
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIodine-mediated synthesis of benzo[a]fluorenones from yne-enones†
Sikkandarkani Akbar,
V. John Tamilarasan and
Kannupal Srinivasan *
*
School of Chemistry, Bharathidasan University, Tiruchirappalli-620024, India. E-mail: srinivasank@bdu.ac.in; Fax: +91-431-2407043; Tel: +91-431-2407053
First published on 30th July 2019
Abstract
The chalcones derived from o-alkynylacetophenones and aromatic aldehydes (yne-enones) when heated under reflux with iodine in acetic acid gave a range of benzo[a]fluorenone derivatives in moderate to good yields. The transformation involves the formation of a vinyl indenone intermediate through regioselective alkyne hydration and intramolecular aldol condensation followed by electrocyclic ring closure and aromatization.
Introduction
The benzo[a]fluorenone core is present in the fluostatin family of natural products which possess potential antibiotic and cytotoxic properties (Fig. 1).1 In addition, few benzo[a]fluorene derivatives serve as active materials in the fabrication of optoelectronic devices.2 Despite their importance in the fields of medicine and materials science, only a handful of methods are available to access benzo[a]fluorene derivatives, especially for benzo[a]fluorenones.3 The recent methods available for the synthesis of benzo[a]fluorenones include thermal cyclization of diaryldiynones,4 Suzuki coupling of functionalized arenes followed by cyclization,5 annulation of arynes with 2-haloarenecarboxaldehydes,6 and gold-catalyzed cyclization of trityl-substituted diynes followed by oxidative photocyclization.7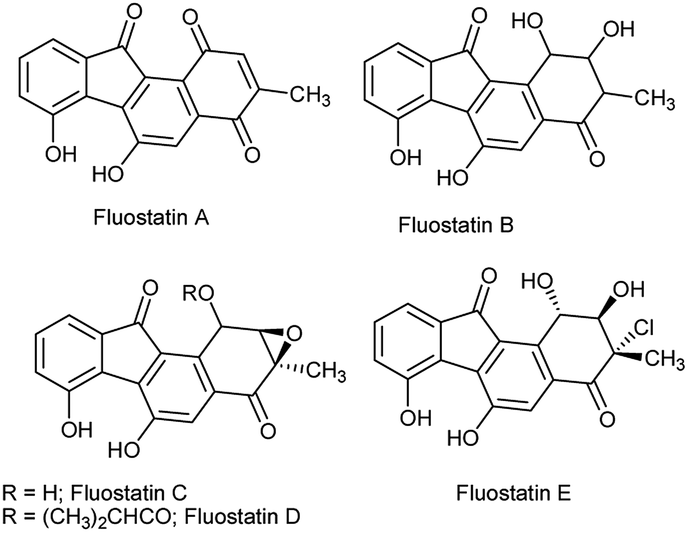 | ||
| Fig. 1 Structures of fluostatin natural products. | ||
While exploring the synthetic potentials of o-alkynylchalcones,8 we discovered that related yne-enones 1 when heated under reflux with iodine in AcOH gave benzo[a]fluorenones 2 through a cascade process (Scheme 1, eqn (1)). It is worthwhile to compare these results with Hu's work,9 in which chalcones 1 have been converted into benzo[b]fluorenols 3 by a Pd-catalyzed intramolecular dehydroaromatization and carbonyl reduction sequence (Scheme 1, eqn (2)) and also with Li's work,10 in which chalcones 1 have been transformed into 1,4-naphthoquinones 4 by a Cu-catalyzed intramolecular oxidative 6-endo-trig cyclization (Scheme 1, eqn (3)). The present work represents yet another proof of the versatility of iodine in mediating complex organic transformations.11
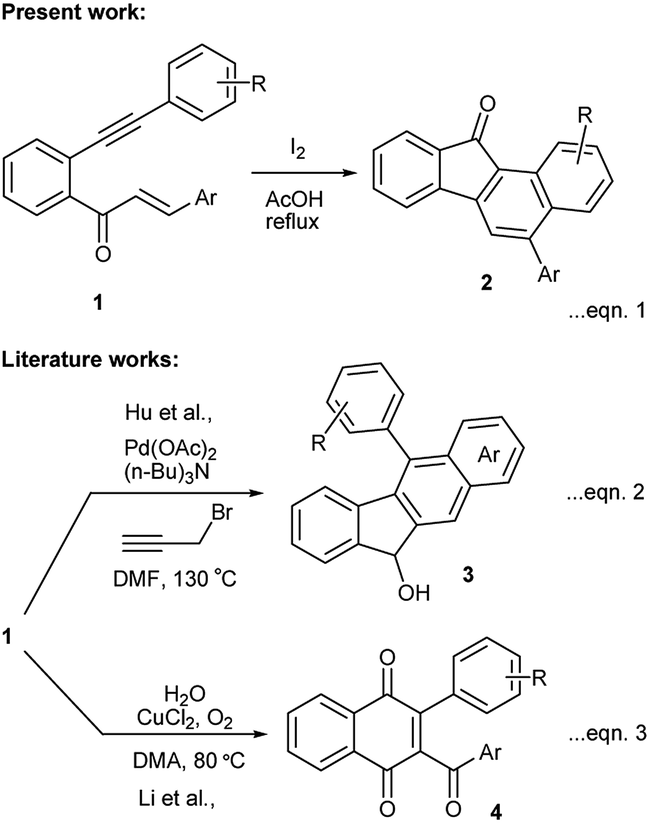 | ||
| Scheme 1 Present synthesis of benzo[a]fluorenones and comparison with literature reports. | ||
Results and discussion
The requisite starting materials 1 for the present study were prepared by the base-mediated condensation of o-alkynylacetophenones 5 (which were in turn prepared by the Sonogashira coupling of the corresponding o-haloacetophenones with terminal arylacetylenes) with aromatic aldehydes 6 (Scheme 2). | ||
| Scheme 2 Preparation of starting materials. | ||
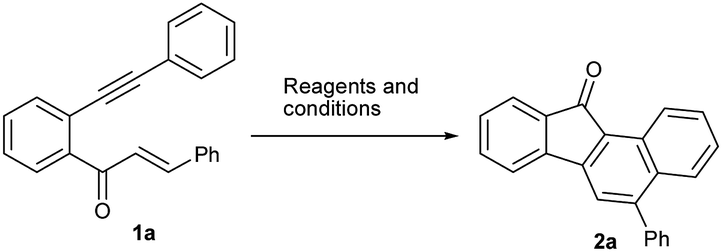
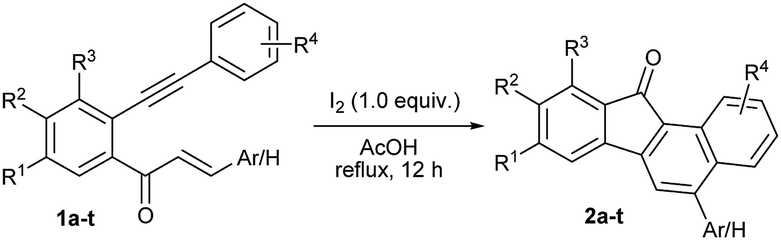
