
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biocatalytic transamination in a monolithic flow reactor: improving enzyme grafting for enhanced performance†
Ludivine van den Biggelaara,
Patrice Soumillionb and
Damien P. Debecker *a
*a
aInstitute of Condensed Matter and Nanosciences, UCLouvain, Place Louis Pasteur 1, 1348 Louvain-la-Neuve, Belgium. E-mail: damien.debecker@uclouvain.be
bLouvain Institute of Biomolecular Science and Technology, UCLouvain, Place Croix du Sud 2, 1348 Louvain-la-Neuve, Belgium
First published on 12th June 2019
Abstract
Transaminases were immobilized onto macrocellular silica monoliths and used for carrying a continuous flow mode transamination reaction. Monoliths were prepared via an emulsion-templated sol–gel method and functionalised by amino-moieties (3-aminopropyl-triethoxysilane, APTES) in order to covalently immobilize the enzymes, using glutaraldehyde as a cross-linking agent. In order to obtain higher performance and improved reproducibility, we investigate the key parameters of APTES functionalisation and of enzyme grafting. Four functionalisation protocols were studied. We show that enhancing the homogeneity of the APTES grafting and controlling the moisture level during functionalisation led to a 3-fold increase in activity as compared to the previously reported data, and greatly improved the reproducibility. Additionally, we report a strong beneficial effect of running the enzyme immobilisation at room temperature instead of 4 °C, further enhancing the obtained activity. Finally, the popular method which consists of stabilizing the covalent attachment of the enzyme by reducing the imine bonds formed between the enzyme and the functionalised surface was investigated. We highlight a strong enzyme deactivation caused by cyanoborohydride, making this strategy irrelevant in this case. The improvements presented here led to more active macrocellular monoliths, of general interest for continuous flow mode biocatalysis.
Introduction
The use of enzymes for catalysing chemical transformations is of increasing interest, especially in the perspective of greener synthesis of high-value chemicals.1–3 Indeed, enzymes often work with high efficiency and (enantio)-selectivity, and in mild conditions.4 In this vibrant field of research, the biocatalytic synthesis of chiral amines using transaminase enzymes is a good example of an application for which tremendous achievements have been made, both at the fundamental and applied levels.5–9In the perspective of practical applications, immobilizing enzymes on a solid carrier allows for their facile recovery (e.g. by filtration or centrifugation) so that the enzymes can then be reused several times.10–12 Among the various methods for enzyme immobilisation,13 covalent anchoring on porous carriers is probably the most successful approach, allowing a robust attachment of the enzyme.11 Importantly, the immobilisation of enzymes onto solid supports allows envisaging continuous flow biocatalytic processes to replace discontinuous batch processes.14–16 Such transfer from batch mode to continuous flow mode usually allows to increase the global productivity and to lower the environmental cost of chemical processes.17–20
Instead of the classical powdery materials that can be used as carriers and packed in a fixed bed reactor, a seducing approach for flow chemistry is to use self-standing porous monoliths.21–23 Inorganic or polymer monoliths containing intricate pore networks can be obtained in various desired shape and provide unique advantages such as fast kinetics and high throughput.21 Porous monolithic materials can have a high void fraction, thereby provoking only low pressure drop during the passage of a fluid.24,25
In particular, macrocellular silica monoliths have been extensively investigated because they can be obtained with tailored texture, structure, and surface functionalities, using bottom up sol–gel chemistry methods.26–28 In flow biocatalysis, for example, Brun et al.29 have reported on the use of macrocellular silica monoliths – coined “Si(HIPE)” – for the flow mode transesterification of crude oil using immobilized lipase. Si(HIPE) monoliths27 are obtained using a concentrated oil-in-water emulsion as a template. Their surface can then be functionalised, for example with epoxide or amine functions which serve as anchoring points for enzyme immobilisation.
Recently, we reported on the use of Si(HIPE) monoliths for the flow mode kinetic resolution of chiral amines using immobilized transaminases.30 The enzyme was effectively anchored using a well-known strategy:31 glutaraldehyde is employed as a coupling agent between the lysine residues and the amino groups brought on the silica surface by functionalisation with 3-aminopropyl-triethoxysilane (APTES). The role of APTES was shown to be decisive in the performance of these functional materials. Yet, neither the APTES functionalisation, nor the enzyme grafting were optimized.
In the present paper, we report significant improvements that can be obtained in the production of monolithic biocatalysts exhibiting enhanced activity. First, we inspect the spatial homogeneity of the APTES functionalisation and we propose a method leading to a more homogeneous distribution of the amino groups throughout the materials and subsequently to enhanced catalytic performance. Second, we address repeatability issues by controlling the water content during functionalisation. Third, we discuss the role of temperature during the enzyme grafting step. Finally, we consider the possibility to reduce the hydrolysable imine bonds formed in the covalent grafting process, to ensure a more robust immobilisation. We discuss the pertinence of these strategies in the context of flow biocatalysis.
Experimental
Materials
Acetone (≥99.9%), (3-aminopropyl)triethoxysilane (APTES; ≥ 98%), 4′-bromoacetophenone (BAP; ≥ 98%), n-dodecane (≥90%), hydrochloric acid (37% wt, aqueous solution), pyridoxal 5′-phosphate hydrate (PLP; ≥ 98%), sodium cyanoborohydride solution (5.0 M in 1 M NaOH; NaBH3CN), sodium hydroxide aqueous solution (50%), sodium pyruvate (≥99%), tetraethyl orthosilicate (TEOS; ≥ 98%), toluene (≥99.8%, anhydrous), trimethyltetradecylammonium bromide (TTAB; ≥ 99%) were purchased from Sigma-Aldrich. 4-Bromo-α-methylbenzylamine (BMBA; ≥ 99%; racemate), dibasic potassium phosphate (≥99%), dimethylsulfoxide (DMSO; ≥ 99.8%) and monobasic potassium phosphate (≥99%) were purchased from Acros Organics. D-Alanine (≥99%) was purchased from CarlRoth. Tetrahydrofuran (THF; ≥ 99.6%) was purchased from VWR Chemicals. Heat-shrinkable polytetrafluoroethylene (PTFE) tube was purchased from RS Components. Codexis generously supplied ATA-117 transaminase. Distilled water was applied for all synthesis and treatment processes.Silica monolith synthesis
Porous monoliths synthesis was performed according to Ungureanu et al.:32 6 g of a concentrated hydrochloric acid solution (37% wt) were introduced in 16 g of a TTAB aqueous solution (35% wt). Then 5 g of TEOS was added. The aqueous phase was stirred until a monophasic hydrophilic medium was obtained. 35 g dodecane was then added dropwise while stirring to form an emulsion. The latter was cast into a polypropylene 10 mL flask, allowed to condense for 1 week at room temperature. Resulting material was washed three times with 50 mL of a THF/acetone mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) (each washing lasted 24 h) and then gently dried in air during 3 days before calcined at 650 °C for 6 h (heating rate of 2 °C min−1 with a first plateau at 180 °C for 6 h). Porous monoliths were stored in a desiccator at room temperature.
