
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstitutional isomers of brominated-functionalized copillar[5]arenes: synthesis, characterization, and crystal structures†
Talal F. Al-Azemi *,
Mickey Vinodh,
Fatemeh H. Alipour and
Abdirahman A. Mohamod
*,
Mickey Vinodh,
Fatemeh H. Alipour and
Abdirahman A. Mohamod
Chemistry Department, Kuwait University, P. O. Box 5969, Safat 13060, Kuwait. E-mail: t.alazemi@ku.edu.kw; Fax: +965-2481-6482; Tel: +965-2498-554
First published on 3rd May 2019
Abstract
We herein report the preparation of constitutional isomers of brominated-functionalized pillar[5]arenes via co-condensation of 1,4-bis(2-bromoethoxy)benzene and 1,4-dimethoxybenzene. The structures of the obtained isomers were then established using single crystal X-ray diffraction. We also found that the isomeric yield distribution of the different constitutional isomers was independent of the monomer's mole feed ratio, as revealed by HPLC analysis of the crude mixture. Finally, further characterization of the separated constitutional isomers indicated that they possess different melting points, NMR spectra, crystal structures, binding constants and stacking patterns in the solid state.
Introduction
Pillar[n]arenes are versatile macrocyclic compounds which have gained increasing interest for their ease of formation, functionalization and their excellent host–guest properties. Different sized pillar[n]arenes have been reported, including pillar[5]arenes (i.e., containing a 5-membered ring),1,2 pillar[6]arenes,3 and pillar[7]arenes.4 In addition, larger pillar[n = 6–15]arenes have been synthesized recently through the ring expansion of pillar[5]arenes.5 Pillar[5]arene and its derivatives have received the most attention to date, due to their ease of formation and functionalization. The electron-rich cavity of pillar[5]arene and its analogs favors the binding of positively charged or electron-deficient guests,6 and have been demonstrated to act as hosts toward a range of organic compounds, including viologens,7 alkanediamines,8 dinitrobenzenes,9 azobenzene derivatives,10 and neutral molecules.11,12Introduction of functional groups on the macrocycles structure is an important target in the receptor design and allows further chemical and physical manipulation to the system. Therefore, different strategies have been utilized including direct deprotection, oxidation-followed-by-reduction and direct cyclization (followed by deprotection) to introduce functional groups such as bromo,13 amino,14 alkyne,15 and hydroxyl moieties16–20 have been reported thus far. In the synthesis of tetrahydroxy functionalized pillar[5]arene using oxidation-followed-by-reduction protocol,21 regioisomers of the quinones derivative were successfully isolated and reduced to its corresponding hydroxy-pillar[5]arenes. Different hydroxylated regioisomers were also synthesized by de-O-methylation of pillar[5]arene using various stoichiometry of the deprotecting reagent (boron tribromide, BBr3).21 The isolation of the hydroxylated derivatives was carried out after the reaction with trifluoromethanesulfonic anhydride to facilitated the chromatographic separation. Other constitutional isomers were synthesized and isolated using the co-cyclization approach by adjusting the feed ratio to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 of 1,4-bis(4-bromobutoxy)benzene and 1,4-dimethoxybenzene respectively.22 The different proximity of functional groups on the macrocycle rim influence the physical, conformation and host–guest properties. This is evident from the different physical, chemical, and complexation behavior of constitutional isomer of pillar[5]arene obtained by condensation of asymmetric hydroquinone derivatives.
:3 of 1,4-bis(4-bromobutoxy)benzene and 1,4-dimethoxybenzene respectively.22 The different proximity of functional groups on the macrocycle rim influence the physical, conformation and host–guest properties. This is evident from the different physical, chemical, and complexation behavior of constitutional isomer of pillar[5]arene obtained by condensation of asymmetric hydroquinone derivatives.
Herein we report the synthesis of constitutional isomers of tetra-and-hexa-bromo-functionalized pillar[5]arenes through the co-condensation of hydroquinone derivatives of 1,4-bis(2-bromoethoxy)benzene and 1,4-dimethoxybenzene. Separation of the constitutional isomers and their resulting isomeric yield distributions with respect to varying the feed ratio are also investigated by HPLC. In addition, the obtained constitutional isomers are characterized by NMR spectroscopy and X-ray single crystal diffraction studies. The complexation behavior of the isolated constitutional isomers toward a quaternary ammonium salt is investigated in details.
Results and discussion
Pillararenes synthesized by co-oligomerization of two different monomers mainly utilize the molar ratio of 1:4 and even more of 1:16 to simplify the chromatographic separation.18 The co-condensation of 1,4-bis(2-bromoethoxy)benzene and 1,4-dimethoxybenzene result in eight possible pillar[5]arenes including regioisomers which exhibit practically identical properties during column chromatography (Fig. 1). Therefore, most of the literature reports of copillar[5]arenes with two repeating units are in 1:4 molar ratio. However, the separation of copillararenes is dependent on the chemical structure of monomers. The syntheses of functionalized copillararenes with repeating units other than the repetitious molar ratio of 1:4, offer control of the receptor design and enhanced its recognition ability.
