DOI:
10.1039/C9RA02525A
(Paper)
RSC Adv., 2019,
9, 13878-13886
Synthesis of novel natural product-like diaryl acetylenes as hypoxia inducible factor-1 inhibitors and antiproliferative agents†
Received
3rd April 2019
, Accepted 26th April 2019
First published on 7th May 2019
Abstract
The selaginellin derivatives are a type of novel natural pigments with an unusual alkynyl phenol skeleton from the genus Selaginella. Some of these natural compounds were previously reported to show important bioactivities, including anticancer activity, cardiovascular protection and phosphodiesterase-4 inhibition. We designed and synthesized fifteen biphenyl-containing diaryl acetylene derivatives mimicking the skeleton of natural alkynyl phenols. In MTT assay in cancer cells, compounds 1c, 2d, 2g, 2h, 2i and 2j exhibited potent antiproliferative activity. The evaluation of Hypoxia Inducible Factor-1 (HIF-1) pathway inhibitory activity in dual luciferase assay demonstrated that most tested compounds exhibited moderate to good activities. Compounds 1a, 2f and 2h displayed high HIF-1 inhibitory activities and relatively low cytotoxicity, demonstrating great potential as HIF-1 inhibitors. These results afford a new strategy for the discovery of new HIF-1 inhibitors and anti-proliferative agents from natural or synthetic diaryl acetylene derivatives.
1. Introduction
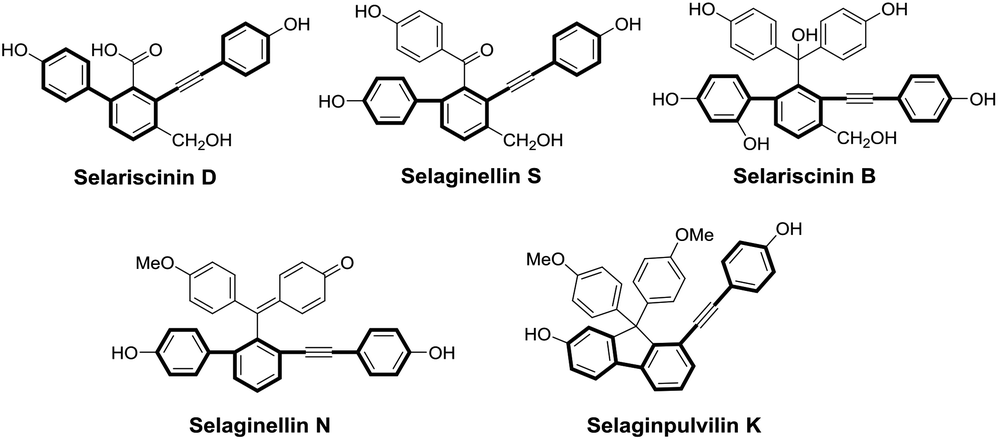
Natural products and their synthetic derivatives have served as a consistent source of valuable new drug leads for centuries, and natural products with bioactive pharmacophores are biologically validated starting points for the development of new drugs.1 In the last decade, a type of novel natural pigment with an unusual alkynyl phenol skeleton has been successively isolated from the genus Selaginella, mainly from S. tamariscina and S. pulvinata which are the two qualified species listed in Chinese Pharmacopoeia and used as traditional Chinese medicines for the treatment of dysmenorrhea, abdominal mass and traumatic injury. The pharmacological investigations of the genus Selaginella have revealed that it has anticancer, antivirus, antioxidant and anti-inflammatory activities.2 The phytochemical investigations of the genus Selaginella have led to the identification of more than forty natural alkynyl phenols hitherto, including selaginellin,3 selaginellins A–W,4–19 selariscinins A–D,14,17 and selaginpulvilins A–L.20–25 All these natural products possess unique skeletons characterized by biphenyl-containing diaryl acetylene, as shown in Fig. 1.
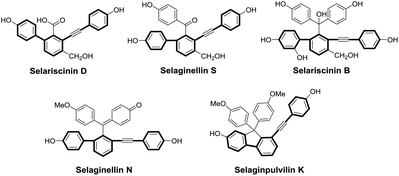 |
| | Fig. 1 Selected examples of natural diaryl acetylene compounds from the genus Selaginella. | |
The biological activities of these alkynyl phenol compounds have been demonstrated to involve hypoglycemic,14 anti-inflammatory,26 and anti-oxidant27 activity, cardiovascular protection,28 anticancer effects,9 and neuroprotective effects.18,29,30 Selaginellin and selaginellins A, C, M, N, O, Q have been reported to be cytotoxic against U251, HeLa, MCF-7 and BGC-823 tumor cells.9–11 Selaginpulvilins A–L, with an unprecedented 9,9-diphenyl-1-(phenylethynyl)-9H-fluorene skeleton, exhibit remarkable phosphodiesterase-4 inhibitory activity with IC50 of 0.011–1.38 μM.20,22,25 The previous work in our laboratory on the bioactive constituents from the genus Selaginella revealed selaginellins A and B were moderately cytotoxic against H322 cell, and selaginellin A displayed inhibitory activity on hypoxia inducible factor-1 (HIF-1) transcription in dual luciferase reporter assay (unpublished).
HIF-1 is a central regulator involved in detection and adaption to cellular oxygen stress through regulation of the hypoxic transcriptional program in angiogenesis, erythropoiesis, and metabolism.31 The HIF-1 pathway is involved in many diseases, including anemia, ischemia, most tumors, and other hypoxic-ischemic diseases, making HIF-1 an attractive target for therapeutic application.32 A considerable proportion of solid tumors are induced by hypoxia which results in overexpression of the HIF-1 pathway. Moreover, overexpression of the HIF-1 pathway has been correlated with poor prognosis, invasive tumor growth, and resistance to radiation for tumor patients.33 Therefore, the inhibition of HIF-1 has been identified as an effective therapeutic strategy for various solid tumors.34
The unique skeletons associated with important bioactivities of this family of alkynyl phenols have attracted great attention of scientists from fields of organic synthesis and medicinal chemistry. Up to now, the reports on the total synthesis of natural alkynyl phenols are rare and all focus on the diarylfluorene derivatives selaginpulvilins,21,23–25 whereas the total synthesis of selaginellins and selariscinins has not been reported yet. In our effort to develop synthetic method for selaginellins, we achieved the precursors of selaginellin N (1a–1d) with the skeleton of selariscinin D and selaginellin S. The difficulty in the synthesis of selaginellins with moiety of benzyl para-quinone methide is considered to result from the large steric hindrance of polycyclic skeleton. Considering the unique diaryl acetylene skeleton as a good privileged structure, we designed and synthesized a series of non-natural diaryl acetylene derivatives (2a–2j) analogous to selaginellins but with less steric hindrance. Herein we report the synthesis of this family of diaryl acetylene derivatives, their cytotoxic activity against tumor cells and HIF-1 inhibitory activity.
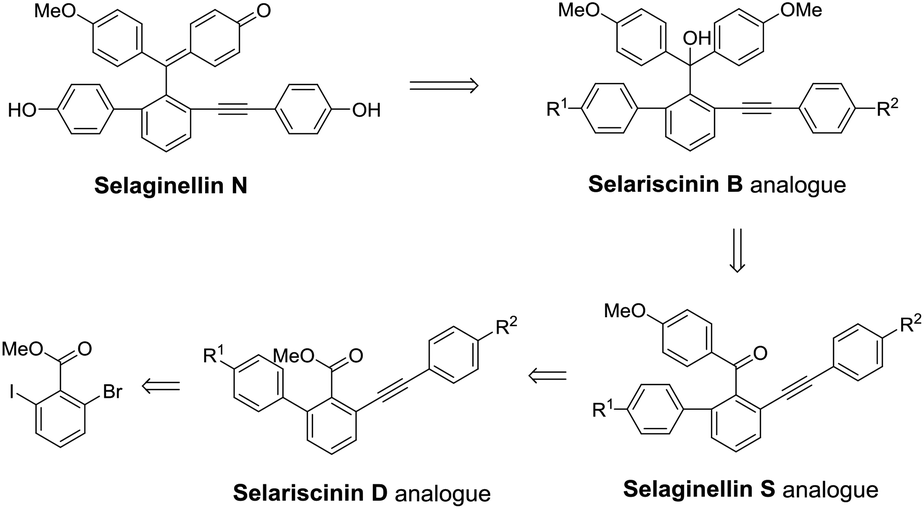
By analyzing the structures of selaginellin and its reported derivatives, P. F. Tu et al. proposed a potential biosynthetic pathway of selaginellin and its derivatives.35 We compared the different skeletons of natural alkynyl phenols as displayed in Fig. 1 and presumed that the formation of selaginellin N analogues might undergo the intermediates of selariscinin D analogues, selaginellin S analogues and selariscinin B analogues successively. Based on this presumption, a retrosynthetic analysis of selaginellin N was established as shown in Scheme 1, where selariscinin D analogues were synthesized starting with halogenated benzoate via the Suzuki coupling and Sonogashira reaction.
