DOI:
10.1039/C9RA02213A
(Paper)
RSC Adv., 2019,
9, 14242-14246
Selective oxidation of 5-hydroxymethylfurfural into 2,5-diformylfuran over VPO catalysts under atmospheric pressure†
Received
22nd March 2019
, Accepted 2nd May 2019
First published on 7th May 2019
Abstract
Vanadium phosphate oxide (VPO) heterogeneous catalysts with different V/P molar ratios were prepared and used for selective oxidation of biomass-derived 5-hydroxymethylfurfural (HMF) to produce 2,5-diformylfuran (DFF) in the liquid phase. It was found that the VPO catalyst with V/P molar ratio 0.25 exhibited the best catalytic performance. Then the VPO catalyst was utilized to catalyze the oxidation of HMF in a batch reactor under different conditions, in terms of type of solvent (water and organic), reaction time and temperature. A high DFF yield of 83.6% with HMF conversion of 100% was obtained under atmospheric pressure.
Introduction
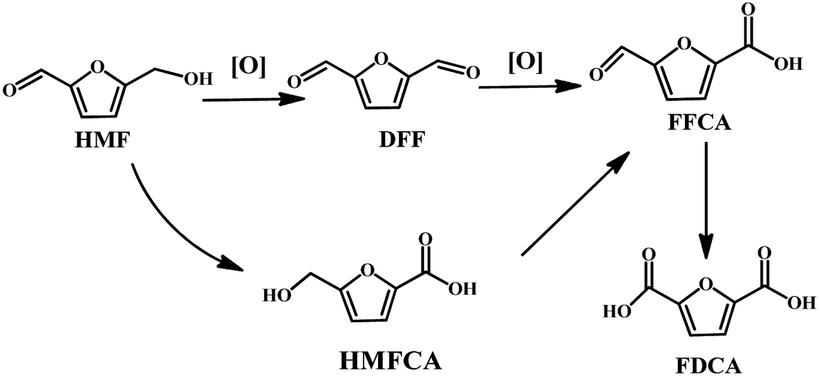
In recent years, the high value-added chemicals and fuels prepared from renewable natural biomass resources have captured intense interest, coined “biorefinery”.1–3 Among them, the dehydration of C6 based carbohydrates can generate an important platform chemical 5-hydroxymethylfurfural (HMF), which is considered to be one of the top value-added chemicals. 2,5-Diformylfuran (DFF) is one of the main products of HMF oxidation, and has been used as a starting material for the synthesis of ligands, drugs, pesticide antifungal agents, fluorescent materials and new polymeric materials.4,5
However, as shown in Scheme 1, the HMF structure contains a hydroxyl and a formyl moiety together with a furan ring, opening an avenue for chemical reactions. Therefore, the oxidation of HMF makes it susceptible to undergo many side reactions such as over oxidation of DFF into 2,5-furandicarboxylic acid (FDCA), the oxidation of aldehyde to 5-hydroxymethyl-2-furancarboxylic acid (HMFCA), decarbonylation, and cross-polymerization to produce unwanted byproducts. Therefore, it is still a challenge to selectively oxidize HMF into DFF.
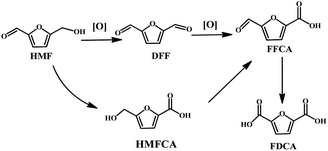 |
| | Scheme 1 Possible oxidation products from the oxidation of HMF. | |
How to achieve selective oxidation of HMF to prepare DFF under mild conditions, it has always been an important and challenging subject. To date, there are plenty of catalysts employed in selective oxidation of HMF into DFF, such as the manganese oxide-based material,6–8 GO-related materials,9 supported Ru catalysts10–12 and vanadium-based catalysts.13–16 Zhang et al.17–19 have been committed to research in this area and developed a series of catalysts with excellent catalytic activity. Due to the high price of noble metal, developing cheap transition metal-based heterogeneous catalysts for the synthesis of DFF from the selective oxidation of HMF is thus highly desirable. Among these catalysts, vanadium-containing catalysts are noticeable. In the past decade, some methods on the aerobic oxidation of alcohols using relatively cheap V based catalysts were reported. Inspired by the excellent catalytic activity of VPO catalysts in the oxidation of alcohols, herein, we prepared a series of VPO catalysts with different V/P molar ratios by using the liquid phase method, and used as a novel heterogeneous catalyst for the selective oxidation of HMF into DFF.
Experimental
Materials
5-Hydroxymethylfurfural (99.5%) and 2,5-diformylfuran were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). NH4VO3 and H3PO4 were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai China). All the chemicals were of analytical grade and used without further purification. Ultrapure water was used for the catalyst preparation and catalytic reactions.
