DOI:
10.1039/C9RA02416F
(Paper)
RSC Adv., 2019,
9, 18670-18677
Novel dermacozine-1-carboxamides as promising anticancer agents with tubulin polymerization inhibitory activity†
Received
31st March 2019
, Accepted 27th May 2019
First published on 13th June 2019
Abstract
In the present study, novel dermacozine-1-carboxamides (8a–8n) were synthesized and screened for their in vitro cytotoxic activity against three different cancer cell lines: MCF-7 (breast cancer), A-549 (lung cancer) and DU145 (prostate cancer). All the compounds showed more efficiency against the DU145 cell line than against the MCF-7 and A-549 cell lines. Furthermore, 8a (CTC50: 7.02 μM) and 8l (CTC50: 6.32 μM) have been found to be more effective against the DU145 cells as they arrest the cell cycle at the G2/M phase by interfering with tubulin polymerization; these results indicate that these compounds act as potential anti-cancer agents by inhibiting tubulin polymerization.
Introduction
According to WHO estimates, cancer is the second leading cause of death globally and was responsible for an estimated 9.6 million deaths in 2018; moreover, in some way, about 1 in 6 deaths is due to cancer. Despite the availability of advanced diagnostic and treatment methods, the burden of cancer is increasing every year.1–3 In addition, the increase in the development of drug resistance is one of the main causes of cancer relapse.4 Therefore, researchers are focused on the identification of novel drugs for the treatment of cancer.
Phenazines are symmetrical heteroaromatic compounds, and the template has been found over 6000 compounds, accounting for various pharmacological activities.5,6 Phenazine and fused aryl phenazine derivatives, such as XR11576, XR5944, NC-190 and NC-182, are under clinical studies for their cytotoxic activity.7
The dermacozines A–J are a class of reduced phenazine-based marine natural products isolated from Dermacoccus abyssi sp. nov., strains MT1.1 and MT1.2, which are highly pigmented aromatic compounds. These compounds have been reported to exhibit various biological activities such as radical scavenging activity, cytotoxicity against the resistant cancer cell line K562 (human chronic myelogenous leukemia), and so on;8,9 moreover, previously, we have reported the first total synthesis of the dermacozine A, B and C.10 In the present study, a new series of dermacozine-1-carboxamides 8a–8n was synthesized, and the cytotoxic activity of these compounds as well as their effect on the cell cycle and associated cyclin and tubulin polymerization were investigated to elucidate their possible mechanism of action.
Results and discussion
Chemistry
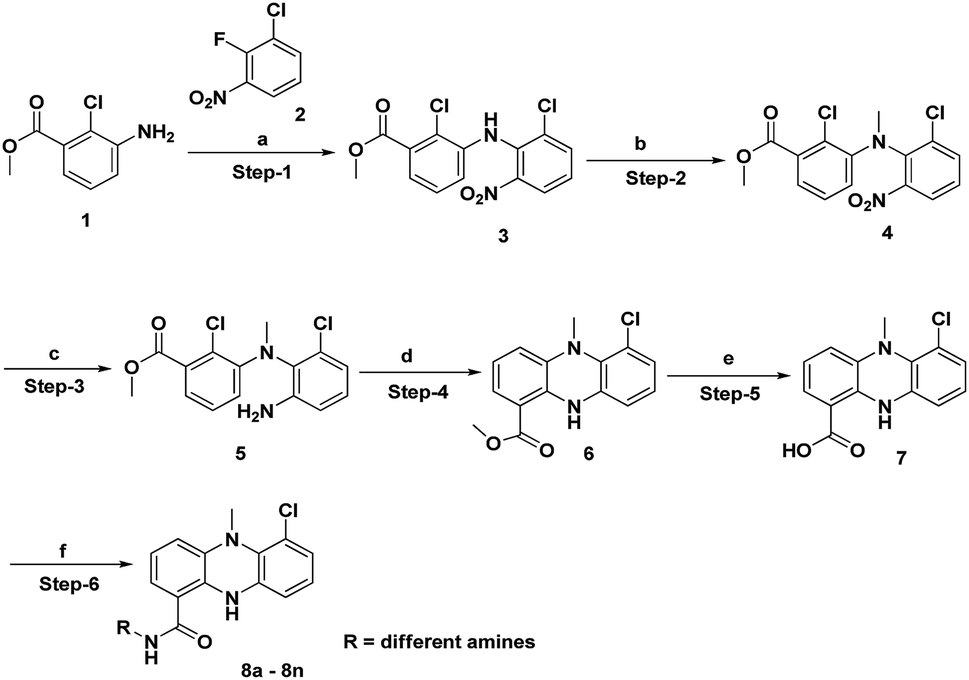
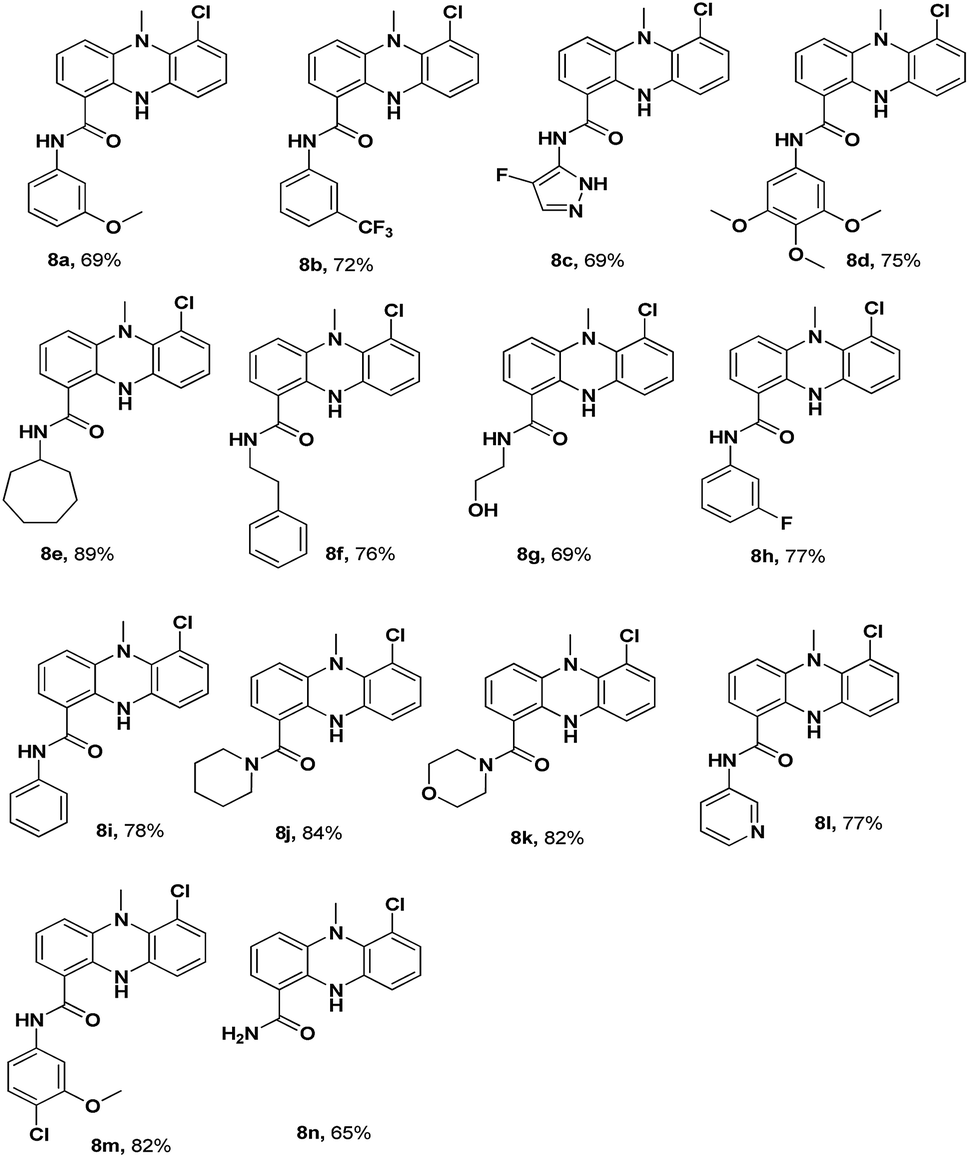
The synthesis of the dermacozine-1-carboxylic acid 7 and its derivatives was achieved in 6 steps, as shown in Scheme 1.10 The commercially available methyl 3-amino-2-chlorobenzoate 1 was treated with 1-chloro-2-fluoro-3-nitrobenzene 2 in the presence of LiHMDS at −78 °C to afford the methyl 2-chloro-3-((2-chloro-6-nitrophenyl)amino)benzoate 3.11 Methylation of 3 with MeI in the presence of NaOH afforded the N-methyl intermediate 4. Then, the reduction of the nitro group was performed using Fe–NH4Cl at reflux temperature to furnish 5. After this, intramolecular cyclisation of 5 was conducted under Buchwald conditions in the presence of Johnphos, pd2 (dba)3 and Cs2CO3, at 120 °C under sealed conditions. Then, hydrolysis of the methyl ester 6 to the acid 7 was performed using 4 N NaOH at room temperature, as shown in Scheme 1. The amide derivatives 8a–8n were synthesized in good yields by treating the acid 7 with 14 different amines using HATU–DIPEA at room temperature (Fig. 1).