We chose the yne-enone 1a as a model substrate for optimizing the reactions conditions for the desired transformation (Table 1). When 1a was heated under reflux with one equiv. of iodine in AcOH for 12 h, benzo[a]fluorenone 2a was produced in 75% yield (entry 1) (the starting material did not undergo any change at room temperature). When substoichiometric amounts of iodine (0.5 or 0.3 equiv.) were used, the reaction did not go to completion even after 24 h and thus gave lower yields of the product (entries 2 and 3). At the same time, the use of 1.5 or 2.0 equiv. of iodine did not alter the yield of the reaction or reaction time noticeably (entries 4 and 5). The reaction did not work when EtOH, 1,2-dichloroethane (1,2-DCE) or THF was used as the solvent (entries 6–8). Likewise, the reaction did not take place when N-iodosuccinimide (NIS) or ICl is employed instead of iodine (entries 9 and 10). We also screened the suitability of the hypervalent iodine reagents, namely PhI(OAc)2 and 2-iodoxybenzoic acid (IBX) for the reaction, but they gave poor yields of the product (entries 11 and 12). Thus, we identified refluxing the substrate with one equiv. of iodine in AcOH as the optimal conditions for the reaction.
|
||
|---|---|---|
| Entry | Reagents (equiv.) and conditionsa | Yield of 2ab (%) |
| a No reaction takes place at room temperature.b Isolated yield.c No reaction. | ||
| 1 | I2 (1.0), AcOH, reflux, 12 h | 75 |
| 2 | I2 (0.5), AcOH, reflux, 24 h | 33 |
| 3 | I2 (0.3), AcOH, reflux, 24 h | 10 |
| 4 | I2 (1.5), AcOH, reflux, 12 h | 75 |
| 5 | I2 (2.0), AcOH, reflux, 12 h | 73 |
| 6 | I2 (1.0), EtOH, reflux, 24 h | NRc |
| 7 | I2 (1.0), 1,2-DCE, reflux, 24 h | NRc |
| 8 | I2 (1.0), THF, reflux, 24 h | NRc |
| 9 | NIS (1.0), AcOH, reflux, 24 h | NRc |
| 10 | ICl (1.0), AcOH, reflux, 24 h | NRc |
| 11 | PhI(OAc)2 (1.0), AcOH, reflux, 24 h | 28 |
| 12 | IBX (1.0), AcOH, reflux, 24 h | 15 |
Next, we focused attention on exploring the scope of the reaction for various yne-enones (Table 2). We first examined substrates having different aryl rings (Ar) bonded to the enone unit. The reaction tolerated the presence of phenyl rings substituted with neutral, electron donating, halogen and electron withdrawing groups, bulky naphthyl ring and Heteroaromatic furyl, pyrrolyl and thienyl rings in the position and furnished the respective benzo[a]fluorenones 2a–k in 50–80% yields (entries 1–11). The yield was low (50%) for the substrate containing the p-nitrophenyl ring due to the formation of an indenone byproduct (intermediate) in 28% yield (discussed later) (entry 6). The attachment of an aryl group to the enonoe unit is not necessary for the success of the reaction as revealed by the formation of benzo[a]fluorenone 2l in 55% yield from a substrate in which Ar = H (entry 12). Next, we tested substrates containing different aryl rings attached to the alkyne unit. Though the reaction tolerated m-tolyl, p-tolyl, m-anisyl and p-chlorophenyl rings in the position and gave the respective products 2m–o and 2q in reasonable to good yields (entries 13–15 and 17), it gave a complicated mixture in the case of p-anisyl ring containing substrate (entry 16). The reason might be due to the oxidation of the p-anisyl ring to a quinone moiety under the reaction conditions. Finally, we investigated substrates having one or two bromo substituents on the main aryl ring and they too gave the expected benzo[a]fluorenones 2r and 2s, in 70 and 61% yields, respectively (entries 18 and 19). For one of the products 2b, the structure was unequivocally confirmed by X-ray analysis (Fig. 2).12
|
|||
|---|---|---|---|
| Entry | R1, R2, R3, R4, Ar/H | Time (h) | Yieldb (%) |
| a The reaction was conducted with 1 (0.25 mmol), I2 (1.0 equiv.) and solvent (5 mL).b Isolated yield.c Complicated mixture.d Contains ca. 7% inseparable impurity. | |||
| 1 | H, H, H, H, Ph (1a) | 12 | 75 (2a) |
