DOI:
10.1039/C9RA02201E
(Paper)
RSC Adv., 2019,
9, 15381-15391
Effect of calcination temperature, pH and catalyst loading on photodegradation efficiency of urea derived graphitic carbon nitride towards methylene blue dye solution
Received
21st March 2019
, Accepted 11th May 2019
First published on 16th May 2019
Abstract
In this study, the photodegradation of methylene blue (MB) dye was performed using urea based graphitic carbon nitride (g-C3N4). Interestingly, it has been observed that the calcination temperature for the synthesis of g-C3N4 along with factors (pH and catalyst loading) influencing the photodegradation process, can make an impactful improvement in its photodegradation activity towards MB dye solution. The concept behind the comparatively improved photoactivity of g-C3N4 prepared at 550 °C was explored using various characterisation techniques like XRD, FTIR, SEM, BET and DRS. The FTIR and XRD patterns demonstrated that synthesis of g-C3N4 took place properly only when the calcination temperature was above 450 °C. The evolution of morphological and optical properties based on calcination temperature led to dramatically increased BET surface area and a decreased optical band gap value of g-C3N4 prepared at 550 °C. The effects of pH conditions and catalyst concentration on the MB dye degradation rate using optimally synthesised g-C3N4 are discussed. The value of the apparent rate constant was found to be 12 times more in the case of photodegradation of the MB dye using g-C3N4 prepared at 550 °C at optimum pH and catalyst loading conditions when compared with g-C3N4 prepared at 450 °C showing the lowest photoactivity potential. Further, high stability of the photocatalyst was observed for four cyclic runs of the photocatalytic reaction. Hence, g-C3N4 can be considered as a potential candidate for methylene blue photodegradation.
1. Introduction
A main focus of researchers these days is establishing easier strategies to abate the complex pollutants harming the environment. In this situation every material or technology which can use solar energy as its driving source is an asset. Recently, a new method of oxidation, the “advanced oxidation processes” (AOP), has been used as an emerging eco-friendly method which leads to almost complete mineralization of pollutants.1,2 In regard to AOPs, the application of photocatalysis has strikingly captivated attention in recent research activities.3,4 A photocatalytic process relying on visible light is more convenient regarding cost aspects and is more harmonious with the environment than one using ultraviolet light. Carbon nitrides are a type of polymeric materials which can be procured from carbon materials by substituting carbon atoms with nitrogen.5 Among carbon nitrides, g-C3N4 has been reported as substantially stable allotrope.6,7 It has been bestowed with generous surface properties that are vital to catalysis, such as, electron-rich properties, basic surface functionalities, bonding motif etc.8 The property of g-C3N4 to use visible light has made it conspicuous among other traditional metal oxides (such as TiO2, ZnO, SnO2) which need ultraviolet light for their functioning as photocatalyst.9–12 There are various perquisites of using g-C3N4 as a photocatalyst. It has a band energy of 2.7 eV, due to which it is active in visible light region without any alterations.13,14 Additionally, for improving its photocatalytic ability band gap can be adjusted by doping or modification of nano-morphology.14,15 The application of g-C3N4 as a photoactive material has also been investigated for various reactions, including oxygen reduction, chemical sensors, humidity sensors, transesterification, photovoltaic solar cells, water splitting, catalysis of organic synthesis reactions, CO2 activation, photodegradation of dyes, etc.13,16–21 However, its effectiveness in visible light is quite low, attributing to inadequate surface area and hindered marginal absorption of light.22,23 In order to improve its efficiency under visible light various modification ideologies have been implied to alter its electronic structure, such as doping with noble metals (Au, Ag, Cu, Pt, Sm) and non-metals (N, S, P), tailoring laminar structure of g-C3N4, metallic oxide composting, copolymerization and dye sensitization.21,24–30 In addition to above mentioned strategies to modify g-C3N4, various studies showed that the calcination temperature, as well as the precursor used for synthesis greatly influence the structure, optical properties and morphology of the as-prepared sample.20,31,32 The above discussion gives us a tremendous opportunity to explore more of the potential of the g-C3N4 as promising photocatalyst towards photodegradation of dyes.
Recently many of the researchers have reported the impact of calcination temperature on g-C3N4 refined photoactivity. Enumerating some of results, Zhang et al. discussed that photoactivity of g-C3N4 (450 to 650 °C) towards H2 production using thiourea got better with the increasing calcination temperature and also Mo et al. reported the same trend for melamine derived g-C3N4 for MB dye degradation.33,34 Gu et al. investigated increment in photoactivity of melamine derived g-C3N4 (470–570 °C) to a certain limit of calcination temperature increment for H2 and O2 evolution experiments and afterwards the photocatalytic activity was found to be decreasing and similar observation was also reported by Fang et al. for urea derived g-C3N4 (400–650 °C) towards rhodamine B dye.35,36 Contrarily, Su et al. reported the increase in catalytic activity with decreasing preparation temperature for urea derived g-C3N4 for CO2 conversion experiment.37 The above findings further suggest that the synthesis temperature and the precursor used for g-C3N4 acts crucially towards performance of its photoactivity. Although, Liu et al. has examined calcination temperature effect over the urea derived g-C3N4 for its structural and optical property evolution but the temperature range in which results has been reported is only up to 550 °C and in particular the effects of calcination temperature on its photocatalytic activity towards MB dye degradation in temperature range of 350 to 700 °C have not been discussed.38 Therefore, we have taken the consideration for synthesizing g-C3N4 using urea at different calcination temperature to find out the extent of effects of calcination temperature along with factors (pH and catalyst loading) influencing photodegradation process, on the photocatalytic activity of g-C3N4 towards MB dye degradation. In this study, the facile preparation of g-C3N4 using urea at various temperatures starting from 350 °C to 700 °C has been performed. The morphological evolutions, textural structures and optical properties based on calcination temperature have been analyzed carefully. The photoactivity of g-C3N4 synthesized at different temperature conditions was evaluated towards MB dye solution separately for each sample. The effect of pH condition and catalyst concentration on MB dye degradation reaction rate using optimally synthesized g-C3N4 has been discussed. Further the recycling of the photocatalyst was done and it was reused to find out its stability as a photocatalyst. The optimally synthesized g-C3N4 will further allow us to find its effectiveness of photocatalytic activity towards various other organic dyes.
2. Experimental
2.1 Preparation of sample
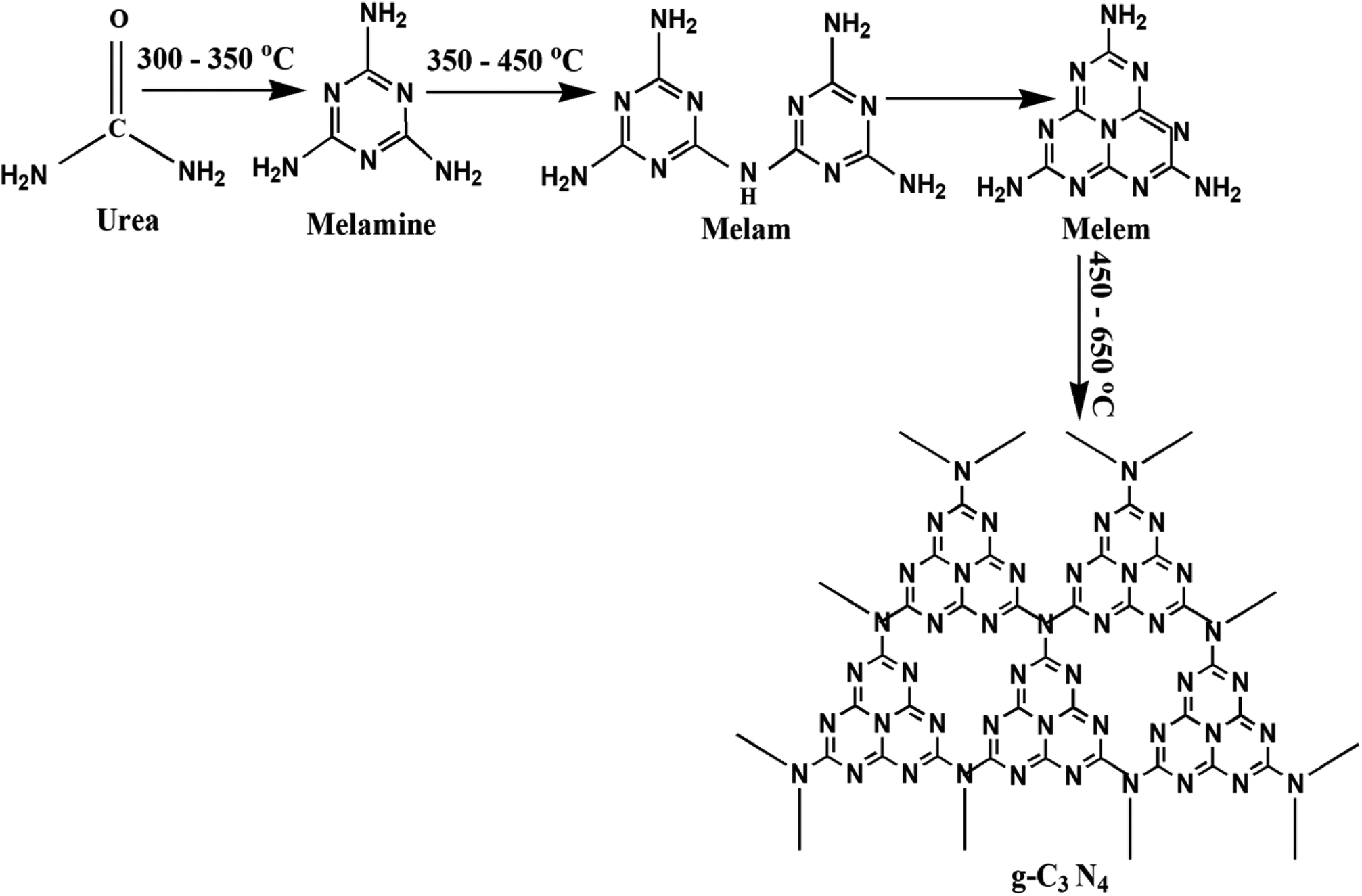
The preparation of g-C3N4 was done by directly heating urea in muffle furnace adequately. 10 g urea was taken in a porcelain covered crucible and wrapped tightly using aluminum foil paper. The calcinations of the samples were done at 350 to 750 °C at 2 °C min−1, for 3 h. The schematic for plausible intermediates at different temperature range has been shown in Fig. 1.38 Finally, the obtained samples were finally powdered and tagged as t (where t represents the corresponding calcination temperature) and further photocatalytic experiments followed by characterizations were performed.
