 1.97 eV. The energy band diagram (Fig. 7), based on the theoretically calculated energy values for different point defects and emission energies deduced after deconvolution of the PL spectra, indicates the most probable defects in the structure of microwave processed zinc oxide particles.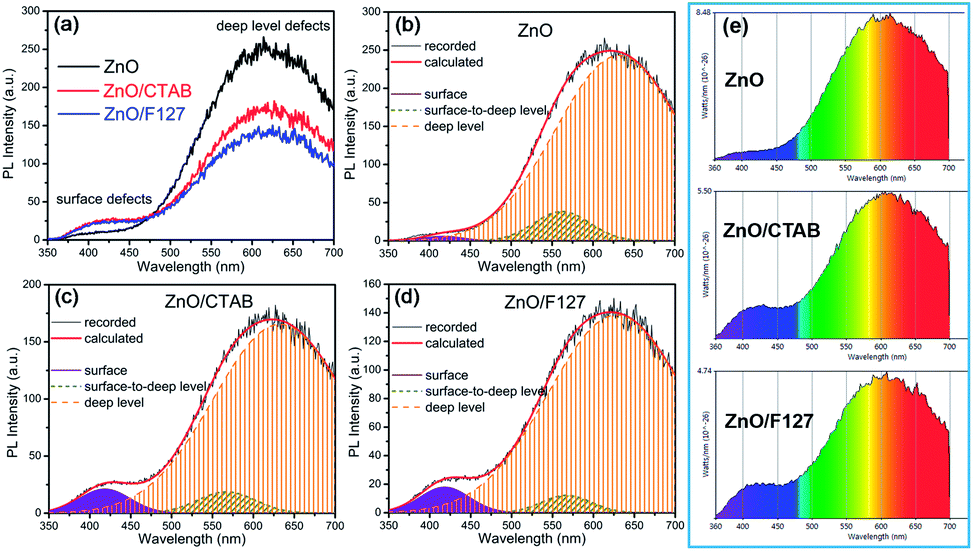
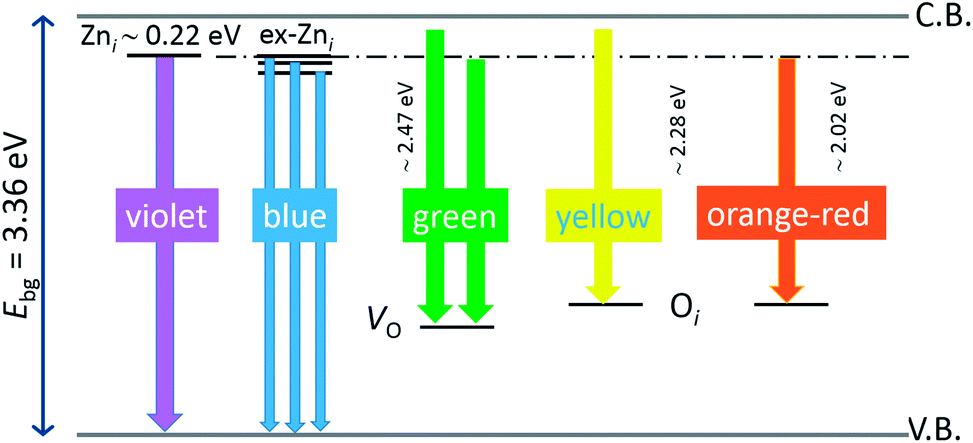
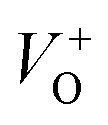 or
or 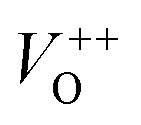 in the ZnO lattice. The PL spectrum of ZnO/F127 is almost the same as the corresponding spectrum of ZnO/CTAB, though the surface-to-bulk area ratio is somewhat different. In this case, oxygen ions of Pluronic F127 were involved in the neutralization of
in the ZnO lattice. The PL spectrum of ZnO/F127 is almost the same as the corresponding spectrum of ZnO/CTAB, though the surface-to-bulk area ratio is somewhat different. In this case, oxygen ions of Pluronic F127 were involved in the neutralization of 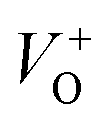 or
or 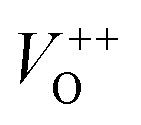 in the ZnO lattice. The recorded PL spectra with additional deconvolution confirm the profound effects of surfactants (used in MW processing) on the relative surface-to-bulk defect ratio. An illustrative representation of the impact of surfactants on the defect chemistry and radiative recombination in the microwave processed zinc oxide particles is given by the chromaticity (CIE 1931) plot in the (x, y) coordinate system, Fig. 8. The chromaticity coordinates are (0.4724, 0.4559), (0.4474, 0.4213), and (0.4393, 0.4202) with correlated color temperatures 2860, 2967, and 3088 K for ZnO, ZnO/CTAB, and ZnO/F127, respectively. This CIE plot displaying ZnO particles with color tunability, indicates great possibilities of the microwave processing of ZnO by merely varying surfactants or polymers. ZnO particles synthesized in this way may be applied as white light emitting devices (WLEDs).73