| 2 | H, H, H, H, 4-MeC6H4 (1b) | 11 | 68 (2b) |
| 3 | H, H, H, H, 4-MeOC6H4 (1c) | 9 | 70 (2c) |
| 4 | H, H, H, H, 4-N(Me)2-C6H4 (1d) | 12 | 65 (2d) |
| 5 | H, H, H, H, 4-BrC6H4 (1e) | 12 | 80 (2e) |
| 6 | H, H, H, H, 4-NO2C6H4 (1f) | 24 | 50 (2f) |
| 7 | H, H, H, H, 1-naphthyl (1g) | 10 | 75 (2g) |
| 8 | H, H, H, H, 2-furyl (1h) | 11 | 57 (2h) |
| 9 | H, H, H, H, 2-pyrrolyl (1i) | 12 | 65 (2i) |
| 10 | H, H, H, H, 1-methyl-2-pyrrolyl (1j) | 12 | 67 (2j) |
| 11 | H, H, H, H, 2-thienyl (1k) | 11 | 60 (2k) |
| 12 | H, H, H, H, H (1l) | 9 | 55 (2l) |
| 13 | H, H, H, 3-Me, Ph (1m) | 10 | 60 (2m) |
| 14 | H, H, H, 4-Me, Ph (1n) | 12 | 73 (2n) |
| 15 | H, H, H, 3-OMe, Ph, (1o) | 12 | 62 (2o) |
| 16 | H, H, H, 4-OMe, Ph, (1p) | 12 | —c (2p) |
| 17 | H, H, H, 4-Cl, Ph (1q) | 12 | 74 (2q) |
| 18 | Br, H, H, H, Ph (1r) | 12 | 70 (2r) |
| 19 | Br, H, Br, H, Ph (1s) | 12 | 61 (2s)d |
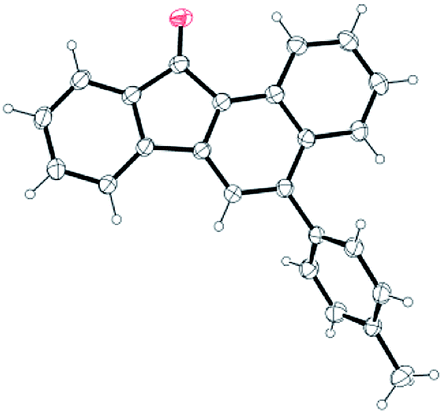 | ||
| Fig. 2 X-ray structure of 2b (30% probability level). | ||
To probe the mechanism of the transformation, we conducted few control experiments (Scheme 3). When the reaction of 1a was carried out under the specified conditions without iodine, the starting material did not undergo any change. Hence iodine is essential for the transformation. When 1a was treated with silver(I) triflate (15 mol%) in the presence of water in acetonitrile,13 it underwent regioselective triple bond hydration to give enone 7, which when heated under reflux with iodine in AcOH afforded benzo[a]fluorenone 2a. Under standard reaction conditions, 1f (a special case) gave a mixture of 2f (50%) and the indenone intermediate 8 (28%). The isolated intermediate 8 was slowly converted into 2f in 55% yield when heated under reflux in AcOH for 24 h. All these reactions clearly indicate that an indenone intermediate formed by regioselective alkyne hydration and intramolecular aldol condensation is involved in the transformation.14 To obtain evidence for the subsequent steps, we stopped the reaction of 1s before completion (in 8 h) and analyzed. The incomplete reaction gave a mixture of 2s (15%) and the corresponding non aromatized product 9 (70%). This indicates that the corresponding indenone intermediate should have undergone 6π electrocyclic ring closure and double bond migration to give 9. The aromatization of 9 to 2s could be effected using DDQ or iodine/AcOH.
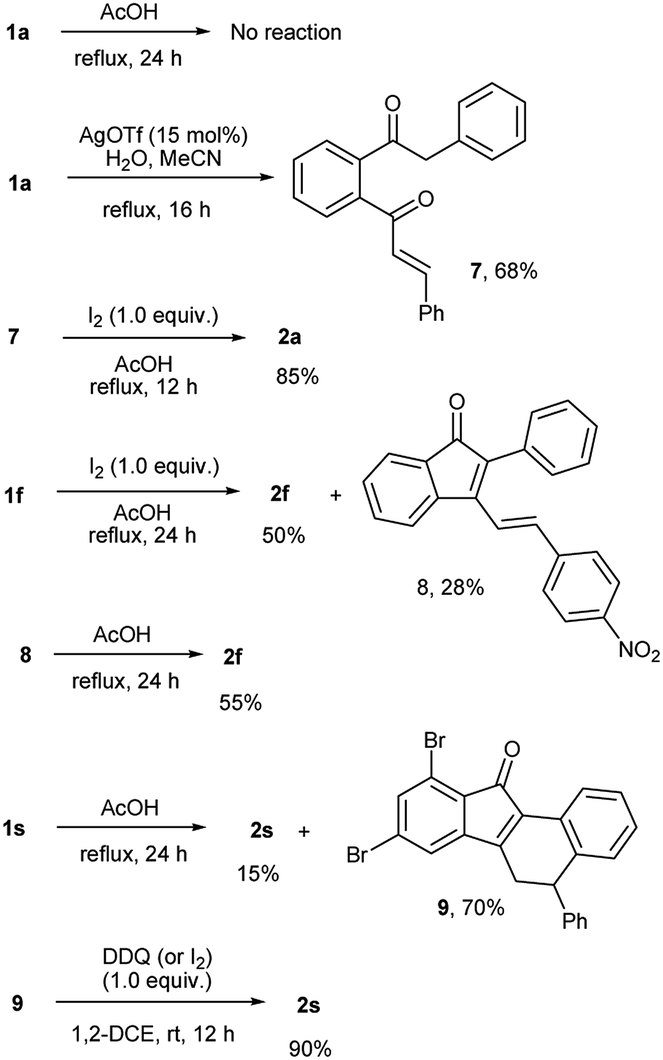 | ||
| Scheme 3 Control experiments. | ||
The above observations indicate that the transformation proceeds through the formation of an indenone intermediate followed by electrocyclic ring closure and aromatization. Accordingly, we propose a tentative mechanism outlined in Scheme 4 for the transformation. The interaction of iodine with acetic acid under refluxing conditions generates HI and HOI.15 The triple bond of yne-enone 1 is activated by HI, which triggers a 5-exo-trig nucleophilic attack by the adjacent carbonyl group to form a five-membered intermediate. Ring opening of the intermediate by a small amount of water present in AcOH (or moisture) gives enone A regioselectively. Acid (HI) promoted intramolecular aldol condensation of A leads to the indenone intermediate B. A 6π disrotatory electrocyclic ring closure of B under thermal conditions and subsequent double bond migration (re-aromatization) forms the dihydrobenzo[a]fluorenone derivative C. Dehydrogenation of C by HOI (or heat in case of 1f) finally yields benzo[a]fluorenone 2.