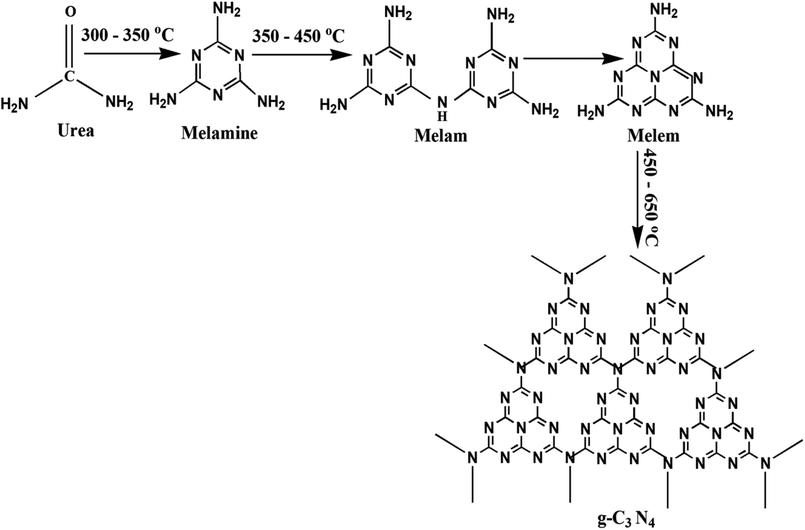 |
| | Fig. 1 Schematic for synthesis of g-C3N4 showing the plausible intermediates at different temperature range. | |
2.2 Photocatalytic activity
Photoactivity evaluation of samples was done by measuring degradation of MB dye in aqueous solutions in simulated solar irradiation. Two 200 W xenon lamps fitted on the two opposite side walls of photocatalytic reactor were used as a source of simulated solar irradiation. Radiant flux was determined by power meter (194.5 W m−2). For each experimental run, 10 ppm of MB aqueous dye solution containing 0.01 g of photocatalyst was taken in 250 mL of conical glass beaker. The solution was kept for stirring for 30 min in dark at room temperature to attain adsorption–desorption equilibrium. At regular time intervals 2 mL of suspensions were drawn out during the irradiation process and centrifuged immediately to separate the photocatalyst. The absorption spectrums of the drawn out samples were taken using UV-vis spectrophotometer in 450 to 800 nm wavelength range of visible spectrum. The change in major absorption peak intensity was used for evaluation of dye degradation. Photocatalytic degradation efficiency was calculated using following equation:
| E = (1 − C/C0) × 100% or E = (1 − A/A0) × 100% |
where, C – concentration of the solution at time t, C0 – adsorption/desorption equilibrium concentration of solution at time t0, A and A0 represents corresponding values.
Simplified pseudo first order kinetic model of Langmuir–Hinshewood (equation) ln(C0/Ct) = kappt = kKt (C – concentration of dye (mg L−1), t – time for degradation (min), k – reaction rate constant (min−1), kapp – apparent rate constant (min−1) and K – adsorption coefficient of dye over catalyst particles), was utilised for estimating apparent rate constant of dye degradation process.
Further, experiments of recycling were carried for four cycles for analysing durability of photocatalytic material. After each cycle, the photocatalyst was recollected with the help of centrifugation at 6000 rpm for duration of 10 min, dried at 100 °C for 2 h and used directly for each test to analyse its reusability aspect.
2.3 Characterization
X-ray diffraction (XRD) analysis was done with Rigaku Ultima IV equipped with CuKα radiation (1.5406 Å). Fourier transform infrared spectroscopy (FTIR, PerkinElmer FRONTIER) (250–8000 cm−1) analysis was carried out with standard KBr disk method. Ultraviolet visible (UV-vis) diffuse reflectance spectrum (DRS) was taken with UV-vis spectrophotometer (Shimadzu UV-2450, Japan) with standard reflectance material – BaSO4. The radiant flux was measured by power meter (Tenmars TM-207). Scanning electron microscope (SEM, EVO LS 10) for investigation of structural morphology. Micromeritics TriStar II 3020 nitrogen adsorption apparatus (USA) for recording Brunauer–Emmett–Teller (BET) specific surface area of samples. Degassing was performed at 180 °C before measurement. BET surface area was calculated through multipoint BET method utilising data of adsorption in range (0.05–0.2) of relative pressure (P/P0).