 |
| | Scheme 1 Synthesis of dermacozine-1-carboxamides. | |
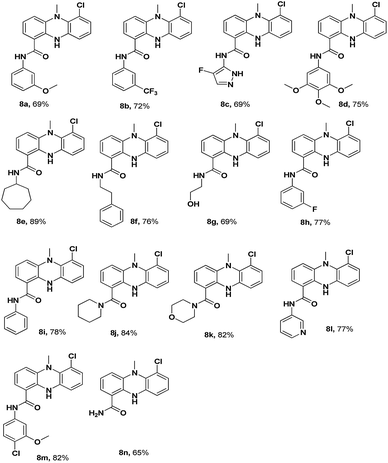 |
| | Fig. 1 Structures of dermacozine-1-carboxamides. | |
The structures of the newly synthesized compounds 8a–8n were confirmed by 1H-NMR, 13C-NMR, FT-IR, ESI-MS and HR-MS analysis data. All the spectral and analytical data of the synthesized compounds were in full agreement with the proposed structures and have also been discussed for the derivative 8f. In the 1H-NMR spectra, the characteristic N-methyl and NH peaks appeared at 3.22 (3H, s, NCH3) and 9.74 (1H, s, NH), all the aromatic protons appeared in the range of 6.65–9.74 (11H, m, ArH), the amide proton appeared at 8.55 (br s, 1H, CONH) and the alkyl proton exhibited a singlet at 2.84 (2H, br t, Ar–CH2) and 3.42–3.51 (2H, m, CONH–CH2), confirming the presence of required protons. In the 13C NMR spectra, the presence of carbon signals at 167 ppm (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 45 ppm (NCH3), 40 ppm (CONH–CH2) and 34 ppm (Ar–CH2) confirmed the presence of characteristic carbon bonds. In the FT-IR spectra, a strong absorption band was observed at 1629 cm−1, which confirmed the formation of the amide bond (CO stretch). Furthermore, a strong absorption peak was observed at 3373 cm−1, which confirmed the N–H bond formation. In the HR-MS of 8f, a peak was found at 378.1367 [M + H]+, which matched the calculated value 378.2137 [M + H]+; this confirmed the molecular weight of the compound 8f (C22H21ON3Cl).
O), 45 ppm (NCH3), 40 ppm (CONH–CH2) and 34 ppm (Ar–CH2) confirmed the presence of characteristic carbon bonds. In the FT-IR spectra, a strong absorption band was observed at 1629 cm−1, which confirmed the formation of the amide bond (CO stretch). Furthermore, a strong absorption peak was observed at 3373 cm−1, which confirmed the N–H bond formation. In the HR-MS of 8f, a peak was found at 378.1367 [M + H]+, which matched the calculated value 378.2137 [M + H]+; this confirmed the molecular weight of the compound 8f (C22H21ON3Cl).
Biology
Cytotoxicity. MTT assay was performed to determine the cytotoxicity of the synthesized dermacozine-1-carboxamides against the MCF-7 (breast cancer), A-549 (lung cancer) and DU145 (prostate cancer) cells. The concentrations at which the test compounds inhibited the growth of 50% of cells (CTC50) are presented in Table 1. The synthesized derivatives 8a–8n were found to be effective in killing the DU145 cells as compared to the case of other cell lines tested herein. Among the various synthesized dermacozine-1-carboxamides, 8a, 8d, 8i, 8l and 8m were found to be more effective, with the CTC50 values of 7.02 ± 0.54, 10.62 ± 0.77, 8.95 ± 0.77, 6.32 ± 0.68 and 9.26 ± 0.57 μM, respectively. Furthermore, 8a (CTC50: 7.02 μM) and 8l (CTC50: 6.32 μM) were selected for mechanistic studies. The derivatives with electron-releasing groups, such as methoxy and chloro, exhibited highest activity as compared to other derivatives.
Table 1 In vitro anticancer activity of dermacozine-1-carboxamides (8a–8n) against the MCF-7, A549 and DU 145 cells (CTC50 in μM)a
| Compound |
MCF7 (μM) |
A549 (μM) |
DU145 (μM) |
| Values are the mean ± SD, n = 3. |
| 8a |
32.31 ± 0.99 |
23.96 ± 0.99 |
7.02 ± 0.54 |
| 8b |
25.95 ± 1.28 |
19.21 ± 0.92 |
12.36 ± 0.45 |
| 8c |
29.23 ± 0.72 |
21.03 ± 1.13 |
15.25 ± 0.29 |
| 8d |
26.22 ± 0.52 |
26.54 ± 1.14 |
10.62 ± 0.77 |
| 8e |
28.12 ± 0.87 |
17.12 ± 0.87 |
15.00 ± 0.80 |
| 8f |
23.18 ± 1.30 |
25.09 ± 1.12 |
16.32 ± 0.68 |
| 8g |
22.26 ± 0.95 |
16.62 ± 2.09 |
18.01 ± 0.32 |
| 8h |
32.03 ± 1.13 |
21.95 ± 1.30 |
16.05 ± 1.86 |
| 8i |
28.36 ± 0.75 |
32.23 ± 0.93 |
8.95 ± 0.77 |
| 8j |
35.26 ± 1.27 |
19.65 ± 1.16 |
13.24 ± 0.60 |
| 8k |
27.04 ± 0.62 |
25.14 ± 1.97 |
18.58 ± 1.11 |
| 8l |
21.05 ± 0.30 |
29.08 ± 2.20 |
6.32 ± 0.68 |
| 8m |
25.82 ± 0.90 |
18.28 ± 1.58 |
9.26 ± 0.57 |
| 8n |
23.12 ± 1.18 |
24.36 ± 0.70 |
19.24 ± 0.93 |
| Dermacozine A |
12.26 ± 0.78 |
22.36 ± 1.69 |
15.62 ± 2.21 |
| Nocodazole |
1.23 ± 0.92 |
0.96 ± 0.65 |
2.62 ± 1.20 |
| Paclitaxel |
2.56 ± 1.53 |
1.61 ± 0.93 |
3.16 ± 0.93 |
Structure–activity relationship (SAR). Based on the CTC50 values, we deduced the SAR of the compounds. Via the SAR studies, we speculated that the compounds 8a–8n bearing aryl, hetero aryl, or aliphatic amide groups exhibited significant anti-cancer activity against the DU145 cell line than against the MCF-7 and A549 cell lines. Moreover, the 3-pyridyl amide compound 8l exhibited highest anti-cancer activity than the other phenyl amide derivatives 8a, 8b, and 8h. Furthermore, the phenyl amide 8a with an electron-donating group (OMe) at the 3rd position showed finer activity as compared to the phenyl amides 8b and 8h with electron-withdrawing groups (CF3 and F) at the third position, respectively. On the other hand, the aliphatic amide derivatives 8e, 8f, 8g, 8k and 8j exhibited moderate anti-cancer activity against the DU145 cell line. Based on the abovementioned observations, the aryl substituents of dermacozine-1-carboxamides are more favourable to the anti-cancer activity.