:1 v/v) (each washing lasted 24 h) and then gently dried in air during 3 days before calcined at 650 °C for 6 h (heating rate of 2 °C min−1 with a first plateau at 180 °C for 6 h). Porous monoliths were stored in a desiccator at room temperature.
Monolith functionalisation
Grafted samples were denoted AxY where ‘A’ stands for APTES grafting, ‘x’ is the APTES concentration in the functionalisation solution (in mM), and ‘Y’ is the functionalisation method used for the monolith (method A, B, C or D).:1 v/v). For each washing, wet monoliths were soaked into the washing solvents by applying dynamic vacuum until effervescence stopped and then static vacuum for 2 h. Functionalised monoliths were dried under vacuum at 60 °C for 24 h and then stored in a desiccator at room temperature.Characterisation
Transamination reaction
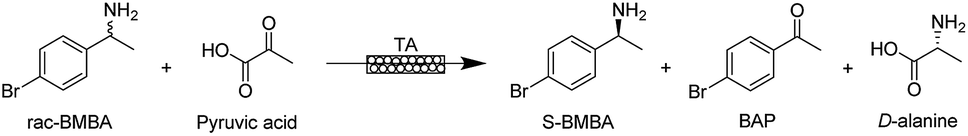 | ||
| Fig. 1 Model reaction: kinetic resolution of S-BMBA. | ||
Conversion was determined on 100 μL samples taken from the reaction medium. 10 μL of sodium hydroxide (2 M) was added and the mixture was vortexed for 1 second. 500 μL of dichloromethane was then added to the aqueous phase and vortexed for 10 seconds to allow extraction of BAP and BMBA into the organic phase. This extraction step was repeated twice and the organic phase was collected and analysed by gas chromatography (Bruker Scion 456 GC with a WCOT fused silica BR-5 column (30 m × 0.32 mm ID × 1.0 μm) and helium as carrier gas (25 mL min−1), oven temperature at 150 °C, split ratio of 20, injector temperature at 250 °C, flame ionisation detector temperature at 300 °C (air flow 300 mL min−1, H2 flow 30 mL min−1)). The yield is defined as the proportion of rac-BMBA converted into 4′-bromoacetophenone (BAP; in %). The maximum yield for the kinetic resolution is 50%.
Results and discussion
Functionalisation (Method A) and APTES dispersion
In our recent work,30 we demonstrated the feasibility of immobilizing a transaminase (ATA-117, from Codexis) on macrocellular silica monoliths for the enantioselective transamination in continuous flow. The enzyme was covalently attached to the amino-functionalised silica monolith (with APTES), using glutaraldehyde as a coupling agent. In the best conditions, using a monolith functionalized according to the protocol here called “Method A” and with a contact time of 10 min, we obtained a stable BAP yield of 6%, with excellent enantiospecificity (only R-BMBA was converted).After the reaction, the monolith was cut lengthwise, and a brownish crust was observed (Fig. 2). This crust was not observed when a blank was used (no APTES in the functionalisation solution). This visually indicates that the APTES dispersion throughout the monolith is not homogeneous. We propose that this colour is linked to the reaction between the aminopropyl groups grafted in excess and the reactant BMBA in the presence of glutaraldehyde (Fig. 2c).
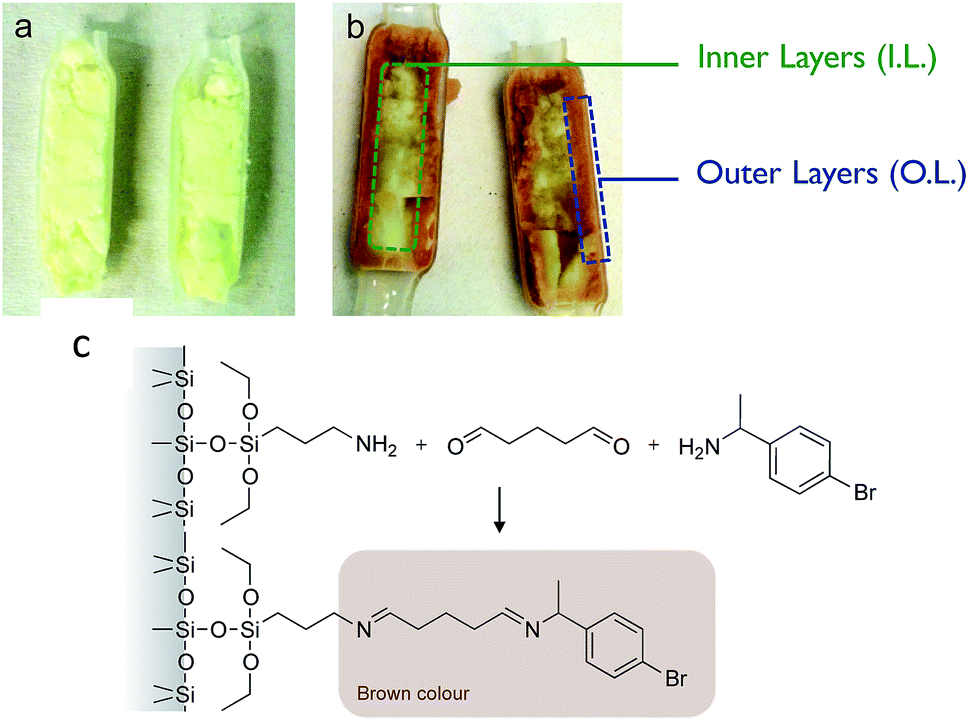 | ||
| Fig. 2 (a) Photograph of a monolith prepared via Method A but without APTES (blank) and cut lengthwise after the flow reaction (TA-A0A-GA), (b) photograph of the TA-A10A-GA cut lengthwise after the flow reaction. For subsequent characterisation, the outer layer (O.L.) was collected by scratching the monolith over 1 mm thickness. The inner layer (I.L.) was the core of the monolith. (c) Proposed reaction between the amino moiety and BMBA in the presence of glutaraldehyde, leading to the brown colour in the APTES-functionalised monoliths. | ||
ATR-FTIR and TGA analyses were performed on the inner and outer layers of the APTES functionalised monolith. TGA analyses (Fig. 3) confirmed an uneven APTES dispersion in the monolith functionalised by Method A. Clearly, the weight loss (associated to grafted aminopropyl moieties) was much higher from the sample scratched from the outer layers than from the inner layer. This was also confirmed by ATR-FTIR analyses as the C–H stretching band was detected around 2900 cm−1 only for the outer layer and not for the inner layer (Fig. 3).