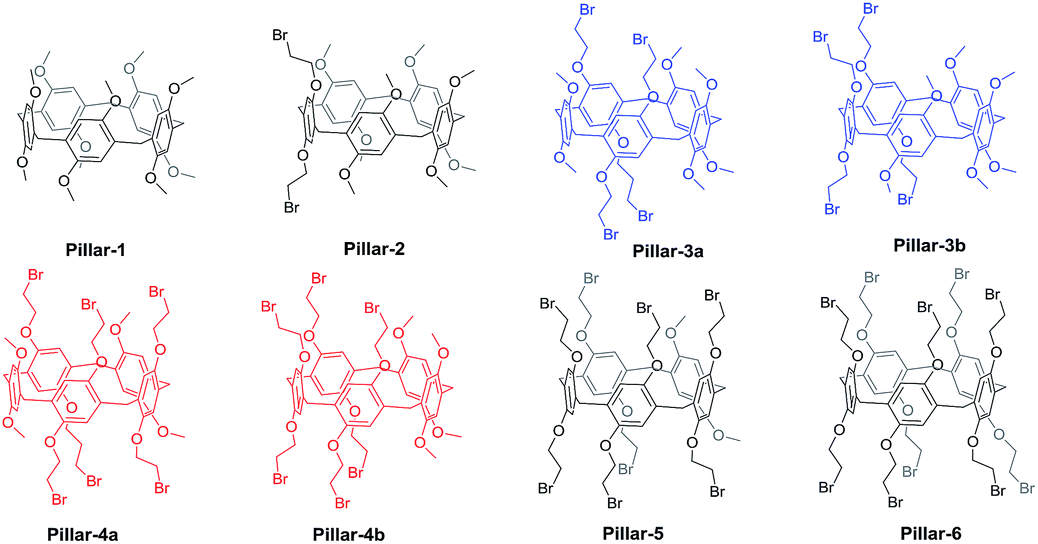 | ||
| Fig. 1 Chemical structures of all possible pillar[5]arenes from the co-cyclization reaction of 1,4-dimethoxybenzene with1,4-bis(2-bromoethoxy)benzene. | ||
Based on such systems, we synthesized and separated all eight pillar[5]arenes shown in Fig. 1 by co-cyclization procedure of 1,4-dimethoxybenzene with1,4-bis(2-bromoethoxy)benzene and paraformaldehyde in the presence of BF3·OEt2. After 30 min the reaction mixture was flush through silica gel plunge and the total yield of all copillar[5]arenes were in range of 57–71%. TLC analysis of the copillar[5]arenes synthesized using the 1,4-dimethoxybenzene with 1,4-bis(2-bromoethoxy)benzene monomers produced eight spots. Column chromatography using a dichloromethane/hexane mixture (60:40 v/v) was then employed to separate the mixture, and gave the eight possible pillar[5]arenes (i.e., Pillars-1–6) as white solids.
After separation, analysis by HRMS were conducted and found the four spots which corresponded to Pillar-3a–3b and Pillar-4a–4b isomers exhibit two set of signals for [M + Na]+ at m/z 1141.0374 and at 1324.8856 respectively. However, the melting points of the four isomers differed significantly (i.e., Pillar-3a = 133 °C; Pillar-3b = 124 °C; Pillar-4a = 125 °C; and Pillar-4b = 130 °C), as did their 1H NMR spectra, as shown in Fig. 2. Inspection of the 1H NMR spectra revealed the complete assignment of their structure is particularly challenging. Even though the relative area of the proton's peaks is similar for Pillar-3a and Pillar-3b and for Pillar-4a and Pillar-4b, the chemical shift and multiplicity are entirely different (Fig. 2). For example, the signals corresponding to methylene (–CH2–Br) shows as one triplet for Pillar-3a, Pillar-3b found to be as two set of triplets. Similar differences also true for the signals corresponding to the phenyl, methylene bridge, and methoxy protons.
