
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonitoring spin coherence of single nitrogen-vacancy centers in nanodiamonds during pH changes in aqueous buffer solutions†
Masazumi Fujiwara *ab,
Ryuta Tsukaharab,
Yoshihiko Serab,
Hiroshi Yukawacde,
Yoshinobu Babacdefg,
Shinichi Shikatab and
Hideki Hashimotob
*ab,
Ryuta Tsukaharab,
Yoshihiko Serab,
Hiroshi Yukawacde,
Yoshinobu Babacdefg,
Shinichi Shikatab and
Hideki Hashimotob
aDepartment of Chemistry, Graduate School of Science, Osaka City University, Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: masazumi@sci.osaka-cu.ac.jp; masazumi@osaka-cu.ac.jp
bSchool of Science and Technology, Kwansei Gakuin University, Sanda, Hyogo 669-1337, Japan
cDepartment of Biomolecular Engineering, Graduate School of Engineering, Nagoya University, Chikusa-ku, Nagoya 464-8603, Japan
dInstitute of Nano-Life-Systems, Institutes of Innovation for Future Society, Nagoya University, Chikusa-ku, Nagoya 464-8603, Japan
eInstitute of Quantum Life Science, National Institutes for Quantum and Radiological Science and Technology, Anagawa 4-9-1, Inage-ku, 263-8555, Chiba, Japan
fHealth Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), Takamatsu 761-0395, Japan
gCollege of Pharmacy, Kaohsiung Medical University, Kaohsiung, 807, Taiwan, Republic of China
First published on 23rd April 2019
Abstract
We report on the sensing stability of quantum nanosensors in aqueous buffer solutions for the two detection schemes of quantum decoherence spectroscopy and nanoscale thermometry. The electron spin properties of single nitrogen-vacancy (NV) centers in 25 nm-sized nanodiamonds have been characterized by observing individual nanodiamonds during a continuous pH change from 4 to 11. We have determined the stability of the NV quantum sensors during the pH change as the fluctuations of ±12% and ±0.2 MHz for the spin coherence time (T2) and the resonance frequency (ω0) of their mean values, which are comparable to the instrument error of the measurement system. We discuss the importance of characterizing the sensing stability during the pH change and how the present observation affects the measurement scheme of nanodiamond-based NV quantum sensing.
1 Introduction
Diamond nitrogen-vacancy (NV) centers have attracted much attention as nanoscale quantum sensors.1–9 NV centers possess unpaired electron spins in diamond lattice structures that can be optically detected (optically detected magnetic resonance: ODMR) with ultra-high sensitivity down to the single electron spin level.10–12 These electron spin properties, such as the resonance frequency and the spin relaxation time, are dependent on multiple physical quantities (magnetic field, electric field, and temperature), so that one can measure their local values around the NV quantum sensors.1–7This attractive multifunctionality of the NV quantum sensors, however, complicates data analysis under most physiological conditions. Among the NV-quantum sensing schemes, quantum decoherence spectroscopy13–16 and nanoscale thermometry17–20 are promising for biological applications. In decoherence spectroscopy, the change in the T2 coherence time is detected, while, in thermometry, the frequency shift of the electron spin resonance is observed. These spin properties (either T2 or resonance frequency ω0) can be simultaneously affected by various parameters, for example, local heat generation in cells17,18,21–24 or the local concentrations of ions13 and pH.25,26 Thus, before proceeding to real biological applications, one needs experiments under controlled conditions to exclude factors, other than the sensing target, that may change the spin properties.
Nanodiamonds are biocompatible and are excellent NV carriers that can be delivered into complex biological structures including cells, organelles, and tissues.20,27–29 This portability provides distinct advantages over bulk-diamond-NV centers in biological sensing. However, the spin properties of nanodiamond NV centers are more sensitive to the surface chemistry than the bulk-diamond NV centers because of the small distance between the NV centers and the surface, which degrades the spin properties.30–33 Moreover, the surface of nanodiamonds is neither uniform nor well defined in contrast to the flat surface of bulk diamond.34–37 The spin properties of nanodiamond NV centers are, thus, thought to be more susceptible in physiological conditions.
One of the most influential parameters in biochemical experiments is the pH. As chemical sensors, nanodiamonds are required to be used in a wide range of pH.38–40 For intracellular applications, nanodiamonds will experience various pH depending on the locations; for example of cellular uptake, endosomes show pH of 6.0–7.0 in the early stage of endocytosis and later around 4.0.41 The pH affects the surface potential of the nanodiamonds and has been demonstrated to convert the charge state of the NV centers between NV0 and NV−.42–44 Very recently there have been several reports that this charge-state instability indeed affects the ODMR measurements, such as compromising the measured spin relaxation time.45–47 Thus properly characterizing the spin coherence of the ND quantum sensors in various pH conditions is required.
Here, we report on the spin-coherence stability of single nanodiamond-NV quantum sensors during a continuous pH change between 4 and 11, the range that is of particular importance for biochemical experiments. During the pH change, the NV spin coherence time (T2) and the spin resonance frequency (ω0) did not show any particular dependence on the pH but fluctuations corresponding to that observed in the steady-pH buffer solutions. We have determined the sensor stability of the NV quantum systems, which provides the fluctuations of ±12% and ±0.2 MHz for T2 and ω0 of their mean values. The observed fluctuations are discussed in relation to the NV-quantum sensing schemes such as decoherence spectroscopy and thermometry.
2 Experiments
2.1 Sample preparation
A commercially available nanodiamond suspension (Microdiamant, MSY 0-0.05, median particle size: 25 nm) was purified by centrifugation and dispersed in distilled water. A small droplet of the suspension was spin-coated on a cleaned coverslip to disperse and immobilize the nanodiamond particles on the coverslip surface. A 25 μm-thin copper wire was placed on the coverslip and both of the ends were soldered with electric connectors. An acrylic chamber with a height of a few millimeters with inlet and outlet tubes was then glued on the coverslip using a UV-curing resin. It was sealed with a glass plate.The topographies of the spin-coated samples were obtained using an atomic force microscope (AFM, Bruker, Edge). The AFM image is shown in Fig. 1a. The peak heights of the distributed nanodiamonds were measured to obtain the particle size distribution, which indicated a mean particle size of 30 nm based on log-normal fitting (Fig. 1b). Dynamic light scattering and zeta-potential measurements (Malvern, Zetasizer Nano ZS) were performed, which provides a mean particle size of 58 nm (log-normal fitting) and zeta-potential of smaller than −35 mV for all the pH region of more than 4 (see Fig. S2†).