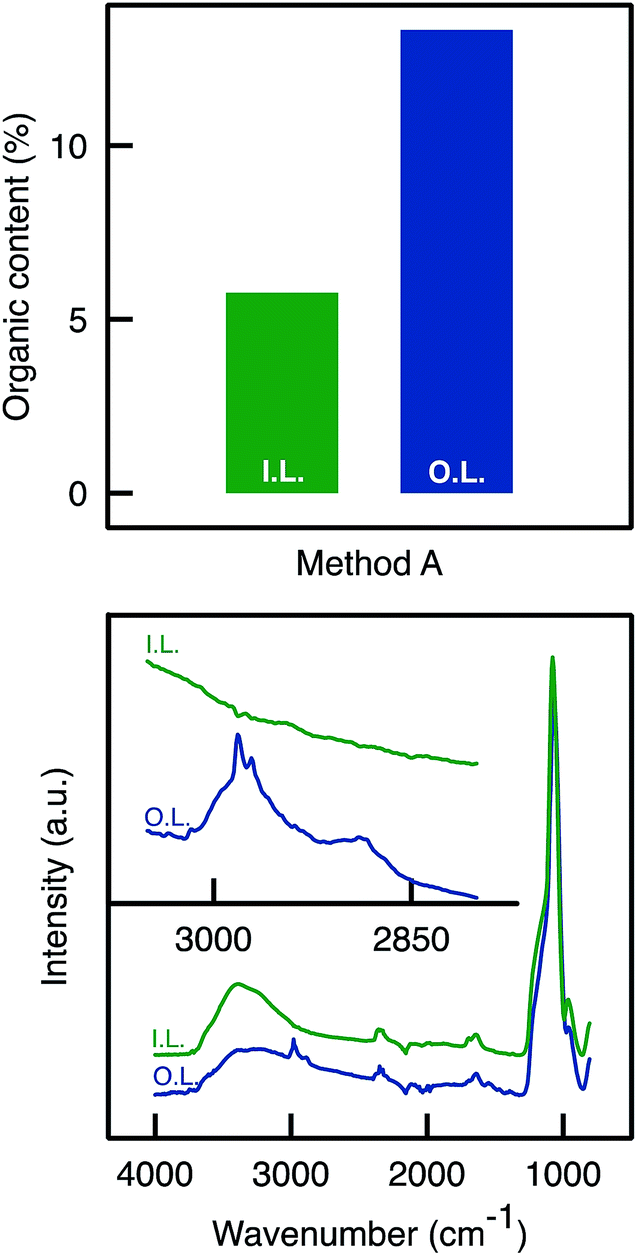 | ||
| Fig. 3 (Top) Total organic content determined by TGA analyses on the outer and inner parts of the A50A monolith. (Bottom) C–H stretching band absorbance was detected around 2900 cm−1 only for the outer layer of the sample A50A. | ||
Our interpretation of the heterogeneity of the APTES grafting was linked to the method of functionalisation. During the functionalisation process with Method A, the monolith was soaked into a large excess of the grafting solution. Thus, the porosity of the monolith was filled with an APTES solution of relatively low concentration, and the external environment was also composed of the same APTES solution. During the 24 h of functionalisation, APTES molecules could continuously diffuse from the excess solution and enter into the monolith, thereby functionalizing mostly the outer part of the monolith. In this way, a radial APTES gradient was established. It appears reasonable to put forward that a more homogeneous APTES functionalisation throughout the monolith would lead to a more effective enzyme immobilisation and possibly, to higher biocatalytic performance. This is why Methods B, C, and D were proposed as attempts to improve the quality of the APTES functionalisation step.