Cell cycle distribution. The results are provided in Table 2 and Fig. 2. The effect of 8a and 8l on the cell cycle of the DU145 cells was analyzed by flow cytometry, and the results showed a profound effect of 8a and 8l on the cell cycle as these compounds arrested the cell cycle at the G2/M phase; 8a and 8l arrested 62.3 and 65.6% of the cells in the G2/M phase, respectively, as compared to the case of the control cells (10.2%).
Table 2 Cell cycle distribution of the DU 145 cells at the Sub G1, G0/G1, S phase and G2/M phase
| Compound |
Sub-G1 |
G0/G1 |
S phase |
G2/M phase |
| Control |
2.8 |
83.3 |
5.6 |
10.2 |
| 8a |
1.6 |
24.6 |
8.9 |
62.3 |
| 8l |
1.2 |
18.9 |
9.1 |
65.6 |
 |
| | Fig. 2 Cell cycle distribution of the DU145 cells. | |
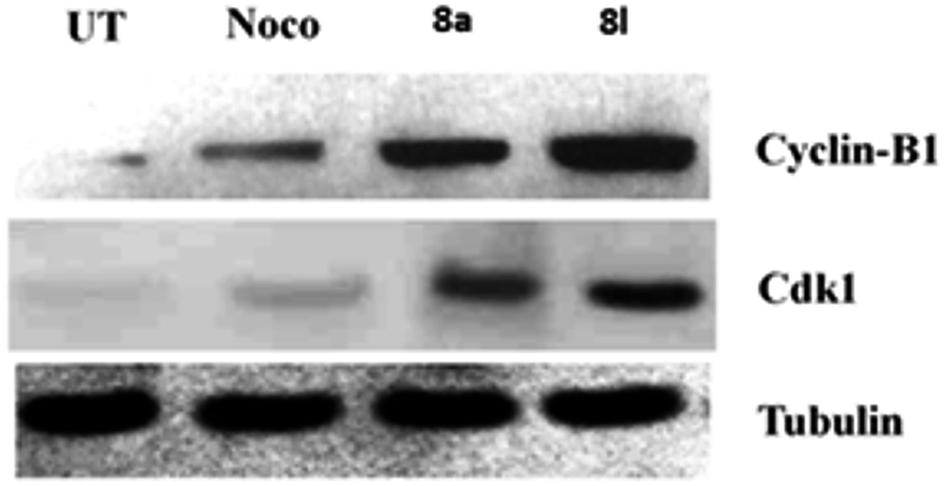
Effects of 8a and 8g on the expressions of cyclin-B1 and Cdk1. The investigation of the effects of 8a and 8l on the expressions of cyclin-B1 and Cdk1 is important as cyclin-B1 and Cdk1 are the key kinases involved in the G2/M phase of cell cycle transition and play an important role in the metaphase by activating the motor proteins required for spindle separation. The activation of Cdk1 primarily involves the expression of cyclin-B1. Hence, in the present study, we studied the expression of Cdk1 and cyclin-B1 after the treatment of the DU 145 cells with the compounds 8a, 8l and nocodazole, which acted as a standard. Tubulin acts as a loading control, and the results of the Western blot analysis are shown in Fig. 3. The uniform expression of tubulin in all the lanes indicated uniform sample loading. However, we observed a difference in the expressions of Cdk1 and cyclin-B1 in the treatment groups. The expression of Cdk1 and cyclin-B1 was observed to be comparatively more in the cells treated with the compounds 8a and 8l, indicating significant G2/M arrest by the inhibition of mitosis and interference with spindle separation. Via the Western blot analysis, it was found that 8a and 8l killed the DU 145 cancer cells by inhibiting the cell cycle at mitosis and interfering with spindle separation.
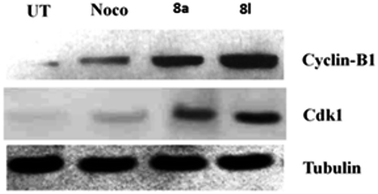 |
| | Fig. 3 Immunoblot analysis for the expression of cyclin-B1 and Cdk1. Tubulin was used as a loading control. | |
Effect of 8a and 8l on tubulin polymerization. Via the cell cycle analysis, it was evident that 8a and 8l arrested the cell cycle at the G2/M phase. These compounds, therefore, might inhibit tubulin polymerization. Hence, we assessed the ability of 8a and 8l to inhibit tubulin polymerization. Nocodazole was used as a positive control. 8a and 8l significantly inhibited tubulin polymerization. They showed potent inhibition of tubulin polymerization with the IC50 values of 2.64 μM and 2.36 μM, respectively, whereas nocodazole showed the IC50 value of 2.51 μM (Table 3).
Table 3 Anti-tubulin polymerization inhibitory activity of 8a and 8l
| Compound |
Anti-tubulin activity (IC50, μM) |
| 8a |
2.64 |
| 8l |
2.36 |
| Nocodazole |
2.51 |
Effect of 8a and 8l on the microtubule network. Antimitotic agents affect the microtubule network. We, therefore, observed the alterations in the microtubule network caused by 8a and 8l in the DU 145 cells via immune fluorescence microscopy. The test results demonstrated a well-organized microtubular network in the control cells; however, the cells treated with the compounds 8a and 8l showed disrupted microtubule organization (Fig. 4), thus demonstrating the inhibition of tubulin polymerization.
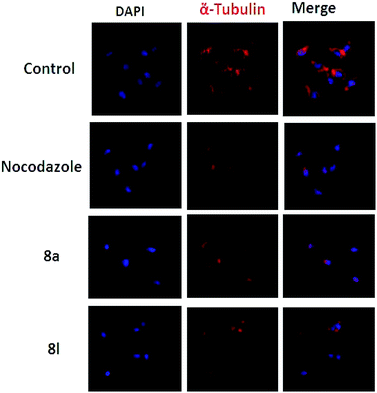 |
| | Fig. 4 Effect of 8a and 8l on the organization of the cellular microtubule network. 8a and 8l altered the tubulin assembly, which showed an irregular morphology. Cells were counterstained with DAPI to stain the nuclei. | |
Experimental
Chemistry
All commercially available reagents used in this study were bought from local suppliers, Sigma-Aldrich and Alfa Aesar, and used as received. Thin-layer chromatography was performed using pre-coated TLC silica gel 60 F254 plates on aluminum (Merck). TLC was visualized via UV-254 at 360 nm using iodine on silica gel. Column chromatography was performed using 100–200 mesh silica gel. Automated purifications were conducted using the Teledyne ISCO Combiflash companion. Unless otherwise stated, all the reactions were carried out under an atmosphere of argon in dried glassware using standard techniques. The 1H and 13C NMR spectra were obtained using the Varian 400 and Varian 300 spectrometers. Chemical shifts are reported in ppm with TMS as the internal standard. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), coupling constants and number of protons. IR spectra were obtained via the FT-IR PerkinElmer Spectrum 2000 using KBr pellets. Melting point (mp) determinations were performed using the Mel Temp apparatus, and the values reported herein are uncorrected.