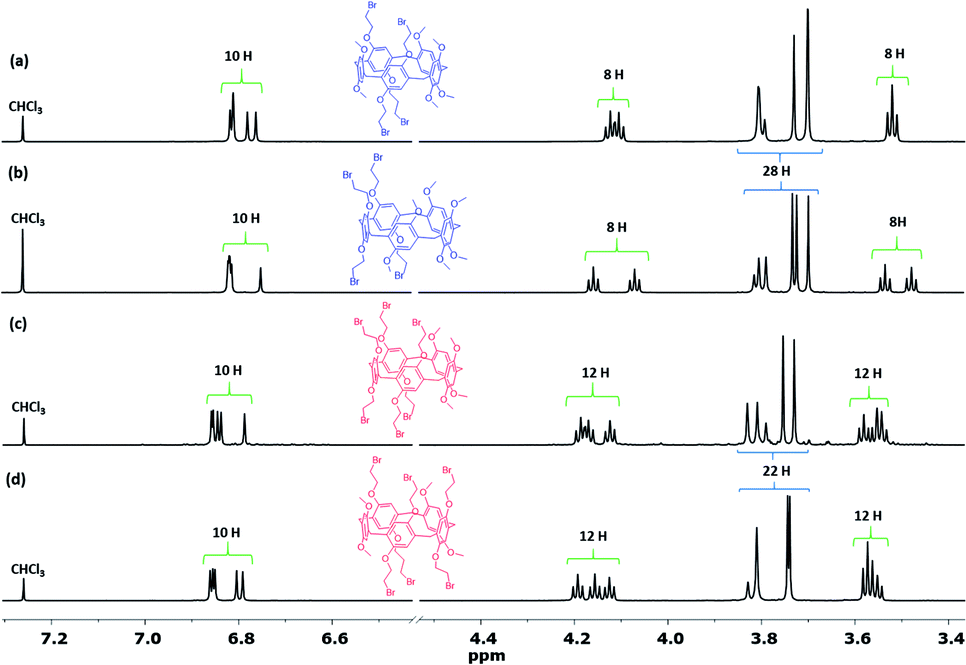 | ||
| Fig. 2 1H NMR spectra (600 MHz, CDCl3) of the constitutional isomers of Pillars-3a–3b and 4a–4b and insets showing the X-ray single crystal diffraction structures. (a) The 1,3-alternate isomer, Pillar-3a, (b) the 1,2-alternate isomer, Pillar-3b, (c) the 1,2-alternate isomer, Pillar-4a, and (d) the 1,3-alternate isomer, Pillar-4b. | ||
Thus, the structures of the different isomers were assigned using single crystal X-ray diffraction, which clearly confirmed their corresponding substitution patterns (Fig. 3). To obtain suitable crystals for analysis, single crystals of Pillars-3a, 3b, 4a, and 4b were grown by solvent diffusion method. In the solid state, these constitutional isomers were found to stack either in the edge-to-edge (registered) style (i.e., Pillars-3a and 4b) or the corner-to-edge style (i.e., Pillars-3b and 4a) (Fig. S5 and S6†). Interestingly, the solvent molecule 1,2-dicholorethane found encapsulate inside the Pillar-3a cavity involved in halogen–halogen bonding with the functional bromine on the adjacent pillar[5]arene resulting in the formation of supramolecular dimer, whereas in Pillar-4b similar halogen–halogen bond induced the formation supramolecular polymeric assembly.
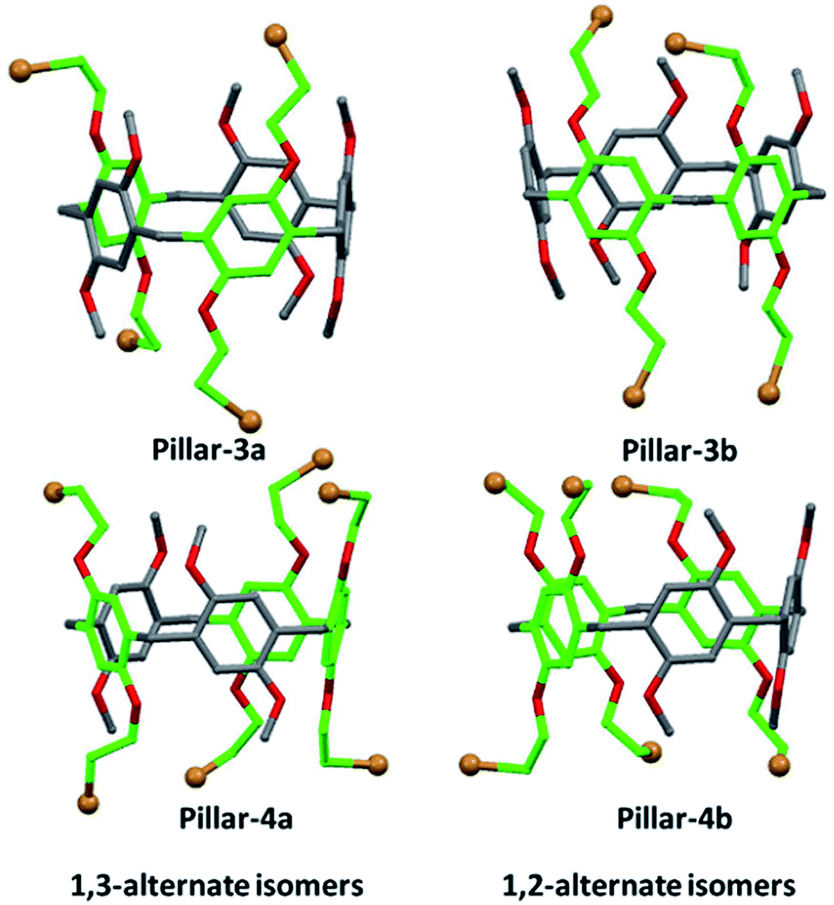 | ||
| Fig. 3 Crystal structures of the constitutional isomers of tetra-and hexabromo-functionalized pillar[5]arenes obtained for single crystal X-ray diffraction analysis. | ||
To gain a clear insight on the copillar[5]arenes reaction, HPLC was used to examine yield distribution when the mole feed ratio were varied of monomer 1 (1,4-dimethoxybenzene) and monomer 2 (1,4-bis(2-bromoethoxy)benzene). HPLC technique is ideal due to small differences between the retention factors (Rf values) of the different copillar[5]arenes especially for the constitutional isomers of Pillar-3 and Pillar-4, which is rather difficult to estimation of their yield distribution following separation by column chromatography. Control experiments were therefore conducted using both monomers and the