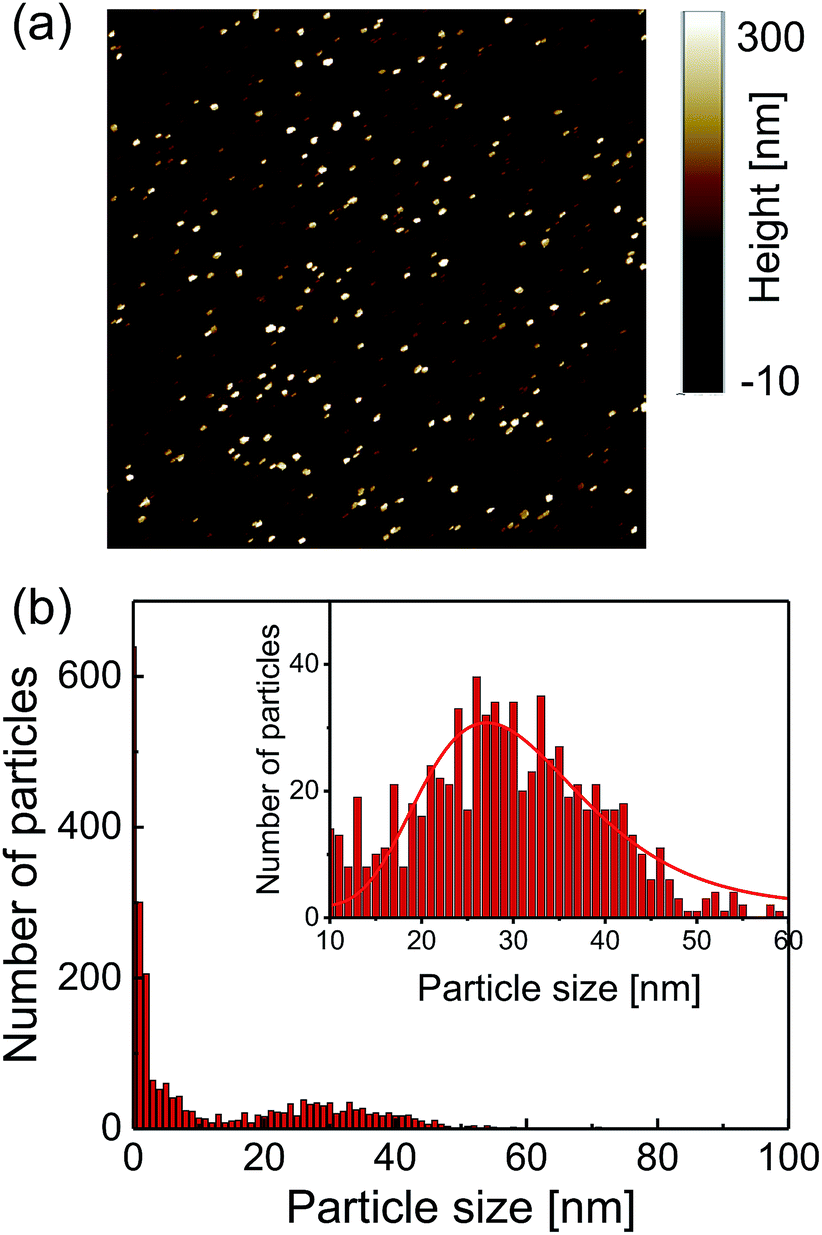 | ||
| Fig. 1 (a) Atomic force microscopy topography image of 8 × 8 μm region and (b) the corresponding particle size distribution. A large number of particles smaller than 10 nm in the region are considered to be debris (not diamond) included in the centrifugation process. | ||
2.2 Optical measurements
The perfusion chamber was mounted on a three-axis piezo stage and observed by a home-built confocal fluorescence microscope (Fig. 2a). A continuous-wave 532 nm laser was used for the excitation with a typical excitation intensity of ca. 90 kW cm−2 for the image scanning and second-order photon correlation measurements, which is near the fluorescence saturation laser intensity (ESI Fig. S1†). An oil-immersion microscope objective with numerical aperture of 1.4 was used for both the excitation and the fluorescence collection. The NV fluorescence was filtered by a dichroic beam splitter (Semrock, FF560-FDi01) and a long pass filter (Semrock, BLP01-561R) to remove the residual green laser scattering. The microscope was then coupled to an optical fiber that acted as a pinhole (Thorlabs, 1550HP, core diameter ca. 10 μm). The fiber-coupled fluorescence was finally guided into a Hanbury-Brown–Twiss (HBT) setup that consisted of two avalanche photodiodes (PerkinElmer SPCM AQRH-14) and a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 beam splitter. For the spectral measurements, the microscope was connected to a fiber-coupled spectrometer equipped with a liquid-nitrogen cooled charge-coupled device (CCD) camera (Princeton, LNCCD). By scanning the sample with the piezo stage, we were able to obtain fluorescence scanning images of the nanodiamonds. A time-correlated single-photon counting module (PicoQuant, TimeHarp-260) was used to obtain second-order photon correlation histograms to identify single NV centers by measuring the antibunching.
:50 beam splitter. For the spectral measurements, the microscope was connected to a fiber-coupled spectrometer equipped with a liquid-nitrogen cooled charge-coupled device (CCD) camera (Princeton, LNCCD). By scanning the sample with the piezo stage, we were able to obtain fluorescence scanning images of the nanodiamonds. A time-correlated single-photon counting module (PicoQuant, TimeHarp-260) was used to obtain second-order photon correlation histograms to identify single NV centers by measuring the antibunching.
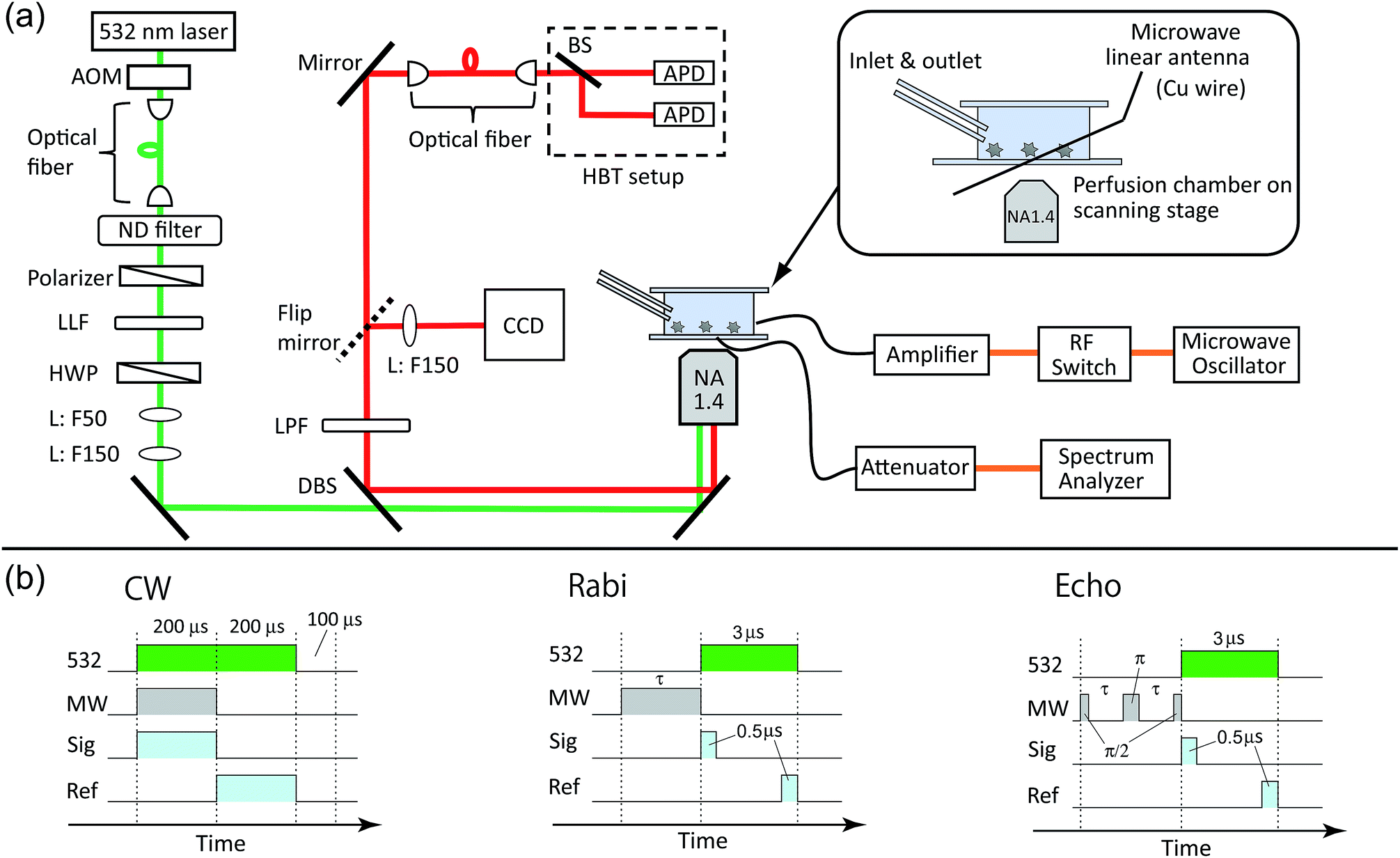 | ||
| Fig. 2 (a) Schematic drawings of the experimental setup for the perfusion chamber, optical layout and microwave circuit. AOM: acousto-optic modulator. ND: neutral density: LLF: laser line filter. HWP: half-wave plate. L: lens. DBS: dichroic beam splitter. LPF: long pass filter. CCD: charge-coupled device camera. BS: beam splitter. APD: avalanche photodiode. (b) The pulse sequences used for the electron spin measurements for CW, Rabi, and echo. 532: green laser. MW: microwave. Sig: signal counter. Ref: reference counter. | ||
The perfusion chamber was first filled with water and then exchanged with the buffer solutions. We used two kinds of mixed buffer solutions to measure the pH range between 4–7 and 7–11. A citric acid (0.1 M)–Na2HPO4 (0.2 M) mixed buffer solution was used to control the pH range to 4–7. For the pH range of 7–11, a Na2CO3 (0.1 M)–HCl (0.5 M) mixed solution was used. The pH of the solutions was varied stepwise by ΔpH ∼ 1 by changing the mixing ratio of the two constituents. During the optical excitation, these solutions were pumped through the perfusion chamber continuously at a rate of 80 μL min−1 to prevent photothermal aggregation of the nanoparticles48 (these nanoparticles may be nanodiamonds detached from other places or ionic salt nanocrystals created by mixing the buffer solutions).
2.3 Electron spin resonance (ESR) measurements