Methyl 2-chloro-3-((2-chloro-6-nitrophenyl)amino)benzoate (3). To a stirred solution of methyl 3-amino-2-chlorobenzoate (1) (1 g, 5.405 mmol) in dry THF, cooled to −78 °C, 2 M LiHMDS (5.9 mL, 11.89 mmol) in THF (20 mL) was added followed by stirring at −78 °C for 30 min. To this mixture, 1-chloro-2-fluoro-3-nitrobenzene (2) (948 mg, 5.405 mmol) dissolved in THF (10 mL) was added followed by stirring at −78 °C for 1 h. After completion of the reaction, the mixture was diluted with a sat. NH4Cl solution (50 mL) and water (50 mL), extracted with EtOAC (2 × 50 mL)-combined organic layers, dried over anhydrous sodium sulphate, filtered, and then concentrated under vacuum. The crude product was purified using a 100–200 silica gel column and then eluted with a gradient of 10% ethyl acetate in pet. ether to afford (1.45 g, 78%) a light orange solid.MR: 117–119 °C; 1H NMR (400 MHz, CDCl3): δ ppm 3.95 (3H, s, COOCH3), 6.67 (1H, d, J = 8.11 Hz, Ar–H), 7.08–7.29 (2H, m, Ar–H), 7.39 (1H, dd, J = 7.67, 1.10 Hz, Ar–H), 7.69 (1H, dd, J = 7.89, 1.10 Hz, Ar–H), 8.02 (1H, dd, J = 8.33, 1.32 Hz, Ar–H), 8.09 (1H, s, NH); 13C NMR (101 MHz, CDCl3): δ ppm 52.4 (OCH3), 119.7, 122.2, 123.6, 123.8, 124.7, 126.2, 130.5, 131.4, 134.2, 136.0, 139.3, 143.9 (Ar–C), 166.2 (COOMe); LCMS (ES): m/z 338.98 (M − H)+; FT-IR (KBr) νmax 3363 (NH), 3098, 1721 (CO), 1577, 1492, 1339, 1280 (Ar–Cl), 746 cm−1.
Methyl 2-chloro-3-((2-chloro-6-nitrophenyl)(methyl)amino)benzoate (4). To a stirred solution of methyl 2-chloro-3-((2-chloro-6-nitrophenyl)amino)benzoate (3) (1.4 g, 4.10 mmol) in DMF (30 mL), KOH pellets (919 mg, 16.42 mmol) were added followed by stirring at room temperature for 1 h. To this, methyl iodide (1.15 g, 8.2 mmol) was added followed by stirring at room temperature for 16 h. The reaction mixture was poured in ice water (100 mL), and a solid was formed, which was filtered under vacuum and dried to afford (1.35 g, 90%) a light yellow solid.MR: 122–124 °C; 1H NMR (400 MHz, CDCl3): δ ppm 3.38 (3H, s, N–CH3), 3.85 (3H, s, COOCH3), 7.20–7.36 (4H, m, Ar–H), 7.56 (1H, dd, J = 8.11, 1.32 Hz, Ar–H), 7.66 (1H, dd, J = 8.00, 1.43 Hz, Ar–H); 13C NMR (101 MHz, CDCl3): δ ppm 41.0 (N–CH3), 52.3 (COOCH3), 123.5, 124.1, 124.4, 124.5, 126.0, 126.7, 133.1, 134.6, 135.8, 139.7, 145.0, 149.0 (Ar–H), 166.6 (COOCH3); LCMS (ES): m/z 355.07 (M + H)+; FT-IR (KBr) νmax 3433 (NH), 2954, 1727 (CO), 1437, 1276.9 (Ar–Cl), 1046, 766 cm−1.
Methyl 3-((2-amino-6-chlorophenyl)(methyl)amino)-2-chlorobenzoate (5). To a stirred solution of methyl 2-chloro-3-((2-chloro-6-nitrophenyl)(methyl)amino)benzoate (4) (1.3 g, 3.66 mmol) in DMF (20 mL), Fe powder (1.0 g, 18.3 mmol) was added followed by NH4Cl (970 mg, 18.3 mmol), and then, the reaction mixture was heated to 90 °C for 5 h. The reaction mixture was then cooled down to room temperature, diluted with EtOAC (80 mL), passed through a Celite bed and washed with EtOAC (10 mL). The filtrate was washed with water (3 × 100 mL) and brine (100 mL), dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude material was purified by silica gel column chromatography using a 10–15% gradient of EtOAC in petroleum ether to afford 5 (800 mg, 67%) as an off-white solid and the cyclized compound 6 (100 mg, 10%) as a fluorescent yellow solid.MR: 122–124 °C; 1H NMR (400 MHz, CDCl3): δ ppm 3.24 (3H, s, N–CH3), 3.87 (3H, s, COOCH3), 4.18 (2H, br s, Ar–NH2), 6.67 (2H, m, Ar–H), 6.92–6.98 (1H, dd, J = 12.72, 8.11, 1.10 Hz, Ar–H), 7.23–7.31 (3H, m, Ar–H); 13C NMR (101 MHz, CDCl3): δ ppm 39.5 (N–CH3), 52.3 (COOCH3), 114.1, 119.7, 122.6, 123.0 (2C), 126.7, 127.8, 131.0, 133.2, 133.5, 146.0, 147.8 (Ar–C), 167.2 (COOCH3); LCMS (ES): m/z 325.08 (M)+; FT-IR (KBr) νmax 3447 (NH), 3358 (NH2), 2899, 1720 (COOCH3), 1257 (Ar–Cl), 1047, 758 cm−1.
Methyl 6-chloro-5-methyl-5,10-dihydrophenazine-1-carboxylate (6). An argon-purged reaction tube was charged with Johnphos (485 mg, 1.63 mmol), Pd2(dba)3 (745 mg, 0.815 mmol), and dry toluene (25 mL) and purged with argon for 10 min. To this, methyl 3-((2-amino-6-chlorophenyl)(methyl)amino)-2-chlorobenzoate (5) (2.65 g, 8.15 mmol) and Cs2CO3 (5.31 g, 16.3 mmol) were added under an inert atmosphere. The reaction mixture was heated to 120 °C and stirred at the same temperature in a sealed tube for 40 h. The reaction mixture was then cooled down to room temperature, diluted with EtOAC (100 mL), passed through a Celite bed and washed again with EtOAC (10 mL). The filtrate was concentrated under reduced pressure, and the obtained residue was purified by methanol (20 mL) washing, filtered and dried to afford 6 (2.1 g, 79%) as a florescent yellow solid.MR: 72–74 °C; 1H NMR (400 MHz, DMSO-d6): δ ppm 3.24 (3H, s, N–CH3), 3.83 (3H, s, COOCH3), 6.60–6.87 (5H, m, Ar–H), 7.27 (1H, br d, J = 7.89 Hz, Ar–H), 9.09 (1H, s, NH); 13C NMR (101 MHz, DMSO-d6) δ ppm 45.2 (N–CH3), 52.3 (COOCH3), 109.8, 113.1, 113.3, 120.3, 122.8, 124.0, 124.3, 124.8, 132.7, 138.7, 139.2, 141.7 (Ar–C), 167.8 (COOCH3); LCMS (ES): m/z 288.23 (M)+.