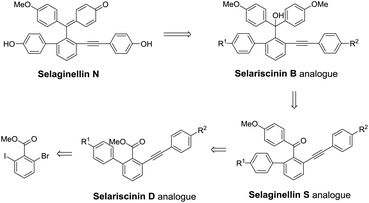 |
| | Scheme 1 Retrosynthetic analysis of diaryl acetylene compounds. | |
2. Results and discussion
2.1. Chemistry
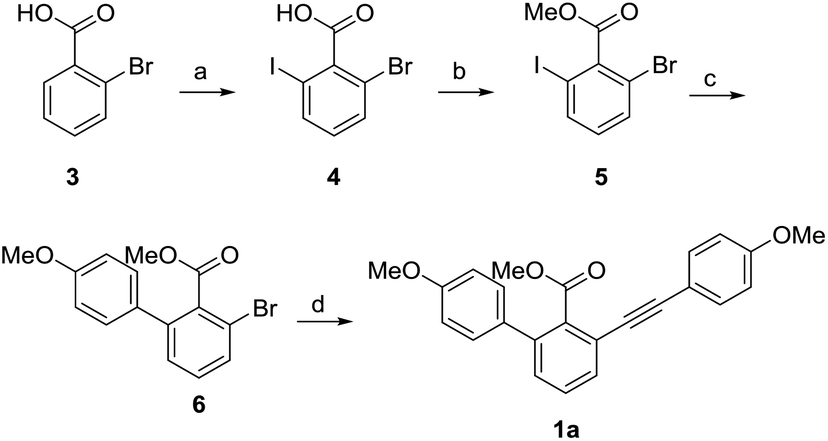
With the aim to synthesize selaginellin N, we first synthesized 1a using o-bromobenzoic acid as the starting material via iodization, methyl esterification, regioselective Suzuki coupling reaction and Sonogashira reaction (Scheme 2). Using THF–H2O as the solvent in Suzuki reaction, compound 6 was obtained under mild condition with a better yield than reported.24 Compound 1a has a skeleton similar to selariscinin D,15 which exhibited stimulatory effect on glucose uptake in 3T3-L1 adipocyte cells and potent inhibitory effect against PTP1B.
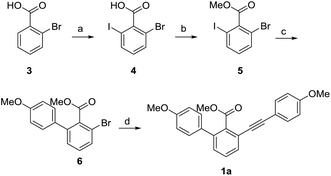 |
| | Scheme 2 Synthesis of compound 1a. Reagents and conditions: (a) Pd(OAc)2, NIS, DMF, 100 °C, 12 h, 90%; (b) K2CO3, CH3I, acetone, 40 °C, 5 h, 100%; (c) Na2CO3, 4-methoxyphenylboronic acid, PdCl2(PPh3)2, THF/H2O = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 60 °C, 8 h, 82%; (d) 4-methoxyphenylacetylene, CuI, PdCl2(PPh3)2, PPh3, Et3N, DMF, 120 °C, 12 h, 48%. :1, 60 °C, 8 h, 82%; (d) 4-methoxyphenylacetylene, CuI, PdCl2(PPh3)2, PPh3, Et3N, DMF, 120 °C, 12 h, 48%. | |
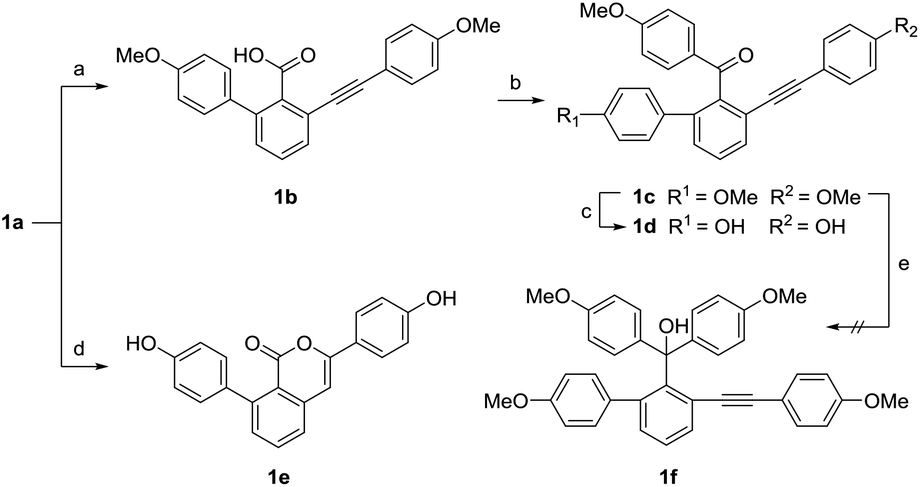
We attempted to synthesize selaginellin N through the demethylation and dehydration of triphenyl methol intermediate 1f. However, the nucleophilic addition of 1a with 4-methoxyphenyl bromide under the presence of n-butyllithium produced the diphenyl ketone 1c instead of triphenyl methol (Scheme 3). The steric hindrance of 1c is presumably the dominated factor to prevent the further reaction to produce triphenyl methol. We tried using the more reactive acyl chloride, which was prepared via the hydrolysis and chlorination of 1a. As a result, 1c was still yielded as the major product and no product of triphenyl methol was detected. Although the efforts to synthesize selaginellin N failed, the diphenyl ketones 1c and 1d are still important products since they bear the same skeleton with selaginellin S, which showed inhibitory effects against hepatitis B virus gene expression and replication.16 During the demethylation of 1a with boron tribromide, an unexpected isocoumarin 1e was obtained as the main product.
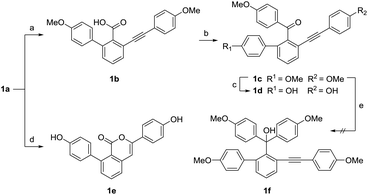 |
| | Scheme 3 Synthesis of compounds 1b–1e. Reagents and conditions: (a) NaOH, CH3OH/H2O = 5:1, 80 °C, 8 h, 77%; (b) SOCl2, CH2Cl2, r.t. 2 h, then n-BuLi, 1-bromo-4-methoxybenzene, THF, −83 °C, 2 h, 38%; (c) BBr3, 0 °C, 30 min, 17%; (d) BBr3, 0 °C, 2 h, 33%. (e) n-BuLi, 1-bromo-4-methoxybenzene, THF, −83–0 °C, 2 h, not detected. | |
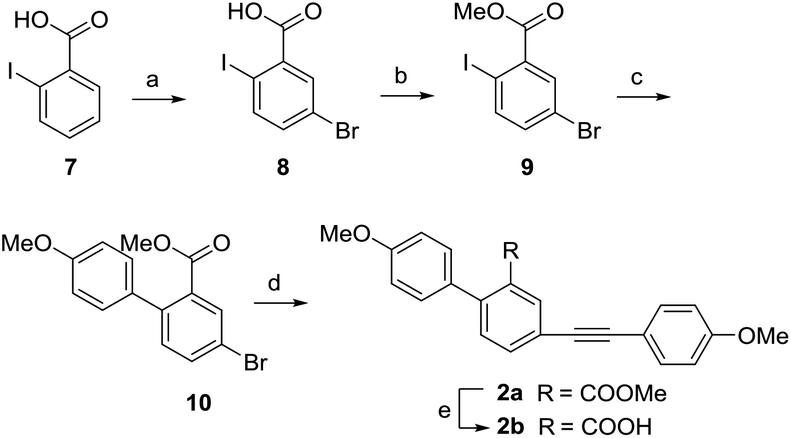
In view of the high steric hindrance in natural selaginellins, we designed and synthesized a series of selaginellin analogs (2a–2j) with a different attached position of 4-methoxyl phenylacetylene group (Scheme 4). Using the similar synthetic approach of 1a, 2a was obtained with a high yield starting with o-iodobenzoic acid via bromination, methyl esterification, regioselective Suzuki coupling and Sonogashira reaction. Compared with the yield of 1a in Sonogashira reaction, the yield of 2a was greatly increased due to less side reaction between meta-substituted carboxylic ester and alkynyl.36
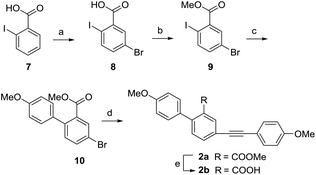 |
| | Scheme 4 Synthesis of compounds 2a and 2b. Reagents and conditions: (a) NBS, H2SO4, 60 °C, 2 h, 86%; (b) K2CO3, CH3I, acetone, 40 °C, 5 h, 100%; (c) Na2CO3, 4-methoxyphenylboronic acid, PdCl2(PPh3)2, THF/H2O = 1:1, 60 °C, 12 h, 80%; (d) 4-methoxyphenylacetylene, CuI, PdCl2(PPh3)2, PPh3, Et3N, DMF, 120 °C, 12 h, 78%; (e) NaOH, CH3OH/H2O = 4:1, 100 °C, 2 h, 99%. | |
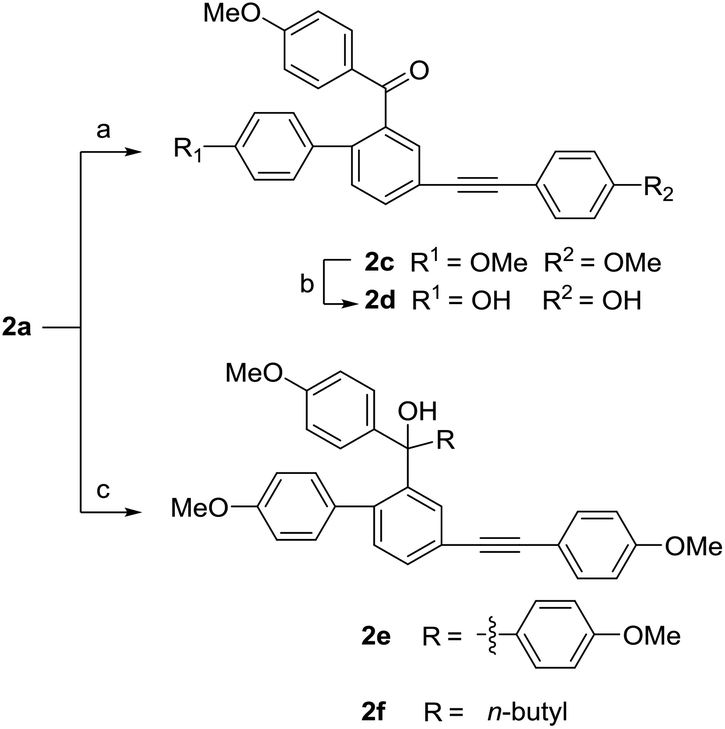
In the next nucleophilic addition of 2a, using 4-methoxy-phenylmagnesium bromide as the nucleophilic regent yielded a diphenyl ketone product 2c, whereas using 4-methoxyphenyl bromide under the presence of n-butyllithium produced the triphenyl methol 2e along with a side product 2f (Scheme 5). As we expected, the less steric hindrance of 2a than that of 1a allowed the production of triphenyl methol under the same conditions.