Microwaves were generated from a microwave source (Rohde & Schwarz, SMB100A) and amplified by 45 dB (Mini-circuit, ZHL-16W-43+). The microwaves were fed to the microwave linear antenna in the perfusion chamber (Fig. 2a). The typical microwave excitation power for the continuous-wave ODMR spectral measurement was 35 dBm (3.2 W). The avalanche photodiode (APD) detection was gated for microwave irradiation ON and OFF by using a radiofrequency (RF) switch (Mini-circuit, ZYSWA-2-50DR-S) and a bit pattern generator (Spincore, PBESR-PRO-300).49 The gate width was 200 μs, common to both gates, followed by a laser shut-off time of 100 μs, giving IONPL and IOFFPL (see Fig. 2b) with a repetition rate of 2 kHz. Note that an external magnetic field was not applied in this study. We selected NV centers that showed naturally single or doubly split (and well separated in frequency) ODMR peaks to excite only the single resonance peak of either of the transitions between |0〉 → | ±1〉 in the following pulsed ODMR measurements.50,51 The Rabi and spin echo measurements were performed on either of |0〉 → | ±1〉 transitions identified from the cw ODMR spectra. The Rabi signal determines the pulse durations of π/2 and π pulses for the subsequent spin echo measurements (see Fig. 2d). The spin echo measurements determine the spin coherence time (T2). Note that we measured both π/2–π–π/2 and π/2–π–3π/2 sequences and subtracted these signals from each other to cancel the common mode noise in the spin echo measurements.523 Results and discussion
Fig. 3a shows a confocal fluorescence scanning image of the nanodiamonds in the citric-acid–Na2HPO4 buffer solution of pH 7.0; there is a fluorescent spot at the center. Fig. 3b and c show the second-order photon correlation histogram and the spectrum of the emitted fluorescence, respectively. By curve fitting to the data based on the equation in ref. 53, we determined the excited-state lifetime to be 13.0 ns and the lifetime of the nearby metastable state to be 95.8 ns. The fluorescence spectrum consists of a zero-phonon line at around 637 nm and a broad phonon sideband ranging from 650 to 750 nm. These observed fluorescence properties allowed us to clearly identify nanodiamonds that incorporate single NV centers for the subsequent measurements. Note that the probability of finding NV centers in nanodiamonds is less than 5% and they were mostly single. | ||
| Fig. 3 (a) A confocal fluorescence scanning image of the nanodiamonds in the citric-acid–Na2HPO4 buffer solution at pH 7.0. (b) A second-order fluorescence photon correlation histogram of the central fluorescence spot in (a). The accumulation time was 100 s. (c) The fluorescence spectrum of the emission. (d) The ODMR spectrum of the NV center. (e) Its Rabi profile and (f) spin-echo profile. The Rabi and spin echo measurements were performed on the |0〉 → | −1〉 transition (indicated by the arrow) identified from the cw-ODMR spectrum in this measurement. | ||
After we had identified single NV centers, we measured the ODMR signals. Fig. 3d shows the ODMR spectrum of a single NV center. The peak splits without an external magnetic field because of the lattice strain in the nanodiamond, which is well known for NV centers in nanodiamonds.50,51 We set the microwave frequency to either of the two peaks (here, the left peak is chosen: 2.8660 GHz). Fig. 3e and f are temporal profiles of the Rabi and spin echo sequences. In the Rabi sequence, the microwave pulse duration was varied and the resultant fluorescence change was recorded. By fitting the damped sinusoidal function to the data, we determined the time duration of the π pulse. With this π pulse (and its half pulse π/2), the spin echo can be measured. The spin echo signal shows exponential decay with a spin coherence time (T2) of 1862 ns. We performed this set of the spin measurements each time that the pH was changed. Note that the first dot (20–40 ns) of the spin echo signal is omitted from the curve fitting because the pulse duration is not as short as designed because of the timing jitter of the RF switch (ca. 10 ns). Note also that we used a single exponential fitting to all the following data because of the short T2 time. It is well known that spin echo profiles show exp[−(2t/T2)α] with α = 1–3 when T2 is long, such as 10 μs.51,54,55 However, the echo profile can be approximated as a single exponential when T2 time is short. We, therefore, use single exponential fitting by taking α = 1 in the present study.