Preparation of catalyst
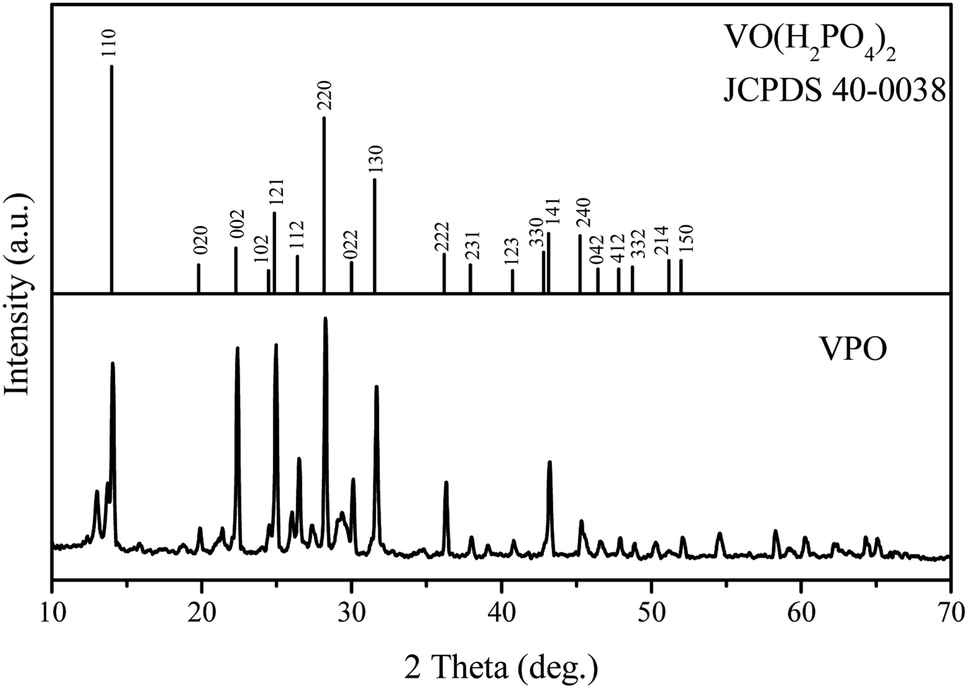
Vanadium phosphate oxide (VPO) was prepared according to the following procedure. V2O5 (1.4 g), derived from the pyrolysis of NH4VO3 at 180 °C, and H3PO4 (85%, 7 g) were refluxed in isobutanol (25 mL) at 120 °C for 12 h. The resulting solid was isolated by vacuum filtration, washed with isobutanol, and dried under vacuum at 80 °C. Powder X-ray pattern of the solid prasinous product corresponded to that found for VO(H2PO4)2.
Synthesis of DFF from HMF
The oxidation of HMF in the air under atmospheric pressure was performed in a 25 mL round bottom flask, which was coupled with a reflux condenser using the following general procedure. Typically, HMF (0.1 mmol, 12.6 mg) was firstly dissolved into DMSO (5 mL) in the flask with a magnetic stirrer. Then, 10 mg VPO catalysts were added into the reaction mixture. The mixture was heated to the desired temperature while stirring under an air atmosphere. After reaction, the reaction solution was quantitatively analyzed by HPLC. According to test results, the oxidation of HMF mainly leads to DFF, HMFCA, 5-formyl-2-furancarboxylic (FFCA) and FDCA.
Quantification of the products
The product analysis was performed using Agilent 1100 HPLC system equipped with Agela Technologies Venusil XBP C18(L) (4.6 × 250 mm) and a UV-Vis (280 nm) detector. The mobile phase was constituted of acetonitrile and 0.1 wt% acetic acid aqueous solution (v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v = 15:85), and the samples was eluted at a rate of 0.5 mL min−1 at 30 °C. The amount of each compound was quantified by external standard calibration curve method.
:v = 15:85), and the samples was eluted at a rate of 0.5 mL min−1 at 30 °C. The amount of each compound was quantified by external standard calibration curve method.
To calculate HMF conversion, DFF selectivity and yield are defined as follow:
| HMF conversion = (1 − moles of HMF/moles of HMF added) × 100% |
| DFF selectivity = moles of DFF/(moles of HMF added − moles of HMF) × 100% |
| DFF yield = moles of DFF/moles of HMF added × 100% |
Results and discussion
Catalyst characterization
The VPO catalyst was characterized using XRD, TEM, TG-DTG, Raman and FI-IR analysis. The XRD pattern in Fig. 1 illustrated the VO(H2PO4)2 crystal structure with JCPDS card 40-0038 for the VPO catalyst, demonstrating that this phase has been successfully synthesized. However, the VPO catalyst demonstrated the largely enhance (002) and (121) facet signal at 22.38° and 24.96°.