results are summarized in Table 1. Prior to HPLC analysis the crude reaction mixtures were passed through silica gel plugs and eluted with dichloromethane. Typical chromatograph is showing in Fig. 4 along with the peak assignments which were based on a comparison with the retention times of the pure isolated corresponding pillar[5]arenes.
| Entry | Mole feed ratio | Pillar[5]arenes relative distributionc (%) | ||||||
|---|---|---|---|---|---|---|---|---|
| Monomer 1b | Monomer 2b | 1 | 2 | 3(a)d + 3(b) | 4(a)d + 4(b) | 5 | 6 | |
| a Reactions were carried out for 30 min at room temperature in dichloroethane.b Monomer 1 = 1,4-dimethoxybenzene; monomer 2 = 1,4-bis(2-bromoethoxy)benzene.c % relative distributions were calculated from HPLC.d Ratio of 3(a):3(b) and 4(a):4(b) are 3:1 and 3:2 respectively.e Not detected.f % relative distributions were calculated after separation from column chromatography. |
||||||||
| 1 | 4.0 | 1.0 | 53 | 30 | 14 | 3 | nde | nde |
| 2 | 3.5 | 1.5 | 44(47)f | 30(28) | 19(11 + 7) | 6(3 + 2) | 1<(2) | 1< |
| 3 | 3.0 | 2.0 | 31 | 31 | 25 | 10 | 2 | 1 |
| 4 | 2.5 | 2.5 | 17 | 27 | 32 | 19 | 4 | 1 |
| 5 | 2.0 | 3.0 | 11(8)f | 21(15) | 32(20 + 7) | 26(19 + 12) | 7(14) | 3(5) |
| 6 | 1.5 | 3.5 | 8(6)f | 18(12) | 30(23 + 6) | 29(23 + 11) | 10(12) | 5(7) |
| 7 | 1.0 | 4.0 | 3 | 10 | 24 | 35 | 19 | 9 |
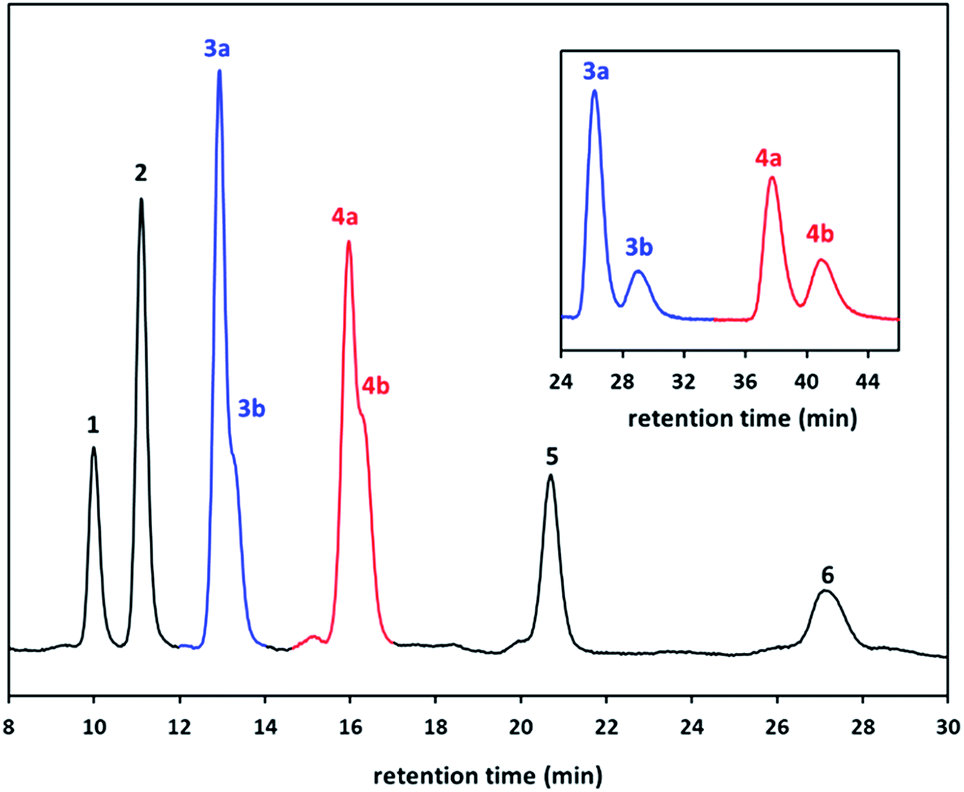 | ||
| Fig. 4 HPLC chromatogram for the crude reaction mixtures of the copillar[5]arenes synthesized from monomers 1,4-dimethoxybenzene and 1,4-bis(2-bromoethoxy)benzene and insets showing the chromatogram of separated constitutional isomers of Pillar-3 and Pillar-4 (Table 1, entry 6). | ||
In column chromatography the elution of the pillar[5]arene derivatives is reversed compare to the order shown in Fig. 4 as the permethylated pillar[5]arene, Pillar-1 eluted last and Pillar-6 eluted first because of the use of LC-CN HPLC based column. The separation was achieved by using hexane: chloroform (85:15 v/v) solvent system with follow rate of 0.7 mL min−1. As for the regioisomers the separation was accomplished by altering the solvent system to hexane:chloroform (95:5 v/v).