6-Chloro-5-methyl-5,10-dihydrophenazine-1-carboxylic acid (7). To a stirred solution of methyl 6-chloro-5-methyl-5,10-dihydrophenazine-1-carboxylate (6) (500 mg, 1.73 mmol) in ethanol–THF (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; 15 mL), 4 N aqueous NaOH solution (173 mg, 4.34 mmol) was added followed by stirring at room temperature for 5 h. The reaction mixture was concentrated under vacuum, diluted with water (20 mL), and adjusted to pH ∼ 5 with a 4 N aqueous HCl solution (2 mL) to obtain a solid, which was filtered and dried. Again the crude was triturated with pentane (15 mL) decant and dried to afford the acid 7 (350 mg, 73%) as a yellowish brown solid.
:1; 15 mL), 4 N aqueous NaOH solution (173 mg, 4.34 mmol) was added followed by stirring at room temperature for 5 h. The reaction mixture was concentrated under vacuum, diluted with water (20 mL), and adjusted to pH ∼ 5 with a 4 N aqueous HCl solution (2 mL) to obtain a solid, which was filtered and dried. Again the crude was triturated with pentane (15 mL) decant and dried to afford the acid 7 (350 mg, 73%) as a yellowish brown solid.MR: 210–212 °C; 1H NMR (400 MHz, DMSO-d6): δ ppm 3.24 (3H, s, N–CH3), 6.54–6.88 (5H, m, Ar–H), 7.28 (1H, dd, J = 8.11, 1.32 Hz, Ar–H), 9.32 (1H, s, Ar–NH), 13.08 (1H, br s, COOH); 13C NMR (101 MHz, DMSO-d6): δ ppm 45.8 (N–CH3), 110.7, 113.0, 122.5, 122.9, 122.9, 123.8, 124.8 (2C), 132.7, 138.6, 139.4, 141.9 (Ar–C), 169.5 (COOH); LCMS (ES): m/z 274.07 (M)+.
General procedure for amide coupling. To a stirred solution of 6-chloro-5-methyl-5,10-dihydro phenazine-1-carboxylic acid 7 (70 mg, 0.25 mmol) in DMF (15 mL), amines (0.379 mmol), HATU (292 mg, 0.5 mmol) and diisopropylethylamine (0.75 mmol) were added at room temperature followed by stirring for 1 h. The reaction mixture was diluted with EtOAC (80 mL) and washed with cold water (2 × 50 mL) and brine (50 mL); then, the organic layer was dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude material was purified by silica gel column chromatography using a 10–40% gradient of EtOAC in petroleum ether to afford the required compound in a yield of 60–90%.
6-Chloro-N-(3-methoxyphenyl)-5-methyl-5,10-dihydrophenazine-1-carboxamide (8a). Appearance: greenish yellow solid; yield: 69%; MR: 147–149 °C; 1H NMR (400 MHz, DMSO-d6): δ ppm 3.25 (3H, s, N–CH3), 3.76 (3H, s, O–CH3), 6.61 (1H, dd, J = 7.13, 1.86 Hz, Ar–H), 6.66–6.84 (5H, m, Ar–H), 7.21–7.33 (3H, m, Ar–H), 7.36–7.39 (1H, m, Ar–H), 9.21 (1H, s, NH), 10.12 (1H, s, CONH); 13C NMR (101 MHz, DMSO-d6): δ ppm 45.3 (N–CH3), 55.0 (OCH3), 106.7, 109.3, 112.4, 113.1, 114.9, 119.6, 121.0, 122.3, 122.5, 122.8, 124.3, 129.2, 132.2, 138.3, 139.6 (2C), 139.7 (Ar–C), 159.3 (Ar–C–OCH3), 166.6 (CONH); LCMS (ES): m/z 379.12 (M)+; FT-IR (KBr) νmax 3356 (NH), 3240 (CONH), 2928, 1638 (CO), 1481, 1273 (Ar–Cl), 717 cm−1.
6-Chloro-5-methyl-N-(3-(trifluoromethyl)phenyl)-5,10-dihydrophenazine-1-carboxamide (8b). Appearance: light green solid; yield: 72%; MR: 187–189 °C; 1H NMR (400 MHz, DMSO-d6): δ ppm 3.25 (3H, s, NCH3) 6.61–6.67 (1H, m, Ar–H), 6.69–6.87 (4H, m, Ar–H), 7.32 (1H, d, J = 7.89 Hz, Ar–H), 7.46 (1H, br d, J = 7.67 Hz, Ar–H) 7.60 (1H, t, J = 8.00 Hz, Ar–H) 7.96 (1H, br d, J = 8.11 Hz, Ar–H) 8.20 (1H, s, ArH) 9.14 (1H, s, NH) 10.44 (1H, s, CONH); 13C NMR (101 MHz, DMSO-d6): δ ppm 45.3 (N–CH3), 112.6, 114.5, 116.8, 116.9, 119.7, 120.0, 121.2, 122.3, 122.4, 122.7, 124.3, 125.4, 129.0, 129.3, 129.6, 132.2, 138.3, 139.4, 139.6 (Ar–C), 166.9 (CONH); LCMS (ES): m/z 417.21 (M)+; FT-IR (KBr) νmax: 3361 (NH), 3282 (CONH), 2923, 1635 (CO), 1447, 1249 (Ar–Cl), 768 cm−1.
6-Chloro-N-(4-fluoro-1H-pyrazol-5-yl)-5-methyl-5,10-dihydrophenazine-1-carboxamide (8c). Appearance: light orange solid; yield: 69%; MR: 229–231 °C; 1H NMR (400 MHz, DMSO-d6): δ ppm 3.24 (3H, s, N–CH3), 5.97 (2H, s, Ar–H), 6.55–6.82 (5H, m, Ar–H), 7.08 (1H, dd, J = 8.00, 1.43 Hz, Ar–H), 8.14 (1H, s, CONH), 8.28 (1H, s, NH); 13C NMR (101 MHz, DMSO-d6): δ ppm 45.3 (N–CH3), 112.5, 114.8, 115.3, 119.2, 120.9, 122.7, 124.2, 125.2, 132.3, 137.8, 138.4, 139.9, 141.2, 143.8, 148.6 (Ar–C), 164.4 (CONH); LCMS (ES): m/z 358.16 (M + H)+; FT-IR (KBr) νmax 3410 (NH), 2919, 1665 (CONH), 1378, 1277 (Ar–Cl), 744 cm−1.