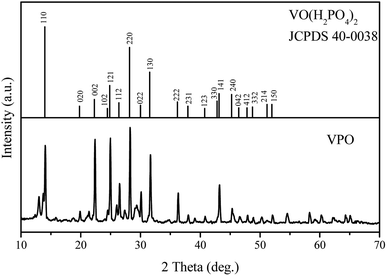 |
| | Fig. 1 XRD patterns of VPO. | |
Fig. 2 showed TEM images of VPO catalyst at different magnifications. The TEM observation in Fig. 2a verified a nanosheet structure of VPO covered by a different phase (Fig. 2b). This new phase seems to be a film forming some aggregates or clusters (Fig. 2c and d) on the nanosheets. From the Fig. 2, it can be clearly distinguished the aggregates with multiple grains of different sizes. In agreement with XRD, this dispersed phase seems to be a mixture of VPO mixed-oxide species.20,21
 |
| | Fig. 2 TEM images of VPO catalysts. | |
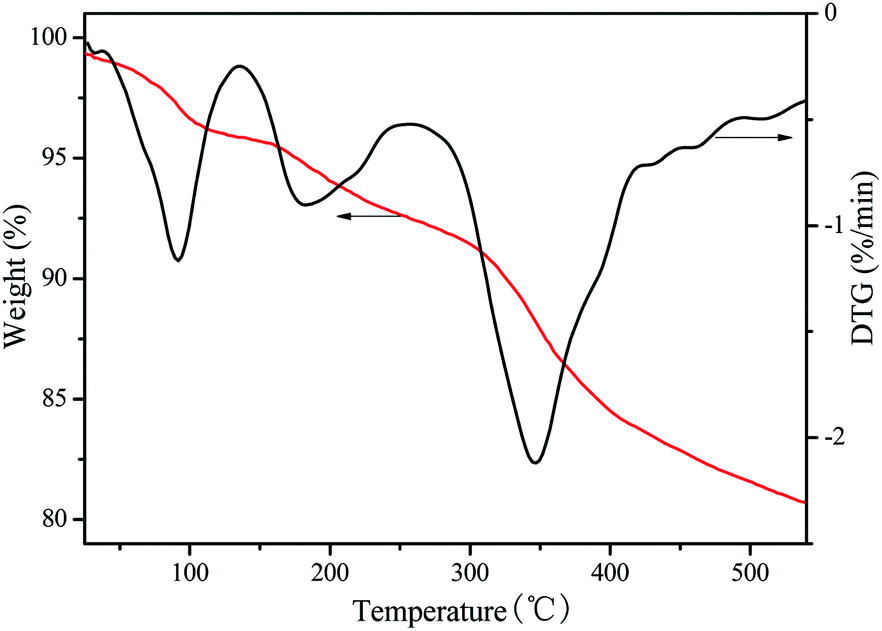
The thermogravimetric analysis for VPO sample carried out in nitrogen flow. It can be seen from the Fig. 3 that the VPO catalyst has three weight loss stages. Firstly, around 5% of mass loss at temperatures lower than 150 °C, due to the loss of absorbed water; second, the mass loss lower than 5% at temperatures range at 150–250 °C, due to the loss of crystal water; then, a flat region up to 300 °C; and then it starts to be decomposed into VO(PO3)2.22 Thus, this catalyst can be used under oxidizing conditions up to 300 °C. As expected, the VPO catalyst profile only presents a mass loss at low temperature due to absorbed water and then it does not present a weight loss when the temperature increased.