 | ||
| Scheme 4 Proposed mechanism of the reaction. | ||
Conclusions
In summary, we have developed a convenient, environmentally benign, metal-free procedure for the synthesis of benzo[a]fluorenone derivatives from the chalcones derived from o-alkynylacetophenones and aromatic aldehydes. The reaction proceeds through the formation of a vinyl indenone intermediate and subsequent electrocyclic ring closure and aromatization. Work is in progress to apply the present methodology for the synthesis of biologically important benzo[a]fluorenones including natural products.Experimental section
General procedure for the synthesis of benzo[a]fluorenones 2
To a solution o-alkynylarene chalcones 1 (0.25 mmol) in acetic acid (5 mL) was added iodine (0.25 mmol) and the reaction mixture was heated under reflux for 10–24 h. After completion of the reaction, the reaction mixture was cooled to room temperature and quenched with aq. sodium thiosulphate. The residue was diluted with water and extracted with ethyl acetate. The combined organic layer was washed with water, satd. sodium bicarbonate solution, dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product obtained was purified by column chromatography (SiO2; EtOAc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane, 1:99 v/v) to afford pure product 2.
:hexane, 1:99 v/v) to afford pure product 2.
5-Phenyl-benzo[a]fluoren-11-one (2a)
Red-orange solid. Yield: 75% (57 mg). Mp: 240–242 °C. IR (KBr): 1695 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). 1H NMR (400 MHz, CDCl3): δ 9.07 (d, J = 8.8 Hz, 1H), 7.80 (d, J = 8.8 Hz, 1H), 7.56–7.58 (m, 3H), 7.55–7.49 (m, 5H), 7.47–7.36 (m, 3H), 7.29 (d, J = 7.2 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ 194.2, 147.4, 144.5, 142.7, 139.2, 133.9, 133.1, 131.6, 129.8, 128.7, 128.3, 128.2, 127.5, 127.1, 125.9, 125.4, 125.0, 123.5, 122.8, 118.9, 118.4 ppm. HRMS (ESI): calcd for C23H14O: 307.1117 [M + H+]; found 307.1120.
O). 1H NMR (400 MHz, CDCl3): δ 9.07 (d, J = 8.8 Hz, 1H), 7.80 (d, J = 8.8 Hz, 1H), 7.56–7.58 (m, 3H), 7.55–7.49 (m, 5H), 7.47–7.36 (m, 3H), 7.29 (d, J = 7.2 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ 194.2, 147.4, 144.5, 142.7, 139.2, 133.9, 133.1, 131.6, 129.8, 128.7, 128.3, 128.2, 127.5, 127.1, 125.9, 125.4, 125.0, 123.5, 122.8, 118.9, 118.4 ppm. HRMS (ESI): calcd for C23H14O: 307.1117 [M + H+]; found 307.1120.
5-p-Tolyl-benzo[a]fluoren-11-one (2b)
Red-orange solid. Yield: 68% (54 mg). Mp: 251–253 °C. IR (KBr): 1692 cm−1 (CO). 1H NMR (400 MHz, CDCl3): δ 9.06 (d, J = 8.4 Hz, 1H), 7.83 (d, J = 8.8 Hz, 1H), 7.63–7.57 (m, 3H), 7.48–7.41 (m, 4H), 7.38–7.34 (m, 3H), 7.29 (d, J = 7.2 Hz, 1H), 2.49 (s, 3H), ppm. 13C NMR (100 MHz, CDCl3): δ 195.2, 148.6, 145.5, 143.7, 138.0, 137.3, 134.9, 134.1, 132.7, 130.9, 129.6, 129.3, 129.2, 129.1, 127.0, 126.3, 125.9, 124.5, 123.8, 119.9, 119.4, 21.3 ppm. HRMS (ESI): calcd for C24H16O: 321.1274 [M + H+]; found 321.1309.
5-(4-Methoxy-phenyl)-benzo[a]fluoren-11-one (2c)
Red-orange solid. Yield: 70% (59 mg). Mp: 275–277 °C. IR (KBr): 1694 cm−1 (CO). 1H NMR (400 MHz, CDCl3): δ 9.06 (d, J = 8.4 Hz, 1H), 7.84 (d, J = 8.8 Hz, 1H), 7.63–7.57 (m, 3H), 7.49–7.43 (m, 4H), 7.41–7.36 (m, 1H), 7.29 (d, J = 7.6 Hz, 1H), 7.07 (d, J = 8.4 Hz, 2H), 3.92 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3): δ 195.2, 159.7, 148.3, 145.5, 143.7, 135.0, 134.1, 132.7, 132.5, 131.0, 129.3, 129.1, 127.0, 126.3, 125.7, 124.6, 123.8, 119.9, 119.4, 114.0, 55.4 ppm. HRMS (ESI): calcd for C24H16O2: 359.1042 [M + Na+]; found 359.1049.