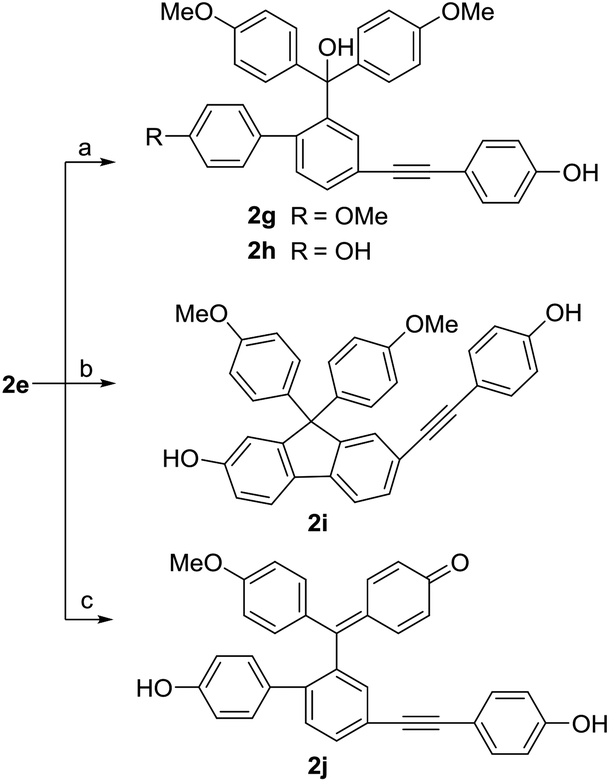
As Scheme 6 described, the demethylation of 2e with BBr3 under different conditions gave three different products. By controlling the reaction temperature at −83 °C and using small equivalents of BBr3, we obtained two demethylation products 2g and 2h. When the reaction temperature was set at 0 °C, the diarylfluorene derivative 2i, the analog of selaginpulvilin K, was yielded, probably from the Friedel–Crafts reaction under the catalysis of boron tribromide. In order to hydrolyze more methyl groups, we used excessive equivalents of BBr3 and slowly raised the reaction temperature from −83 °C to 0 °C. As we expected, the demethylation and dehydration occurred simultaneously to produce the benzyl para-quinone methide derivative 2j which resembles selaginellin N in molecular skeleton.
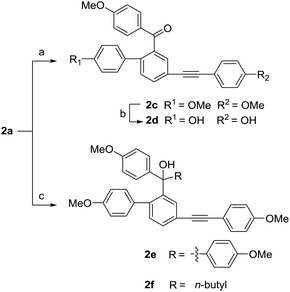 |
| | Scheme 5 Synthesis of compounds 2c–2f. Reagents and conditions: (a) 4-methoxyphenylmagnesium bromide, THF, −83 °C → r.t. 6 h, 36%; (b) BBr3, 0 °C, 30 min, 61%; (c) n-BuLi, 1-bromo-4-methoxybenzene, THF, −83 °C → 0 °C, 4 h, 2e: 82%, 2f: 18%. | |
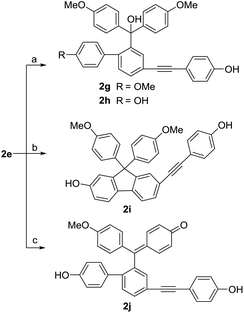 |
| | Scheme 6 Synthetic route to compounds 2g–j. Reagents and conditions: (a) BBr3, −83 °C, 1 h, 2g: 30%, 2h: 50%; (b) BBr3, 0 °C, 30 min, 27%; (c) BBr3, −83 → 0 °C, 4 h, 48%. | |
2.2. Biological results
2.2.1. MTT assay for tumor cell growth inhibition. The in vitro cytotoxic activity of the synthesized compounds against MCF-7, HepG2 and L-O2 cells was tested using MTT assay with cisplatin as a positive control. From the experiment data listed in Table 1, the tested compounds with different skeletons exhibited diverse cytotoxic effects. Compounds 1a–1e showed weak cytotoxic effects with the exception of 1c (IC50 = 7.18 μM against MCF-7). Among compounds 2a–2j, 2a–2f showed weak cytotoxic effects with the exception of 2d, while 2g–2j showed moderate cytotoxic effects. The cytotoxicity of 2d was more potent than that of 1d, suggesting that the attached position of phenylacetylene greatly influenced their cytotoxicity. Compounds 2g and 2h showed much more cytotoxic effects than their methylated precursor 2e, suggesting the presence of phenolic hydroxyl favors the antitumor activity of these compounds. Compounds 2h, 2i and 2j showed similar cytotoxic effects although they possess different scaffolds. Interestingly, compound 2j with a non-natural skeleton showed more cytotoxic effects than its natural analogs selaginellins, enabling 2j to be a more promising anti-tumor agent due to its advantage of easier synthesis.
Table 1 Anti-proliferative activity of synthesized compounds
| Compounds |
IC50a (μM) |
| MCF-7 |
HepG2 |
L-O2 |
| Data are mean values of three independent experiments. Errors represent standard deviation. The IC50 value was not determined owing to the nonlinear correlation between inhibition rates and concentrations. |
| Cisplatin |
7.24 ± 0.3 |
9.66 ± 0.1 |
3.64 ± 0.1 |
| 1a |
>100 |
>100 |
>100 |
| 1b |
>100 |
>100 |
>100 |
| 1c |
7.18 ± 1.2 |
—b |
—b |
| 1d |
51.16 ± 0.6 |
54.75 ± 1.9 |
64.00 ± 1.0 |
| 1e |
95.57 ± 0.4 |
55.51 ± 1.9 |
92.51 ± 2.0 |
| 2a |
>100 |
>100 |
>100 |
| 2b |
>100 |
>100 |
>100 |
| 2c |
>100 |
>100 |
>100 |
| 2d |
6.49 ± 0.1 |
24.99 ± 0.1 |
7.83 ± 1.2 |
| 2e |
>100 |
>100 |
>100 |
| 2f |
>100 |
>100 |
>100 |
| 2g |
12.98 ± 0.7 |
15.13 ± 0.6 |
16.36 ± 1.3 |
| 2h |
11.90 ± 0.5 |
11.81 ± 0.8 |
10.58 ± 1.1 |
| 2i |
9.23 ± 0.0 |
8.96 ± 0.4 |
10.91 ± 1.0 |
| 2j |
12.10 ± 0.1 |
11.63 ± 0.6 |
11.07 ± 0.7 |
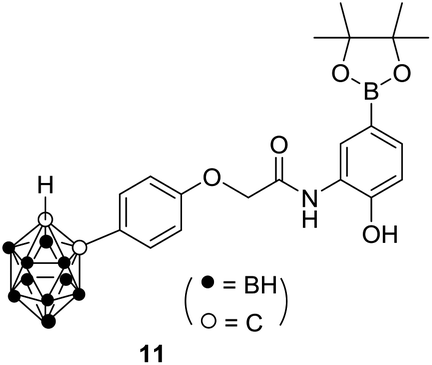
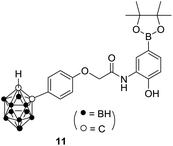
2.2.2. Cell-based reporter assays for hypoxia-inducible factor-1 (HIF-1) inhibition. Almost all solid tumors have a common feature, that is, hypoxia, resulting in overexpression of the HIF pathway in the tumor microenvironment.37 Therefore, HIF inhibition has been suggested to be an attractive and promising therapeutic strategy for various solid tumors. To date, the mechanism underlying the anticancer effects of alkynyl phenol derivatives has not been investigated. We here explored the effect of synthesized alkynyl phenol derivatives on HIF pathway by dual luciferase reporter system. The carborane derivative 11, an HIF-1 inhibitor reported by Nakamura,38 was used as a reference substance. As the data listed in Table 2, all tested compounds exhibited moderate to strong HIF inhibitory activities except 2j. Compounds 1a, 2f and 2h displayed comparative HIF inhibitory activities and low cytotoxicity compared with compound 11. Different from the rest synthetic derivatives, compound 2j showed potent anti-proliferative effect, but little HIF inhibitory activity was determined, suggesting a distinct effect resulting from its para-quinone methide moiety.