3. Results and discussion
3.1 XRD analysis
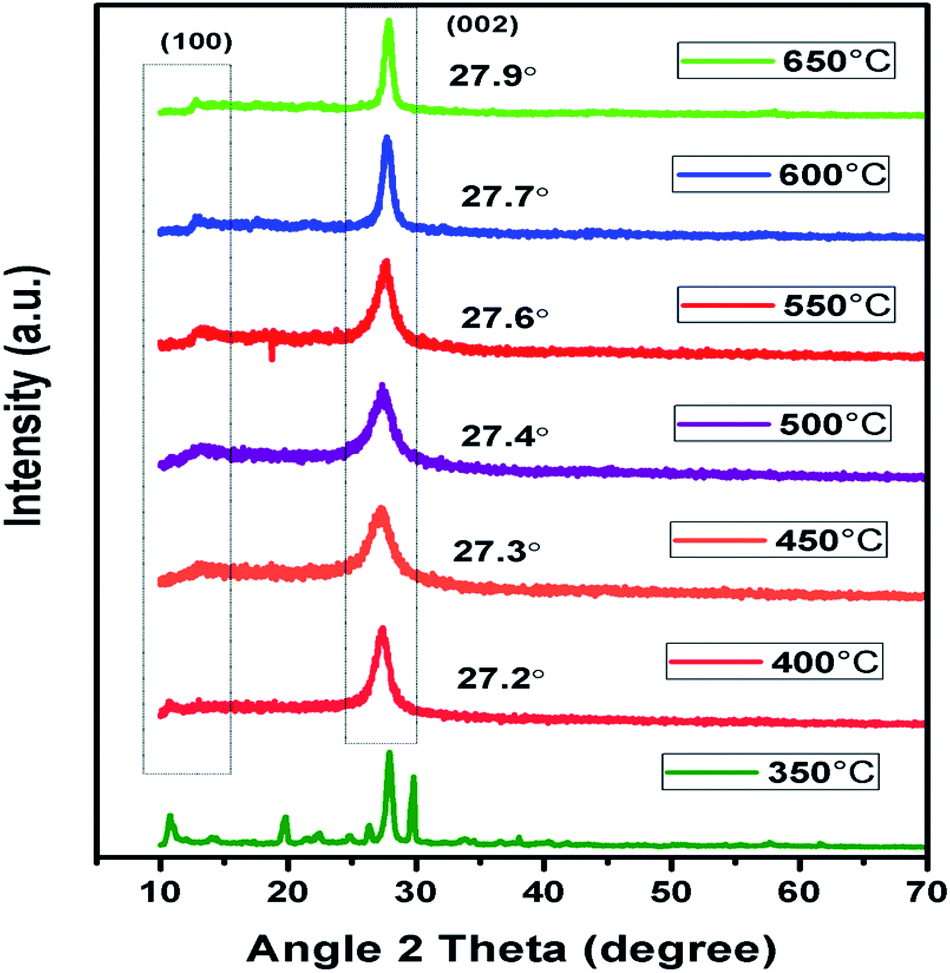
In context to identify structural aspects of g-C3N4 synthesised at different temperatures, XRD patterns were analysed. Two major peaks of diffraction nearly 27.6° and 13.1° were spotted in the XRD patterns of g-C3N4 prepared at 450 °C onwards as shown in Fig. 2. The peak around 27.6° can be indexed to (002) plane resulting due to interplanar stacking of aromatic systems with an interlayer distance of d = 0.326 nm.39,40 It was observed that with rise in calcination temperature, peak (002) shifted to a higher angle (27.3° to 27.9°) depicting the corresponding change in distance of interlayer from 0.328 to 0.319 nm. This leads to a deduction that with the increasing calcination temperature the interlayer stacking order of g-C3N4 can be improved but heating above 550 °C will lead to a denser packing among conjugated aromatic systems resulting in formation of more compact g-C3N4 with an interlayer distance of d = 0.322 to 0.319 nm.41 In addition, it can be clearly seen that the (002) peaks becomes more intense and narrower with increasing calcination temperature, indicating towards increase in crystallinity of g-C3N4 samples with increasing temperature.42 The other smaller peak around 13.1° could be indexed to (100) plane which corresponds to in-planar structural packing motif.43–45 The (100) peak was found to be intense and narrow for g-C3N4 samples at 550 to 650 °C, indicative of higher degree of polymerisation. The broader peak for samples at 450 and 500 °C is indicative of low degree of polymerization of g-C3N4.42 It has been observed that no clear peak around 13.1° was found in the case of g-C3N4 prepared at 400 °C which reveals that in-planar structural packing did not take place properly below 400 °C. Also, at 350 °C the obtained XRD pattern was not similar to the other samples which probably depict some intermediate of urea resulting due to low polycondensation condition.34 XRD analysis revealed that the synthesis of urea derived g-C3N4 takes place at temperature higher than 450 °C which is further confirmed by FTIR analyses. Also, the g-C3N4 polycondensation could be optimized in the range of calcination temperature (450 to 550 °C), but the excessive thermal heating could induce the decomposition of carbon nitride polymer. Whereas at 700 °C no sample was obtained which implies that g-C3N4 got fully decomposed at calcinations temperature higher than 650 °C.
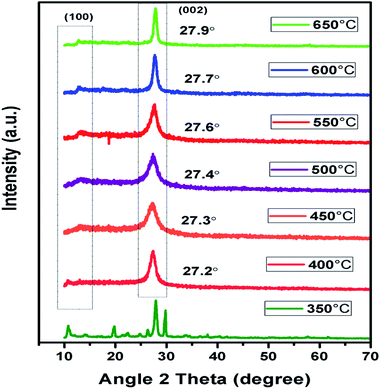 |
| | Fig. 2 XRD pattern of g-C3N4 prepared at different calcination temperature. | |
3.2 FTIR analysis
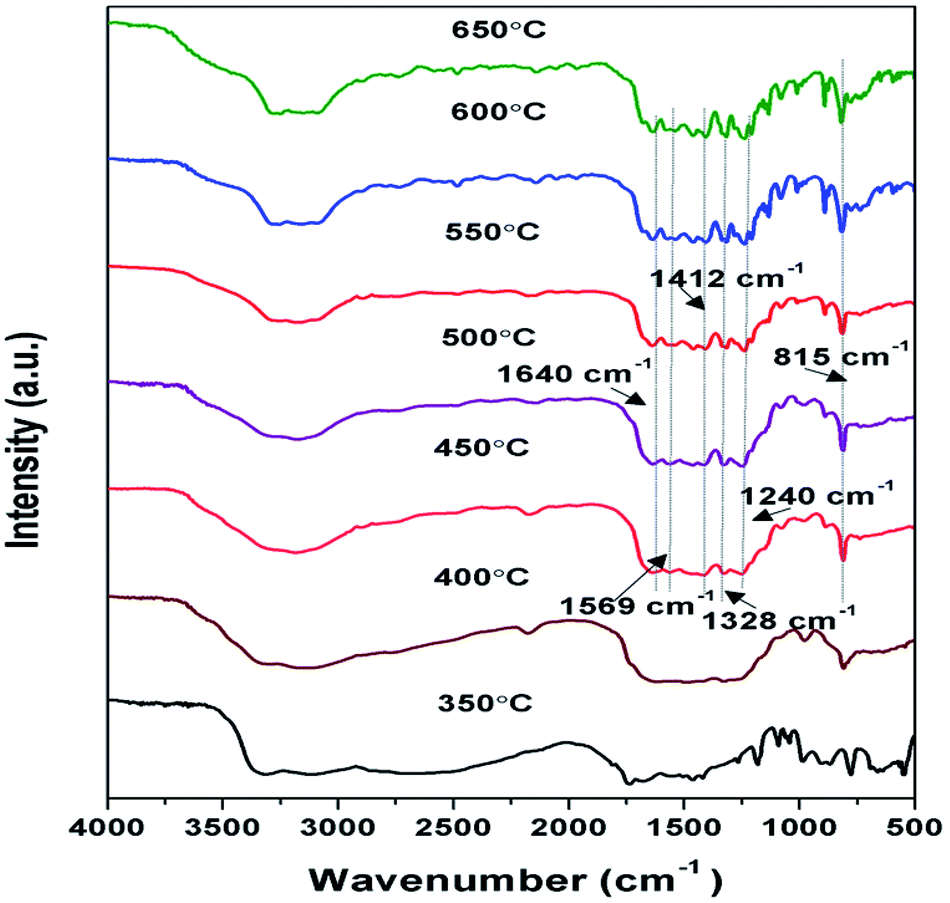
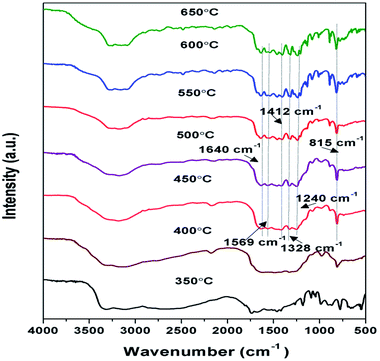
FTIR spectroscopy analysis of samples prepared at 350 to 650 °C as shown in Fig. 3, were performed to further analyse their chemical structure. The principal absorption band in FTIR spectra appeared in 1200 to 1650 cm−1 which is similar to earlier reporting. Peaks observed at 1640, 1569, 1326, 1412, and 1240 cm−1 correspond to stretching vibrational mode of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–C heterocycles.38,46 Intensity of these peaks was found to be increasing towards higher temperature, suggesting the increase in the degree of polymerisation. The broad band observed between 3000 to 3400 cm−1 were due to N–H stretching and minute adsorbed H2O molecules.40,47 The intensity of the peaks in this region decreases with the corresponding increase in the synthesis temperature conveying the reduction in the number of –NH or –NH2 groups for the corresponding samples and increase of degree of polymerisation.37 A strong peak observed at 815 cm−1 was designated to s-triazine units breathing mode.45,48 Whereas in case of sample prepared at 350 and 400 °C the formation of characteristic peaks of g-C3N4 couldn't be observed representing possibly the formation of urea intermediate and incomplete polymerisation respectively. The FTIR patterns of the samples further confirmed that the synthesis of g-C3N4 takes place at temperature higher than 450 °C which is in agreement with results of XRD.