5-(4-Dimethylamino-phenyl)-benzo[a]fluoren-11-one (2d)
Red-orange solid. Yield: 65% (57 mg). Mp: 235–237 °C. IR (KBr): 1629 cm−1 (CO). 1H NMR (400 MHz, CDCl3): δ 9.06 (d, J = 8.4 Hz, 1H), 7.96 (d, J = 8.8 Hz, 1H), 7.62–7.52 (m, 3H), 7.48–7.20 (m, 5H), 7.29–7.25 (m, 1H), 6.88 (d, J = 8.8 Hz, 2H), 3.07 (s, 6H), ppm. 13C NMR (100 MHz, CDCl3): δ 195.2, 150.4, 149.1, 145.7, 143.8, 135.1, 134.0, 132.7, 131.1, 130.8, 129.2, 129.0, 127.9, 127.3, 126.1, 125.1, 124.5, 123.7, 119.8, 119.2, 112.2, 40.5 ppm. HRMS (ESI): calcd for C25H19NO: 350.1539 [M + H+]; found 350.1537.
5-(4-Bromo-phenyl)-benzo[a]fluoren-11-one (2e)
Red-orange solid. Yield: 80% (77 mg). Mp: 267–269 °C. IR (KBr): 1690 cm−1 (CO). 1H NMR (400 MHz, CDCl3): δ 9.07 (d, J = 8.4 Hz, 1H), 7.73 (d, J = 8.8 Hz, 1H), 7.67 (d, J = 8.0 Hz, 2H), 7.64–7.57 (m, 3H), 7.49–7.44 (m, 2H), 7.42–7.36 (m, 3H), 7.29 (t, J = 7.0 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ 195.1, 146.9, 145.4, 143.5, 139.1, 134.8, 134.2, 132.4, 131.7, 131.3, 130.8, 129.5, 129.3, 126.64, 126.55, 126.3, 124.7, 123.9, 122.5, 119.9, 119.3 ppm. HRMS (ESI): calcd for C23H13BrO: 385.0223 [M + H+]; found 385.0220.
5-(4-Nitro-phenyl)-benzo[a]fluoren-11-one (2f)
Red-orange solid. Yield: 50% (44 mg). Mp: 243–245 °C. 1H NMR (400 MHz, CDCl3): δ 9.10 (d, J = 8.4 Hz, 1H), 8.42 (d, J = 8.8 Hz, 2H), 7.71 (d, J = 8.8 Hz, 2H), 7.67–7.62 (m, 3H), 7.61 (s, 1H), 7.51–7.47 (m, 2H), 7.45–7.40 (m, 1H), 7.32–7.30 (m, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ 195.0, 147.8, 146.9, 145.4, 145.3, 143.3, 134.6, 134.4, 132.1, 130.8, 129.7, 129.6, 127.1, 127.0, 126.1, 124.9, 124.1, 123.8, 120.0, 119.4 ppm. HRMS (ESI): calcd for C23H13NO3: 374.0788 [M + Na+]; found 374.0794.5-Naphthalen-1-yl-benzo[a]fluoren-11-one (2g)
Yellow solid. Yield: 75% (67 mg). Mp: 211–213 °C. 1H NMR (400 MHz, CDCl3): δ 9.08 (s, 1H), 8.77 (d, J = 8.4 Hz, 1H), 7.86 (d, J = 7.6 Hz, 1H), 7.73–7.69 (m, 3H), 7.62 (d, J = 6.4 Hz, 4H), 7.44–7.42 (m, 3H), 7.25–7.13 (m, 2H), 6.28 (d, J = 7.2 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ 193.6, 145.0, 137.8, 137.4, 136.3, 135.9, 135.4, 134.6, 132.5, 131.9, 131.4, 130.7, 129.82, 129.76, 129.31, 128.7, 128.4, 127.5, 127.3, 124.8, 124.2, 123.6, 123.2, 119.3 ppm. HRMS (ESI): calcd for C27H16O: 357.1274 [M + H+]; found 357.1271.5-Furan-2-yl-benzo[a]fluoren-11-one (2h)
Yellow solid. Yield: 57% (42 mg). Mp: 221–223 °C. 1H NMR (400 MHz, CDCl3): δ 7.82 (s, 1H), 7.68 (d, J = 2.0 Hz, 1H), 7.66–7.63 (m, 1H), 7.63–7.51 (m, 3H), 7.49 (d, J = 4.0 Hz, 2H), 7.16–7.14 (m, 2H), 6.67–6.63 (m, 1H), 6.51 (d, J = 2.0 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ 193.2, 154.7, 148.4, 144.9, 136.8, 135.9, 135.5, 134.4, 134.0, 132.6, 130.9, 129.2, 129.0, 128.5, 128.0, 124.1, 122.7, 108.0, 107.2 ppm. HRMS (ESI): calcd for C21H12O2: 297.0910 [M + H+]; found 297.0908.5-(1H-Pyrrol-2-yl)-benzo[a]fluoren-11-one (2i)
Yellow solid. Yield: 65% (48 mg). Mp: 227–228 °C. 1H NMR (400 MHz, CDCl3): δ 8.18 (s, 1H), 7.90 (d, J = 10.0 Hz, 2H), 7.84 (d, J = 8.4 Hz, 1H) 7.77–7.72 (m, 3H), 7.59–7.54 (m, 3H), 7.48 (t, J = 8.0 Hz, 2H), 7.36 (t, J = 8.0 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3): 193.1, 144.9, 138.5, 137.0, 136.2, 135.0, 133.7, 132.9, 130.9, 129.2, 129.0, 128.8, 127.0, 125.7, 124.5, 121.0, 119.1 ppm. HRMS (ESI): calcd for C21H13NO: 318.0889 [M + H+]; found 318.0899.5-(1-Methyl-1H-pyrrol-2-yl)-benzo[a]fluoren-11-one (2j)
Yellow solid. Yield: 67% (52 mg). Mp: 199–201 °C. 1H NMR (400 MHz, CDCl3): δ 8.44 (s, 1H), 8.08 (d, J = 8.4 Hz, 1H), 7.63–7.58 (m, 3H), 7.55–7.53 (m, 2H), 7.48–7.44 (m, 1H), 7.30 (d, J = 7.6 Hz, 1H) 7.15–7.11 (m, 1H), 7.07–7.03 (m, 1H), 6.04 (d, J = 7.6 Hz, 1H), 3.27 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3): δ 193.1, 145.1, 142.6, 142.5, 140.2, 136.9, 136.7, 134.0, 130.4, 129.1, 128.8, 128.1, 126.4, 123.6, 123.0, 121.7, 120.7, 120.2, 117.0, 109.3, 32.1 ppm. HRMS (ESI): calcd for C22H15NO: 310.1226 [M + H+]; found 310.1227.5-Thiophen-2-yl-benzo[a]fluoren-11-one (2k)