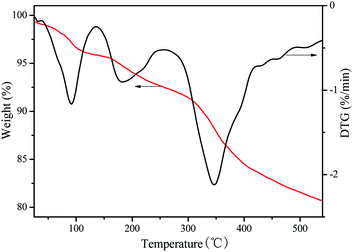 |
| | Fig. 3 TG-DTG curves of VPO. | |
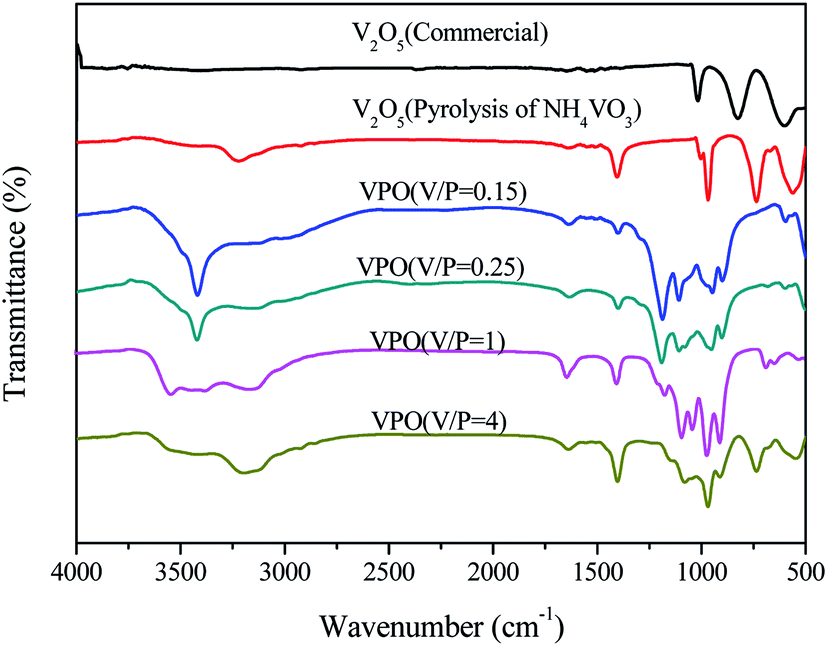
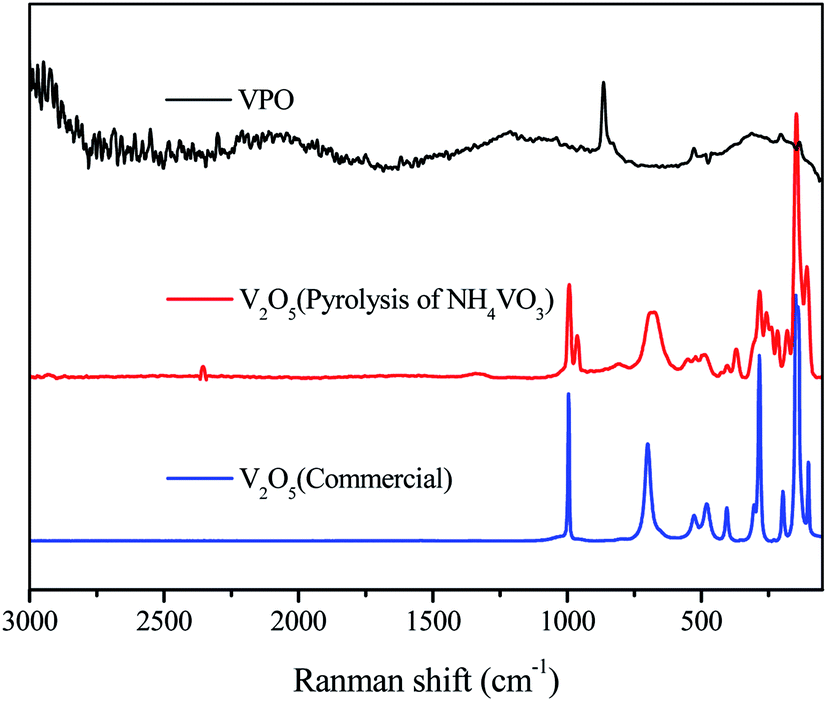
The FT-IR spectra of different V2O5 and a series of the VPO catalyst with different V/P molar ratios were analyzed (Fig. 4). The FT-IR spectra of different V2O5 illustrated the typical V2O5 crystal structure for both the commercial bulk V2O5 and the V2O5 derived from the pyrolysis of NH4VO3. However, the V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the V2O5, derived from the pyrolysis of NH4VO3, demonstrated the smaller wave number (below 1000 cm−1). And the adsorption peaks appearing at around 3200 cm−1 and 1400 cm−1 correspond to the N–H. For four curve of VPO, with the increase of V2O5 content in VPO, the characteristic absorption intensity of VO (below 1000 cm−1) increases continuously, and the vibration absorption intensity of two PO3 groups (1000–1300 cm−1) decreases, which differ by the type of symmetry.23 The broad peaks around 3600 cm−1 and 1640 cm−1 are attributed to the stretching and bending modes of the O–H of surface hydroxyl groups and physically adsorbed water. The peak at 3420 cm−1 arises from the O–H stretching vibration of V–O–H.
O of the V2O5, derived from the pyrolysis of NH4VO3, demonstrated the smaller wave number (below 1000 cm−1). And the adsorption peaks appearing at around 3200 cm−1 and 1400 cm−1 correspond to the N–H. For four curve of VPO, with the increase of V2O5 content in VPO, the characteristic absorption intensity of VO (below 1000 cm−1) increases continuously, and the vibration absorption intensity of two PO3 groups (1000–1300 cm−1) decreases, which differ by the type of symmetry.23 The broad peaks around 3600 cm−1 and 1640 cm−1 are attributed to the stretching and bending modes of the O–H of surface hydroxyl groups and physically adsorbed water. The peak at 3420 cm−1 arises from the O–H stretching vibration of V–O–H.