Table 2 HIF-1 inhibition in dual luciferase-reporter assay and cell growth inhibition of synthesized compounds
|
| Compounds |
HRE IC50a (μM) |
GI50a (μM) |
| Data are mean values of three independent experiments. Errors represent standard deviation. The IC50 value was not determined. |
| 11 |
4.46 ± 0.2 |
10.80 ± 1.9 |
| 1a |
5.41 ± 1.1 |
>100 |
| 1b |
20.69 ± 2.5 |
>100 |
| 1c |
2.36 ± 0.4 |
3.75 ± 0.2 |
| 1d |
9.52 ± 0.4 |
44.81 ± 0.2 |
| 1e |
13.66 ± 0.4 |
86.31 ± 0.5 |
| 2a |
18.30 ± 1.6 |
>100 |
| 2b |
15.57 ± 1.6 |
>100 |
| 2c |
39.93 ± 3.1 |
>100 |
| 2d |
4.47 ± 0.6 |
3.24 ± 0.1 |
| 2e |
20.81 ± 0.8 |
>100 |
| 2f |
1.89 ± 0.4 |
>100 |
| 2g |
8.10 ± 1.4 |
15.48 ± 0.1 |
| 2h |
2.52 ± 0.4 |
10.78 ± 0.3 |
| 2i |
5.55 ± 1.3 |
7.58 ± 0.1 |
| 2j |
—b |
4.47 ± 0.5 |
3. Experimental section
3.1. Chemistry
All chemicals and solvents were commercially available, at least reagent grade. 1H NMR and 13C NMR spectra were recorded on 400 MHz NMR Spectrometer instrument (Bruker Avance II). HRMS data were detected with LTQ Orbitrap XL ETD Mass Spectrometer instrument (Thermo Scientific). The binary solvent system (A/B) of HPLC was as follows: water (A) and CH3OH (B); gradient condition: from 60% B to 100% B in 60 min. The absorbance was detected at 254 nm. Reactions were monitored by the thin layer chromatography (TLC) from Qingdao Haiyang Chemical Co. Ltd (200 × 200 mm, 0.2–0.25 mm). Chromatograms were visualized by 254 nm UV light.
3.1.1. 2-Bromo-6-iodobenzoic acid (4). Compound 3 (6.03 g, 30.16 mmol), N-iodosuccinimide (8.10 g, 36 mmol), palladium acetate (673.5 mg, 3 mmol) and 50 mL DMF were added to a 100 mL sealed tube. Then the reaction mixture was stirred at 100 °C for 12 hours. The mixture was cooled down to room temperature and diluted with ethyl acetate. The solution was washed with 0.5 M HCl and water. The combined extract was dried by anhydrous sodium sulfate. Ethyl acetate was evaporated under vacuum and the residue was purified by silica gel column chromatography to afford compound 4 (8.84 g, 90%). White solid; mp 153–155 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.09 (dd, J = 7.8, 7.9 Hz, 1H), 7.68 (d, J = 7.9 Hz, 1H), 7.87 (d, J = 7.8 Hz, 1H). HRMS (ESI) (m/z): [M − H]− calcd for C7H4BrIO2, 324.8367; found, 324.8369.
3.1.2. Methyl 2-bromo-6-iodobenzoate (5). Compound 4 (7.80 g, 23.93 mmol), potassium carbonate (6.66 g, 48.22 mmol), 50 mL acetone and excess methyl iodide were added to a 100 mL round bottom flask. Then the reaction mixture was stirred at 40 °C for 5 h. Acetone was evaporated under vacuum. The residue was dissolved with ethyl acetate and washed with saturated brine. The combined extract was dried by anhydrous sodium sulfate. Ethyl acetate was evaporated under vacuum to afford compound 5 (8.13 g, 100%). White solid; mp 87–89 °C; 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 7.9 Hz, 1H), 7.56 (d, J = 8.1 Hz, 1H), 6.96 (t, J = 8.0 Hz, 1H), 3.99 (s, 3H). HRMS (ESI) (m/z): [M + H]+ calcd for C8H6BrIO2, 340.8669; found, 340.8675.
3.1.3. Methyl 3-bromo-4′-methoxy-[1,1′-biphenyl]-2-carboxylate (6). Compound 5 (8.13 g, 23.93 mmol), p-methoxyphenylboronic acid (4.36 g, 28.72 mmol), 50 mL THF and 50 mL water were added to a 250 mL three-necked flask and stirred at room temperature. Bis-triphenylphosphine palladium dichloride (842.3 mg, 1.2 mmol) was added under nitrogen atmosphere. When the temperature reached 60 °C, sodium carbonate (5.07 g, 47.86 mmol) was added. Then the reaction mixture was refluxed for 12 hours. The mixture was cooled to room temperature and diluted with ethyl acetate. The solution was washed with saturated brine. The combined extract was dried by anhydrous sodium sulfate. Ethyl acetate was evaporated under vacuum. The residue was purified by silica gel column chromatography to afford compound 6 (6.28 g, 82%). White solid; mp 89–91 °C; 1H NMR (400 MHz, acetone-d6) δ 7.64 (dd, J = 7.0, 1.9 Hz, 1H), 7.47–7.42 (t, 1H), 7.41 (d, J = 6.0 Hz, 1H), 7.33 (d, J = 8.7 Hz, 2H), 7.00 (d, J = 8.7 Hz, 2H), 3.83 (s, 3H), 3.69 (s, 3H). HRMS (ESI) (m/z): [M + H]+ calcd for C15H13BrO3, 321.0121; found, 321.0126.
3.1.4. Methyl 4′-methoxy-3-((4-methoxyphenyl)ethynyl)-[1,1′-biphenyl]-2-carboxylate (1a). Compound 6 (5.08 g, 15.88 mmol), triphenylphosphine (416.5 mg, 1.59 mmol), cuprous iodide (151.2 mg, 0.79 mmol) and 30 mL DMF were added to a 250 mL three-necked flask and stirred at room temperature. Bis-triphenylphosphine palladium dichloride (557.3 mg, 0.79 mmol) and 4-ethynylanisole (8.24 mL, 63.52 mmol) were added under nitrogen atmosphere. Then 30 mL triethylamine was added and refluxed at 120 °C for 12 hours. The reaction mixture was cooled to room temperature and diluted with ethyl acetate. The solution was washed with saturated ammonium chloride solution. The combined extract was dried by anhydrous sodium sulfate. Ethyl acetate was evaporated under vacuum. The residue was purified by silica gel column chromatography to afford compound 1a (3.0 g, 48%). White solid; mp 98–99 °C; the purity of the compound was detected by analytical HPLC to be over 95%. 1H NMR (400 MHz, CDCl3) δ 7.51 (dd, J = 7.7, 1.1 Hz, 1H), 7.44 (d, J = 8.8 Hz, 2H), 7.40 (d, J = 7.7 Hz, 1H), 7.33 (d, J = 8.7 Hz, 2H), 7.32–7.30 (m, 1H), 6.94 (d, J = 8.7 Hz, 2H), 6.87 (d, J = 8.8 Hz, 2H), 3.84 (s, 3H), 3.83 (s, 3H), 3.74 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 168.57, 159.88, 159.11, 138.87, 135.18, 133.03, 131.29, 129.99, 129.58, 129.17, 120.65, 114.56, 114.16, 113.66, 92.89, 85.36, 55.34, 55.18, 52.21. HRMS (ESI) (m/z): [M + H]+ calcd for C24H20O4, 373.1434; found, 373.1439.
3.1.5. 4′-Methoxy-3-((4-methoxyphenyl)ethynyl)-[1,1′-biphenyl]-2-carboxylic acid (1b). Compound 1a (722.0 mg, 1.94 mmol), 50 mL methanol and 10 mL 60% sodium hydroxide solution were added to a 100 mL round bottom flask. Then the reaction mixture was refluxed at 80 °C for 8 hours. The mixture was cooled to room temperature. The solvent was evaporated under vacuum. The residue was diluted with ethyl acetate and washed with dilute hydrochloric acid and saturated brine. The combined extract was dried by anhydrous sodium sulfate. Ethyl acetate was evaporated under vacuum. The residue was purified by silica gel chromatography to afford compound 1b (538.0 mg, 77%). White solid; mp 145–147 °C; the purity of the compound was detected by analytical HPLC to be over 95%. 1H NMR (400 MHz, acetone-d6) δ 7.55 (dd, J = 7.7, 1.3 Hz, 1H), 7.52–7.45 (m, 3H), 7.45–7.41 (m, 2H), 7.38 (dd, J = 7.6, 1.3 Hz, 1H), 7.01–6.94 (m, 4H), 3.83 (s, 6H); 13C NMR (101 MHz, acetone-d6) δ 169.79, 161.07, 160.45, 140.02, 137.66, 133.87, 133.11, 130.90, 130.45, 130.36, 129.97, 121.94, 115.67, 115.05, 114.66, 93.48, 86.65, 55.71, 55.56. HRMS (ESI) (m/z): [M − H]− calcd for C23H18O4, 357.1132; found, 357.1138.