Upon inspection of results obtained for the pillar[5]arenes yield distribution in Table 1, it's clear that monomer 1 (1,4-dimethoxybenzene) more reactive than monomer 2 (1,4-bis(2-bromoethoxy)benzene) toward electrophilic aromatic substitution due to the presence of electron withdrawing group (bromine) as evidence of relative high yield distributions of the permethylated pillar[5]arene (Pillar-1). In addition, the low formation of Pillar-6 even with the use of higher feed ratio of monomer 2 of 3.5 and 4.0 mole (Table 1, entry 6 and 7). When monomer 1 to 2 mole feed ratio was 4:1 respectively, Pillar-5 and Pillar-6 were not detected in the HPLC chromatogram (Table 1, entry 1). In all reactions in Table 1, the relative ratio of the constitutional isomers 1,3-alternate and 1,2-alternate kept constant. The tetrabromo-functionalized pillar[5]arene isomers Pillar-3a to Pillar-3b and for the heaxbromo-functionalized pillar[5]arene isomers Pillar-4a and Pillar-4b ratios were 3:1 and 3:2 respectively.
The noncovalent interaction of the obtained constitutional isomers Pillar-(3a–3b) and Pillar-(4a–4b) with n-octyltrimethyl ammonium hexafluorophosphate (OMA) was investigated using a 1H NMR titration method. OMA is widely used as a guest species for pillar[5]arenes,19 1H NMR spectra of mixtures of OMA (8.4 mM) and varying concentrations of Pillar-3a in CDCl3 at 25 °C confirmed host–guest complexation, as evidenced by an upfield shift of resonances for the trimethyl protons (Ha) of OMA (Fig. S25†). At higher concentration of the host, the guest was completely encapsulated and negative region were observed indicating the linear guest OMA is threaded through the pillararene cavity as shown in Fig. 5b. Interestingly, the terminal methyl group (Hi) and the methylene protons (Hh) were shifted downfield due to their location in deshielding region of the macrocyclic aromatic system (Fig. S26†). Similar effect was observed in 1H NMR titrations carried out for all pillararene isomers.