C and C–C heterocycles.38,46 Intensity of these peaks was found to be increasing towards higher temperature, suggesting the increase in the degree of polymerisation. The broad band observed between 3000 to 3400 cm−1 were due to N–H stretching and minute adsorbed H2O molecules.40,47 The intensity of the peaks in this region decreases with the corresponding increase in the synthesis temperature conveying the reduction in the number of –NH or –NH2 groups for the corresponding samples and increase of degree of polymerisation.37 A strong peak observed at 815 cm−1 was designated to s-triazine units breathing mode.45,48 Whereas in case of sample prepared at 350 and 400 °C the formation of characteristic peaks of g-C3N4 couldn't be observed representing possibly the formation of urea intermediate and incomplete polymerisation respectively. The FTIR patterns of the samples further confirmed that the synthesis of g-C3N4 takes place at temperature higher than 450 °C which is in agreement with results of XRD.
 |
| | Fig. 3 FTIR pattern of g-C3N4 prepared at different calcination temperature. | |
3.3 SEM and TEM analysis
SEM images of samples calcined at 450 to 650 °C can be observed in Fig. 4(a–e). The morphology of samples obtained at 450 °C and 500 °C appeared to be consisting of lumps. Sample prepared at 550 °C showed the stacked layered morphology as observed from SEM and TEM images shown in Fig. 4(c and f).32 Samples prepared at 600 °C and 650 °C resulted in contraction of layered flakes of g-C3N4 suggesting the possibility of reduction of the surface area at calcination temperature above 550 °C.
 |
| | Fig. 4 SEM images of g-C3N4 prepared at (a) 450 °C (b) 500 °C (c) 550 °C (d) 600 °C (e) 650 °C (f) TEM image of g-C3N4 prepared at 550 °C. | |
3.4 BET analysis
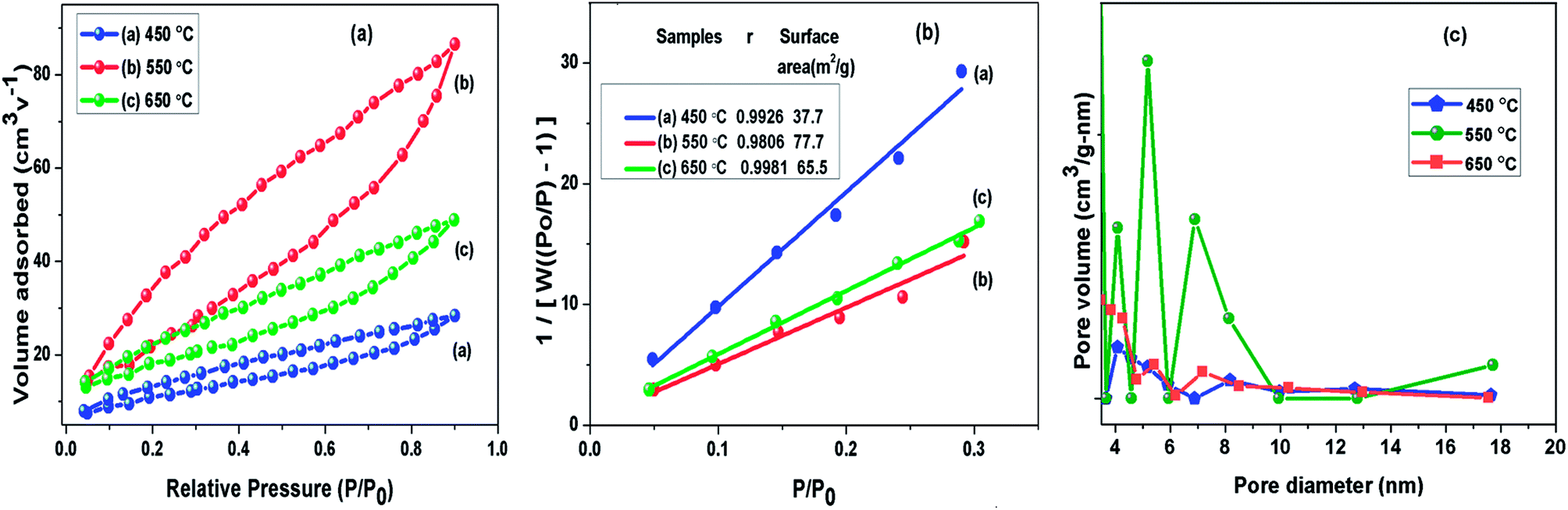
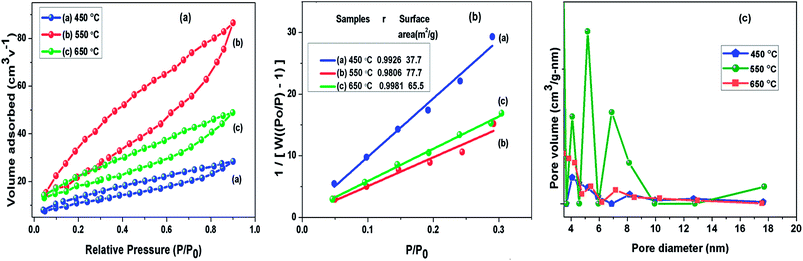
To understand the calcination temperature effect towards specific surface area of samples, nitrogen adsorption–desorption was utilised as presented in Fig. 5(a) together with their corresponding multipoint BET plots presented in Fig. 5(b) and Barret–Joyner–Halenda (BJH) pore size distribution curves presented in Fig. 5(c). The results for BET surface area, pore size and volume of these samples are presented in Table 1. The adsorption–desorption isotherms for all the samples under consideration showed characteristic type III curve.49,50 The BET specific surface areas were estimated to be 37.8 m2 g−1, 73.7 m2 g−1 and 65.6 m2 g−1 for g-C3N4 samples prepared at 450, 550 and 650 °C, respectively. The most probable pore distribution in case of g-C3N4 samples obtained at 450, 550 and 650 °C were found to be centred at around 3.97 nm, 4.13 nm and 3.93 nm, which can be attributed to presence of mesopores. It can be observed in Fig. 5(c) that sample prepared at 450 °C exhibits a small peak near 4.08 nm, with rise in temperature to 550 °C, peak at 4.08 nm grows up gradually and simultaneous peak appears at 6.91 nm and other more pronounced peak appears at 5.3 nm suggesting the increase in the number of pores during heating process in wide pore diameter distribution and volume of pore appeared as largest for the g-C3N4 prepared at 550 °C. However, at 650 °C number of pores decreases. The analysis showed that calcination temperature may affect specific surface area of g-C3N4. The sample obtained at 550 °C had nearly 1.12 and 2 times increased surface area than samples at 650 and 450 °C respectively. Analysis revealed that the surface area of g-C3N4 improved effectively at 550 °C, also showing conformity with observed morphology in SEM analysis.