3.1.6. (4′-Methoxy-3-((4-methoxyphenyl)ethynyl)-[1,1′-biphenyl]-2-yl)(4-methoxyphenyl)methanone (1c). Compound 1b (365.4 mg, 1.02 mmol) was dissolved in 3 mL dichloromethane. Then the reaction mixture was stirred in an ice water bath and 0.2 mL thionyl chloride was added, followed by a drop of DMF. After being stirred for 2 hours, the solvent was evaporated under vacuum. The residue was dissolved in 2 mL THF under nitrogen atmosphere for use. Parabromoanisole (308 μL, 2.4 mmol) and 3 mL anhydrous THF were added to a 50 mL dry round bottom flask under nitrogen atmosphere. Then the reaction mixture was stirred at −83 °C and 1.7 mL nBuLi in n-hexane solution (1.6 M L–1) was added. After being stirred for 30 min, the prepared acid chloride was added to the reaction mixture and stirred for 2 h. The reaction mixture was quenched by adding saturated ammonium chloride solution. The reaction mixture was diluted with ethyl acetate and washed with saturated brine. The combined extract was dried by anhydrous sodium sulfate. Ethyl acetate was evaporated under vacuum. The residue was purified by silica gel chromatography to afford compound 1c (170.3 mg, 38%). White solid; mp 48–49 °C; the purity of the compound was detected by analytical HPLC to be over 95%. 1H NMR (400 MHz, DMSO-d6) δ 7.65 (d, J = 8.9 Hz, 2H), 7.61–7.56 (m, 2H), 7.46 (dd, J = 6.1, 2.9 Hz, 1H), 7.20 (d, J = 8.7 Hz, 2H), 7.03–6.97 (m, 4H), 6.89–6.83 (m, 4H), 3.80 (s, 3H), 3.74 (s, 3H), 3.70 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 195.25, 163.39, 159.65, 158.73, 141.09, 139.22, 132.50, 131.49, 131.41, 129.99, 129.92, 129.80, 129.33, 120.45, 114.30, 114.12, 113.78, 113.59, 93.82, 86.17, 55.55, 55.25, 55.02. HRMS (ESI) (m/z): [M + H]+ calcd for C30H24O4, 449.1747; found, 449.1744.
3.1.7. (4′-Hydroxy-3-((4-hydroxyphenyl)ethynyl)-[1,1′-biphenyl]-2-yl)(4-methoxyphenyl)methanone (1d). Compound 1c (150.8 mg, 0.34 mmol) was dissolved in 2 mL dry dichloromethane in a 10 mL chicken heart flask. Then the reaction mixture was stirred in an ice water bath and boron tribromide (320 μL, 3.38 mmol) was added. After being stirred for 30 min, 3 mL diethyl ether was added. The reaction mixture was stirred for 20 min and saturated sodium bicarbonate solution was slowly added. Then the reaction mixture was diluted with ethyl acetate and washed with saturated brine. The combined extract was dried by anhydrous sodium sulfate. Ethyl acetate was evaporated under vacuum. The residue was purified by silica gel chromatography to afford compound 1d (24.5 mg, 17%). White solid; mp 91–93 °C; the purity of the compound was detected by analytical HPLC to be over 95%. 1H NMR (400 MHz, acetone-d6) δ 8.73 (s, 1H), 8.39 (s, 1H), 7.70 (d, J = 8.8 Hz, 2H), 7.57–7.51 (m, 2H), 7.42 (dd, J = 5.9, 3.0 Hz, 1H), 7.18 (d, J = 8.5 Hz, 2H), 7.01 (d, J = 8.5 Hz, 2H), 6.93 (d, J = 8.8 Hz, 2H), 6.75 (d, J = 8.5 Hz, 2H), 6.73 (d, J = 8.5 Hz, 2H), 3.82 (s, 3H); 13C NMR (101 MHz, acetone-d6) δ 195.31, 163.70, 158.01, 157.02, 141.74, 140.12, 132.86, 131.63, 131.06, 130.77, 130.21, 129.81, 129.55, 128.90, 121.49, 115.47, 115.06, 113.75, 113.53, 94.09, 85.79, 55.02. HRMS (ESI) (m/z): [M − H]− calcd for C28H20O4, 449.1289; found, 419.1293.
3.1.8. 3,8-Bis(4-hydroxyphenyl)-1H-isochromen-1-one (1e). Compound 1a (186.5 mg, 0.50 mmol) was dissolved in 2 mL dry dichloromethane in a 10 mL chicken heart flask. Then the reaction mixture was stirred in an ice water bath and boron tribromide (470 μL, 4.97 mmol) was added. After being stirred for 2 hours, 3 mL diethyl ether was added. The reaction mixture was stirred for 20 min and saturated sodium bicarbonate solution was slowly added. The reaction mixture was diluted with ethyl acetate and washed with saturated brine. The combined extract was dried by anhydrous sodium sulfate. Ethyl acetate was evaporated under vacuum. The residue was purified by silica gel chromatography to afford compound 1e (54.9 mg, 33%). Light yellow solid; mp 297–299 °C; the purity of the compound was detected by analytical HPLC to be over 95%. 1H NMR (400 MHz, acetone-d6) δ 8.87 (s, 1H), 8.36 (s, 1H), 7.80 (d, J = 8.7 Hz, 2H), 7.74 (t, J = 7.7 Hz, 1H), 7.58 (d, J = 7.8 Hz, 1H), 7.29 (d, J = 7.5 Hz, 1H), 7.21 (d, J = 8.4 Hz, 2H), 7.16 (s, 1H), 6.98 (d, J = 8.7 Hz, 2H), 6.87 (d, J = 8.4 Hz, 2H); 13C NMR (101 MHz, acetone-d6) δ 160.51, 160.13, 157.68, 154.57, 146.79, 140.77, 134.62, 133.82, 131.82, 130.85, 127.65, 126.18, 124.49, 117.92, 116.66, 115.25, 100.76. HRMS (ESI) (m/z): [M − H]− calcd for C21H14O4, 329.0819; found, 329.0829.
3.1.9. 5-Bromo-2-iodobenzoic acid (8). Compound 7 (8.44 g, 34.03 mmol) and 60 mL H2SO4 were added to a 250 mL round bottom flask. NBS (4.82 g, 38.06 mmol) was then added at 60 °C and the mixture was stirred for 2 hours. Then the reaction mixture was cooled to room temperature and dropped into ice water. The mixture was diluted with ethyl acetate and washed with saturated brine. The combined extract was dried by anhydrous sodium sulfate. Ethyl acetate was evaporated under vacuum. The residue was purified by silica gel chromatography to afford compound 8 (9.54 g, 86%). White solid; mp 159–161 °C; 1H NMR (400 MHz, CDCl3) δ 8.14 (d, J = 2.4 Hz, 1H), 7.90 (d, J = 8.4 Hz, 1H), 7.33 (dd, J = 8.4, 2.4 Hz, 1H). HRMS (ESI) (m/z): [M − H]− calcd for C7H4BrIO2, 324.8367; found, 324.8369.
3.1.10. Methyl 5-bromo-2-iodobenzoate (9). Compound 8 (9.54 g, 29.4 mmol), potassium carbonate (8.13 g, 58.8 mmol), 50 mL acetone and excess methyl iodide were successively added to a 100 mL round bottom flask. Then the reaction mixture was stirred at 40 °C for 5 h. Acetone was evaporated under vacuum. The residue was diluted with ethyl acetate and washed with saturated brine. The combined extract was dried by anhydrous sodium sulfate. Ethyl acetate was evaporated under vacuum to afford compound 9 (9.99 g, 100%). White solid; mp 48–49 °C; 1H NMR (400 MHz, acetone-d6) δ 7.98 (d, J = 8.4 Hz, 1H), 7.91 (d, J = 2.4 Hz, 1H), 7.48 (dd, J = 8.4, 2.4 Hz, 1H), 3.92 (s, 3H). HRMS (ESI) (m/z): [M + H]+ calcd for C8H6BrIO2, 340.8669; found, 340.8675.
3.1.11. Methyl 4-bromo-4′-methoxy-[1,1′-biphenyl]-2-carboxylate (10). Compound 9 (9.99 g, 29.4 mmol), p-methoxyphenylboronic acid (5.36 g, 35.28 mmol), 50 mL THF and 50 mL water were successively added to a 250 mL three-necked flask and stirred at room temperature. Bis-triphenylphosphine palladium dichloride (1.03 g, 1.47 mmol) was then added under nitrogen atmosphere. When the temperature reached 60 °C, sodium carbonate (6.23 g, 58.8 mmol) was added. Then the reaction mixture was refluxed for 12 hours. The mixture was cooled to room temperature and diluted with ethyl acetate. The solution was washed with saturated brine. The combined extract was dried by anhydrous sodium sulfate. Ethyl acetate was evaporated under vacuum. The residue was purified by silica gel chromatography to afford compound 10 (7.53 g, 80%). White solid; mp 76–77 °C; 1H NMR (400 MHz, acetone-d6) δ 7.86 (d, J = 2.2 Hz, 1H), 7.75 (dd, J = 8.3, 2.2 Hz, 1H), 7.38 (d, J = 8.3 Hz, 1H), 7.28–7.21 (m, 2H), 7.02–6.95 (m, 2H), 3.84 (s, 3H), 3.66 (s, 3H). HRMS (ESI) (m/z): [M + H]+ calcd for C15H13BrO3, 321.0121; found, 321.0126.