Fig. 4 shows the ODMR spectra and the corresponding echo profiles of the single NV center at pH 4 to 7. In the spin echo measurements, we excited the left peak. The ODMR spectra are almost the same during the pH change and the echo profiles basically show single exponential decay. The T2 times are 2510, 2138, 2440, and 1862 ns at pH 4.1, 5.0, 6.0, and 7.0, respectively. Note that Fig. 3f was measured at pH 7.0. There was a fluctuation in the T2 coherence time, but there is no clear dependence on the pH. We, therefore, measured the echo profiles at different pH values for a number of single NV centers in the nanodiamonds to assess the pH dependence by statistical means. We measured the T2 coherence time at each pH and normalized the value to the mean value (TM2) to show the relative deviation of the measurement to the mean (T2/TM2). The results are graphically summarized in Fig. 5a. Analogously, the resonance frequency was measured to determine the fluctuating peak shift from its mean value (Fig. 5b). Both the T2 time and the resonance frequency show a certain range of fluctuations with typical errors of 5–15% for T2 and 0.06–0.18 MHz for ω0, but there is no clear dependence on the pH. The optical excitation power used in these measurements were 18–50 kW cm−2.
 | ||
| Fig. 4 The CW-ODMR spectra (left panel) and the corresponding spin-echo profiles (right panel) of the single NV center during a pH change from 4.1 to 7.0. | ||
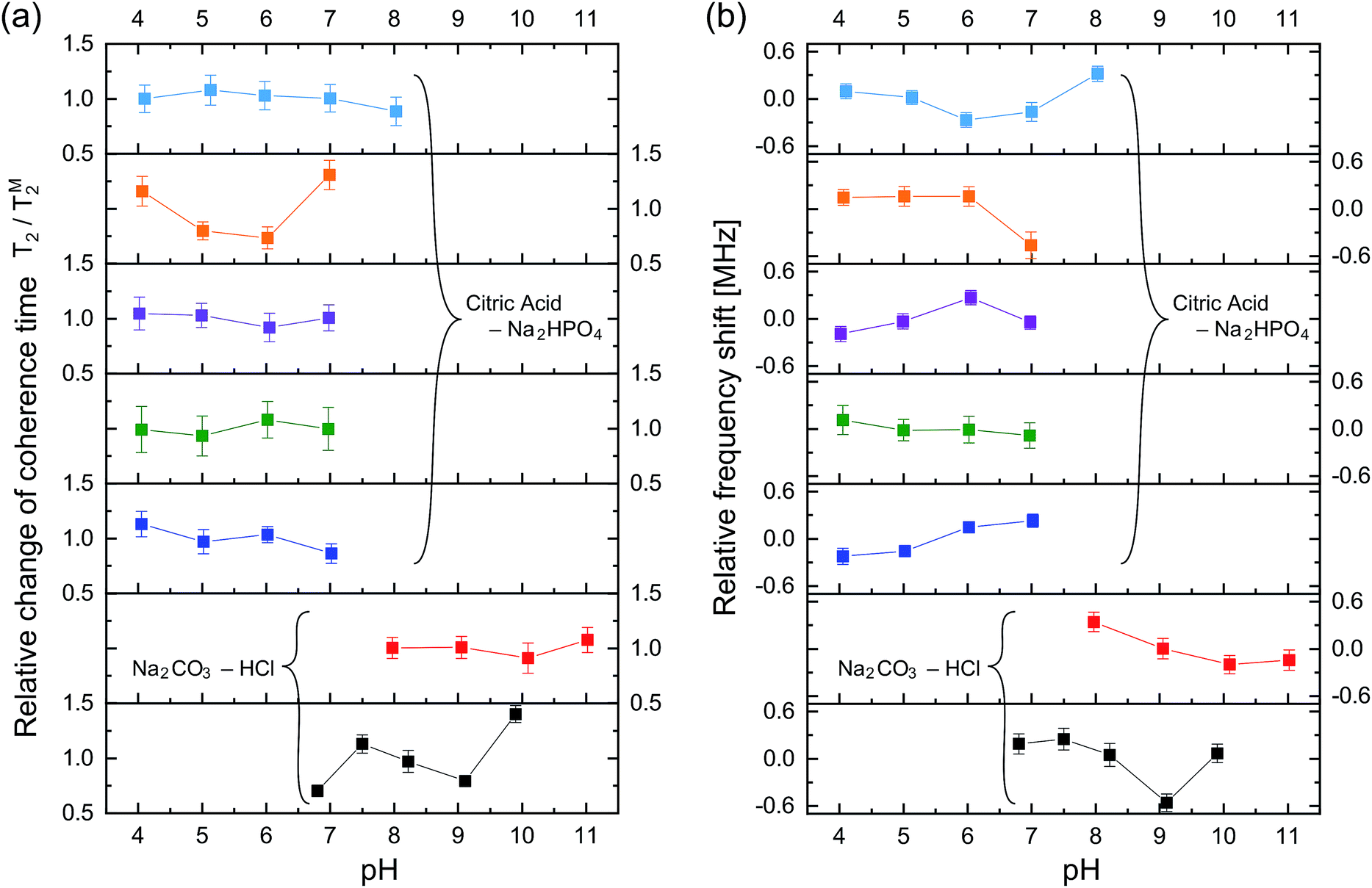 | ||
| Fig. 5 (a) The fluctuation of each T2 measurement value relative to its mean (TM2) as a function of pH. In total, seven nanodiamonds were investigated. All the nanodiamonds contain single NV centers. The upper-left five nanodiamonds were measured in the phosphate-based buffer solution and the lower right two were in the carbonate-based buffer solution. The solid lines are for ease of visualization. (b) The corresponding ω0 fluctuation of the respective NV center to its mean over the pH range. The error bars are the fitting errors. | ||
In addition to these statistical measurements, we measured the NV sensing stability and repeatability in the same pH range with lower optical excitation intensity of 5.4 kW cm−2 to avoid associated effects of the high optical excitation intensity, such as optical decoherence (laser power broadened linewidth) and photoionization of the NV charge state, since these effects might affect the NV sensing stability determined in the above experiments. Fig. 6a and b show T2 and ω0 of a single NV center during the repeated pH change from 6.1 → 5.1 → 4.1 → 5.1 → 6.0 → 7.0 → 8.0 → 7.0 → 6.0 with a step size of ΔpH = 1. The data again show no significant dependence of the NV sensing ability on the pH change but show a certain range of fluctuations. While T2 shows almost constant value within the error range, ω0 exhibits a relatively large shift compared with its error on the order of hundreds of kHz up to 1 MHz.