Improving APTES-functionalisation
 | ||
| Fig. 4 Functionalisation Method B led to a homogeneous APTES dispersion into monoliths (TA-A500B-GA sample). | ||
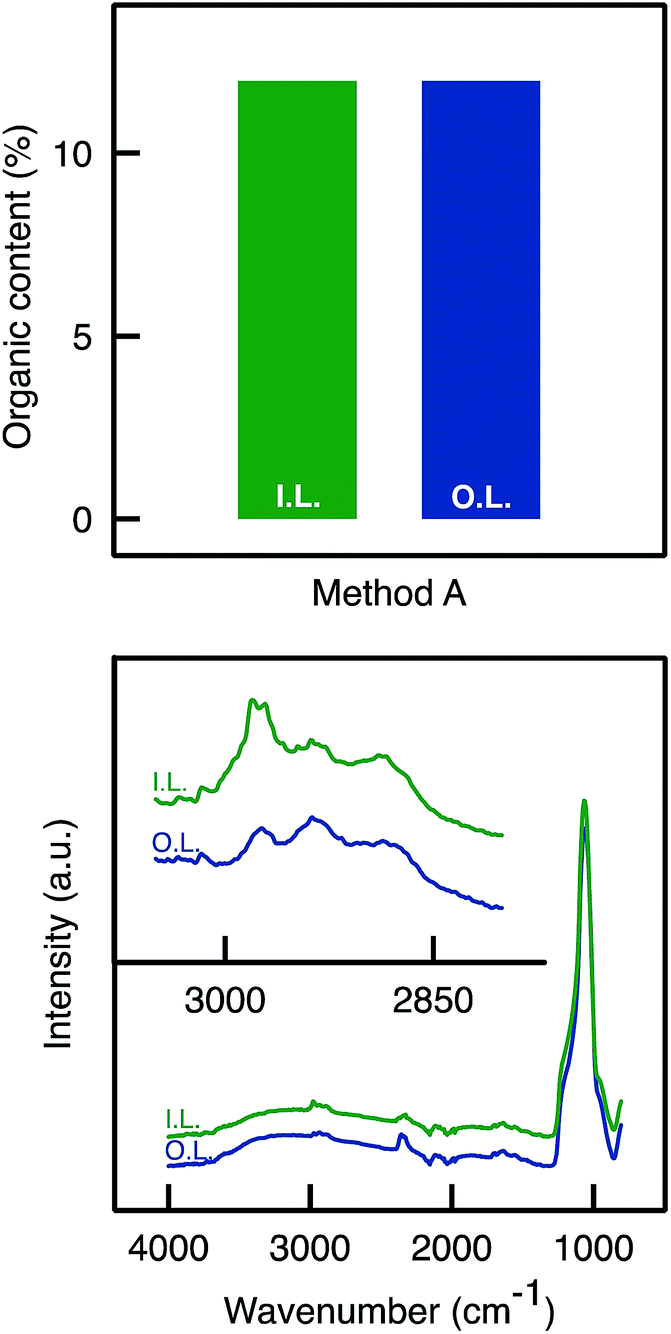 | ||
| Fig. 5 Characterisation of a monolith functionalised via Method B (A500B). (Top) TGA Analysis on the outer and inner layers (Method B allowed a homogeneous APTES dispersion; same organic content in both layers). (Bottom) ATR-FTIR: C–H stretching band absorbance was detected around 2900 cm−1 in both outer and inner layers with similar intensity. | ||
Eight TA-A500B-GA monoliths were prepared independently following strictly the same procedure used to load the enzyme and were tested in the kinetic resolution of BMBA. The catalysts showed on average a yield of 11%, significantly higher than the 6% yield obtained with Method A samples (Fig. 6). A 2 mg loading was obtained in average for A500B-GA samples (standard deviation, RSD, was 57% for the loading measurement on the eight monoliths).
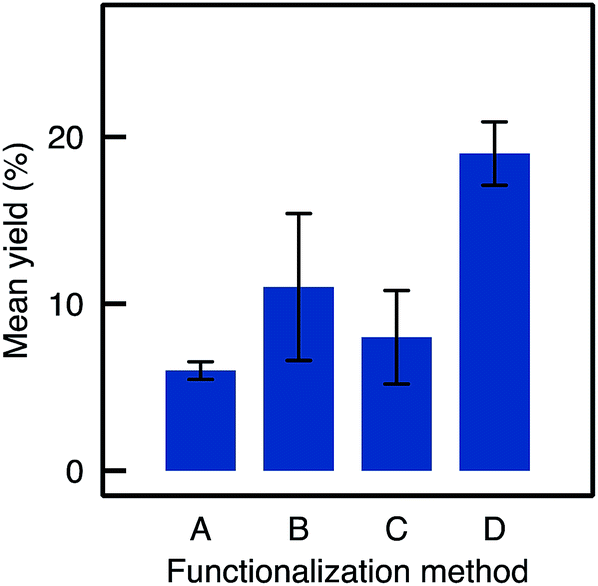 | ||
| Fig. 6 Mean yields and RSD for flow reactions depending on the functionalisation method used (samples are TA-A50A-GA, TA-A500B-GA, TA-A500C-GA, TA-A500D-GA). | ||
Thus, the better dispersion of APTES throughout the monoliths seems to be beneficial for the enzyme anchoring. However, the repeatability was poor (RSD was 40% for the final yield obtained with the eight independent monoliths prepared via Method B, see error bar in Fig. 6). This may be assigned to a relatively high variability of the silanol content on Si(HIPE) monoliths from batch-to-batch. Indeed, according to Zhuravlev,33 dehydroxylation of the silica surface should be expected to occur during monolith calcination. In contrast, water from air humidity could slowly re-hydroxylate the silica surface before the functionalisation procedure is applied. This rehydroxylation step was not controlled in Method B and may have varied from one experiment to the other. Yet, those silanols are the anchoring points for the grafting of the aminopropyl groups. Thus, Method C was implemented in order to increase the silanol surface density and to control the rehydroxylation step.