3.1.12. Methyl 4′-methoxy-4-((4-methoxyphenyl)ethynyl)-[1,1′-biphenyl]-2-carboxylate (2a). Compound 10 (7.53 g, 23.535 mmol), triphenylphosphine (617.3 mg, 2.35 mmol), cuprous iodide (224.1 mg, 1.18 mmol) and 40 mL DMF were added to a 250 mL three-necked flask and stirred at room temperature. Bis-triphenylphosphine palladium dichloride (826.0 mg, 1.18 mmol) and 4-ethynylanisole (9.5 mL, 73.25 mmol) were added under nitrogen atmosphere. Then 40 mL triethylamine was added and refluxed at 120 °C for 8 hours. The reaction mixture was cooled to room temperature and diluted with ethyl acetate. The solution was washed with saturated ammonium chloride solution. The combined extract was dried by anhydrous sodium sulfate. Ethyl acetate was evaporated under vacuum. The residue was purified by silica gel chromatography to afford compound 2a (6.84 g, 78%). White solid; mp 138–139 °C; the purity of the compound was detected by analytical HPLC to be over 95%. 1H NMR (400 MHz, acetone-d6) δ 7.83 (d, J = 1.8 Hz, 1H), 7.69 (dd, J = 8.0, 1.8 Hz, 1H), 7.57–7.50 (m, 2H), 7.46 (d, J = 8.0 Hz, 1H), 7.32–7.26 (m, 2H), 7.03–6.96 (m, 4H), 3.86 (s, 3H), 3.85 (s, 3H), 3.67 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 168.05, 159.75, 158.97, 140.26, 133.48, 133.09, 131.73, 131.70, 131.26, 130.81, 129.31, 121.33, 114.45, 113.86, 90.81, 86.79, 55.30, 55.14, 52.14. HRMS (ESI) (m/z): [M + H]+ calcd for C24H20O4, 373.1434; found, 373.1442.
3.1.13. 4′-Methoxy-4-((4-methoxyphenyl)ethynyl)-[1,1′-biphenyl]-2-carboxylic acid (2b). Compound 2a (372.1 mg, 1 mmol), sodium hydroxide (1.0 g), 20 mL methanol and 5 mL water were added to a 100 mL round bottom flask. Then the reaction mixture was refluxed at 100 °C for 2 hours. The mixture was cooled to room temperature. The solvent was evaporated under vacuum. The residue was diluted with ethyl acetate and washed with dilute hydrochloric acid and saturated brine. The combined extract was dried by anhydrous sodium sulfate. Ethyl acetate was evaporated under vacuum. The residue was purified by silica gel chromatography to afford compound 2b (354.5 mg, 99%). White solid; mp 191–193 °C; the purity of the compound was detected by analytical HPLC to be over 95%. 1H NMR (400 MHz, DMSO-d6) δ 12.98 (s, 1H), 7.79 (d, J = 1.7 Hz, 1H), 7.66 (dd, J = 8.0, 1.8 Hz, 1H), 7.56–7.51 (m, 2H), 7.41 (d, J = 8.0 Hz, 1H), 7.33–7.27 (m, 2H), 7.03–6.96 (m, 4H), 3.81 (s, 3H), 3.80 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 169.65, 160.20, 159.38, 140.51, 133.55, 133.38, 133.31, 132.63, 131.97, 131.24, 129.92, 121.66, 114.92, 114.44, 114.21, 91.06, 87.49, 55.77, 55.61. HRMS (ESI) (m/z): [M − H]− calcd for C23H18O4, 357.1132; found, 357.1139.
3.1.14. (4′-Methoxy-4-((4-methoxyphenyl)ethynyl)-[1,1′-biphenyl]-2-yl)(4-methoxyphenyl)methanone (2c). Compound 2a (744.3 mg, 2 mmol) and 5 mL of anhydrous THF were added to a 50 mL dry round bottom flask under nitrogen atmosphere. Then the reaction mixture was stirred at −83 °C and 2.5 mL of p-methoxyphenylmagnesium bromide (1.0 M L−1 in THF) was added. The temperature was naturally raised to room temperature. After being stirred for 6 hours, the reaction mixture was quenched by adding saturated ammonium chloride solution. The reaction mixture was diluted with ethyl acetate and washed with saturated brine. The combined extract was dried by anhydrous sodium sulfate. Ethyl acetate was evaporated under vacuum. The residue was purified by silica gel chromatography to afford compound 2c (319.2 mg, 36%). White solid; mp 153–155 °C; the purity of the compound was detected by analytical HPLC to be over 95%. 1H NMR (400 MHz, DMSO-d6) δ 7.72 (dd, J = 8.0, 1.8 Hz, 1H), 7.64–7.59 (m, 2H), 7.52 (m, J = 4.8, 3.9 Hz, 4H), 7.23–7.16 (m, 2H), 7.01–6.97 (m, 2H), 6.97–6.93 (m, 2H), 6.88–6.82 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 195.37, 163.36, 159.70, 158.83, 139.10, 139.06, 133.05, 132.40, 131.97, 131.31, 130.38, 130.28, 129.68, 129.32, 121.13, 114.43, 113.99, 113.95, 90.74, 87.15, 55.56, 55.29, 55.06. HRMS (ESI) (m/z): [M + H]+ calcd for C30H24O4, 449.1747; found, 449.1741.
3.1.15. (4′-Hydroxy-4-((4-hydroxyphenyl)ethynyl)-[1,1′-biphenyl]-2-yl)(4-methoxyphenyl)methanone (2d). Compound 2c (319.2 mg, 0.71 mmol) was dissolved in 5 mL dry dichloromethane added in a 50 mL round bottom flask. Then the reaction mixture was stirred in an ice water bath and boron tribromide (340 μL, 3.6 mmol) was added. After being stirred for 30 min, 10 mL diethyl ether was added. The reaction mixture was stirred for 20 min and saturated sodium bicarbonate solution was slowly added. Then the reaction mixture was diluted with ethyl acetate and washed with saturated brine. The combined extract was dried by anhydrous sodium sulfate. Ethyl acetate was evaporated under vacuum. The residue was purified by silica gel chromatography to afford compound 2d (180.7 mg, 61%). White solid; mp 229–231 °C; the purity of the compound was detected by analytical HPLC to be over 95%. 1H NMR (400 MHz, DMSO-d6) δ 9.97 (s, 1H), 9.54 (s, 1H), 7.68 (dd, J = 8.0, 1.7 Hz, 1H), 7.58 (d, J = 8.8 Hz, 2H), 7.49 (d, J = 8.1 Hz, 1H), 7.46 (d, J = 1.6 Hz, 1H), 7.39 (d, J = 8.6 Hz, 2H), 7.08 (d, J = 8.5 Hz, 2H), 6.93 (d, J = 8.9 Hz, 2H), 6.80 (d, J = 8.6 Hz, 2H), 6.65 (d, J = 8.5 Hz, 2H), 3.79 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 195.60, 163.25, 158.23, 157.08, 139.30, 138.94, 133.13, 132.31, 131.88, 130.34, 130.03, 129.75, 129.70, 129.35, 121.12, 115.77, 115.34, 113.89, 112.24, 91.16, 86.60, 55.51. HRMS (ESI) (m/z): [M + H]+ calcd for C28H20O4, 421.1434; found, 421.1432.
3.1.16. (4′-Methoxy-4-((4-methoxyphenyl)ethynyl)-[1,1′-biphenyl]-2-yl)bis(4-methoxyphenyl)methanol (2e); 1-(4′-methoxy-4-((4-methoxyphenyl)ethynyl)-[1,1′-biphenyl]-2-yl)-1-(4-methoxyphenyl)pentan-1-ol (2f). Parabromoanisole (2.03 g, 10.84 mmol) and 5 mL anhydrous THF were added to a 100 mL dry round bottom flask under nitrogen atmosphere. Then the reaction mixture was stirred at −83 °C and 7.45 mL n-BuLi in n-hexane solution (1.6 M L−1) was added. After being stirred for 1 hour, 15 mL solution of compound 2a (2.02 g, 5.42 mmol) in THF was added to the reaction mixture. The temperature was naturally raised to 0 °C. The reaction mixture was stirred for 4 hours and quenched by adding saturated ammonium chloride solution. The reaction mixture was diluted with ethyl acetate and washed with saturated brine. The combined extract was dried by anhydrous sodium sulfate. Ethyl acetate was evaporated under vacuum. The residue was purified by silica gel chromatography to afford compound 2e (2.47 g, 82%) and compound 2f (493.9 mg, 18%). The purity of the two compounds was detected by analytical HPLC to be over 95%. Compound 2e: pink solid; mp 179–181 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.09 (d, J = 1.5 Hz, 1H), 7.51 (d, J = 8.6 Hz, 2H), 7.34 (dd, J = 7.7, 1.4 Hz, 1H), 6.98 (d, J = 8.6 Hz, 2H), 6.85–6.89 (m, 3H), 6.68 (d, J = 8.7 Hz, 2H), 6.61 (d, J = 8.4 Hz, 2H), 6.44 (d, J = 8.2 Hz, 2H), 4.79 (s, 1H), 3.81 (s, 3H), 3.72 (s, 6H), 1.99–1.84 (m, 2H), 1.30–1.11 (m, 3H), 1.03–0.88 (m, 1H), 0.79 (t, J = 7.2 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 159.51, 157.76, 157.67, 145.84, 140.71, 140.39, 134.13, 132.95, 132.33, 129.64, 128.64, 127.71, 120.74, 114.42, 114.37, 112.71, 112.17, 89.16, 88.54, 75.70, 55.28, 55.05, 54.98, 38.47, 25.73, 22.60, 14.11. ESI MS (m/z): [M − OH]+ calcd for C34H34O4, 489.24; found, 489.36. Compound 2f: white solid; mp 113–114 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.48–7.41 (m, 3H), 7.17 (d, J = 1.7 Hz, 1H), 7.03 (d, J = 7.8 Hz, 1H), 6.95–6.99 (m, 6H), 6.77 (d, J = 8.8 Hz, 4H), 6.64 (d, J = 8.7 Hz, 2H), 6.55 (d, J = 8.7 Hz, 2H), 5.76 (s, 1H), 3.78 (s, 3H), 3.72 (s, 6H), 3.67 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 159.54, 157.69, 157.31, 146.30, 142.35, 139.86, 134.99, 133.21, 132.94, 131.57, 130.02, 129.47, 128.87, 120.18, 114.36, 114.04, 112.66, 111.94, 89.35, 88.05, 80.72, 55.26, 54.99, 54.90. HRMS (ESI) (m/z): [M − OH]+ calcd for C37H32O5, 539.2222; found, 539.2209.