6-Chloro-5-methyl-N-(3,4,5-trimethoxyphenyl)-5,10-dihydrophenazine-1-carboxamide (8d). Appearance: light yellow solid; yield: 75%; MR: 235–237 °C; 1H NMR (400 MHz, DMSO-d6): δ ppm 3.25 (3H, s, N–CH3), 3.65 (3H, s, OCH3), 3.77 (6H, s, OCH3), 6.61 (1H, dd, J = 7.13, 1.86 Hz, Ar–H), 6.66–6.88 (4H, m, Ar–H), 7.13 (2H, s, Ar–H) 7.22–7.35 (1H, m, Ar–H), 9.22 (1H, s, NH), 10.04 (1H, s, CONH); 13C NMR (101 MHz, DMSO-d6): δ ppm 45.2 (N–CH3), 55.7 (2C, OCH3), 60.7 (OCH3), 98.8, 112.4, 114.9, 119.6, 121.0, 122.2, 122.5 (2C), 122.8, 124.3, 132.2, 134.0, 134.6, 138.3, 139.5 (Ar–C), 139.7 (Ar–C–OCH3), 152.5 (2C, Ar–C–OCH3), 166.4 (CONH); LCMS (ES): m/z 440.21 (M + H)+; FT-IR (KBr) νmax 3392 (NH), 2938, 1641 (CONH), 1449, 1231 (Ar–Cl), 763 cm−1; HR-MS (ESI): m/z calcd for C23H22ClN3O4 ([M + H]+): 440.1377 found: 440.1372.
6-Chloro-N-cycloheptyl-5-methyl-5,10-dihydrophenazine-1-carboxamide (8e). Appearance: light green solid; yield: 89%; MR: 216–218 °C; 1H NMR (400 MHz, DMSO-d6): δ ppm 1.35–1.71 (10H, m, CH2), 1.78–1.87 (2H, m, CH2), 3.21 (3H, s, N–CH3), 3.90–3.99 (1H, m, CONH–CH), 6.53 (1H, dd, J = 6.25, 2.96 Hz, Ar–H), 6.64–6.79 (4H, m, Ar–H), 7.20 (1H, d, J = 7.67 Hz, Ar–H), 8.18–8.24 (1H, m, CONH), 9.73 (1H, s, NH); 13C NMR (101 MHz, DMSO-d6): δ ppm 23.9 (2C, CH2) 27.6 (2C, CH2) 34.1 (2C, CH2) 45.2 (NCH3) 50.2 (CONH–CH) 112.2, 114.2, 119.4, 120.7, 121.9, 122.4, 122.6, 124.4, 132.1, 138.1, 139.7, 139.8 (Ar–C), 166.5 (CONH); LCMS (ES): m/z 370.39 (M + H)+; FT-IR (KBr) νmax 3406 (NH), 2936, 1630 (CO), 1474, 1276 (Ar–Cl), 741 cm−1.
6-Chloro-5-methyl-N-phenethyl-5,10-dihydrophenazine-1-carboxamide (8f). Appearance: yellowish green solid; yield: 76%; MR: 147–149 °C; 1H NMR (400 MHz, DMSO-d6): δ ppm 2.84 (2H, br t, J = 7.34 Hz, Ar–CH2), 3.22 (3H, s, NCH3), 3.42–3.51 (2H, m, CONH–CH2), 6.52 (1H, dd, J = 6.36, 2.85 Hz, Ar–H), 6.65–6.78 (4H, m, Ar–H), 7.13 (1H, br d, J = 7.23 Hz, Ar–H), 7.18–7.33 (5H, m, Ar–H), 8.55 (1H, br s, CONH), 9.74 (1H, s, NH); 13C NMR (101 MHz, DMSO-d6): δ ppm 34.9 (Ar–CH2), 40.5 (CONH–CH2), 45.2 (N–CH3), 112.2, 113.8, 119.6, 120.6, 121.5, 122.5, 122.7, 124.4, 126.0, 128.2, 128.3, 128.5, 128.9, 132.1, 133.6, 138.3, 139.3, 139.8 (Ar–C), 167.7 (CONH); LCMS (ES): m/z 378.20 (M + H)+; FT-IR (KBr) νmax 3373 (NH), 2924, 1629 (CO), 1459, 1274 (Ar–Cl), 740 cm−1; HR-MS (ESI): m/z calcd for C22H21ClN3O ([M + H]+): 378.1373 found: 378.1367.
6-Chloro-N-(2-hydroxyethyl)-5-methyl-5,10-dihydro phenazine-1-carboxamide (8g). Appearance: yellowish green solid; yield: 69%; MR: 143–145 °C; 1H NMR (400 MHz, DMSO-d6): δ ppm 3.22 (3H, s, N–CH3), 3.26–3.36 (2H, m, CONH–CH2), 3.51 (2H, q, J = 5.99 Hz, O–CH2), 4.70 (1H, t, J = 5.59 Hz, OH), 6.51 (1H, dd, J = 6.36, 2.63 Hz, Ar–H), 6.62–6.81 (4H, m, Ar–H), 7.19 (1H, d, J = 7.67 Hz, Ar–H), 8.40 (1H, br t, J = 4.93 Hz, CONH), 9.77 (1H, s, NH); 13C NMR (101 MHz, DMSO-d6): δ ppm 41.8 (CONH–CH2), 45.2 (N–CH3), 59.4 (CH2–OH), 112.1, 113.9, 119.5, 120.6, 121.6, 122.5, 122.7, 124.4, 132.1, 138.2, 139.7, 139.7 (Ar–C), 167.8 (CONH); LCMS (ES): m/z 318.13 (M + H)+; FT-IR (KBr) νmax 3330 (NH), 2922, 1622 (CONH), 1487, 1450, 1278 (Ar–Cl), 756 cm−1; HR-MS (ESI): m/z calcd for C16H16ClN3O2 ([M + H]+): 318.1009 found: 318.1005.
6-Chloro-N-(3-fluorophenyl)-5-methyl-5,10-dihydrophenazine-1-carboxamide (8h). Appearance: yellowish green solid; yield: 77%; MR: 143–145 °C; 1H NMR (400 MHz, DMSO-d6): δ ppm 3.25 (3H, s, N–CH3), 6.62 (1H, dd, J = 7.23, 1.75 Hz, Ar–H), 6.69–6.87 (3H, m, Ar–H), 6.94 (1H, td, J = 8.39, 2.30 Hz, Ar–H), 7.20–7.43 (1H, m, Ar–H), 7.27–7.31 (1H, m, Ar–H), 7.34–7.43 (1H, m, Ar–H), 7.50 (1H, d, J = 8.33 Hz, Ar–H), 7.69 (1H, dt, J = 11.73, 2.14 Hz, Ar–H), 9.14 (1H, s, NH) 10.32 (1H, s, CONH); 13C NMR (101 MHz, DMSO-d6): δ ppm 45.3 (NCH3), 107.6, 110.1, 110.3, 112.5, 114.7, 116.5, 119.7, 121.2, 122.3, 122.5, 122.9, 124.4, 130.0, 130.1, 132.2, 138.3, 139.6, 140.4 (Ar–C), 166.8 (CONH); LCMS (ES): m/z 367.22 (M)+; FT-IR (KBr) νmax 3303 (NH), 2920, 1634 (CONH), 1604, 1486, 1275 (Ar–Cl), 716 cm−1.
6-Chloro-5-methyl-N-phenyl-5,10-dihydrophenazine-1-carboxamide (8i). Appearance: light yellow solid; yield: 78%; MR: 196–198 °C; 1H NMR (400 MHz, DMSO-d6): δ ppm 3.25 (3H, s, N–CH3), 6.60 (1H, dd, J = 7.23, 1.97 Hz, Ar–H), 6.68–6.86 (4H, m, Ar–H), 7.07–7.15 (1H, m, Ar–H), 7.26–7.41 (3H, m, Ar–H), 7.65–7.75 (2H, m, Ar–H), 9.25 (1H, s, NH), 10.16 (1H, s, CONH); 13C NMR (101 MHz, DMSO-d6): δ ppm 45.3 (NCH3), 112.4, 114.8, 119.7, 120.9, 121.1, 122.3, 122.5, 122.8, 123.8, 124.4, 128.4 (3C), 132.2, 138.3, 138.5, 139.6, 139.7 (Ar–C), 166.6 (CONH); LCMS (ES): m/z 350.05 (M + H)+; FT-IR (KBr) νmax 3338 (NH), 3050, 1631 (CONH), 1480, 1270 (Ar–Cl), 760 cm−1; HR-MS (ESI): m/z calcd for C20H16ClN3O ([M − H]+): 348.0904 found: 348.0914.