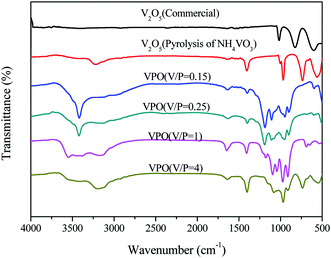 |
| | Fig. 4 FT-IR spectra of different V2O5 and a series of the VPO catalysts with different V/P molar ratios. | |
The Raman spectra of different V2O5 samples in Fig. 5 illustrated the V2O5 crystal structure. However, the V2O5 derived from the pyrolysis of NH4VO3 gave a new intense Raman band at 964 cm−1 compared with the commercial bulk V2O5. In agreement with FT-IR, it is characteristic of V–OH. There is no V2O5 Raman bands in Raman spectrum of VPO, and Raman bands near 1034 cm−1 redshifts upon hydration and it is characteristic of the VO bond stretching mode,24,25 confirming the presence of dispersed vanadium oxide species (VOX) and well dispersed. The intense Raman band at 940 cm−1 has been assigned to VO(H2PO4)2,26 same as XRD and FT-IR result. Thus, the VO(H2PO4)2 is certainly achieved.
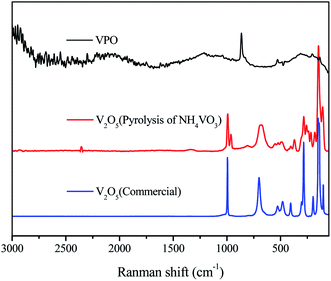 |
| | Fig. 5 Raman spectra of VPO and different V2O5. | |
Effect of V/P molar ratios on the selective oxidation of HMF
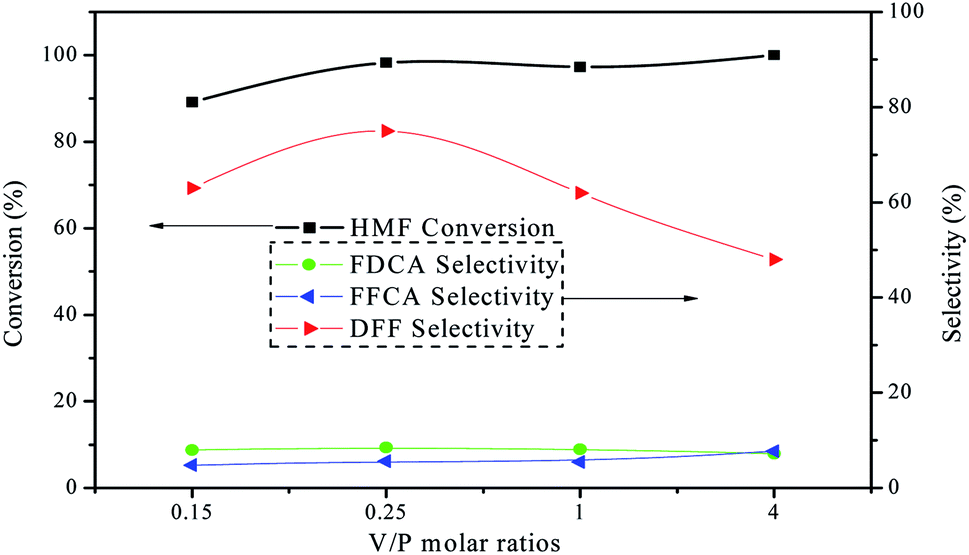
The catalytic activity of the as-prepared VPO catalysts with different V/P molar ratios was evaluated by the oxidation of HMF in DMSO under atmospheric air pressure at 120 °C. Fig. 6 depicted the results of HMF conversion and products selectivity using various VPO catalysts. HMF conversion and selectivity of DFF were all changed greatly with the increase of vanadium content in the VPO catalysts. When V/P molar ratio of the VPO catalyst was 4, the HMF was fully transformed, but the selectivity and yield of the DFF were relatively low. This implies an increase in the V/P ratio can maintain the oxidation properties of VPO catalyst and increases in turn the formation of side products, the lower DFF selectivity accordingly.27,28 In addition, V2O5 was also active for HMF oxidation with conversion of 89.5% under the same reaction conditions, yet with the lower selectivity toward DFF of 69.7%, since further oxidation to form FFCA (10.2%) and FDCA (9.9%) easily occurred. While the VPO catalyst with V/P molar ratio 0.25, HMF was almost quantitatively converted, and the selectivity of DFF reached the maximum 75.0%. This enlightens that the VPO catalyst exhibits excellent catalytic activity at room pressure, compared to 5% conversion in the previous work.29 Therefore, all subsequent experiments used of the VPO catalyst with V/P molar ratio 0.25.