Yellow solid. Yield: 60% (47 mg). Mp: 233–235 °C. 1H NMR (400 MHz, CDCl3): δ 8.19 (s, 1H), 7.66 (d, J = 6.8 Hz, 1H), 7.60–7.55 (m, 3H), 7.52 (d, J = 5.6 Hz, 1H), 7.74–7.45 (m, 2H), 7.20–7.13 (m, 2H), 6.98 (d, J = 5.6 Hz, 1H), 6.49 (d, J = 7.2 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ 193.2, 144.9, 144.8, 139.8, 137.7, 136.4, 135.8, 134.5, 132.9, 131.9, 130.6, 129.3, 129.1, 128.43, 128.38, 124.3, 124.1, 123.1, 118.8 ppm. HRMS (ESI): calcd for C21H12SO: 313.0682 [M + H+]; found 313.0708.Benzo[a]fluoren-11-one (2l)3b
Red-orange solid. Yield: 55% (32 mg). Mp: 132–134 °C [Lit. 132 °C]. 1H NMR (400 MHz, CDCl3): δ 8.95 (d, J = 8.4 Hz, 1H), 7.98 (d, J = 8.4 Hz, 1H), 7.78 (d, J = 8.4 Hz, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.61–7.56 (m, 2H), 7.49–7.41 (m, 3H), 7.28–7.24 (m, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ 195.4, 146.1, 143.9, 135.9, 134.6, 134.4, 134.2, 130.2, 129.4, 129.3, 128.5, 126.9, 126.4, 124.3, 123.8, 119.9, 118.1 ppm.4-Methyl-5-phenyl-benzo[a]fluoren-11-one (2m)
Red-orange solid. Yield: 60% (48 mg). Mp: 255–257 °C. 1H NMR (400 MHz, CDCl3): δ 8.96 (d, J = 7.6 Hz, 1H), 8.0 (d, J = 8.0 Hz, 1H), 7.79 (d, J = 8.4 Hz, 1H), 7.66 (d, J = 8.4 Hz, 1H), 7.62–7.57 (m, 2H), 7.50 (d, J = 7.6 Hz, 2H), 7.46–7.42 (m, 3H), 7.29–7.25 (m, 1H), 7.17 (d, J = 7.2 Hz, 1H), 2.33 (s, 3H), ppm. 13C NMR (100 MHz, CDCl3): δ 195.3, 148.7, 145.7, 143.8, 138.2, 137.4, 135.0, 134.3, 132.8, 131.0, 129.8, 129.4, 129.34, 129.29, 127.2, 126.5, 126.0, 124.7, 123.9, 120.0, 119.5 ppm. HRMS (ESI): calcd for C24H16O: 321.1274 [M + H+]; found 321.1308.3-Methyl-5-phenyl-benzo[a]fluoren-11-one (2n)
Red-orange solid. Yield: 73% (58 mg). Mp: 219–221 °C. 1H NMR (400 MHz, CDCl3): δ 8.96 (d, J = 8.4 Hz, 1H), 7.61 (d, J = 7.2 Hz, 1H), 7.56–7.50 (m, 6H), 7.46–7.40 (m, 3H), 7.28–7.24 (m, 2H), 2.40 (s, 3H), ppm. 13C NMR (100 MHz, CDCl3): δ 195.3, 147.6, 144.6, 143.9, 140.4, 136.3, 134.9, 134.1, 132.9, 131.5, 129.7, 129.1, 129.0, 128.5, 128.0, 126.0, 125.8, 124.3, 123.7, 119.7, 119.5, 22.0 ppm. HRMS (ESI): calcd for C24H16O: 321.1274 [M + H+]; found 321.1300.2-Methoxy-5-phenyl-benzo[a]fluoren-11-one (2o)
Red-orange solid. Yield: 62% (52 mg). Mp: 189–191 °C. 1H NMR (400 MHz, CDCl3): δ 8.45 (d, J = 2.4 Hz, 1H),7.68 (d, J = 9.2 Hz, 1H), 7.62 (d, J = 7.2 Hz, 1H), 7.55–7.48 (m, 5H), 7.47–7.41 (m, 3H), 7.29 (dd, J = 7.4, 3.8 Hz, 1H), 7.00 (dd, J = 6.0, 4.6 Hz, 1H), 4.0 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3): δ 194.3, 158.8, 147.4, 144.6, 142.8, 134.1, 133.2, 131.8, 131.6, 130.1, 128.4, 128.2, 126.1, 125.4, 123.7, 122.9, 119.0, 118.5, 113.1, 54.5 ppm. HRMS (ESI): calcd for C24H16O2: 337.1223 [M + H+]; found 337.1233.3-Chloro-5-phenyl-benzo[a]fluoren-11-one (2q)
Red-orange solid. Yield: 74% (63 mg). Mp: 215–217 °C. 1H NMR (400 MHz, CDCl3): δ 9.00 (d, J = 9.2 Hz, 1H), 7.76 (d, J = 9.2 Hz, 1H), 7.62 (d, J = 6.8 Hz, 2H), 7.58–7.53 (m, 3H), 7.52–7.48 (m, 3H), 7.46–7.42 (m, 2H), 7.31–7.27 (m, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ 194.8, 147.5, 145.5, 143.4, 139.5, 134.6, 134.4, 133.4, 132.7, 130.0, 129.6, 129.0, 128.7, 128.4, 126.1, 126.0, 125.8, 124.0, 120.5, 120.1 ppm. HRMS (ESI): calcd for C23H13ClO: 341.0728 [M + H+]; found 341.0695.8-Bromo-5-phenyl-benzo[a]fluoren-11-one (2r)
Orange solid. Yield: 70% (67 mg). Mp: 237–239 °C. 1H NMR (400 MHz, CDCl3): δ 9.04 (d, J = 8.4 Hz, 1H), 7.82 (d, J = 8.8 Hz, 1H), 7.63–7.59 (m, 2H), 7.57 (s, 1H), 7.57–7.47 (m, 6H), 7.44–7.38 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3): δ 193.8, 148.7, 145.5, 144.1, 139.9, 133.5, 132.9, 132.1, 130.7, 129.7, 129.4, 129.0, 128.5, 128.3, 127.0, 126.8, 126.3, 125.0, 124.6, 123.5, 119.4 ppm. HRMS (ESI): calcd for C23H13BrO: 385.0223 [M + H+]; found 385.0220.8,10-Dibromo-5-phenyl-benzo[a]fluoren-11-one (2s)