(6-Chloro-5-methyl-5,10-dihydrophenazin-1-yl)(piperidin-1-yl)methanone (8j). Appearance: light green gummy; yield: 84%; 1H NMR (400 MHz, DMSO-d6): δ ppm 1.43–1.67 (6H, m, CH2), 3.21 (3H, s, N–CH3) 3.32–3.61 (4H, m, N–CH2), 6.59 (1H, dd, J = 5.92, 3.07 Hz, Ar–H), 6.66–6.75 (5H, m, Ar–H), 7.59 (1H, s, NH); 13C NMR (101 MHz, DMSO-d6): δ ppm 23.9 (CH2), 25.3 (2C, CH2), 28.9 (2C, CONH–CH2), 45.2 (NCH3), 112.2, 119.1, 119.7, 120.4, 121.4, 122.2, 122.6, 124.2, 132.2, 134.8, 137.8, 140.8 (Ar–C), 166.4 (CONH); LCMS (ES): m/z 342.06 (M + H)+.
(6-Chloro-5-methyl-5,10-dihydrophenazin-1-yl)(morpholino)methanone (8k). Appearance: light green gummy; yield: 82%; 1H NMR (400 MHz, DMSO-d6): δ ppm 3.22 (3H, s, N–CH3), 3.35–3.45 (4H, m, N–CH2), 3.48–3.70 (4H, m, O–CH2), 6.59–6.64 (1H, m, Ar–H), 6.65–6.76 (5H, m, Ar–H) 7.70 (1H, s, NH); 13C NMR (101 MHz, DMSO-d6): δ ppm 28.8 (2C, CONH–CH2) 45.2 (N–CH3), 65.9 (2C, O–CH2), 112.1, 118.9, 119.3, 120.5, 121.7, 122.3, 122.6, 124.2, 132.2, 135.0, 137.9, 140.7 (Ar–C), 166.7 (CONH); LCMS (ES): m/z 344.06 (M + H)+.
6-Chloro-5-methyl-N-(pyridin-3-yl)-5,10-dihydrophenazine-1-carboxamide (8l). Appearance: dark yellow solid; yield: 77%; MR: 204–206 °C; 1H NMR (400 MHz, DMSO-d6): δ ppm 3.23–3.27 (3H, m, N–CH3), 6.61 (1H, dd, J = 7.23, 1.97 Hz, Ar–H), 6.67–6.88 (4H, m, Ar–H), 7.28–7.35 (1H, m, Ar–H), 7.37–7.44 (1H, m, Ar–H), 8.11 (1H, ddd, J = 8.33, 2.63, 1.53 Hz, Ar–H), 8.32 (1H, dd, J = 4.71, 1.42 Hz, Ar–H), 8.87 (1H, d, J = 2.19 Hz, Ar–H), 9.20 (1H, s, NH), 10.34 (1H, s, CONH); 13C NMR (101 MHz, DMSO-d6): δ ppm 45.3 (N–CH3), 112.5, 114.3, 119.7, 121.2, 122.3, 122.5, 122.9, 123.3, 124.3, 128.0, 132.2, 135.2, 138.3, 139.5, 139.7, 142.5, 144.7 (Ar–C), 167.0 (CONH); LCMS (ES): m/z 351.01 (M + H)+; FT-IR (KBr) νmax 3448 (NH), 2923, 1650 (CONH), 1472, 1278 (Ar–Cl), 708 cm−1.
6-Chloro-N-(4-chloro-3-methoxyphenyl)-5-methyl-5,10-dihydrophenazine-1-carboxamide (8m). Appearance: dark yellow solid; yield: 82%; MR: 166–168 °C; 1H NMR (400 MHz, DMSO-d6): δ ppm 3.25 (3H, s, N–CH3), 3.85 (3H, s, O–CH3), 6.61 (1H, dd, J = 7.23, 1.75 Hz, Ar–H), 6.69–6.85 (4H, m, Ar–H), 7.29 (1H, dd, J = 7.89, 1.10 Hz, Ar–H), 7.38 (2H, m, Ar–H), 7.57 (1H, s, Ar–H), 9.18 (1H, s, NH), 10.24 (1H, s, CONH); 13C NMR (101 MHz, DMSO-d6): δ ppm 45.2 (N–CH3), 55.8 (O–CH3), 105.5, 112.5, 113.5, 114.7, 115.5, 119.6, 121.1, 122.2, 122.5, 122.9, 124.3, 129.4, 132.2, 138.3, 138.8, 139.6 (2C) (Ar–C), 154.2 (Ar–C–OCH3), 166.6 (CONH); LCMS (ES): m/z 413.03 (M)+; FT-IR (KBr) νmax 3331, 2923, 1633, 1594, 1489, 1277 (Ar–Cl), 744 cm−1.
6-Chloro-5-methyl-5,10-dihydrophenazine-1-carboxamide (8n). Appearance: yellow solid; yield: 65%; MR: 192–194 °C; 1H NMR (400 MHz, DMSO-d6): δ ppm 3.22 (3H, s, NCH3), 6.49 (1H, dd, J = 6.58, 2.63 Hz, Ar–H), 6.55–6.83 (4H, m, Ar–H), 7.21 (1H, dd, J = 8.00, 1.21 Hz, Ar–H), 7.40 (1H, br s, CONH), 7.97 (1H, br s, CONH), 10.01 (1H, s, NH); 13C NMR (101 MHz, DMSO-d6): δ ppm 45.2 (N–CH3), 112.1, 113.1, 119.4, 120.8, 122.3, 122.5, 122.7, 124.4, 132.1, 138.2, 139.7, 140.3 (Ar–C), 170.2 (CONH2); LCMS (ES): m/z 273.09 (M + H)+; FT-IR (KBr) νmax 3460 (NH2), 3326 (NH), 2923, 1637 (CONH), 1615, 1456, 1276 (Ar–Cl), 756 cm−1; HR-MS (ESI): m/z calcd for C14H12ClN3O ([M + H]+): 274.0747 found: 274.0742.
Biology
Cell and culture conditions. The MCF-7 (breast cancer), A-549 (lung cancer) and DU145 (prostate cancer) cell lines (ATCC) were maintained as a continuous culture in the Dulbecco's Modified Eagle's Medium (DMEM; Sigma-Aldrich, Inc.), supplemented with 10% fetal bovine serum (FBS; Himedia, Mumbai, India), 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin. The cells were grown at 37 °C under the humidified atmosphere of 5% CO2.
In vitro anticancer activity assessment. The cells were maintained in DMEM supplemented with 10% FBS under the humidified atmosphere of 5% CO2 at 37 °C. Then, the cells were seeded in 96-well plates at the density of 10000 cells per well and incubated for 24 h to allow cell attachment. After this, the cells were treated with test compounds at 0.1, 1, 10, 100 μM concentration (all the test compounds were tested below their cytotoxic (CTC50) concentrations (CTC50 > 200 μM) obtained against Vero cell lines). After 24 h treatment and incubation period, fresh DMEM containing MTT (250 μg mL−1) was added to replace the medium, and again, the cells were incubated for an additional 3 h. MTT was aspirated, and DMSO was added to dissolve the formazan crystals. Absorbance was measured at 570 nm using a micro plate reader (Infinite® 200 Pro-Tecan, Switzerland).