 |
| | Fig. 5 (a) Nitrogen adsorption–desorption isotherm; (b) BET adsorption isotherm (c) BJH pore size distribution curve of g-C3N4 prepared at different calcination temperature. | |
Table 1 Summary of BET obtained parameters of g-C3N4 prepared at different calcination temperature
| g-C3N4 prepared at |
SBET (cm2 g−1) |
Total pore vol. (cm3 g−1) |
Avg. pore radius (nm) |
| 450 °C |
37.8 |
0.0439 |
3.97 |
| 550 °C |
73.7 |
0.1338 |
4.13 |
| 650 °C |
65.6 |
0.0756 |
3.93 |
3.5 UV-vis spectroscopic study
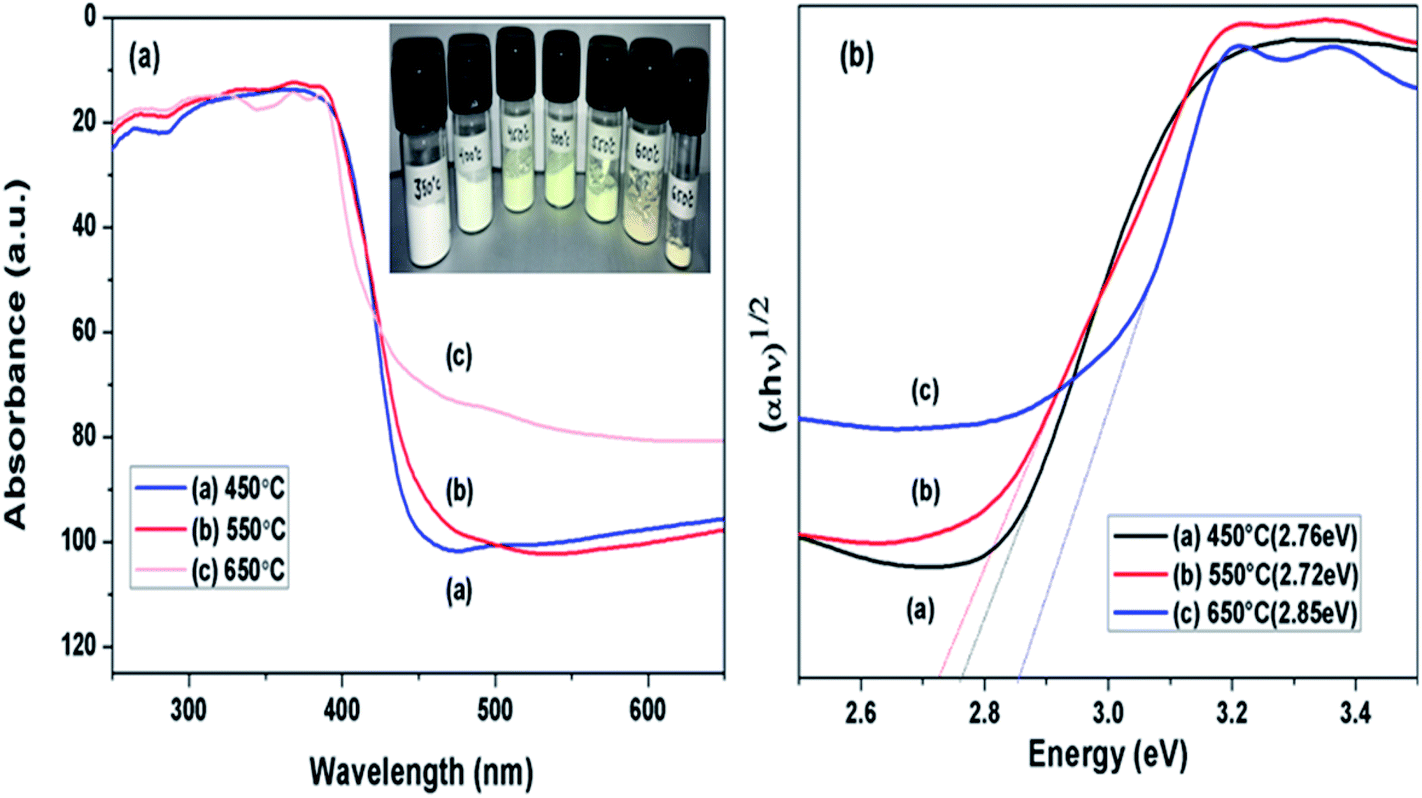
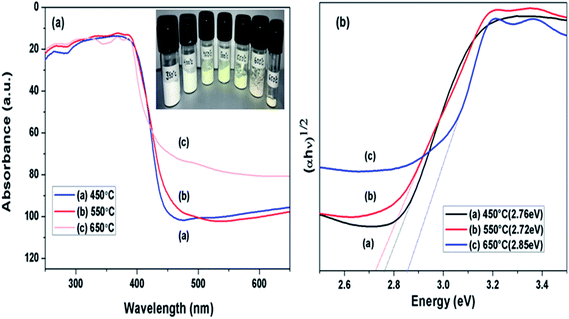
DRS spectra of samples calcined at different temperature were analysed as presented in Fig. 6(a), towards effect of change in calcination temperature on optical property of prepared samples. Variation in the absorption edges was observed with the change in calcination temperature. Further the band gap energy was calculated using equation: α = A(hν − Eg)n/hν, where α is coefficient of absorption coefficient.51 The band gap energy of samples obtained at 450, 550 and 650 °C were estimated to be 2.77, 2.72 and 2.83 eV, respectively as shown in Fig. 6(b). Firstly, when calcination temperature was raised from 450 to 550 °C, the band gap energy showed a decreasing trend from 2.77 to 2.72 eV, this could be because of increased degree of polymerisation of g-C3N4, resulting in increase of π-plane conjugation degree of heptazine rings via N2 atoms.42 When temperature was further raised to 650 °C, a blue shift in absorption edge was observed and band gap energy increased to 2.83 eV. The blue shift could be a possible result of strong quantum confinement effect and the possible reason for better photoactivity of sample obtained at 650 °C than 450 °C may be assigned to increased surface area as discussed earlier.33 This revealed that band gap in case of g-C3N4 prepared at 550 °C was narrower than samples prepared at 450 and 650 °C. It can be concluded that g-C3N4 prepared at 550 °C had higher ability of absorbing visible light which can lead to better photoactivity in comparison to other samples in consideration.33
 |
| | Fig. 6 (a) UV-vis diffused absorbance spectra (b) estimated band gaps of g-C3N4 prepared at different calcination temperature. | |
3.6 Effect of calcination temperature on the photocatalytic activity
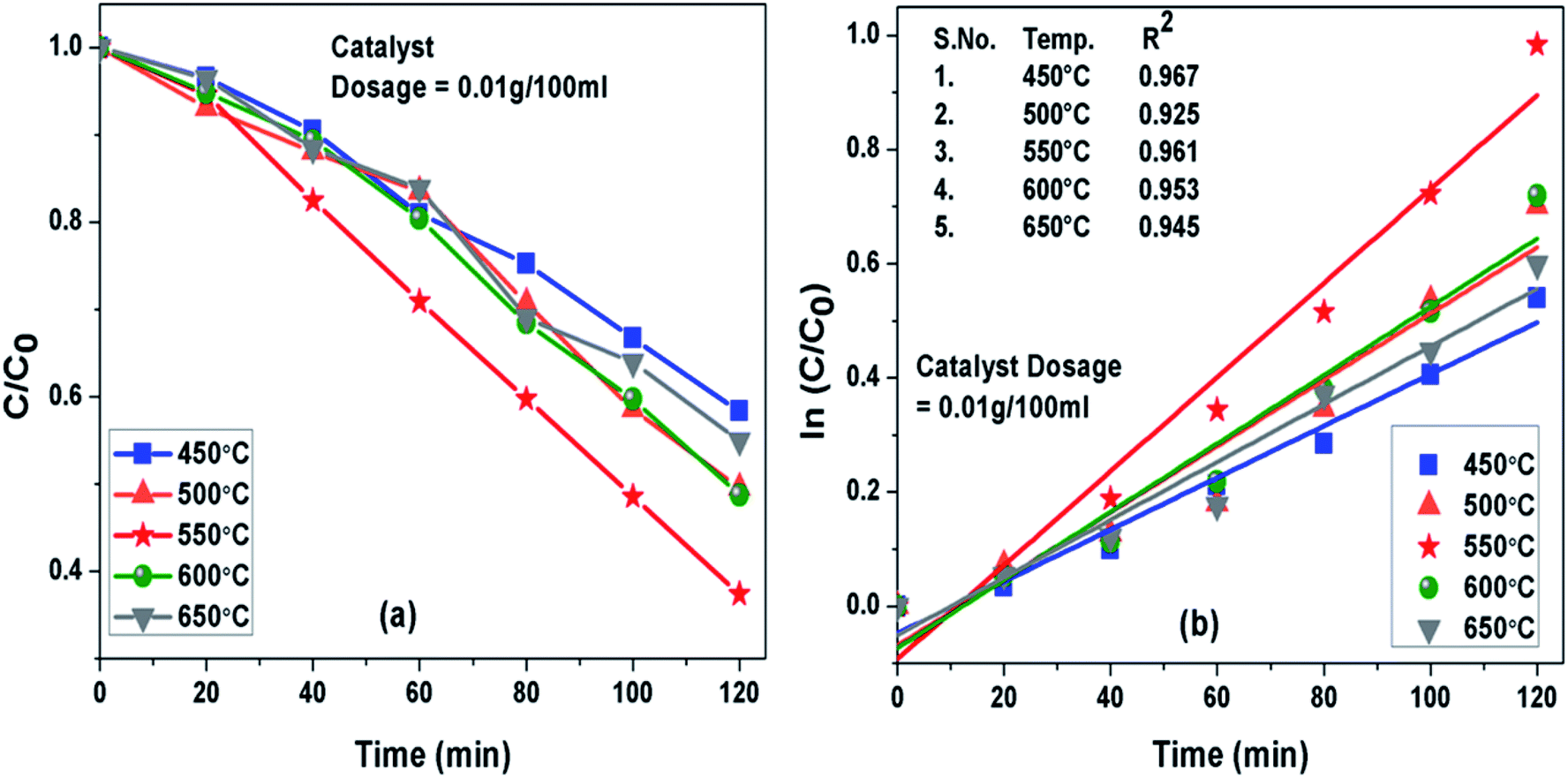
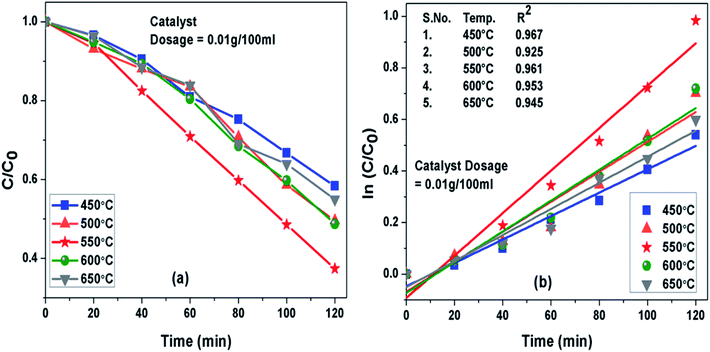
Photoactivity of g-C3N4 was assessed by performing photodegradation of MB dye under simulated solar irradiation separately for all the samples synthesized at different temperature conditions. Interestingly, the calcination temperature affected photodegradation activity of g-C3N4 noticeably towards MB dye solution. The adsorption efficiencies of g-C3N4 prepared at 450, 500, 550, 600 and 650 °C were about 18%, 26.8%, 34.4%, 27.7% and 21.7% for MB dye solution, respectively. The g-C3N4 sample calcined at 550 °C displayed highest adsorption ability and order of adsorption ability observed in case of sample prepared at 450![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 550 and 650 °C was found to be consistent well with their surface area as discussed earlier in this study. The degradation efficiency and the apparent rate constant measured for all the prepared samples of g-C3N4 at different temperatures towards MB dye for the duration of 120 min have been compared in Fig. 7(a) and (b) respectively and are represented in Table 2. Photoactivity of g-C3N4 was observed to be increasing corresponding to rise in calcination temperature from 450 to 550 °C and after that photocatalytic activity was found to follow a decreasing trend from 550 to 650 °C. Sample obtained at 550 °C exhibited the maximum degradation efficiency of 62.6% in 120 min for MB dye. The results suggested that g-C3N4 photoactivity may be greatly refined by raising calcination temperature to some an extent and further increment of temperature may lead to decline in the photoactivity as well. The g-C3N4 prepared at 550 °C showed largest rate of reaction constant (k) of 0.0081 which is nearly 2 and 1.6 times higher than the g-C3N4 synthesized at 450 and 650 °C. The increased reaction rate of g-C3N4 calcined at 550 °C could be due to its higher efficiency of absorbing visible light which can lead to better generation of photo-generated electrons causing more efficient separation of charge and hence better photoactivity.