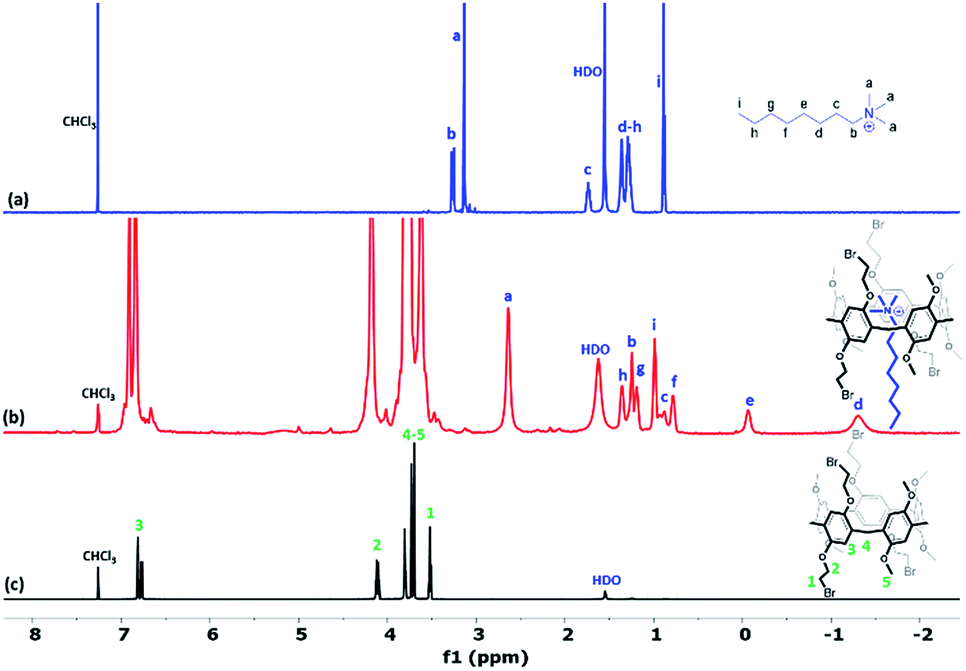 | ||
| Fig. 5 1H NMR (600 MHz, CDCl3 at 298 K) spectra of (a) 8.4 mM OMA, (b) 3.4 × 10−2M of Pillar-3a and 8.4 mM OMA, and (c) 8.4 mM of Pillar-3a. | ||
The stoichiometry of complexation was established by the method of continuous variations (Job's method). All Job's plot between the mole fraction of the guest (χ) and the observed chemical shift change of N-trimethyl protons on the OMA guest in 1H NMR multiplied by the guest mole fraction (χ) show maxima at a mole fraction of 0.5 which indicates a 1:1 host-to-guest stoichiometric ratio of complexation (Fig. S23†). Further evidence of the formation of a 1:1 complex were obtained for the ESI-MS spectrum where relevant molecular peaks at m/z = 1294.26 and 1482.05 were found corresponding to [(Pillar-(3a–3b) + OMA) − PF6]+ and [(Pillar-(4a–4b) + OMA) − PF6]+, in agreement with the results obtained from 1H NMR titration experiments (Fig. S24†). The association constant for complexation for all pillar[5]arene isomers were determined from chemical shift changes of the trimethyl groups of the guest OMA at 3.16 ppm and the data fitted to 1:1 binding isotherm.24 The calculated association constant Ka were affected by the different spatial arrangement of the pillar[5]arene substituents. The isomer based on 1,2-alternate of tetrabromo-functionalized Pillar-3b shows the highest Ka value of 140.4 ± 9.4 M−1, which is approximately three folds the binding constant of the 1,3-alternate isomer (Pillar-3a) (58.9 ± 2.6 M−1). On the other hand, the constitutional isomer based on the hexa-bromo-pillar[5]arenes, the 1,2-alternate isomer Pillar-4b displays the lowest binding constant of 34.9 ± 1.9 M−1, where the 1,3-alternate Pillar-4a shows Ka of 50.7 ± 2.2 M−1 which is 46% increased compare to 1,2 alternate isomer. From the binding study, it is well evident that these pillararene constitutional isomers have different degree of affinity towards guest molecules (OMA) and hence could effectively employed for molecular recognition by properly tuning their peripheral motifs.
Experimental
Materials and methods
NMR spectroscopy was carried out by on Bruker Avance II 600 MHz (Bruker, Germany). Electron impact (EI) mass spectrometry was performed using a DFS High Resolution GC/MS (Thermo Scientific, Germany). Electrospray ionization was carried out in high resolution mode using a Waters Xevo G2-S QTOF LC-MS/MS (Waters, Germany). As detailed later, single crystal X-ray diffraction was carried out using an R-AXIS RAPID II diffractometer (Rigaku, Japan), and the data were collected at −123 °C (Oxford Cryosystems, UK). LC was carried out using an instrument equipped with a photodiode array detector (LC-MS/MS, Thermo Scientific, Germany). Flash column chromatography was performed using silica gel (silica gel 60, 40–60 mesh ASTM, EMD Millipore, Merck KGaA, Germany). Dimethylformamide (DMF), dichloroethane, and acetonitrile were distilled prior to use. All other reagents and solvents were of reagent grade purity and were used without further purification. n-Octyltrimethyl ammonium hexafluorophosphate (OMA) was synthesized according to literature procedure.19 HPLC was carried out using a Waters HPLC system equipped with a 1525 binary pump, a 2487 dual absorbance detector, a 717 plus autosampler, and Breeze software (Waters, German). Pillararenes were separated using a Waters SUPELCOSIL™ 5 μm LC-CN HPLC column using hexane: chloroform (85:15 v/v) as the eluent, with a run time of 30 min and a flow rate of 0.7 mL min−1. The constitutional isomer Pillar-3–4(a, b) were separated using hexane:isopropanol (94:6 v/v) as the eluent, with a run time of 45 min and a flow rate of 0.7 mL min−1.