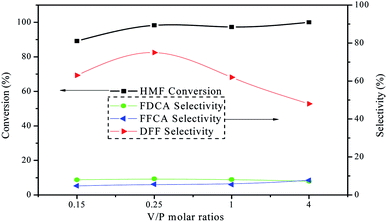 |
| | Fig. 6 The results of HMF oxidation over the VPO catalysts with different V/P molar ratios under atmospheric pressure. Reaction conditions: 0.1 mmol HMF, 10 mg catalyst, 5 mL DMSO, 120 °C, 10 h, in the air. | |
Effect of solvents on the selective oxidation of HMF
The selective oxidation of HMF was further carried out in a variety of solvents. As shown in Table 1, the solvent showed a remarkable effect on the oxidation of HMF. DMSO with high polarity and high boiling point was found to be the best solvent. HMF completely converted and the selectivity of DFF reached 83.6% when using DMSO as the solvent (Table 1 Entry 5). Moderate HMF conversions around 30–40% were obtained in MIBK and DMF, as much more DFF was further oxidized into FDCA. The selectivity of DFF in DMF was the lowest of all testing organic solvent. Reaction carried in toluene and water produced low HMF conversion (Table 1 Entry 1 and Entry 2). Yang et al.30 also reported that polar solvent like DMSO shows better HMF conversion and DFF selectivity as compared to non-polar solvent such as toluene. Hence, further study was done by taking DMSO as solvent for the selective oxidation of HMF into DFF.
Table 1 Effect of solvents on the selective oxidation of HMF into DFFa
| Entry |
Solvent |
HMF conversion (%) |
Selectivity (%) |
DFF yield (%) |
| FDCA |
FFCA |
DFF |
| Reaction condition: 0.1 mmol HMF, 10 mg VPO, 5 mL solvent, 120 °C, 10 h, in the air. |
| 1 |
H2O |
22.0 |
— |
— |
— |
— |
| 2 |
Toluene |
15.0 |
— |
— |
100 |
15.0 |
| 3 |
MIBK |
43.1 |
28.9 |
25.8 |
45.2 |
19.5 |
| 4 |
DMF |
30.5 |
27.4 |
— |
37.1 |
11.3 |
| 5 |
DMSO |
100 |
9.2 |
6.1 |
83.6 |
83.6 |
Effect of time on the selective oxidation of HMF
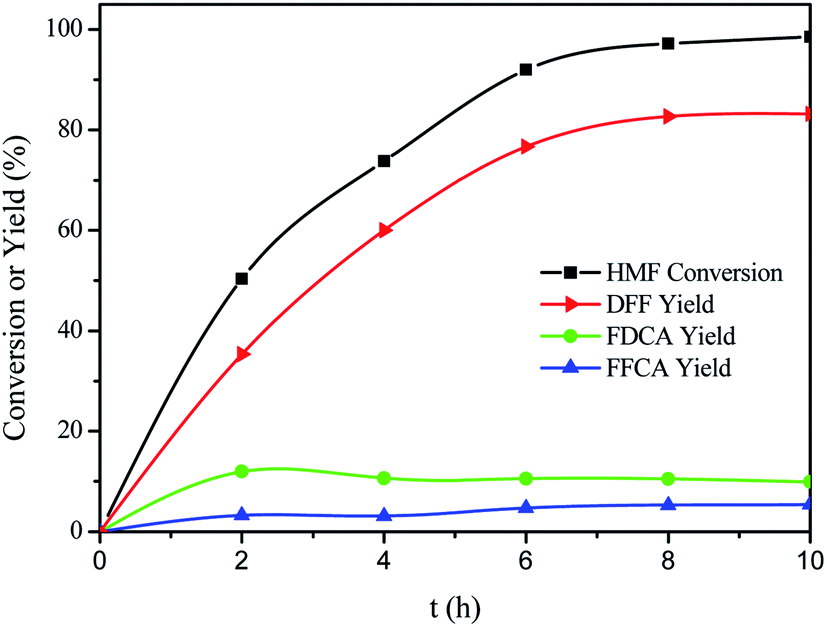
Fig. 7 showed time course for each product in the VPO-catalyzed oxidation of HMF to DFF. As shown in Fig. 7, the oxidation of HMF was very fast, and then slowed down. The fast reaction rate in the initial stage should be due to the high concentration of HMF at an early reaction stage.31 HMF was fully converted after 10 h, and DFF was obtained in a highest yield of 83.2%. FDCA was obtained in a highest yield of 12.0% at 2 h, and the yield of FFCA increased slowly and reached 5.4%. HMFCA was not detected during this reaction. It is therefore expected that HMF is oxidized into DFF, and that FDCA formed though FFCA derived from DFF.