550 and 650 °C was found to be consistent well with their surface area as discussed earlier in this study. The degradation efficiency and the apparent rate constant measured for all the prepared samples of g-C3N4 at different temperatures towards MB dye for the duration of 120 min have been compared in Fig. 7(a) and (b) respectively and are represented in Table 2. Photoactivity of g-C3N4 was observed to be increasing corresponding to rise in calcination temperature from 450 to 550 °C and after that photocatalytic activity was found to follow a decreasing trend from 550 to 650 °C. Sample obtained at 550 °C exhibited the maximum degradation efficiency of 62.6% in 120 min for MB dye. The results suggested that g-C3N4 photoactivity may be greatly refined by raising calcination temperature to some an extent and further increment of temperature may lead to decline in the photoactivity as well. The g-C3N4 prepared at 550 °C showed largest rate of reaction constant (k) of 0.0081 which is nearly 2 and 1.6 times higher than the g-C3N4 synthesized at 450 and 650 °C. The increased reaction rate of g-C3N4 calcined at 550 °C could be due to its higher efficiency of absorbing visible light which can lead to better generation of photo-generated electrons causing more efficient separation of charge and hence better photoactivity.
 |
| | Fig. 7 (a) Comparison of photocatalytic activity of g-C3N4 prepared at different temperature (b) ln(C0/C(t)) for MB degradation with g-C3N4 prepared at different temperature as a function of simulated solar irradiation time. | |
Table 2 Degradation efficiency and the apparent rate constant (k) values measured for all the prepared samples of g-C3N4 at different temperatures
| S. no. |
Calcination temp. (°C) |
Degradation efficiency (%) |
Apparent rate constant (k) in min−1 |
| 1 |
450 |
41.6 |
0.0044 |
| 2 |
500 |
50.4 |
0.0058 |
| 3 |
550 |
62.6 |
0.0081 |
| 4 |
600 |
51.2 |
0.0059 |
| 5 |
650 |
45 |
0.0050 |
3.7 Optimization of photodegradation process of MB dye solution
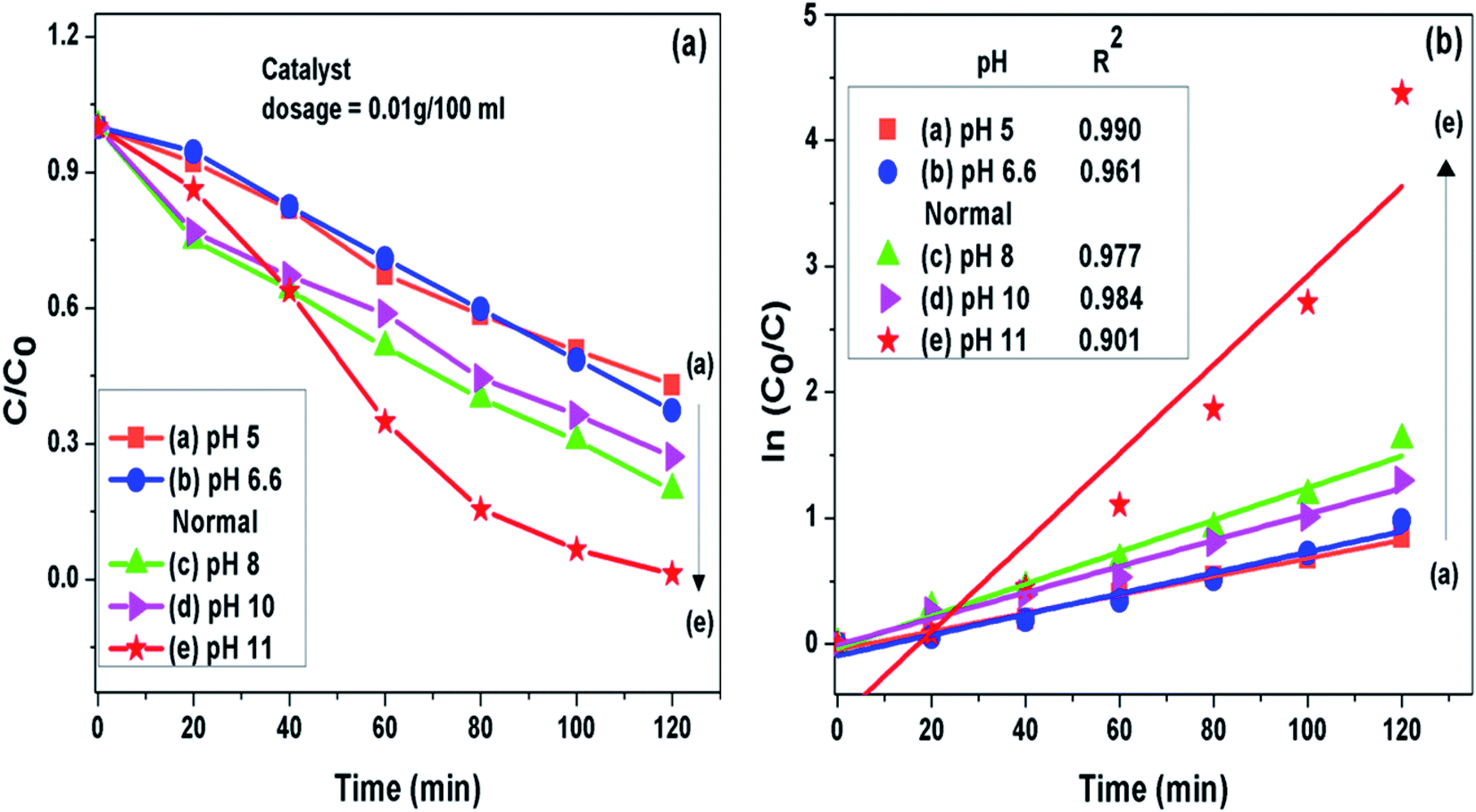
3.7.1 pH of the solution. It was noted that the photocatalytic degradation process of pollutants got pronouncedly affected by pH of the solution. The g-C3N4 prepared at 550 °C was further analyzed to find the effect of pH on photodegradation process of MB dye. Initial pH of dye solution was set in the range of pH 5 to pH 11 by adjusting with dil. solution of HCl and NaOH. It was seen that when the pH value was set lesser than the normal pH in the acidic range of the MB dye solution, the fall in g-C3N4 photoactivity was observed where as on increasing pH value from the normal in the basic range the rise in g-C3N4 activity could be seen. This observation might be related to the fact that at pH value greater than 6.8 the MB dye molecules are negatively charged in alkaline medium which may result in strong adsorption of dye on the positively charged g-C3N4 particles because of electrostatic attraction among the two.40,52,53 This also implies that the formation of reactive intermediates like super oxide radicals is significantly enhanced in the alkaline medium ultimately enhancing the reaction rate. The g-C3N4 prepared at 550 °C showed highest degradation efficiency of 98.7% for the MB dye solution set at pH 11 which is 1.57 times higher than the degradation efficiency at the normal pH condition. The degradation efficiency and the apparent rate constant measured for all the photocatalytic reactions for the duration of 120 min at different pH conditions are presented in Fig. 8(a) and (b) respectively and are represented in Table 3.