in the ZnO lattice. The recorded PL spectra with additional deconvolution confirm the profound effects of surfactants (used in MW processing) on the relative surface-to-bulk defect ratio. An illustrative representation of the impact of surfactants on the defect chemistry and radiative recombination in the microwave processed zinc oxide particles is given by the chromaticity (CIE 1931) plot in the (x, y) coordinate system, Fig. 8. The chromaticity coordinates are (0.4724, 0.4559), (0.4474, 0.4213), and (0.4393, 0.4202) with correlated color temperatures 2860, 2967, and 3088 K for ZnO, ZnO/CTAB, and ZnO/F127, respectively. This CIE plot displaying ZnO particles with color tunability, indicates great possibilities of the microwave processing of ZnO by merely varying surfactants or polymers. ZnO particles synthesized in this way may be applied as white light emitting devices (WLEDs).73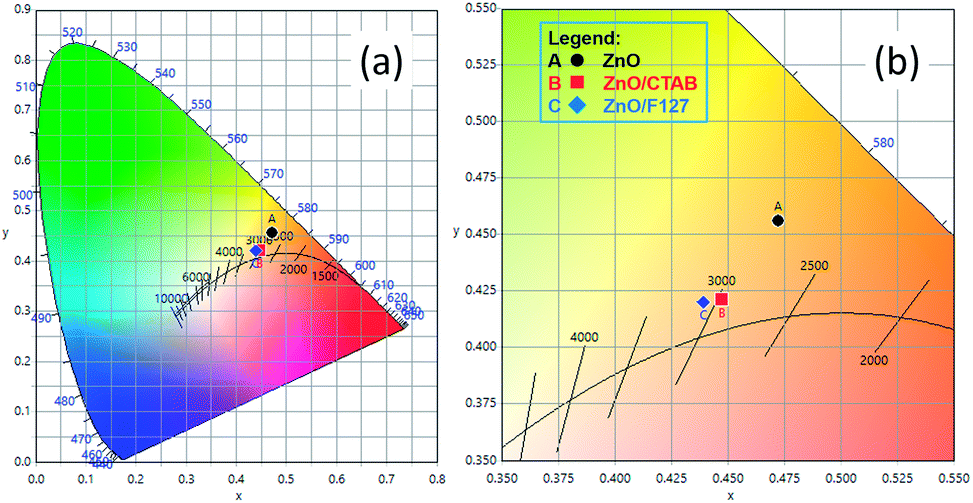
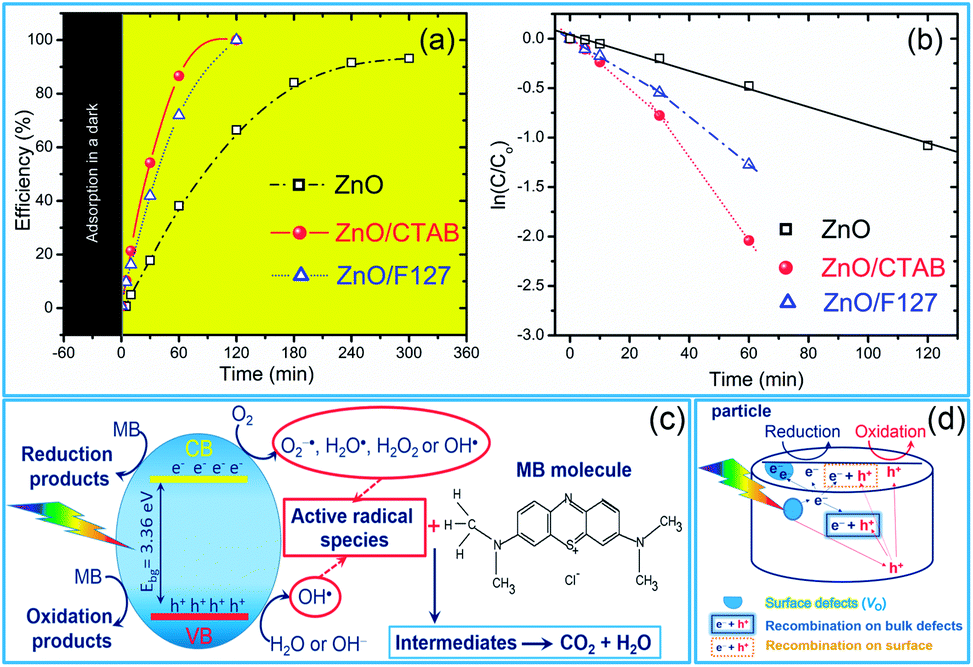
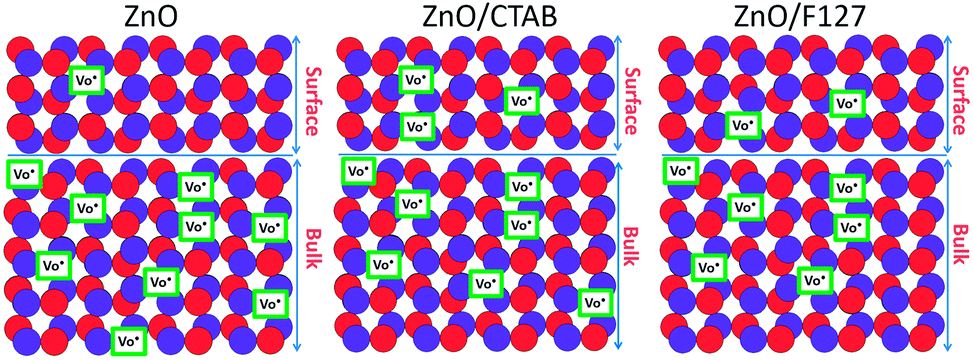
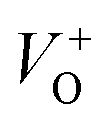 and
and 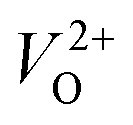 are low, ∼2.0 eV and ∼2.3 eV, respectively.89 The photo-ionized oxygen vacancies can be neutralized to VO by a recombination process with the photo-generated electron or intrinsic electrons, eqn (12) and (13):85
are low, ∼2.0 eV and ∼2.3 eV, respectively.89 The photo-ionized oxygen vacancies can be neutralized to VO by a recombination process with the photo-generated electron or intrinsic electrons, eqn (12) and (13):85