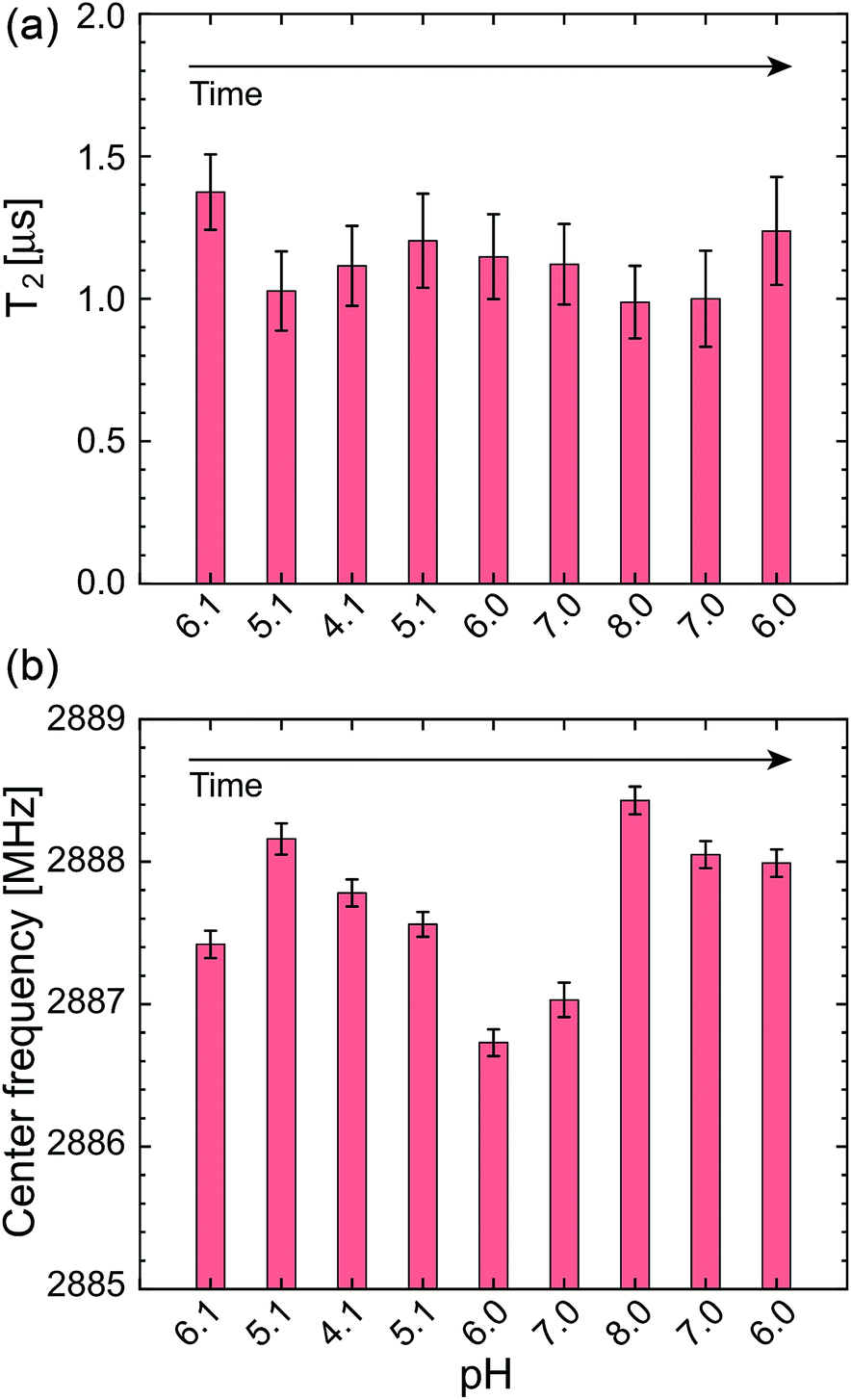 | ||
| Fig. 6 (a) T2 and (b) ω0 of a single NV center during the repeated pH change of 6.1 → 4.1 → 8.0 → 6.0, measured with a lower optical excitation intensity of 5.4 kW cm−2. The error bars are the fitting errors. | ||
To clarify the origin of the fluctuation, we quantified the experimental error of the present quantum spin measurement scheme. We measured the fluctuations of the T2 time and resonance frequency over 19 h for single NV centers in the same buffer solution at constant pH of 6.1 with the similar optical excitation intensity of 5.4 kW cm−2. It was observed that ω0 shows sudden shifts on the order of hundreds of kHz as can be seen in Fig. 6. The standard deviation of this 19 h measurement is ±6.2% of the mean T2 time and ±0.24 MHz of the mean ω0 (see Table 1). Nevertheless, most of the data points shown in Fig. 5 are located within the range of this measurement fluctuation, indicating that the pH does not affect the T2 and ω0 within the error range of the present spin measurement parameters (number of accumulation, step size, etc.…). It is therefore concluded that the effect of pH to T2 and ω0 is smaller than the instrument measurement error of the present experimental system. Note that the stability of the NV spin properties measured in the buffer solutions is almost the same (or even better) as that in an air environment (see Table 2).
| Time [h] | ωR0 [GHz] | ΔωR0 [MHz] | T2 [ns] | ΔT2 [ns] |
|---|---|---|---|---|
| 0 | 2.88757 | 0.10 | 1381 | 188 |
| 2.03 | 2.88766 | 0.09 | 1251 | 143 |
| 5.8 | 2.88898 | 0.12 | 1060 | 120 |
| 13.4 | 2.88856 | 0.09 | 1343 | 142 |
| 17.4 | 2.88837 | 0.10 | 937 | 158 |
| 19.1 | 2.88742 | 0.10 | 1374 | 133 |
| Mean | 2.88809 | 1224 | ||
| Std. err. | 0.24 [MHz] | 76 [ns] |
| Time [h] | ω0 [GHz] | Δω0 [MHz] | T2 [ns] | ΔT2 [ns] |
|---|---|---|---|---|
| 0 | 2.88758 | 0.06 | 620 | 81 |
| 4.42 | 2.88763 | 0.11 | 531 | 47 |
| 17.2 | 2.88830 | 0.12 | 978 | 102 |
| 20.3 | 2.88857 | 0.14 | 916 | 92 |
| 23.25 | 2.88916 | 0.09 | 504 | 33 |
| 24.95 | 2.88934 | 0.01 | 420 | 42 |
| Mean | 2.88843 | 662 | ||
| Std. err. | 0.30 [MHz] | 94 (14%) |
It should be noted that the fluctuations of T2 and ω0 mainly come from the fluctuations of the fluorescence photon counts and the ODMR spectral shape. The pH change and aqueous environment affect the NV charge stability and hence fluorescence emission properties. Since the NV spin measurement relies on the fluorescence detection, these fluorescence fluctuation significantly affects the sensitivity and precision of the NV quantum sensors, i.e. making noise. While we have not observed any particular pH dependence of the NV quantum sensors, this noise can be regarded as the effect of pH change or buffer solutions to the NV quantum sensing.