Orange solid. Yield: 61% (70 mg). Mp: 269–271 °C. 1H NMR (400 MHz, CDCl3): δ 9.02 (d, J = 8.4 Hz, 1H), 7.81 (d, J = 8.4 Hz, 1H), 7.61–7.59 (m, 1H), 7.57–7.54 (m, 2H), 7.52–7.50 (m, 2H), 7.51–7.49 (m, 1H), 7.47–7.45 (m, 1H), 7.42–7.39 (m, 2H), 7.40–7.36 (m, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ 193.5, 148.3, 145.1, 143.6, 139.5, 133.1, 132.5, 131.7, 130.3, 129.3, 129.0, 128.7, 128.6, 128.3, 128.1, 127.9, 126.6, 126.4, 125.9, 124.6, 124.2, 123.1, 119.0 ppm. MS (ESI): m/z 485 [M + Na+]. Anal. calcd for C23H12Br2O: C 59.52, H 2.61; found: C 59.63, H 2.55.3-Phenyl-1-(2-phenylacetyl-phenyl)propenone (7)10
Compound 7 was obtained by heating under reflux 1a (0.25 mmol) with silver(I) triflate (10 mg, 15 mol%) in the presence of water (0.25 mL) in acetonitrile (5 mL). Pale yellow solid. Yield: 68% (56 mg). Mp: 90–92 °C [Lit. 91.8–93.6 °C]. 1H NMR (400 MHz, CDCl3): δ 8.01–7.99 (m, 2H), 7.72 (dd, J = 7.6 Hz, 1.2 Hz, 1H), 7.57–7.35 (m, 10H), 7.30 (d, J = 7.6 Hz, 1H), 7.24 (s, 1H), 7.20 (s, 1H), 4.62 (s, 2H) ppm. 13C NMR (100 MHz, CDCl3): δ 197.3, 195.1, 145.9, 138.7, 137.0, 134.7, 134.7, 133.1, 132.5, 131.2, 130.6, 129.1, 128.9, 128.6, 128.5, 128.3, 126.9, 125.6, 43.6 ppm. Compound 7 when heated under reflux with iodine in AcOH afforded benzo[a]fluorenone 2a in 85% yield.3-[2-(4-Nitro-phenyl)-vinyl]-2-phenyl-inden-1-one (8)
Compound 8 was obtained as a byproduct in the reaction of yne-enone 1f. Orange solid. Yield: 28% (25 mg). Mp: 115–117 °C. 1H NMR (400 MHz, CDCl3): δ 8.24 (d, J = 8.8 Hz, 2H), 7.63–7.61 (m, 4H), 7.55 (s, 1H), 7.55–7.47 (m, 5H), 7.46–7.40 (m, 2H), 7.35 (t, J = 8.0 Hz, 1H), ppm. 13C NMR (100 MHz, CDCl3): δ 195.4, 149.3, 147.7, 143.2, 142.7, 135.9, 134.1, 133.5, 131.5, 130.8, 130.2, 129.2, 128.7, 128.5, 127.7, 125.7, 124.3, 123.3, 121.5 ppm. HRMS (ESI): calcd for C23H15NO3: 354.1125 [M + H+]; found 354.1121. Compound 8 was converted into 2f in 55% yield when heated under reflux in AcOH for 24 h.8,10-Dibromo-5-phenyl-5,6-dihydro-benzo[a]fluoren-11-one (9)
Compound 9 was obtained from the incomplete the reaction (8 h) of yne-enone 1t. Brown semisolid. Yield: 70% (81 mg). 1H NMR (400 MHz, CDCl3): δ 7.92 (s, 1H), 7.91 (s, 1H), 7.44–7.42 (m, 2H), 7.35 (t, J = 7.2 Hz, 2H), 7.26 (t, J = 7.2 Hz, 1H), 7.17–7.10 (m, 2H), 7.05–7.04 (m, 1H), 7.00 (d, J = 7.6 Hz, 1H), 4.39 (d, J = 4.8 Hz, 1H), 3.12 (dd, J = 18.8, 10.4 Hz, 1H), 2.89 (dd, J = 18.6, 9.4 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ 194.4, 144.0, 143.6, 142.2, 140.6, 139.0, 135.6, 128.9, 128.8, 128.6, 128.5, 127.8, 127.0, 124.2, 122.9, 103.6, 45.9, 44.0 ppm. HRMS (ESI): calcd for C23H14Br2O: 464.9484 [M + H+]; found 464.9476. This compound (46 mg, 0.1 mmol) was mixed with DDQ (23 mg, 0.1 mmol) in 1,2-dichloroethane (2 mL) and stirred at room temperature for 12 h to give 2t (41 mg, 90%).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Council of Scientific and Industrial Research (CSIR), Science and Engineering Research Board (SERB), India for financial support; DST-FIST for instrumentation facilities at School of Chemistry, Bharathidasan University. V. J. T. thanks University Grants Commission (UGC) for a BSR-RFSMS fellowship.Notes and references
- (a) S. Baur, J. Niehaus, A. D. Karagouni, E. A. Katsifas, K. Chalkou, C. Meintanis, A. L. Jones, M. Goodfellow, A. C. Ward, W. Beil, K. Schneider, R. D. Sussmuth and H. P. Fiedler, J. Antibiot., 2006, 59, 293–297 CrossRef CAS PubMed; (b) Z. Y. Feng, J. H. Kim and S. F. Brady, J. Am. Chem. Soc., 2010, 132, 11902–11903 CrossRef CAS PubMed; (c) G. Y. Zhang, W. M. Zhang, J. S. Liu and C. S. Zhang, J. Nat. Prod., 2012, 75, 1937–1943 CrossRef PubMed.