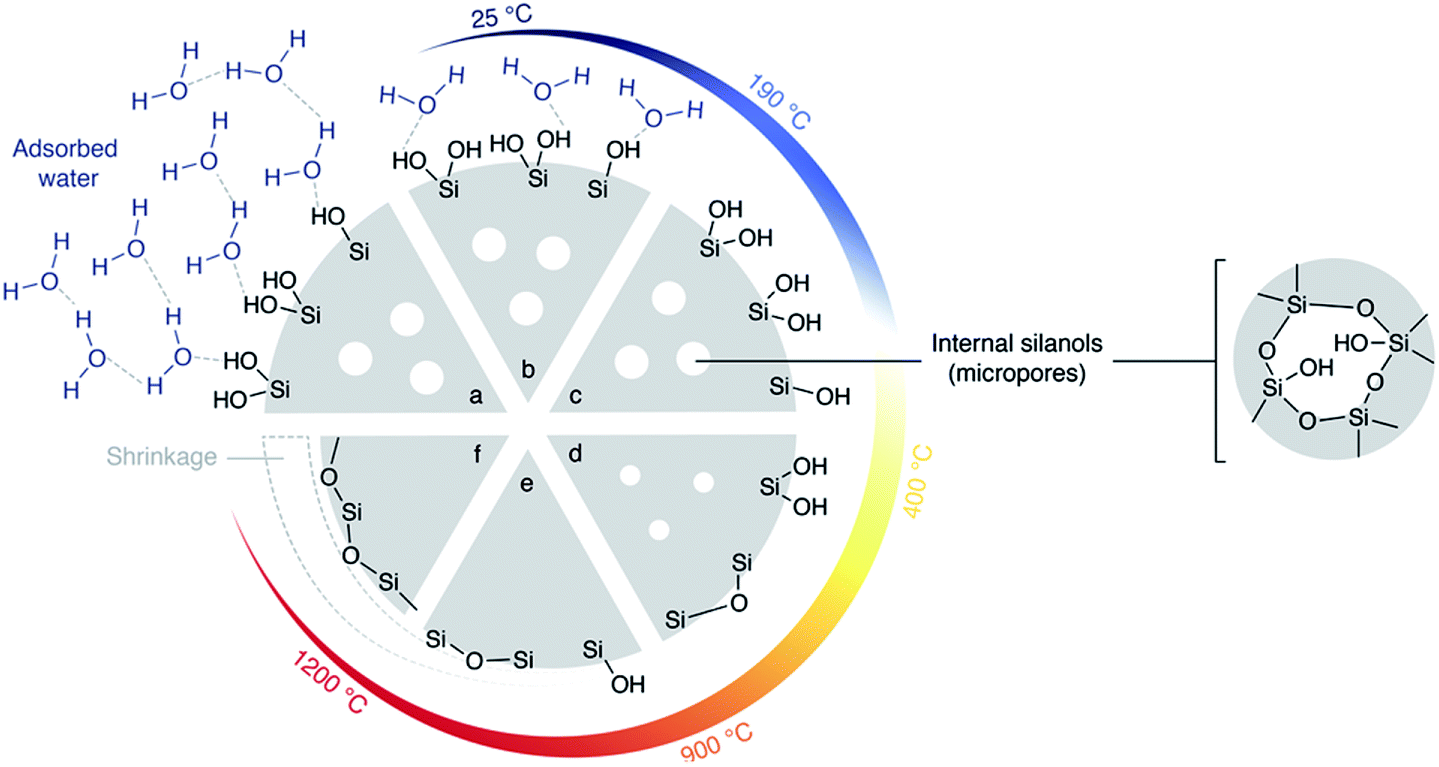 | ||
| Fig. 7 Silica heated under vacuum: Zhuravlev model.33 (a) Hydrated silica at 25 °C. Many silanols are present at the surface as well as internal silanols (in micropores). Water is in excess and forms multilayers. When heated under vacuum (b) silica gradually loses surface water. (c) At the key temperature of 190 °C, silica is totally dehydrated but still contains many silanols. (d) Beyond 190 °C, silanols condensation into siloxane bridges gradually occurs at the surface. Over 400 °C, this condensation is irreversible. (e) Over 900 °C, condensation internal silanols and condensation of most of the surface silanols occurs. (f) Finally, by heating in vacuum up to 1200 °C, silica surface loses its last silanols into siloxanes. | ||
Benefiting from a higher surface density of hydroxyl groups, it was expected that more APTES molecules could be grafted. One the one hand, TGA analyses confirmed the presence of a larger organic content on the samples that have been rehydroxylated (A500C versus A500B, see Fig. S3†). One other hand, ATR-FTIR analyses did not allow evidencing a higher intensity for organic moieties in the sample prepared by Method C as compared to Method B (not shown).
In the flow mode transamination reaction, the TA-A500C-GA biocatalysts showed a slightly lower yield (8.4%) as compared to A500B-GA (Fig. 6). The method still showed a low repeatability (RSD on the yield was 35% when three different monoliths functionalised by Method C were tested independently). Thus, although Method C seems to allow increasing the silanol surface density on the silica monoliths, it did not allow to increase the efficiency of the APTES functionalisation, and the biocatalytic performance obtained after subsequent enzyme grafting were not improved either. Moreover, the reproducibility on the final yield remained poor.
On the one hand, Si(HIPE) monoliths were dried at 120 °C under vacuum for 24 h in order to remove excess water from the monolith. After cooling under vacuum, functionalisation of the monolith was applied without further delay. On the second hand, the APTES functionalisation solution was prepared using water-saturated toluene as the solvent, so that the water input was strictly the same for all samples. The preparation was repeated independently on three different silica monoliths.
TGA analyses of samples functionalised with APTES by Method D confirmed that the organic content was higher than in the blank sample (which has undergone the same functionalisation procedure but without APTES; Fig. 8). However, the organic content did not increase markedly with the APTES concentration and instead seemed to level off, suggesting that the monoliths are saturated, already upon impregnation with 100 mM APTES solution. The organic content was similar as that obtained with Method C (∼15%). ATR-FTIR analyses (Fig. 9) showed a slight increase in C–H vibration band absorbance with the presence of APTES. Moreover, similar to Method C, the primary amine scissoring band appeared (at 1570 cm−1)39 with APTES functionalisation.