Indeed, during the long-term optical excitation, some NV centers were destabilized. These NV centers showed stable fluorescence initially but were later destabilized, resulting in fluorescent bursts or blinking, as shown in Fig. S2.† Such an instability is caused by the optical excitation in aqueous solutions and has not been observed in air, which causes the measurement noise in the end. We believe that this destabilization is related to the surface adsorption of water or solvent molecules, causing the photoionization of NV charge states. This point is important when performing the spin measurements in aqueous environment, and the continuous irradiation by the laser no longer guarantees the stability of the NV-fluorescent probes, particularly at the single NV level. The effect of the photoionization needs to be reduced by decreasing the laser power in case of the long-term tracking of nanodiamonds, though robustness of the NV centers is different particle by particle (see Fig. S3† for example). Increasing the surface oxidation coverage can also improve the photostability. The photoionization is related to the charge instability of NV centers, which switches between NV0 and NV−. Such charge state conversion is strongly related to the surface termination of nanodiamonds, as has been demonstrated in relation to the charge conversion between these two states by applying an electric potential and surface termination.42–45,56 The surface-oxidized diamonds do not show charge conversion under the electric field because the band is lower than the potential of the electrolyte. The surfaces of our present nanodiamonds could be oxidized further by acid cleaning or high-temperature treatment to prevent destabilization in aqueous solutions.57,58 Note that measurement of the zeta potential of the nanodiamonds in the present experimental conditions is not possible because high ionic strength of the present buffer solutions causes the sedimentation of nanodiamonds.59–62 Zeta potential measurements in diluted suspensions are possible and performed as described in Fig. S4.†
We also note that the present experiment does not provide information on the relationship between the surface pH and NV spin properties. The surface inhomogeneity by which the nanodiamond-NV centers might have some pH dependence could result in significant differences between the surface pH and the bulk pH. The surface pH is an important parameter for nanoparticle science63 in biological applications, and it may be necessary to study the effect of the surface pH in the future.
The present results concerning the fluctuations of T2 and ω0 in the quantum spin measurement schemes provide an important direction for the real implementation of nanoscale thermometry17–20 and quantum decoherence spectroscopy.13,14,16 For example, NV-nanodiamond thermometry is promising for biological analysis and is expected to allow the observation of cells in activated conditions, such as neuronal excitation20,64,65 and mitochondrial activity.22 The resonance frequency of ODMR has a temperature dependence of −74 kHz K−1 and, through this change, one can measure the local temperature around the nanodiamonds. A realistic temperature range monitored during mammalian cellular activities is 34–42 °C with a standard temperature of 38 °C.21–24 This means that only a temperature change of ±4 °C (±0.3 MHz), at largest, is expected.
Since our measurement method based on the curve fitting to the whole spectral profile is useful as a first choice given our experimental error of ω0 to be ±0.2 MHz over the 16 h measurement, more stable and high precision schemes to determine ω0 with a fast acquisition time should be implemented. While the present method based on curve-fitting to the whole spectral profile is useful as a first choice in the biological applications, it takes a long measurement time to reach the sufficient precision. With the data acquisition time of 12 min, it can reach an error of ±35 kHz for ω0 (Fig. S4†), though it is not realistic to spend more than 12 min to determine the parameters. Ratiometric analysis of the ODMR spectral shape may be a good choice to simultaneously obtain the precision and the fast acquisition speed. Another technique that may overcome this measurement error is a use of statistical analysis of number of NV nanodiamonds to obtain high precision.20 The same discussion also applies to the decoherence spectroscopy where the T2 relaxation time is fitted to the whole profile of the exponential decay of the spin coherence time. In case of the decoherence spectroscopy, single point analysis has been proposed and demonstrated for T1 measurements.66 Implementing these ratiometric analyses or statistical analysis seem necessary for the real biological applications.
4 Conclusion
In conclusion, we have reported the effects of aqueous buffer solutions on the electron spin properties of single NV centers in 25 nm-sized nanodiamonds by tracking individual nanodiamonds during continuous pH changes in the range of pH 4 to 11. During the pH changes, the T2 time and the spin resonance frequency did not show any particular pH dependence but did show fluctuations that correspond to the experimental errors observed in the steady-pH buffer solutions over 19 h (±23% and ±0.2 MHz for T2 and ω0 of their mean values). The observed fluctuations are significant when performing nanoscale thermometry and quantum decoherence spectroscopy in various biological contexts and more stable and faster measurement schemes should be necessary such as ratio-metric analyses.Measuring the stability of the spin properties of single nanodiamond-NV centers during the pH change is important to the future development of nanodiamond-based NV quantum sensing, because the measured spin coherence can be compromised by the charge-state instability of NV centers as recently reported.47 Furthermore, bare nanodiamonds cannot be dispersed in high-ionic-strength buffer solutions59,60 that include most biochemical buffers, such as phosphate-buffered-saline cell culture media. Consequently, it is important to functionalize the nanodiamond surface to prevent aggregation or binding with other biological molecules.67,68 Surface functionalization is also used for nanodiamond-based pH sensors.38 However, after the surface functionalization, nanodiamonds must maintain the original spin properties at various pH values. The present results and measurement scheme can provide a way to evaluate the stability of such quantum sensors in the stage of material development before the use, thus allowing the exploration of the applications of NV quantum sensors for biological and biochemical applications.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank Prof. Noboru Ohtani for the AFM measurements and Ms. Kaori Kobayashi and Mr Yuta Ueda for the support in zeta-potential measurements. MF acknowledges financial support from JSPS-KAKENHI (No. 26706007, 26610077, and 17H02741), MEXT-LEADER program, and Osaka City University (OCU-Strategic Research Grant 2017 for young researchers). MF, HY thank JSPS-KAKENHI (No. 16K13646). SS acknowledges financial support from JSPS-KAKENHI (No. 26220602). HH thanks JSPS KAKENHI, Grant-in-Aids for Basic Research (B) (No. 16H04181) and Scientific Research on Innovative Areas “Innovations for Light-Energy Conversion (I4LEC)” (No. 17H06433, 17H0637) for financial support.References
- G. Balasubramanian, I. Chan, R. Kolesov, M. Al-Hmoud, J. Tisler, C. Shin, C. Kim, A. Wojcik, P. R. Hemmer, A. Krueger, H. Tobias, A. Leitenstorfer, R. Bratschitsch, F. Jelezko and J. Wrachtrup, Nature, 2008, 455, 648–651 CrossRef CAS PubMed.
- M. W. Doherty, N. B. Manson, P. Delaney, F. Jelezko, J. Wrachtrup and L. C. Hollenberg, Phys. Rep., 2013, 528, 1–45 CrossRef CAS.
- J. Maze, P. Stanwix, J. Hodges, S. Hong, J. Taylor, P. Cappellaro, L. Jiang, M. G. Dutt, E. Togan, A. Zibrov, A. Yacoby, R. L. Walsworth and M. D. Lukin, Nature, 2008, 455, 644–647 CrossRef CAS PubMed.
- T. Plakhotnik, H. Aman, S. Zhang and Z. Li, J. Phys. Chem. C, 2015, 119, 20119–20124 CrossRef CAS.
- S. Hemelaar, P. de Boer, M. Chipaux, W. Zuidema, T. Hamoh, F. P. Martinez, A. Nagl, J. Hoogenboom, B. Giepmans and R. Schirhagl, Sci. Rep., 2017, 7, 5862 CrossRef PubMed.