Effect on cell cycle. The distribution of cells at different phases of the cell cycle was analysed using flow cytometry. Briefly, the cells were seeded in 6-well plates at the density of 1 × 105 cells per well and incubated for 24 h. Cells were then treated with 8a, 8l and nocodazole (5 μM) for 48 h to determine their effect on the cell cycle. After treatment, the cells were washed with PBS, harvested using trypsin EDTA, fixed in ice cold ethanol (70%) and stored overnight at 2–8 °C. The fixed cells were pelleted, resuspended and treated with a staining solution containing propidium iodide, RNAse and Triton X for 30 min at 37 °C to stain the nuclei and get rid of RNA. Herein, 10000 events were analyzed after doublet discrimination module using a flow cytometer (DAKO CYTOMATION, Beckman Coulter, Brea, CA). The Summit Software was used to analyse the histogram.
Western blotting. RIPA buffer (comprising 20 mM NaH2PO4, 50 mM NaF, 2 mM EDTA, 150 mM NaCl, 1% deoxycholic acid, and 0.1% SDS, pH 7.2) was used to prepare whole cell lysates for Western blot analysis. Protease inhibitor cocktail (Sigma Aldrich, Mumbai) was used for the Western blot analysis. The protein (40 mg) was taken and resolved using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Antibodies against tubulin, cyclin-B1 and CDK1 were obtained from Santa cruz biotech, and horseradish peroxidase-conjugated goat anti-rabbit IgG was used as a secondary antibody. This was later detected using chemiluminescence reagents.
Tubulin polymerization inhibition assay. In vitro inhibition of tubulin polymerization was studied using a fluorescence-based tubulin polymerization assay kit (BK011, Cytoskeleton, Inc.). The procedure was according to the manufacturer's protocol. The reaction mixture in a final volume of 10 μL in PEM buffer (80 mM PIPES, 0.5 mM EGTA, 2 mM MgCl2, pH 6.9) in 384 well plates contained 2 mg mL−1 bovine brain tubulin, 10 μM fluorescent reporter, and 1 mM GTP in the presence or absence of test compounds at 37 °C. Tubulin polymerization was observed by monitoring the fluorescence enhancement due to the incorporation of a fluorescence reporter into the microtubules as the polymerization proceeded. Fluorescence emission at 420 nm (excitation wavelength of 360 nm) was measured for 1 h at 1 min intervals using a multimode plate reader (Tecan Pro M200). Nocodazole was the positive control used under similar experimental conditions. The IC50 values against tubulin polymerization were calculated by pre-incubating the test compounds with tubulin at five different concentrations.
Immunohistochemistry. Cells were seeded on a glass cover slip and incubated for 24 h in the presence or absence of test compounds (5 μM). Cells grown on cover slips were fixed in 3.5% formaldehyde in phosphate-buffered saline (PBS) (pH 7.4) for 10 minutes at room temperature. Cells were permeabilized for 6 minutes in PBS containing 0.5% Triton X-100 (Sigma) and 0.05% Tween-20 (Sigma). The permeabilized cells were blocked with 2% BSA (Sigma) in PBS for 1 h. Later, the cells were incubated with a primary antibody for tubulin obtained from Sigma at a 1:200 dilution in a blocking solution for 4 h at room temperature. Subsequently, the antibodies were removed, and the cells were washed three times with PBS. Cells were then incubated with the FITC-labeled anti-mouse secondary antibody (1:500) for 1 h at room temperature. The cells were washed three times with PBS and mounted in a medium containing DAPI. Images were obtained using the Olympus confocal microscope.
Conclusions
In conclusion, a set of novel dermacozine-1-carboxamides (8a–8n) was synthesized and well characterized by 1H NMR, 13C NMR, FT-IR, mass and HR-MS analysis. The synthesized compounds were subjected to in vitro cytotoxic activity tests against the selected cancer cell lines MCF7, A549 and DU145 by the MTT assay. The dermacozine derivatives 8a and 8l showed significant anticancer potential comparable to the standard drug paclitaxel, especially against the DU-145 cells. Mechanistic investigations showed that these molecules arrested the cell cycle at the G2/M phase by affecting Cdk1 and cyclin-B1. It was also found that these compounds effectively inhibited tubulin polymerization. Hence, these compounds have the potential to be anticancer agents.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are thankful to the GVK Biosciences Private Limited for providing financial support and encouragement. Support received from the analytical Department GVK Biosciences Private Limited and biological support received from the Department of Pharmacology, JSS College of Pharmacy, are acknowledged. S. K. S. S. Pindiprolu thanks the Indian Council of Medical Research (ICMR), New Delhi, for providing Senior Research Fellowship.
References
- L. A. Torre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortet-Tieulent and A. Jemal, Global cancer statistics, 2012, Ca-Cancer J. Clin., 2015, 65(2), 87–108 CrossRef.
- F. Bray, A. Jemal, N. Grey, J. Ferlay and D. Forman, Global cancer transitions according to the human development index (2008–2030): a population-based study, Lancet Oncol., 2012, 13(8), 790–801 CrossRef PubMed.
- F. Biemar and M. Foti, Global progress against cancer—challenges and opportunities, Cancer Biol. Med., 2013, 10(4), 183 Search PubMed.
- S. Vinogradov and X. Wei, Cancer stem cells and drug resistance: the potential of nanomedicine, Nanomedicine, 2012, 7(4), 597–615 CrossRef CAS.
- N. HariNarayanaMoorthy, C. Karthikeyan and P. Trivedi, Synthesis, cytotoxic evaluation and in silico pharmacokinetic prediction of some benzo [a] phenazine-5-sulfonic acid derivatives, Med. Chem., 2009, 5(6), 549–557 CrossRef CAS.
- J. B. Laursen and J. Nielsen, Phenazine natural products: biosynthesis, synthetic analogues, and biological activity, Chem. Rev., 2004, 104(3), 1663–1686 CrossRef CAS PubMed.
- N. HariNarayanaMoorthy, V. Pratheepa, M. J Ramos, V. Vasconcelos and P. A Fernandes, Fused aryl-phenazines: scaffold for the development of bioactive molecules, Curr. Drug Targets, 2014, 15(7), 681–688 CrossRef CAS.
- W. M. Abdel-Mageed, B. F. Milne, M. Wagner, M. Schumacher, P. Sandor, W. Pathom-aree, M. Goodfellow, A. T. Bull, K. Horikoshi and R. Ebel, Dermacozines, a new phenazine family from deep-sea dermacocci isolated from a Mariana Trench sediment, Org. Biomol. Chem., 2010, 8(10), 2352–2362 RSC.
- M. Schumacher, M. Kelkel, M. Dicato and M. Diederich, A survey of marine natural compounds and their derivatives with anti-cancer activity reported in 2010, Molecules, 2011, 16(7), 5629–5646 CrossRef CAS PubMed.
- V. R. Ghanta, A. Pasula, L. Soma and B. Raman, Synthetic studies on dermacozines: first synthesis of dermacozines A, B and C, ChemistrySelect, 2016, 1(6), 1296–1299 CrossRef CAS.
- R. Heald, S. Price, B. Safina, P. P. A. Savy, E. M. Seward, D. P. Sutherlin and B. Waszkowycz, US Pat. US20120202785 A1, 2012.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02416f |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Kumara Swamy Battulad,
Praveen T. Krishnamurthy
c,
Kumara Swamy Battulad,
Praveen T. Krishnamurthy