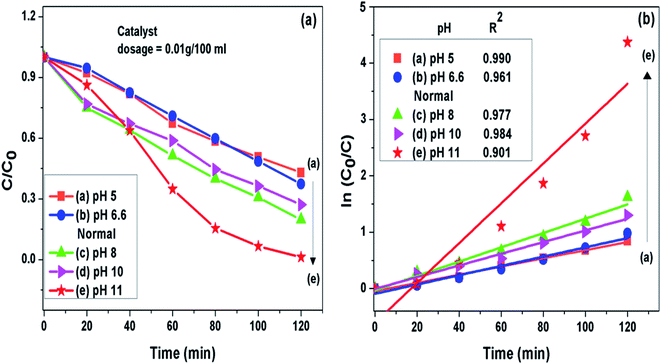 |
| | Fig. 8 (a) Comparison of photocatalytic activity of g-C3N4 at different pH condition (b) ln(C0/C(t)) for MB degradation with g-C3N4 at different pH condition as a function of simulated solar irradiation time. | |
Table 3 Degradation efficiency and the apparent rate constant (k) values measured for all the photocatalytic reactions for the duration of 120 min at different pH conditions
| S. no. |
pH value |
Degradation efficiency in percent (%) |
Apparent rate constant (k) in min−1 |
| 1 |
pH 5 |
57 |
0.00735 |
| 2 |
pH 6.6 (normal) |
62.6 |
0.00811 |
| 3 |
pH 8 |
72.7 |
0.01022 |
| 4 |
pH 10 |
80.2 |
0.01265 |
| 5 |
pH 11 |
98.7 |
0.03398 |
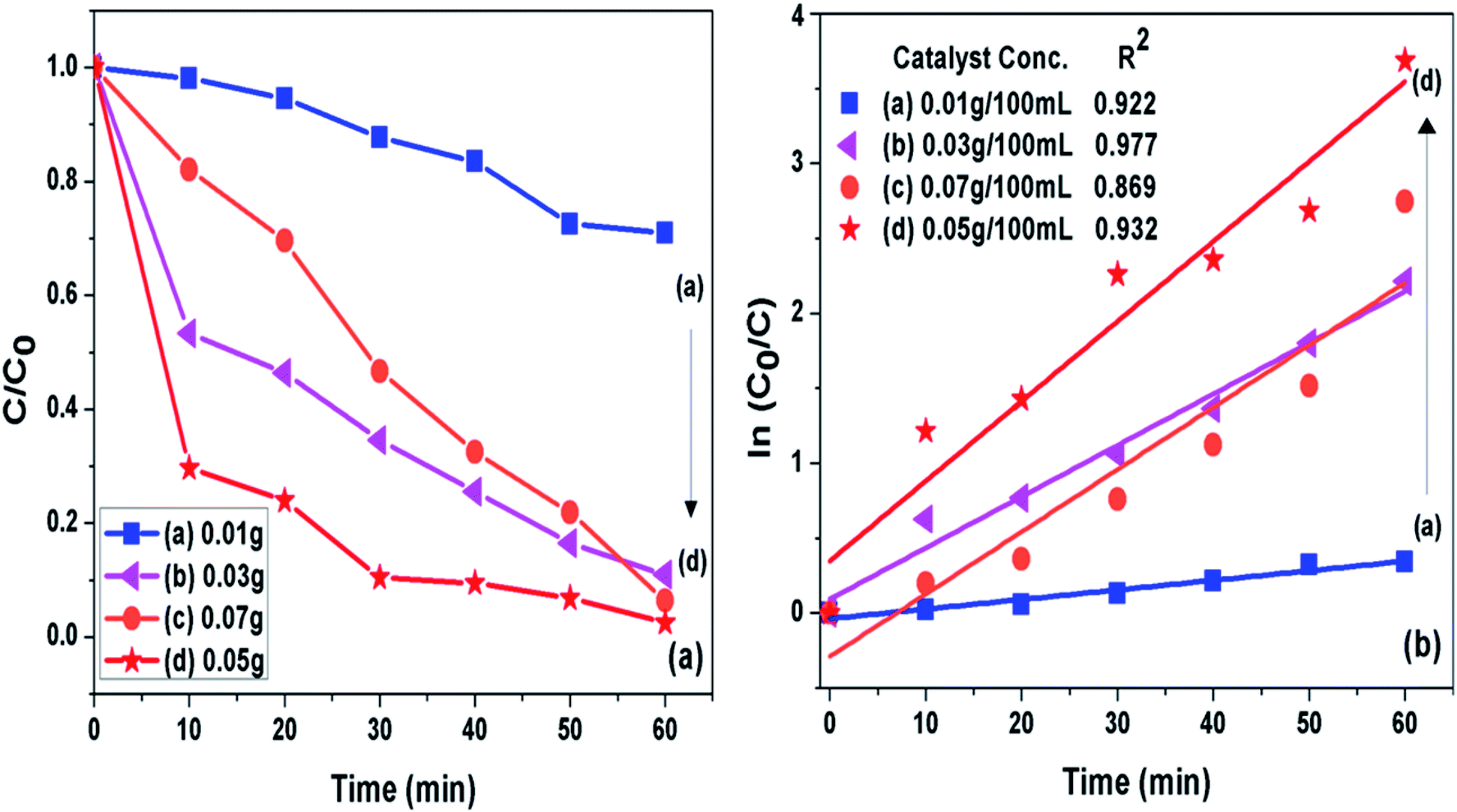
3.7.2 Catalyst loading. The concentration of photocatalyst (g-C3N4 prepared at 550 °C) was varied from 0.01 to 0.07 g per 100 mL of 10 ppm MB dye solution at pH 11 to analyse effect of catalyst concentration on photoactivity of g-C3N4 towards MB dye as presented in Fig. 9(a and b). It was seen that when amount of photocatalyst was increased from 0.01 g to 0.05 g the photo catalytic activity was found to be increasing which could be caused by increased active sites on catalyst surface. The further increase in catalyst amount above 0.05 g might decreased the activity due to hindrance caused to light pathway to reach dye molecules, which could be due to phenomenon of light scattering dominating at higher catalyst loading.54 The 0.05/100 g mL−1 concentration g-C3N4 prepared at 550 °C showed highest degradation efficiency of 100% in 60 min for the MB dye solution set at pH 11. The percent degradation efficiency and the apparent rate constant measured for all the photocatalytic reactions for the duration of 60 min at different photocatalyst concentration are represented in Table 4.