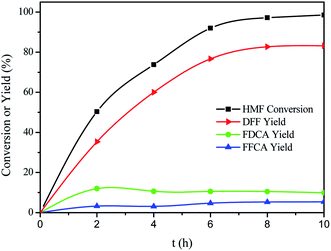 |
| | Fig. 7 Time courses of HMF oxidation over the VPO catalysts under atmospheric pressure. Reaction conditions: 0.15 mmol HMF, 10 mg VPO, 10 mL DMSO, 120 °C, in the air. | |
Effect of reaction temperature on the selective oxidation of HMF
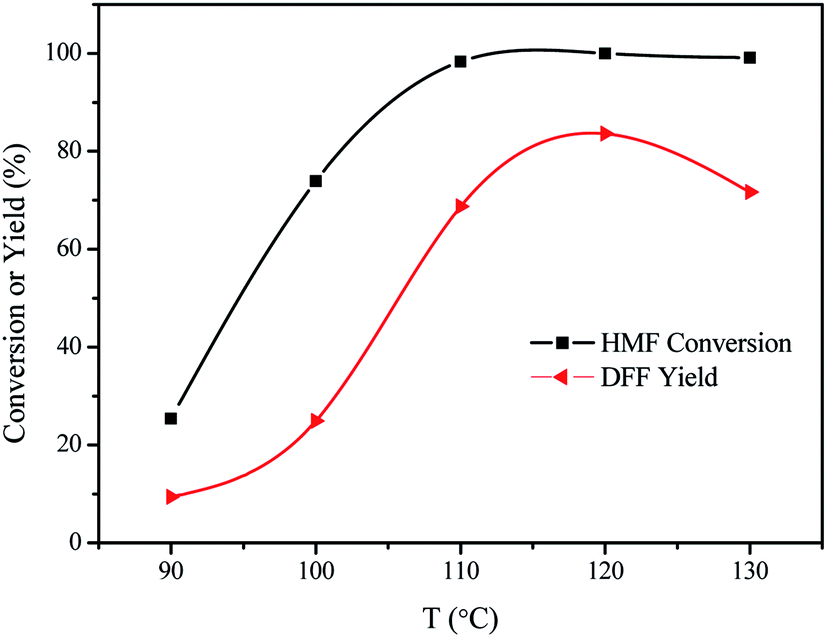
The effect of the reaction temperature was also studied and the results are shown in Fig. 8. HMF conversion and DFF yield increased with the increase of reaction temperature between 90 °C and 110 °C. HMF conversion was obtained in 25.4% when the reaction was carried out at the reaction temperature of 90 °C. Furthering increasing the reaction temperature to 110 °C, HMF conversion increased to 98.3% after 10 h. These results indicated that high reaction temperature promoted the selective oxidation of HMF into DFF. Quantitative HMF conversion was reached when the reaction was carried out at 120 °C and 130 °C. As far as the products yield, the yield of DFF was also greatly affected by the reaction temperature. The yield of DFF gradually increased with the increase of the reaction temperature. The highest yield of DFF was obtained in 83.6% at 120 °C and it slightly decreased to 71.7% at 130 °C.
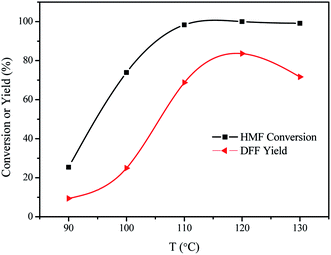 |
| | Fig. 8 The results of HMF oxidation over the VPO catalysts at different reaction temperatures under atmospheric pressure. Reaction conditions: 0.1 mmol HMF, 10 mg VPO, 5 mL DMSO, 10 h, in the air. | |
Conclusions
In conclusion, a new and environmentally friendly method was developed for the synthesis of DFF from the selective oxidation of HMF into DFF. The prepared VPO catalysts showed an excellent catalytic performance with DFF yield of 83.6% and high HMF conversion of 100% under atmospheric pressure in the air. Several important parameters were studied for the oxidation of HMF into DFF. It was found that the solvents played a crucial role in the conversion of HMF and the selectivity of DFF. This work opens up a new way to apply VPO as heterogeneous catalyst in the sustainable conversion of biomass derived compounds into valuable chemicals.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the financial support of the National Natural Science Foundation of China (21606082 and 21776068), Hunan Provincial Natural Science Foundation of China (2018JJ3334), the Scientific Research Fund of Hunan Provincial Education Department (17C0951) and Hunan Provincial Innovation Foundation for Postgraduate (CX2018B295).
Notes and references
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS PubMed
 .
. - M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed .
- J. C. Colmenares and R. Luque, Chem. Soc. Rev., 2014, 43, 765–778 RSC .
- X. X. Liu, J. F. Xiao, H. Ding, W. Z. Zhong, Q. Xu, S. P. Su and D. L. Yin, Chem. Eng. J., 2016, 283, 1315–1321 CrossRef CAS .
- B. Liu and Z. Zhang, ChemSusChem, 2016, 9, 2015–2036 CrossRef CAS PubMed .