- S. Ong, M. Chipaux, A. Nagl and R. Schirhagl, Phys. Chem. Chem. Phys., 2017, 19, 10748–10752 RSC.
- M. Fujiwara, K. Yoshida, T. Noda, H. Takashima, A. W. Schell, N. Mizuochi and S. Takeuchi, Nanotechnology, 2016, 27, 455202 CrossRef PubMed.
- M. Fujiwara, H.-Q. Zhao, T. Noda, K. Ikeda, H. Sumiya and S. Takeuchi, Opt. Lett., 2015, 40, 5702–5705 CrossRef CAS PubMed.
- M. Fujiwara, O. Neitzke, T. Schröder, A. W. Schell, J. Wolters, J. Zheng, S. Mouradian, M. Almoktar, S. Takeuchi and D. Englund, et al., ACS Omega, 2017, 2, 7194–7202 CrossRef CAS.
- F. Jelezko and J. Wrachtrup, Phys. Status Solidi A, 2006, 203, 3207–3225 CrossRef CAS.
- R. Hanson, O. Gywat and D. D. Awschalom, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 1–4 CrossRef.
- L. Childress, M. G. Dutt, J. Taylor, A. Zibrov, F. Jelezko, J. Wrachtrup, P. Hemmer and M. Lukin, Science, 2006, 314, 281–285 CrossRef CAS PubMed.
- L. T. Hall, C. D. Hill, J. H. Cole, B. Städler, F. Caruso, P. Mulvaney, J. Wrachtrup and L. C. Hollenberg, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18777–18782 CrossRef CAS PubMed.
- J. H. Cole and L. C. Hollenberg, Nanotechnology, 2009, 20, 495401 CrossRef PubMed.
- L. P. McGuinness, Y. Yan, A. Stacey, D. A. Simpson, L. T. Hall, D. Maclaurin, S. Prawer, P. Mulvaney, J. Wrachtrup, F. Caruso, R. E. Scholten and L. C. L. Hollenberg, Nat. Nanotechnol., 2011, 6, 358 CrossRef CAS PubMed.
- L. Luan, M. S. Grinolds, S. Hong, P. Maletinsky, R. L. Walsworth and A. Yacoby, Sci. Rep., 2015, 5, 8119 CrossRef CAS PubMed.
- G. Kucsko, P. Maurer, N. Yao, M. Kubo, H. Noh, P. Lo, H. Park and M. Lukin, Nature, 2013, 500, 54–58 CrossRef CAS PubMed.
- P. Neumann, I. Jakobi, F. Dolde, C. Burk, R. Reuter, G. Waldherr, J. Honert, T. Wolf, A. Brunner, J. H. Shim, D. Suter, H. Sumiya, J. Isoya and J. Wrachtrup, Nano Lett., 2013, 13, 2738–2742 CrossRef CAS PubMed.
- Y.-K. Tzeng, P.-C. Tsai, H.-Y. Liu, O. Y. Chen, H. Hsu, F.-G. Yee, M.-S. Chang and H.-C. Chang, Nano Lett., 2015, 15, 3945–3952 CrossRef CAS PubMed.
- D. A. Simpson, E. Morrisroe, J. M. McCoey, A. H. Lombard, D. C. Mendis, F. Treussart, L. T. Hall, S. Petrou and L. C. Hollenberg, ACS Nano, 2017, 11, 12077–12086 CrossRef CAS PubMed.
- J. S. Donner, S. A. Thompson, M. P. Kreuzer, G. Baffou and R. Quidant, Nano Lett., 2012, 12, 2107–2111 CrossRef CAS PubMed.
- K. Okabe, N. Inada, C. Gota, Y. Harada, T. Funatsu and S. Uchiyama, Nat. Commun., 2012, 3, 705 CrossRef PubMed.
- H. Liu, Y. Fan, J. Wang, Z. Song, H. Shi, R. Han, Y. Sha and Y. Jiang, Sci. Rep., 2015, 5, 14879 CrossRef CAS PubMed.
- T. Tsuji, K. Ikado, H. Koizumi, S. Uchiyama and K. Kajimoto, Sci. Rep., 2017, 7, 12889 CrossRef PubMed.
- C.-J. Tsou, C.-H. Hsia, J.-Y. Chu, Y. Hung, Y.-P. Chen, F.-C. Chien, K. C. Chou, P. Chen and C.-Y. Mou, Nanoscale, 2015, 7, 4217–4225 RSC.
- Y. V. Morimoto, N. Kami-Ike, T. Miyata, A. Kawamoto, T. Kato, K. Namba and T. Minamino, mBio, 2016, 7, e01911–e01916 CrossRef CAS PubMed.
- J. Beranová, G. Seydlová, H. Kozak, O. Benada, R. Fišer, A. Artemenko, I. Konopásek and A. Kromka, FEMS Microbiol. Lett., 2014, 351, 179–186 CrossRef PubMed.
- K. Turcheniuk and V. Mochalin, Nanotechnology, 2017, 28, 252001 CrossRef CAS PubMed.
- N. Mohan, C.-S. Chen, H.-H. Hsieh, Y.-C. Wu and H.-C. Chang, Nano Lett., 2010, 10, 3692–3699 CrossRef CAS PubMed.
- Y. Romach, C. Müller, T. Unden, L. Rogers, T. Isoda, K. Itoh, M. Markham, A. Stacey, J. Meijer and S. Pezzagna, et al., Phys. Rev. Lett., 2015, 114, 017601 CrossRef CAS PubMed.
- T. Ishikawa, K.-M. C. Fu, C. Santori, V. M. Acosta, R. G. Beausoleil, H. Watanabe, S. Shikata and K. M. Itoh, Nano Lett., 2012, 12, 2083–2087 CrossRef CAS PubMed.
- L. Rondin, G. Dantelle, A. Slablab, F. Grosshans, F. Treussart, P. Bergonzo, S. Perruchas, T. Gacoin, M. Chaigneau and H.-C. Chang, et al., Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 115449 CrossRef.
- X. Song, J. Zhang, F. Feng, J. Wang, W. Zhang, L. Lou, W. Zhu and G. Wang, AIP Adv., 2014, 4, 047103 CrossRef.
- R. Kaur and I. Badea, Int. J. Nanomed., 2013, 8, 203 CrossRef PubMed.
- A. Nagl, S. R. Hemelaar and R. Schirhagl, Anal. Bioanal. Chem., 2015, 407, 7521–7536 CrossRef CAS PubMed.
- H. Girard, T. Petit, S. Perruchas, T. Gacoin, C. Gesset, J. Arnault and P. Bergonzo, Phys. Chem. Chem. Phys., 2011, 13, 11517–11523 RSC.
- J. T. Paci, H. B. Man, B. Saha, D. Ho and G. C. Schatz, J. Phys. Chem. C, 2013, 117, 17256–17267 CrossRef CAS.
- T. Rendler, J. Neburkova, O. Zemek, J. Kotek, A. Zappe, Z. Chu, P. Cigler and J. Wrachtrup, Nat. Commun., 2017, 8, 14701 CrossRef CAS PubMed.