3.1.17. 4-((2-(Hydroxybis(4-methoxyphenyl)methyl)-4′-methoxy-[1,1′-biphenyl]-4-yl)ethynyl)phenol (2g); 2′-(hydroxybis(4-methoxyphenyl)methyl)-4′-((4-hydroxyphenyl)ethynyl)-[1,1′-biphenyl]-4-ol (2h). Compound 2e (139.1 mg, 0.25 mmol) was dissolved in 2 mL dry dichloromethane in a 10 mL chicken heart flask. Then the reaction mixture was stirred at −83 °C and boron tribromide (140 μL, 1.4 8 mmol) was added. After being stirred for 1 hour, 5 mL diethyl ether was added. The reaction mixture was stirred for 20 min and saturated sodium bicarbonate solution was slowly added. Then the reaction mixture was diluted with ethyl acetate and washed with saturated brine. The combined extract was dried by anhydrous sodium sulfate. Ethyl acetate was evaporated under vacuum. The residue was purified by silica gel chromatography to afford compound 2g (40.6 mg, 30%) and compound 2h (66.0 mg, 50%). The purity of the two compounds was detected by analytical HPLC to be over 95%. Compound 2g: pink solid; mp 199–201 °C; 1H NMR (400 MHz, acetone-d6) δ 8.79 (s, 1H), 7.44 (dd, J = 7.8, 1.5 Hz, 1H), 7.34 (d, J = 8.5 Hz, 2H), 7.09–7.11 (m, 2H), 7.06 (d, J = 8.8 Hz, 4H), 6.82–6.86 (m, 6H), 6.68–6.74 (m, 4H), 3.87 (s, 1H), 3.79 (s, 6H), 3.76 (s, 3H); 13C NMR (101 MHz, acetone-d6) δ 158.67, 158.65, 157.93, 146.40, 141.46, 139.97, 134.23, 133.13, 133.02, 132.11, 130.40, 129.59, 129.10, 121.43, 115.60, 113.74, 112.80, 89.68, 87.29, 82.03, 54.58. HRMS (ESI) (m/z): [M − H]− calcd for C36H30O5, 541.2020; found, 541.2011. Compound 2h: pink solid; mp 119–121 °C; 1H NMR (400 MHz, acetone-d6) δ 8.76 (s, 1H), 8.37 (s, 1H), 7.43 (d, J = 8.3 Hz, 1H), 7.33 (d, J = 8.5 Hz, 2H), 7.05–7.11 (m, 6H), 6.83–6.86 (m, 6H), 6.64 (q, J = 8.7 Hz, 4H), 3.78 (s, 6H), 3.75 (s, 1H); 13C NMR (101 MHz, acetone-d6) δ 158.70, 157.90, 156.46, 146.40, 141.52, 140.03, 133.17, 133.01, 132.87, 132.13, 130.50, 129.59, 129.09, 121.35, 115.60, 114.43, 113.77, 112.82, 89.65, 87.32, 82.17, 54.59. HRMS (ESI) (m/z): [M − H]− calcd for C35H28O5, 527.1864; found, 527.1866.
3.1.18. 7-((4-Hydroxyphenyl)ethynyl)-9,9-bis(4-methoxy-phenyl)-9H-fluoren-2-ol (2i). Compound 2e (155.7 mg, 0.28 mmol) was dissolved in 2 mL dry dichloromethane in a 10 mL chicken heart flask. Then the reaction mixture was stirred in an ice water bath and boron tribromide (110 μL, 1.16 mmol) was added. After being stirred for 30 min, 5 mL diethyl ether was added. The reaction mixture was stirred for 20 min and saturated sodium bicarbonate solution was slowly added. Then the reaction mixture was diluted with ethyl acetate and washed with saturated brine. The combined extract was dried by anhydrous sodium sulfate. Ethyl acetate was evaporated under vacuum. The residue was purified by silica gel chromatography to afford compound 2i (38.8 mg, 27%). White solid; mp 125–127 °C; the purity of the compound was detected by analytical HPLC to be over 95%. 1H NMR (400 MHz, acetone-d6) δ 8.75 (s, 1H), 8.53 (s, 1H), 7.71–7.75 (m, 2H), 7.46 (d, J = 8.1 Hz, 2H), 7.37 (d, J = 8.5 Hz, 2H), 7.12 (d, J = 8.8 Hz, 4H), 6.91–6.80 (m, 8H), 3.74 (s, 6H); 13C NMR (101 MHz, acetone-d6) δ 158.61, 157.97, 157.77, 154.32, 151.69, 140.25, 137.69, 132.94, 130.90, 130.68, 129.01, 128.52, 121.54, 121.04, 119.21, 115.58, 115.05, 114.12, 113.57, 113.02, 89.44, 88.09, 63.93, 54.56. HRMS (ESI) (m/z): [M − H]− calcd for C35H26O4, 509.1758; found, 509.1753.
3.1.19. 4-((4′-Hydroxy-4-((4-hydroxyphenyl)ethynyl)-[1,1′-biphenyl]-2-yl)(4-methoxyphenyl)methylene)cyclohexa-2,5-dien-1-one (2j). Compound 2e (2.23 g, 4.0 mmol) was dissolved in 40 mL dry dichloromethane in a 250 mL round bottom flask. Then the reaction mixture was stirred at −83 °C and boron tribromide (7.6 mL, 80.39 mmol) was added. The temperature was slowly raised to 0 °C. After being stirred for 4 hours, 30 mL diethyl ether was added. The reaction mixture was stirred for 20 min and saturated sodium bicarbonate solution was slowly added. Then the reaction mixture was diluted with ethyl acetate and washed with saturated brine. The combined extract was dried by anhydrous sodium sulfate. Ethyl acetate was evaporated under vacuum. The residue was purified by silica gel chromatography to afford compound 2j (943.4 mg, 48%). Red solid; mp 191–193 °C; the purity of the compound was detected by analytical HPLC to be over 95%. 1H NMR (400 MHz, DMSO-d6) δ 10.01 (s, 1H), 9.49 (s, 1H), 7.66 (dd, J = 8.0, 1.6 Hz, 1H), 7.43 (d, J = 8.0 Hz, 1H), 7.41–7.33 (m, 4H), 7.22 (dd, J = 9.9, 2.6 Hz, 1H), 6.85–6.94 (m, 6H), 6.81 (d, J = 8.6 Hz, 2H), 6.59 (d, J = 8.5 Hz, 2H), 6.33 (dd, J = 10.0, 1.9 Hz, 1H), 6.29 (dd, J = 9.9, 2.0 Hz, 1H), 3.75 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 185.55, 160.71, 159.69, 158.27, 156.77, 142.07, 139.27, 138.91, 138.67, 134.37, 133.29, 133.09, 132.20, 130.86, 130.66, 129.96, 129.59, 128.95, 128.06, 127.90, 121.07, 115.78, 115.01, 113.62, 112.16, 91.18, 86.62, 55.36. HRMS (ESI) (m/z): [M − H]− calcd for C34H24O4, 495.1602; found, 495.1618.
3.2. Biological assays
3.2.1. MTT assay for cell growth inhibition. The inhibition of compounds 1a–1e and 2a–2j against MCF-7, HepG2, HeLa and L-O2 cells were evaluated using a standard MTT-based colorimetric assay. Three thousand corresponding cells per well were seeded into 96-well plates and incubated at 37 °C, 5% CO2 for 24 h. Then 100 μL drug-containing medium with a series of concentration were dispensed into wells to maintain the final concentration as 100, 50, 25, 12.5, 6.25 and 3.125 μM, respectively. Each concentration was in triplicate. After 48 hours of incubation, cell survival was determined by the addition of 20 μL MTT (Sigma-Aldrich, St. Louis, USA) work solution (5 mg mL−1 MTT dissolved in phosphate buffer solution). After post-incubation at 37 °C for 4 hours, the medium was discarded followed by adding 150 μL DMSO (Sigma-Aldrich, St. Louis, USA). The plates were then vortexed for 10 min for complete dissolution. The optical absorbance was measured at 570 nm. The data represented the mean of three independent experiments in triplicate and were expressed as mean ± SD. The IC50 value was defined as the concentration at which 50% of the cells could survive.