- L. Chen, W. Yang, Z. Gui, S. Saravanamurugan, A. Riisager, W. Cao and Z. Qi, Catal. Today, 2019, 319, 105–112 CrossRef CAS .
- B. Liu, Z. Zhang, K. Lv, K. Deng and H. Duan, Appl. Catal., A, 2014, 472, 64–71 CrossRef CAS .
- X. Tong, L. Yu, H. Chen, X. Zhuang, S. Liao and H. Cui, Catal. Commun., 2017, 90, 91–94 CrossRef CAS .
- G. Lv, H. Wang, Y. Yang, T. Deng, C. Chen, Y. Zhu and X. Hou, ACS Catal., 2015, 5, 5636–5646 CrossRef CAS .
- C. A. Antonyraj, J. Jeong, B. Kim, S. Shin, S. Kim, K. Y. Lee and K. C. Jin, Ind. Eng. Chem., 2013, 19, 1056–1059 CrossRef CAS .
- J. Nie, J. Xie and H. Liu, J. Catal., 2013, 301, 83–91 CrossRef CAS .
- A. Takagaki, M. Takahashi, S. Nishimura and K. Ebitani, ACS Catal., 2011, 1, 1562–1565 CrossRef CAS .
- Y. Yan, K. Li, J. Zhao, W. Cai, Y. Yang and J.-M. Lee, Appl. Catal., B, 2017, 207, 358–365 CrossRef CAS .
- C. A. Antonyraj, B. Kim, Y. Kim, S. Shin, K. Y. Lee, I. Kim and K. C. Jin, Catal. Commun., 2014, 57, 64–68 CrossRef CAS .
- N. T. Le, P. Lakshmanan, K. Cho, Y. Han and H. Kim, Appl. Catal., A, 2013, 464–465, 305–312 CrossRef CAS .
- X. Jia, J. Ma, M. Wang, Z. Du, F. Lu, F. Wang and J. Xu, Appl. Catal., A, 2014, 482, 231–236 CrossRef CAS .
- Y. Ren, Z. Yuan, K. Lv, J. Sun, Z. Zhang and Q. Chi, Green Chem., 2018, 20, 4946–4956 RSC .
- B. Liu, Y. Ren and Z. Zhang, Green Chem., 2015, 17, 1610–1617 RSC .
- Z. Yuan, B. Liu, P. Zhou, Z. Zhang and Q. Chi, Catal. Sci. Technol., 2018, 8, 4430–4439 RSC .
- R. Berenguer, J. Fornells, F. J. García-Mateos, M. O. Guerrero-Pérez, J. Rodríguez-Mirasol and T. Cordero, Catal. Today, 2016, 277, 266–273 CrossRef CAS .
- D. B. Williams and C. B. Carter, Transmission Electron Microscopy. Part 2: Diffraction, Springer, New York, 2nd ed., 2009, pp. 293–295 Search PubMed .
- V. Caignaert, M. S. Kishore, V. Pralong, B. Raveau, N. Creon and H. Fjellvaag, J. Solid State Chem., 2007, 180, 2437–2442 CrossRef CAS .
- Y. H. Taufiq-Yap and C. K. Goh, Pertanika J. Sci. Technol., 2003, 11, 293–300 Search PubMed .
- M. A. Bañares and I. E. Wachs, J. Raman Spectrosc., 2002, 33, 359–380 CrossRef .
- I. E. Wachs, J. M. Jehng, G. Deo, B. M. Weckhuysen, V. V. Guliants, J. B. Benziger and S. Sundaresan, J. Catal., 1997, 170, 75–88 CrossRef CAS .
- F. B. Abdelouahab, R. Olier, N. Guilhaume, F. Lefebvre and J. C. Volta, J. Catal., 1992, 134, 151–167 CrossRef CAS .
- A. Datta, S. Sakthivel, M. Kaur, A. M. Venezia, G. Pantaleo and A. Longo, Microporous Mesoporous Mater., 2010, 128, 213–222 CrossRef CAS .
- F. L. Grasset, B. Katryniok, S. Paul, V. Nardello-Rataj, M. Pera-Titus, J. M. Clacens, F. De Campo and F. Dumeignil, RSC Adv., 2013, 3, 9942–9948 RSC .
- C. Carlini, P. Patrono, A. M. R. Galletti, G. Sbrana and V. Zima, Appl. Catal., A, 2005, 289, 197–204 CrossRef CAS .
- Z. Z. Yang, J. Deng, T. Pan, Q. X. Guo and Y. Fu, Green Chem., 2012, 14, 2986–2989 RSC .
- G. D. Yadav and R. V. Sharma, Appl. Catal., B, 2014, 147, 293–301 CrossRef CAS .
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02213a |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Kai Liu‡a,
Shuolin Zhou
a,
Kai Liu‡a,
Shuolin Zhou