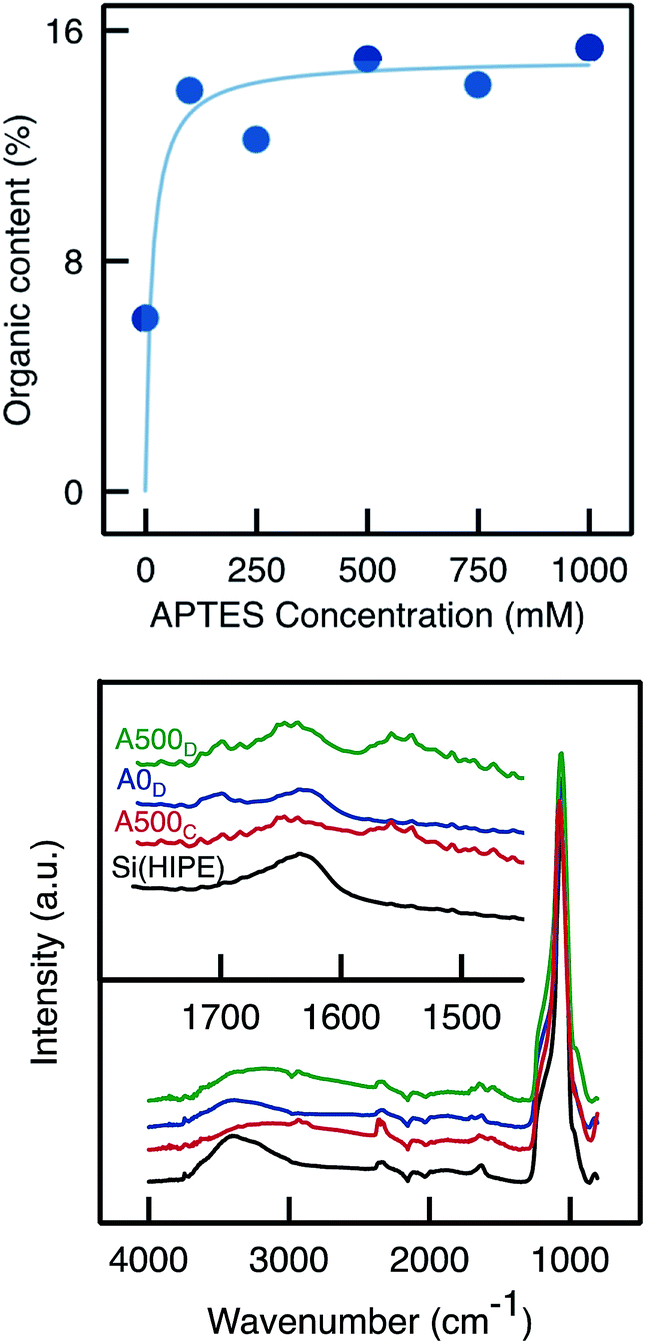 | ||
| Fig. 8 (Top) Total organic content as measured by TGA on monoliths functionalised via Method D with APTES solution of various concentration. (Bottom) ATR-FTIR spectra of Si(HIPE), A500C, A0D (blank) and A500D. | ||
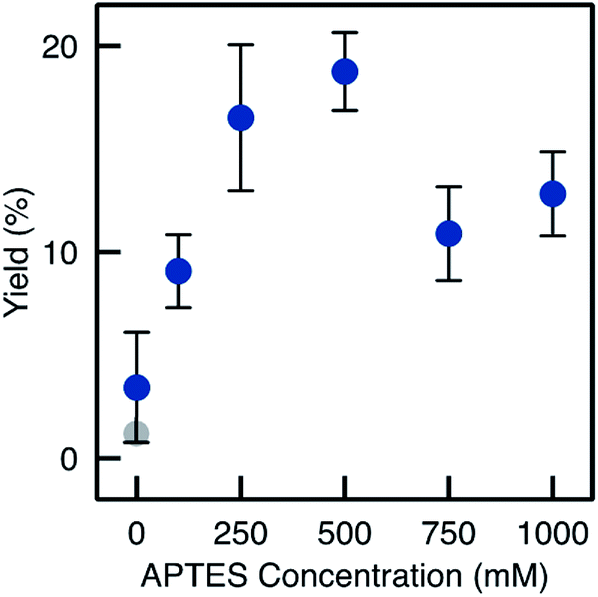 | ||
| Fig. 9 BAP yields with the monoliths prepared by method D (AxD, x = 0 to 1000 mM). The grey dot represents the yield and loading for TA-Si(HIPE) sample. Error bars correspond to the RSD on three independent experiments with freshly prepared monoliths for each APTES concentration. | ||
Despite relatively low enzyme loading (0.7 mg per monolith maximum, see Fig. S4†), the biocatalysts prepared from monoliths functionalised via Method D showed high performance. The yield reached 19% on average with a much improved repeatability (e.g. RSD on the final yield was 9.2% for three different TA-A500D-GA monoliths, see Fig. 6). Yields increased quite linearly with the APTES concentration in the functionalisation solution, up to 250 mM (Fig. 9). At higher APTES concentration, the catalytic yield dropped. This trend was related to the loading of enzyme that is immobilised, depending on the APTES concentration (Fig. S4†). The loading also reached a maximum for TA-A500D-GA and then decreased. From these measurement, it appears interesting to notice that the enzyme loading was high both for the pristine TA-Si(HIPE) sample and for the TA-A0D-GA (i.e. the blank without APTES). This suggests that the enzyme was immobilized in large amounts by simple adsorption, but that it was markedly deactivated upon such simple physisorption. Indeed, it is well documented that enzyme conformation – crucial for catalytic properties – could be altered during simple adsorption.12,13 In contrast, APTES grafting is suggested to confer a favourable environment to preserve enzyme activity (even if the total loading is lower). Hydrophobic effects brought by APTES grafting have been already highlighted in the literature to help preventing the deformation of the structure.40
When comparing the yields obtained with monoliths grafted by Method C and D, we observed that Method D allowed a higher yield as A500C and A500D exhibit respectively 8.4% and 19% yield. Thus, despite similar results in IR and TGA, Method D provides monoliths with higher biocatalytic activity. This may be due to a better control on the APTES dispersion throughout the monolith with Method D. This confirms the crucial role of water in such silanisation reaction. With this method in hand, it is possible to obtain macrocellular monoliths, which exhibit much higher biocatalytic yield, together with a highly improved repeatability. An APTES concentration of 250–500 mM was identified as the best compromise to ensure that enough organic moieties are grafted and to optimize both the enzyme loading and the catalytic yield in the flow reaction. Table 1 summarizes the main improvements obtained by adapting the functionalisation method.