3.2.2. Cell-based reporter assays for hypoxia inducible factor-1 (HIF-1) inhibition. HeLa cells expressing HRE-dependent firefly luciferase reporter construct (HRE-Luc) and constitutively expressing CMV-driven Renilla luciferase reporter with SureFECT transfection reagent were established with Cignal™ Lenti Reporter (SABiosciences, Frederick, MD) according to the manufacturer's instructions. The consensus sequence of HRE was 5′-TACGTGCT-3′ from the erythropoietin gene. Cells stably expressing the HRE-reporter gene were selected with puromycin. The cells were incubated for 12 hours with or without drugs under the normoxic or hypoxic condition (1% O2). After incubation, the luciferase assay was performed using a Luciferase Assay System (Promega, Madison, WI) according to the manufacturer's instructions. The drug concentration required to inhibit the relative light units by 50% (IC50) was determined from semi-logarithmic dose–response plots, and the results represent means ± SD of triplicate samples.
4. Conclusion
In conclusion, we designed and synthesized a class of natural product-like biphenyl-containing diaryl acetylenes mimicking natural alkynyl phenols from the genus Selaginella. In MTT assay in cancer cells, compounds 1c, 2d, 2g, 2h, 2i and 2j exhibited potent cytotoxic activity. The evaluation of HIF-1 inhibitory activity demonstrated that all tested compounds exhibited moderate to good activities except 2j. Compounds 1a, 2f and 2h displayed high HIF-1 inhibitory activities and relatively low cytotoxicity, demonstrating their great potential as HIF-1 inhibitors. These results afford us a new strategy for the discovery of new HIF-1 inhibitors and anti-proliferative agents from natural or synthetic diaryl acetylene derivatives.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (Grant No. 21606036) and the Fundamental Research Funds for the Central Universities (DUT16RC(3)007, DUT18LK11).
Notes and references
- C. Sergiu, Mini-Rev. Med. Chem., 2009, 9, 560–571 CrossRef.
- Y. Xie, K. P. Xu, Z. X. Zou and G. S. Tan, Cent. South Pharm., 2017, 15, 129–142 Search PubMed.
- L. Zhang, Y. Liang, X. Wei and D. Cheng, J. Org. Chem., 2007, 72, 3921–3924 CrossRef CAS PubMed.
- Y. Cao, J. Chen, N. Tan, L. Oberer, T. Wagner, Y. Wu, G. Zeng, H. Yan and Q. Wang, Bioorg. Med. Chem. Lett., 2010, 20, 2456–2460 CrossRef CAS PubMed.
- Y. Cao, J. Chen, N. Tan, Y. Wu, J. Yang and Q. Wang, Magn. Reson. Med., 2010, 48, 656–659 CAS.
- X. L. Cheng, S. C. Ma, J. D. Yu, S. Y. Yang, X. Y. Xiao, J. Y. Hu, Y. Lu, P. C. Shaw, P. H. But and R. C. Lin, Chem. Pharm. Bull., 2008, 7, 982–984 CrossRef.
- K. P. Xu, H. Zou, Q. Tan, F. S. Li, J. F. Liu, H. L. Xiang, Z. X. Zou, H. P. Long, Y. J. Li and G. S. Tan, J. Asian Nat. Prod. Res., 2011, 13, 93–96 CrossRef CAS PubMed.
- K. Xu, H. Zou, G. Liu, H. Long, J. Li, F. Li, Z. Zou, J. Kuang, X. Xie and G. Tan, J. Asian Nat. Prod. Res., 2011, 13, 1051–1055 CrossRef CAS PubMed.
- G. G. Zhang, J. Ying, H. M. Zhang, E. L. Ma and X. Y. Sun, Planta Med., 2012, 78, 390–392 CrossRef CAS PubMed.
- C. Yang, Y. Shao, K. Li and W. Xia, Beilstein J. Org. Chem., 2012, 8, 1884–1889 CrossRef CAS PubMed.
- Y. Cao, Y. Yao, X. Huang, L. Oberer, T. Wagner, J. Guo, W. Gu, W. Liu, G. Lv, Y. Shen and J. Duan, Tetrahedron, 2015, 71, 1581–1587 CrossRef CAS.
- C. Yuan, Y. P. Wu and J. A. Duan, Acta Pharm. Sin., 2015, 50, 199–202 Search PubMed.
- K. Xu, J. Li, G. Zhu, X. He, F. Li, Z. Zou, L. Tan, F. Cheng and G. Tan, J. Asian Nat. Prod. Res., 2015, 17, 819 CrossRef CAS PubMed.
- P. Nguyen, B. Zhao, M. Y. Ali, J. Choi, D. Rhyu, B. Min and M. Woo, J. Nat. Prod., 2014, 78, 34–42 CrossRef PubMed.
- P. Nguyen, D. Ji, Y. Han, J. Choi, D. Rhyu, B. Min and M. Woo, Bioorg. Med. Chem. Lett., 2015, 23, 3730–3737 CrossRef CAS PubMed.
- B. Zhu, T. Wang, L. Hou, H. Lv, A. Liu, P. Zeng and A. Li, Chem. Nat. Compd., 2016, 52, 624–627 CrossRef CAS.
- D. D. Le, D. H. Nguyen, B. T. Zhao, S. H. Seong, J. S. Choi, S. K. Kim, J. A. Kim, B. S. Min and M. H. Woo, Bioorg. Chem., 2017, 72, 273–281 CrossRef CAS PubMed.
- Q. Zhu, Y. Bao, Z. Zhang, J. Su, L. Shao and Q. Zhao, R. Soc. Open Sci., 2017, 4, 170352 CrossRef PubMed.
- G. Tan, K. Xu, F. Li, C. Wang, T. Li, C. Hu, J. Shen, Y. Zhou and Y. Li, J. Asian Nat. Prod. Res., 2009, 11, 1001–1004 CrossRef CAS PubMed.
- X. Liu, H. Luo, Y. Huang, J. Bao, G. Tang, Y. Chen, J. Wang and S. Yin, Org. Lett., 2013, 16, 282–285 CrossRef PubMed.
- B. S. Chinta and B. Baire, Org. Biomol. Chem., 2017, 28, 5857–6060 Search PubMed.
- Y. Huang, X. Liu, D. Wu, G. Tang, Z. Lai, X. Zheng, S. Yin and H. Luo, Biochem. Pharmacol., 2017, 130, 51–59 CrossRef CAS PubMed.
- R. Karmakar and D. Lee, Org. Lett., 2016, 18, 6105–6107 CrossRef CAS PubMed.
- M. J. Sowden and M. S. Sherburn, Org. Lett., 2017, 19, 636–637 CrossRef CAS PubMed.
- J. S. Zhang, X. Liu, J. Weng, Y. Q. Guo, Q. J. Li, A. Ahmed, G. H. Tang and S. Yin, Org. Chem. Front., 2017, 4, 170–177 RSC.
- X. Liu, H. Luo, Y. Huang, J. Bao, G. Tang, Y. Chen, J. Wang and S. Yin, Org. Lett., 2013, 16, 282–285 CrossRef PubMed.
- C. Wang, C. Hu, K. Xu, G. Tan and Y. Li, J. Cardiovasc. Pharmacol., 2010, 55, 560–566 CrossRef CAS PubMed.
- J. Kim, C. Cho, B. Tai, S. Yang, G. Choi, J. Kang and Y. Kim, Molecules, 2015, 20, 21405–21414 CrossRef CAS PubMed.
- C. Wang, C. Hu, K. Xu, Q. Yuan, F. Li, H. Zou, G. Tan and Y. Li, Naunyn-Schmiedeberg's Arch. Pharmacol., 2010, 381, 73–81 CrossRef CAS PubMed.
- W. Zhang, Y. Xu, K. Xu, W. Wu, G. Tan, Y. Li and C. Hu, Eur. J. Pharmacol., 2012, 694, 60–68 CrossRef CAS PubMed.
- G. L. Semenza, Annu. Rev. Phytopathol., 2014, 9, 47–71 CrossRef CAS PubMed.
- K. Lisy and D. J. Peet, Cell Death Differ., 2008, 15, 642–649 CrossRef CAS PubMed.
- B. J. Moeller, Y. Cao, C. Y. Li and M. W. Dewhirst, Cancer Cell, 2004, 5, 429–441 CrossRef CAS PubMed.
- D. Bhattarai, X. Xu and K. Lee, Biochem. Syst. Ecol., 2018, 38, 1404–1442 Search PubMed.
- S. P. Shi, Y. Z. Wang, X. K. Zheng, W. S. Feng and P. F. Tu, Biochem. Syst. Ecol., 2012, 45, 151–154 CrossRef CAS.
- M. Sun, L. Su, J. Dong, L. Liu, Y. Zhou and S. Yin, Tetrahedron Lett., 2017, 58, 2433–2437 CrossRef CAS.
- G. N. Masoud and L. Wei, Acta Pharm. Sin. B, 2015, 5, 378–389 CrossRef.
- G. Z. Li, S. Azuma, H. Minegishi and H. Nakamura, J. Organomet. Chem., 2015, 798, 189–195 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data of synthetic compounds. See DOI: 10.1039/c9ra02525a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Liqiang Liua,
Xiuhan Guo
ab,
Liqiang Liua,
Xiuhan Guo