:2 v/v, 3 × 100 mL), recrystallized from methanol and dried under vacuum to afford the compound as white crystals (14.5 g, 94%). 1H NMR (600 MHz, CDCl3) δ: 3.62 (t, J = 6.0 Hz, 4H), 4.25 (t, J = 6.6 Hz, 4H), 6.88 (s, 4H). 13C NMR (150 MHz, CDCl3), δ: 29.5, 68.9, 116.3, 153.0. HRMS: (m/z): calcd for C10H12O2Br2: 321.9199; found 321.9199.:40 v/v)) to give a mixture of macrocycles Pillar-1–6 (2.95 g, 57%). The pillar[5]arenes are organized according to their order in elution from column chromatography.Preparation of single crystals for X-ray diffraction
Single crystals of the synthesized pillar[5]arenes and their inclusion complexes were grown using either the slow solvent evaporation method or by the diffusion method using dichloromethane and n-hexane or acetonitrile. The single crystal data collections were made on Rigaku R-AXIS RAPID II diffractometer by filtered Mo-Kα radiation. The data were collected under liquid nitrogen (Oxford cryosystems). ‘Crystalclear’ software package was employed to generate hkl and p4p files. The structure was then solved by direct methods using Rigaku's ‘CrystalStructure’ crystallographic software package except for refinement, which was performed using SHELXL-2017/1. The crystallographic data for all structures reported in this paper have been deposited at the Cambridge Crystallographic Data Centre as supplementary publications (CCDC 1880766–1880769).1H NMR titrations
A 0.5 mL sample of the OMA guest solutions was prepared at a concentration of 8.4 mM in chloroform-d. A sample of the host solutions (2 mL) was prepared at a concentration of 0.1 M in a chloroform-d solvent. All titration experiments were carried out in an NMR tube at 298 K, and 1H-NMR spectra were recorded upon successive addition of aliquots of the stock solution of the appropriate host with a syringe. The 1H-NMR spectral changes were fitted to 1:1 binding isotherms by nonlinear least-squares treatment using Microsoft Excel to determine the association constant, Ka.24
Conclusions
In conclusion, constitutional isomers of tetra-and hexabromo-functionalized pillar[5]arenes were synthesized by co-cyclization from hydroquinone derivatives of 1,4-dimethoxybenzene and 1,4-bis(2-bromoethoxy)benzene with paraformaldehyde in presence of BF3·OEt2. Separation of the constitutional brominated-pillar[5]arene isomers by column chromatography was attempted, the yields of the regioisomers dependent on nature and the monomer mole feed ratio. The 1,4-dimethoxybenzene monomer 1 is more reactive relative to 1,4-bis(2-bromoethoxy)benzene toward the electrophilic condensation reaction. HPLC analysis of the crude pillararene mixture, revealed that the isomeric distribution of tetra-and hexabromo-functionalized pilla[5]arene were 3:1 and 3:2 respectively. Characterization of the prepared and isolated constitutional isomers revealed that they exhibited different melting points, NMR spectra, crystal structures, and stacking patterns in the solid state. The binding study carried out for pillar[5]arane constitutional isomers with OMA guest indicate that the association constant of complexation was affected by the spatial arrangement of the substituents on the pillararene rim. Our future work will focus on the modification and application of the synthesized brominated-functionalized pillar[5]arenes isomers in the receptor design.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The support received from the Kuwait Foundation of Advancement Science (KFAS), made available through research grant no. PR17-14SC-07, in addition to the facilities of RSPU (grant no. GS01/01, GS01/03, and GS03/08) is gratefully acknowledged.Notes and references
- (a) T. Ogoshi, S. Kanai, S. Fujinami, T. A. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed; (b) Z. Zhang, Y. Luo, J. Chen, S. Dong, Y. Yu, Z. Ma and F. Huang, Angew. Chem., Int. Ed., 2011, 50, 1397–1401 CrossRef CAS PubMed.