| Sample | Dispersion | Mean yieldb (RSD) | Mean organic contenta | Estimated enzyme loading |
|---|---|---|---|---|
| a TGA on entire functionalised monolith (before enzyme loading).b RSD is calculated based on three independent experiments carried out on freshly prepared monoliths (except for A500B for which eight independent experiments were carried out and used to calculate the yield and RSD). | ||||
| A50A | Poor | 6% (9%) | 11% | 1.2 mg |
| A500B | Good | 11% (40%) | 7% | 2.0 mg |
| A500C | Good | 8% (35%) | 12% | 1.7 mg |
| A500D | Good | 19% (9.2%) | 15% | 0.7 mg |
Improving the enzyme immobilisation efficiency
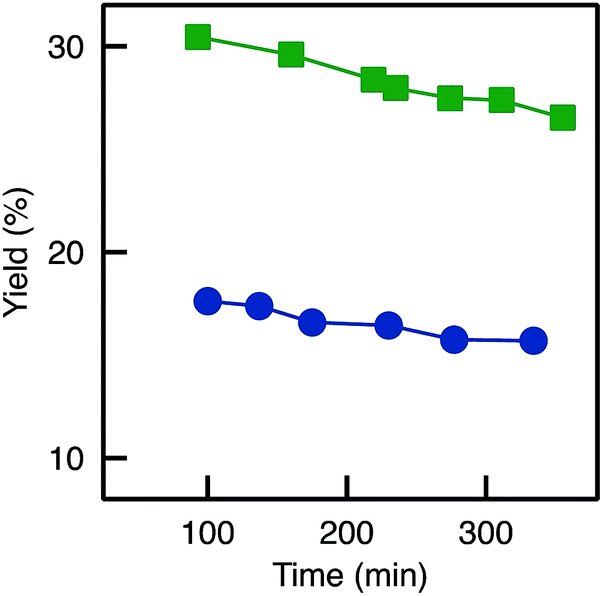 | ||
Fig. 10 Evolution of the BAP yield with time for TA-A250D-GA-30d: ( ) reference (no reduction); ( ) reference (no reduction); ( ) reduction with NaBH3CN (Red3). ) reduction with NaBH3CN (Red3). | ||
This result may find two explanations. First, the kinetics of the reactions that led to the grafting of the enzyme (imine bonds formation between the lysine and the aldehyde) was probably enhanced at higher temperature, leading to a more efficient grafting. Indeed, a higher enzyme loading was measured for TA-A250D-GA-30d (0.7 mg) as compared to the TA-A250D-GA (0.2 mg). Second, the lower activity obtained after immobilisation at 4 °C can tentatively be explained by the so-called “cold dissociation” phenomenon.42 Indeed, it is important to note that transaminases are homo-dimeric enzymes, with the active site formed only when two monomers assemble (Fig. S5†).43 According to Privalov,42 under low temperatures, some multimeric enzymes may be dissociated to the monomers. In this case, a higher proportion of the inactive monomers would have been immobilized on the monolith instead of the active dimers. Once two monomers are immobilized on different grafting sites, reforming the active dimer is impossible. Moreover, the immobilisation of monomer could have occurred through the lysine residue located on the active site, thereby locking the enzyme with the active site towards the support surface, blocking the access for the reactants. Thus, it is clear that enzyme grafting should be done at 30 °C instead of 4 °C and we suggest that further optimisation of the grafting temperature could lead to further increase in the performance.
In a first attempt (Red1), the reducing agent was injected right after the enzyme immobilisation (after washing). In this case, a poor activity level was obtained as compared to the reference experiment without reduction (Fig. S7†). As already reported for PLP-dependent enzymes,46,47 deactivation by cyanoborohydride was suspected. In a control batch reaction, we indeed observed that NaBH3CN strongly deactivates the transaminases (Fig. S8†). It should be noted that the first step of the transamination reaction mechanism (Fig. S9†) is the formation of an imine between the aldehyde group of the cofactor (PLP) and the essential primary amine of the enzyme active site (from a lysine residue). The reducing agent could therefore attack the imine bond, leading to the irreversible formation of a secondary amine, permanently locking the cofactor into the active site, and leading to deactivation.
In a second attempt (Red2) the transaminase solution (containing PLP in the buffer) was injected through the reactor for enzyme immobilisation, and then, the PLP was washed prior to the reduction with NaBH3CN. By doing so, it was hypothesized that the locking of PLP into the active site could be minimized. Before starting the reaction by injecting the reactants together with PLP, the reducing agent was washed. However, this procedure also led to poor yield (Fig. S7†). It can be put forward that PLP remained in the vicinity of the active site, despite the washing step.
Looking back at the batch experiments (Fig. S8†), we observed that the reaction medium could act as a protective agent for the active site: no deactivation occurred when the reducing agent was added directly in the reaction medium. One possible explanation is that the reactants (having a strong affinity for the active site and being present in relatively high concentrations, well above the KM) tend to protect the active sites simply by keeping them less accessible for the reducing agent. Thus, in a last attempt to reduce imine bond while keeping the biocatalysts active (Red3), the reducing agent was fed to the flow reactor during the flow transamination. This system showed some activity (Fig. S8†); yet, the yield was still much lower than for the reference sample (no reduction). Thus, the negative impact of the reducing agent on the active sites was still present in this case. In relative terms, however, the reduction (Red3) could allow to very slightly decrease the deactivation on stream since the decrease in yield as a function of time was slightly slower (Fig. 10).
Conclusions
Starting with the proof-of-concept for carrying flow mode transamination reactions in a silica macrocellular monoliths loaded with transaminase enzymes, we here identify the essential parameters which allow reaching high catalytic performance; (i) functionalisation mode (dry versus wet), (ii) water availability during the functionalisation process and (iii) immobilisation temperature. In practice, briefly dipping the monolith in the functionalisation solution followed by aging in a solvent-saturated atmosphere allowed to obtain a homogeneous APTES dispersion throughout the entire monolith (confirmed by IR and TGA). Total water control during the process (by using dried monoliths and water-saturated solvent) allowed to minimize the batch-to-batch variability which affects enzyme immobilisation and biocatalysts efficiency. An attempt to reduce the imine bond formed upon enzyme immobilisation with APTES and glutaraldehyde was performed in order to minimize any putative enzyme leaching. It appeared that transaminases were irreversibly deactivated by the reducing agent, which disqualifies this strategy. Finally, we highlighted a strong beneficial effect of carrying out the immobilisation at 30 °C instead of 4 °C. This immobilisation temperature could advantageously be optimized in a future study. All in all, the optimized method presented here allowed to reach a 5-fold increase in activity for the studied transamination reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge the Communauté française de Belgique for the financial support – including the PhD fellowship of L. van den Biggelaar – through the ARC programme (15/20-069). F. Devred is acknowledged for the technical and logistic support.References
- T. Narancic, R. Davis, J. Nikodinovic-Runic and K. E. O’ Connor, Biotechnol. Lett., 2015, 37, 943 CrossRef CAS.
- F. Rudroff, M. D. Mihovilovic, H. Gröger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Nat. Catal., 2018, 1, 12 CrossRef.
- R. C. Simon, F. G. Mutti and W. Kroutil, Drug Discovery Today: Technol., 2013, 10, e37 CrossRef PubMed.
- H. Kohls, F. Steffen-Munsberg and M. Höhne, Curr. Opin. Chem. Biol., 2014, 19, 180 CrossRef CAS PubMed.