- (a) T. A. Crowell, C. A. Jones and H. U. Bryant, US 5958917 A, September 28, 1999; (b) S. Zheng, K. M. Vaeth and G. A. Bennett, US 6849348 B2, February 1, 2005.
- For some old methods, see: (a) R. L. Letsinger and J. D. Jamison, J. Am. Chem. Soc., 1961, 83, 193–198 CrossRef CAS; (b) A. Streitwieser Jr and S. M. Brown, J. Org. Chem., 1988, 53, 904–906 CrossRef.
- C. Atienza, C. Mateo, O. de Frutos and A. M. Echavarren, Org. Lett., 2001, 3, 153–155 CrossRef CAS PubMed.
- (a) R. S. Laufer and G. I. Dmitrienko, J. Am. Chem. Soc., 2002, 124, 1854–1855 CrossRef CAS PubMed; (b) W. Liu, M. Buck, N. Chen, M. Shang, N. J. Taylor, J. Asoud, X. Wu, B. B. Hasinoff and G. I. Dmitrienko, Org. Lett., 2007, 9, 2915–2918 CrossRef CAS PubMed; (c) Y.-B. Zhao, B. Mariampillai, D. A. Candito, B. Laleu, M. Li and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 1849–1852 CrossRef CAS PubMed.
- J. P. Waldo, X. Zhang, F. Shi and R. C. Larock, J. Org. Chem., 2008, 73, 6679–6685 CrossRef CAS PubMed.
- (a) P. Nosel, S. Moghimi, C. Hendrich, M. Haupt, M. Rudolph, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2014, 356, 3755–3760 CrossRef. For other comparisons of iodine and other electrophilic activators with other substrates, see also: (b) J. P. Weyrauch, A. S. K Hashmi, A. Schuster, T. Hengst, S. Schetter, A. Littmann, M. Rudolph, M. Hamzic, J. Visus, F. Rominger, W. Frey and J. W. Bats, Chem.–Eur. J., 2010, 16, 956–963 CrossRef CAS PubMed; (c) P. Nosel, T. Lauterbach, M. Rudolph, F. Rominger and A. S. K. Hashmi, Chem.–Eur. J., 2013, 19, 8634–8641 CrossRef PubMed; (d) T. Wang, S. Shi, M. Rudolph and A. S. K. Hashmi, Adv. Synth. Catal., 2014, 356, 2337–2342 CrossRef CAS; (e) For gold catalysis in synthesis, see: D. Pflasterer and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 1331–1367 RSC.
- (a) S. Akbar and K. Srinivasan, Eur. J. Org. Chem., 2013, 1663–1666 CrossRef CAS; (b) S. Akbar and K. Srinivasan, RSC Adv., 2015, 5, 5542–5545 RSC; (c) S. Akbar and K. Srinivasan, Eur. J. Org. Chem., 2015, 7652–7655 CrossRef CAS; (d) S. Akbar and K. Srinivasan, J. Org. Chem., 2016, 81, 1229–1236 CrossRef CAS PubMed.
- Q. Zhao, Q. Hu, L. Wen, M. Wu and Y. Hu, Adv. Synth. Catal., 2012, 354, 2113–2116 CrossRef CAS.
- Z.-Q. Wang, W.-W. Zhang, L.-B. Gong, R.-Y. Tang, X. H. Yang, Y. Liu and J.-H. Li, Angew. Chem., Int. Ed., 2011, 50, 8968–8973 CrossRef CAS PubMed.
- For reviews on iodine mediated organic transformations, see: (a) H. Togo, Synlett, 2006, 2159–2175 CrossRef CAS; (b) M. Jereb, D. Vrazic and M. Zupan, Tetrahedron, 2011, 67, 1355–1387 CrossRef CAS; (c) S. Hummel and S.-F. Kirsch, Beilstein J. Org. Chem., 2011, 7, 847–859 CrossRef CAS PubMed; (d) S. V. Tekale, S. S. Kauthale, S. A. Dake, S. R. Sarda and R. P. Pawar, Curr. Org. Chem., 2012, 16, 1485–1501 CrossRef CAS; (e) T. Aggarwal, S. Kumar and A. K. Verma, Org. Biomol. Chem., 2016, 14, 7639–7653 RSC.
- CCDC 1501649 for compound 2b. See the ESI† for details.
- (a) D. Zheng, S. Li, Y. Luo and J. Wu, Org. Lett., 2011, 13, 6402–6405 CrossRef CAS PubMed; (b) R. Das and D. Chakraborty, Appl. Organomet. Chem., 2012, 26, 722–726 CrossRef CAS.
- For a similar transformation, see: C. González-Rodríguez, L. Escalante, J. A. Varela, L. Castedo and C. Saá, Org. Lett., 2009, 11, 1531–1533 CrossRef PubMed.
- H. Hibbert, J. Am. Chem. Soc., 1915, 37, 1748–1763 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for all products, including copies of 1H and 13C NMR spectra and X-ray structural information of 2b. CCDC 1501649. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra02376c |
| This journal is © The Royal Society of Chemistry 2019 |