- F. Q. Schafer and G. R. Buettner, Free Radicals Biol. Med., 2001, 30, 1191–1212 CrossRef CAS PubMed.
- J. Slegerova, M. Hajek, I. Rehor, F. Sedlak, J. Stursa, M. Hruby and P. Cigler, Nanoscale, 2015, 7, 415–420 RSC.
- A. Sorkin and M. von Zastrow, Nat. Rev. Mol. Cell Biol., 2002, 3, 600 CrossRef CAS PubMed.
- S. Karaveli, O. Gaathon, A. Wolcott, R. Sakakibara, O. A. Shemesh, D. S. Peterka, E. S. Boyden, J. S. Owen, R. Yuste and D. Englund, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3938–3943 CrossRef CAS PubMed.
- C. Schreyvogel, V. Polyakov, R. Wunderlich, J. Meijer and C. Nebel, Sci. Rep., 2015, 5, 12160 CrossRef CAS.
- P. Ji and M. G. Dutt, Phys. Rev. B: Condens. Matter Mater. Phys., 2016, 94, 024101 CrossRef.
- H. Yamano, S. Kawai, K. Kato, T. Kageura, M. Inaba, T. Okada, I. Higashimata, M. Haruyama, T. Tanii, K. Yamada, S. Onoda, W. Kada, O. Hanaizumi, T. Teraji, J. Isoya and H. Kawarada, Jpn. J. Appl. Phys., 2017, 56, 04CK08 CrossRef.
- A. Stacey, N. Dontschuk, J.-P. Chou, D. A. Broadway, A. K. Schenk, M. J. Sear, J.-P. Tetienne, A. Hoffman, S. Prawer, C. I. Pakes, A. Tadich, N. P. de Leon, A. Gali and L. C. L. Hollenberg, Adv. Mater. Interfaces, 2018, 1801449 Search PubMed.
- D. Bluvstein, Z. Zhang and A. C. B. Jayich, Phys. Rev. Lett., 2019, 122, 076101 CrossRef PubMed.
- Y. Nishimura, K. Nishida, Y. Yamamoto, S. Ito, S. Tokonami and T. Iida, J. Phys. Chem. C, 2014, 118, 18799–18804 CrossRef CAS.
- M. Geiselmann, M. L. Juan, J. Renger, J. M. Say, L. J. Brown, F. J. G. De Abajo, F. Koppens and R. Quidant, Nat. Nanotechnol., 2013, 8, 175–179 CrossRef CAS PubMed.
- X. Liu, J. Cui, F. Sun, X. Song, F. Feng, J. Wang, W. Zhu, L. Lou and G. Wang, Appl. Phys. Lett., 2013, 103, 143105 CrossRef.
- J. Tisler, G. Balasubramanian, B. Naydenov, R. Kolesov, B. Grotz, R. Reuter, J.-P. Boudou, P. A. Curmi, M. Sennour, A. Thorel, M. Börsch, K. Aulenbacher, R. Erdmann, P. R. Hemmer, F. Jelezko and J. Wrachtrup, ACS Nano, 2009, 3, 1959–1965 CrossRef CAS PubMed.
- S. V. Bolshedvorskii, V. V. Vorobyov, V. V. Soshenko, V. A. Shershulin, J. Javadzade, A. I. Zeleneev, S. A. Komrakova, V. N. Sorokin, P. I. Belobrov, A. N. Smolyaninov and A. V. Akimov, Opt. Mater. Express, 2017, 7, 4038–4049 CrossRef CAS.
- M. Berthel, O. Mollet, G. Dantelle, T. Gacoin, S. Huant and A. Drezet, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 035308 CrossRef.
- H. S. Knowles, D. M. Kara and M. Atatüre, Nat. Mater., 2014, 13, 21–25 CrossRef CAS PubMed.
- L. McGuinness, L. Hall, A. Stacey, D. Simpson, C. Hill, J. Cole, K. Ganesan, B. Gibson, S. Prawer, P. Mulvaney, F. Jelezko, J. Wrachtrup, R. E. Scholten and L. C. L. Hollenberg, New J. Phys., 2013, 15, 073042 CrossRef.
- M. Hauf, B. Grotz, B. Naydenov, M. Dankerl, S. Pezzagna, J. Meijer, F. Jelezko, J. Wrachtrup, M. Stutzmann and F. Reinhard, et al., Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 081304 CrossRef.
- S. Stehlik, M. Varga, M. Ledinsky, V. Jirasek, A. Artemenko, H. Kozak, L. Ondic, V. Skakalova, G. Argentero and T. Pennycook, et al., J. Phys. Chem. C, 2015, 119, 27708–27720 CrossRef CAS PubMed.
- A. Wolcott, T. Schiros, M. E. Trusheim, E. H. Chen, D. Nordlund, R. E. Diaz, O. Gaathon, D. Englund and J. S. Owen, J. Phys. Chem. C, 2014, 118, 26695–26702 CrossRef CAS PubMed.
- N. Gibson, O. Shenderova, T. Luo, S. Moseenkov, V. Bondar, A. Puzyr, K. Purtov, Z. Fitzgerald and D. Brenner, Diamond Relat. Mater., 2009, 18, 620–626 CrossRef CAS.
- L. Zhao, T. Takimoto, M. Ito, N. Kitagawa, T. Kimura and N. Komatsu, Angew. Chem., Int. Ed., 2011, 50, 1388–1392 CrossRef CAS PubMed.
- Y. Zhang and A. Clapp, Sensors, 2011, 11, 11036–11055 CrossRef PubMed.
- Â. Barreto, L. G. Luis, A. V. Girão, T. Trindade, A. M. Soares and M. Oliveira, J. Nanopart. Res., 2015, 17, 493 CrossRef.
- S. Bellucci, Nanoparticles and Nanodevices in Biological Applications: The INFN Lectures, Springer Science & Business Media, 2008, vol. 1 Search PubMed.
- J. F. Barry, M. J. Turner, J. M. Schloss, D. R. Glenn, Y. Song, M. D. Lukin, H. Park and R. L. Walsworth, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14133–14138 CrossRef CAS PubMed.
- R. Tanimoto, T. Hiraiwa, Y. Nakai, Y. Shindo, K. Oka, N. Hiroi and A. Funahashi, Sci. Rep., 2016, 6, 22071 CrossRef CAS PubMed.
- D. A. Simpson, R. G. Ryan, L. T. Hall, E. Panchenko, S. C. Drew, S. Petrou, P. S. Donnelly, P. Mulvaney and L. C. Hollenberg, Nat. Commun., 2017, 8, 458 CrossRef PubMed.
- T. Genjo, S. Sotoma, R. Tanabe, R. Igarashi and M. Shirakawa, Anal. Sci., 2016, 32, 1165–1170 CrossRef CAS PubMed.
- S. Sotoma, J. Iimura, R. Igarashi, K. M. Hirosawa, H. Ohnishi, S. Mizukami, K. Kikuchi, T. K. Fujiwara, M. Shirakawa and H. Tochio, Nanomaterials, 2016, 6, 56 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02282a |
| This journal is © The Royal Society of Chemistry 2019 |