- C. E. Paul, M. Rodríguez-Mata, E. Busto, I. Lavandera, V. Gotor-Fernández, V. Gotor, S. García-Cerrada, J. Mendiola, Ó. de Frutos and I. Collado, Org. Process Res. Dev., 2014, 18, 788 CrossRef CAS.
- M. Höhne and U. T. Bornscheuer, ChemCatChem, 2009, 1, 42 CrossRef.
- F. Guo and P. Berglund, Green Chem., 2017, 19, 333 RSC.
- M. D. Patil, G. Grogan, A. Bommarius and H. Yun, Catalysts, 2018, 8, 254 CrossRef.
- D. Ghislieri and N. J. Turner, Top. Catal., 2014, 57, 284 CrossRef CAS.
- D. N. Tran and K. J. Balkus, ACS Catal., 2011, 1, 956 CrossRef CAS.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223 RSC.
- T. Jesionowski, J. Zdarta and B. Krajewska, Adsorption, 2014, 20, 801 CrossRef CAS.
- R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289 CrossRef CAS.
- J. Britton, S. Majumdar and G. A. Weiss, Chem. Soc. Rev., 2018, 47, 5891 RSC.
- M. L. Contente and F. Paradisi, Nat. Catal., 2018, 1, 452 CrossRef CAS.
- M. P. Thompson, I. Peñafiel, S. C. Cosgrove and N. J. Turner, Org. Process Res. Dev., 2019, 23, 9 CrossRef CAS.
- X. Y. Mak, P. Laurino and P. H. Seeberger, Beilstein J. Org. Chem., 2009, 5, 19 Search PubMed.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688 CrossRef CAS PubMed.
- M. Planchestainer, M. L. Contente, J. Cassidy, F. Molinari, L. Tamborini and F. Paradisi, Green Chem., 2017, 19, 372 RSC.
- R. Gérardy, R. Morodo, J. Estager, P. Luis, D. P. Debecker and J.-C. M. Monbaliu, Top. Curr. Chem., 2018, 377, 1 CrossRef PubMed.
- F. Svec and J. M. J. Fréchet, Science, 1996, 273, 205 CrossRef CAS PubMed.
- D. P. Debecker, C. Boissière, G. Laurent, S. Huet, P. Eliaers, C. Sanchez and R. Backov, Chem. Commun., 2015, 51, 14018 RSC.
- V. Smeets, L. van den Biggelaar, T. Barakat, E. M. Gaigneaux and D. P. Debecker, ChemCatChem, 2019, 11, 1593 CrossRef CAS.
- J. Wegner, S. Ceylan and A. Kirschning, Chem. Commun., 2011, 47, 4583 RSC.
- G. Jas and A. Kirschning, Chem.–Eur. J., 2003, 9, 5708 CrossRef CAS PubMed.
- C. P. Haas, T. Müllner, R. Kohns, D. Enke and U. Tallarek, React. Chem. Eng., 2017, 2, 498 RSC.
- N. Brun, S. Ungureanu, H. Deleuze and R. Backov, Chem. Soc. Rev., 2011, 40, 771 RSC.
- D. P. Debecker, Chem. Rec., 2018, 18, 662 CrossRef CAS PubMed.
- N. Brun, A. B. Garcia, H. Deleuze, M. F. Achard, C. Sanchez, F. Durand, V. Oestreicher and R. Backov, Chem. Mater., 2010, 22, 4555 CrossRef CAS.
- L. van den Biggelaar, P. Soumillion and D. P. Debecker, Catalysts, 2017, 7, 54 CrossRef.
- I. Migneault, C. Dartiguenave, M. J. Bertrand and K. C. Waldron, BioTechniques, 2004, 37, 790 CrossRef CAS PubMed.
- S. Ungureanu, M. Birot, G. Laurent, H. Deleuze, O. Babot, B. Julian-Lopez, M. F. Achard, M. I. Popa, C. Sanchez and R. Backov, Chem. Mater., 2007, 19, 5786 CrossRef CAS.
- L. T. Zhuravlev, Colloids Surf., A, 2000, 173, 1 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248 CrossRef CAS PubMed.
- T. Zor and Z. Selinger, Anal. Biochem., 1996, 236, 302 CrossRef CAS PubMed.
- E. T. Vandenberg, L. Bertilsson, B. Liedberg, K. Uvdal, R. Erlandsson, H. Elwing and I. Lundström, J. Colloid Interface Sci., 1991, 147, 103 CrossRef CAS.
- K. M. R. Kallury, P. M. Macdonald and M. Thompson, Langmuir, 1994, 10, 492 CrossRef CAS.
- N. Gartmann, C. Schütze, H. Ritter and D. Brühwiler, J. Phys. Chem. Lett., 2010, 1, 379 CrossRef CAS.
- R. M. Pasternack, S. Rivillon Amy and Y. J. Chabal, Langmuir, 2008, 24, 12963 CrossRef CAS.
- O. Barbosa, R. Torres, C. Ortiz and R. Fernandez-Lafuente, Process Biochem., 2012, 47, 1220 CrossRef CAS.
- L. Yang, J. Shi, C. Chen, S. Wang, L. Zhu, W. Xie and L. Guo, Electrophoresis, 2009, 30, 3527 CrossRef CAS PubMed.
- P. L. Privalov, Crit. Rev. Biochem. Mol. Biol., 1990, 25, 281 CrossRef CAS.
- I. C. R. Costa, R. O. M. A. de Souza and U. T. Bornscheuer, Tetrahedron: Asymmetry, 2017, 28, 1183 CrossRef CAS.
- R. F. Borch, M. D. Bernstein and H. D. Durst, J. Am. Chem. Soc., 1971, 93, 2897 CrossRef CAS.
- E. T. Hwang and M. B. Gu, Eng. Life Sci., 2013, 13, 49 CrossRef CAS.
- B. Golinelli-Pimpaneau and B. Badet, Eur. J. Biochem., 1991, 201, 175 CrossRef CAS.
- C. López, S. D. Ríos, J. López-Santín, G. Caminal and G. Álvaro, Biochem. Eng. J., 2010, 49, 414 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02433f |
| This journal is © The Royal Society of Chemistry 2019 |