- (a) C. Han, F. Ma, Z. Zhang, B. Xia, Y. Yu and F. Huang, Org. Lett., 2010, 12, 4360–4363 CrossRef CAS PubMed; (b) D. Cao, Y. Kou, J. Liang, Z. Chen, L. Wang and H. Meier, Angew. Chem., Int. Ed., 2009, 48, 9721–9723 CrossRef CAS PubMed; (c) L. Liu, D. Cao, Y. Jin, H. Tao, Y. Koua and H. Meier, Org. Biomol. Chem., 2011, 9, 7007–7010 RSC.
- Z. Li, J. Yang, J. He, Z. Abliz and F. Huang, Org. Lett., 2014, 16, 2065–2069 Search PubMed.
- T. Ogoshi, K. Masaki, R. Shiga, K. Kitajima and T.-A. Yamagishi, Org. Lett., 2011, 13, 1264–1266 CrossRef CAS PubMed.
- T. Ogoshi, N. Ueshima, F. Sakakibara, T. Yamagishi and T. Haino, Org. Lett., 2014, 16, 2896–2899 CrossRef CAS PubMed.
- T. Ogoshi, Y. Nishida, T.-A. Yamagishi and Y. Nakamoto, Macromolecules, 2010, 43, 3145–3147 CrossRef CAS.
- N. L. Strutt, R. S. Forgan, J. M. Spruell, Y. Y. Botros and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 5668–5671 CrossRef CAS PubMed.
- N. Strutt, D. Fairen-Jimenez, J. Iehl, M. Lalonde, R. Snurr, O. Farha, J. Hupp and F. Stoddart, J. Am. Chem. Soc., 2012, 134, 17436–17439 CrossRef CAS PubMed.
- T. Ogoshi, K. Kida and T. Yamagishi, J. Am. Chem. Soc., 2012, 134, 20146–20150 CrossRef CAS PubMed.
- C. Li, S. Chen, J. Li, K. Han, M. Xu, B. Hu, Y. Yu and X. Jia, Chem. Commun., 2011, 47, 11294–11296 RSC.
- C. Li, K. Han, J. Li, H. Zhang, J. Ma, X. Shu, Z. Chen, L. Weng and X. Jia, Org. Lett., 2012, 14, 42–45 CrossRef CAS PubMed.
- (a) T. Ogoshi, K. Demachi, K. Kitajima and T.-A. Yamagishi, Chem. Commun., 2011, 47, 10290–10292 RSC; (b) Y. Chen, D. Cao, L. Wang, M. He, L. Zhou, D. Schollmeyer and H. Meier, Chem.–Eur. J., 2013, 19, 7064–7070 CrossRef CAS PubMed; (c) H. Huang, L. Liu, W. Duan, Y. Huang and G. Lin, Chin. J. Chem., 2015, 33, 384–388 CrossRef CAS.
- T. Ogoshi, R. Shiga, M. Hashizume and T. Yamagishi, Chem. Commun., 2011, 47, 6927–6929 RSC.
- W. B. Hu, W. J. Hu, X. L. Zhao, Y. A. Liu, J. S. Li, B. Jiang and K. Wen, J. Org. Chem., 2016, 81, 3877–3881 CrossRef CAS PubMed.
- G. Yu, Y. Ma, C. Han, T. Yao, G. Tang, Z. Mao, C. Gao and F. Huang, J. Am. Chem. Soc., 2013, 135, 10310–10313 CrossRef CAS PubMed.
- T. Al-Azemi, M. Vinodh, F. Alipour and A. Mohamod, J. Org. Chem., 2017, 82, 10945–10952 CrossRef CAS PubMed.
- T. Al-Azemi, A. Mohamod, M. Vinodh and F. Alipour, Org. Chem. Front., 2018, 5, 10–18 RSC.
- T. Al-Azemi, M. Vinodh, F. Alipour and A. Mohamod, Org. Biomol. Chem., 2018, 7513–7517 RSC.
- T. Ogoshi, T. Aoki, K. Kitajima, S. Fujinama, T. A. Yamagishi and Y. Nakamoto, J. Org. Chem., 2011, 76, 328–331 CrossRef CAS PubMed.
- T. Ogoshi, D. Yamafuji, D. Kotera, T. Aoki, S. Fujinami and T.-A. Yamagishi, J. Org. Chem., 2012, 77, 11146–11152 CrossRef CAS PubMed.
- J. Han, X. Hou, C. Ke, H. Zhang, N. L. Strutt, C. L. Stern and J. F. Stoddart, Org. Lett., 2015, 17, 3260–3263 CrossRef CAS PubMed.
- J. P. Wei, X. Yan, J. Li, Y. Ma and F. Huang, Chem. Commun., 2013, 49, 1070–1072 RSC.
- Y. Yao, M. Xue, X. Chi, Y. Ma, J. He, Z. Abliz and F. Huang, Chem. Commun., 2012, 48, 6505–6507 RSC.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1880766–1880769. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra02313e |
| This journal is © The Royal Society of Chemistry 2019 |