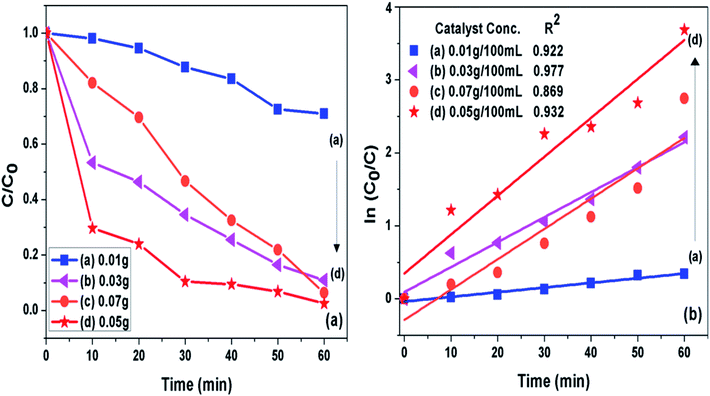 |
| | Fig. 9 (a) Comparison of photocatalytic activity of g-C3N4 at different catalyst concentration (b) ln(C0/C(t)) for MB degradation with g-C3N4 at different catalyst concentration as a function of simulated solar irradiation time. | |
Table 4 Degradation efficiency and the apparent rate constant (k) values measured for all the photocatalytic reactions for the duration of 60 min at different photocatalyst concentration
| S. no. |
Catalyst conc. (g/100 mL of 10 ppm MB dye solution at pH 11) |
Degradation efficiency in percent (%) |
Apparent rate constant (k) in min−1 |
| 1 |
0.01 |
60 |
0.01653 |
| 2 |
0.03 |
89 |
0.03422 |
| 3 |
0.07 |
93 |
0.04156 |
| 4 |
0.05 |
100 |
0.05331 |
3.8 Recycle and reusability experiment
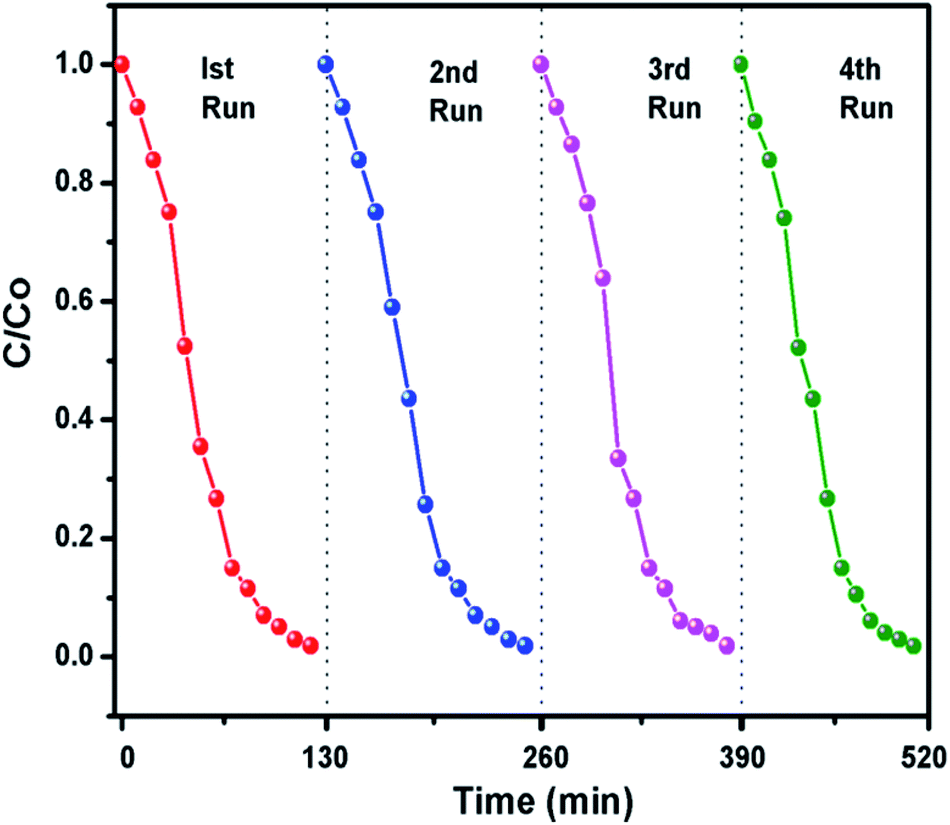
The g-C3N4 sample prepared at 550 °C was checked for its stability for four cyclic runs. From Fig. 10 it can be seen that the photocatalyst showed almost same photodegradation activity towards 10 mg L−1 MB dye solution for all four experiments. A minute decrease in degradation efficiency could be seen which is possibly due to loss in catalyst amount during recollection process after each run. The cycling test support the fact that g-C3N4 has fine stability and can be used for dye degradation application.
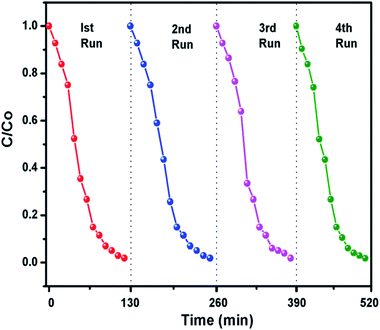 |
| | Fig. 10 Recyclability experiment of the photocatalytic degradation of dyes using g-C3N4 prepared at 550 °C. | |
3.9 Factors influencing the photocatalytic activity of g-C3N4 towards MB dye degradation
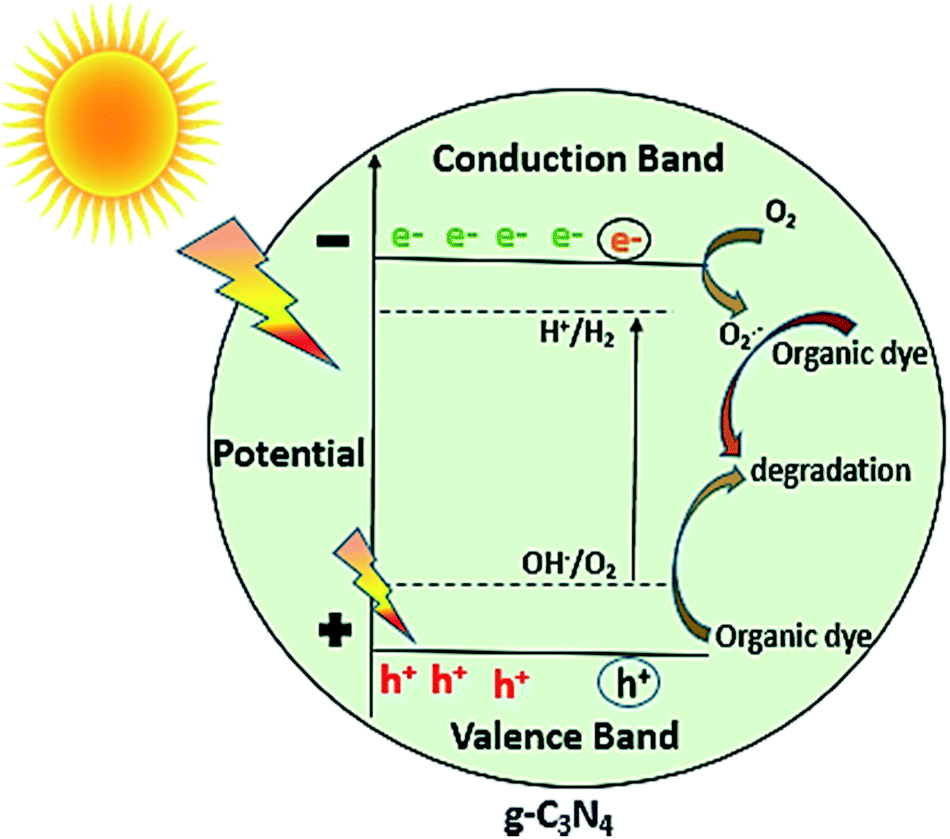
In general, as shown in Fig. 11, when the light of suitable photon energy i.e. higher than band gap energy of g-C3N4, falls over its surface, valence band electrons on absorbing light get excited thereby moving to conduction band, generating holes in valence band. The electrons can join oxygen to generate super oxide radicals, and then superoxide radicals can lead to photo degradation of organic dye or other organic pollutants. The results of photocatalytic reaction of samples prepared at varied calcination temperature towards MB dye showed that the calcination temperature played a major role in improvement of photocatalytic ability of g-C3N4 sample. As observed from results photoactivity of g-C3N4 towards MB dye increases with rise in temperature up to 550 °C, further increment in temperature could result in decrease in activity. Some of the similar studies by Xu et al. and Mo et al. reported the highest activity for sample prepared at 600 and 650 °C respectively, in range of 450 to 650 °C.34,42 The variation in the result could be possibly based on the factors like the duration for which calcination was being carried out and precursor used for synthesising g-C3N4 which can probably affect the degree of polymerisation. The XRD analysis revealed that the calcination temperature had a huge impact on polymerization process of prepared g-C3N4 samples. Increasing temperature to a limit can result in an improved structure with appropriate interlayer distance of aromatic systems. Whereas the higher temperature condition can induce the bulking as well as thermal decomposition of the polymeric material, as can also be observed in the inset of Fig. 6(a) that the amount of sample obtained got reduced with the rising calcination temperature and striking decrement in amount was observed for sample obtained at 650 °C. In case of g-C3N4, protonation depends primarily on degree of polymerization, but also get influenced by degree of condensation. In case of g-C3N4, “underpolymerization” (as for sample obtained at 450 °C), leads to incomplete coupling of heptazine, resulting in excessive hydrogen-passivating N3c′ nitrogen sites, thereby hindering activity. The “overpolymerization” (as in case of sample obtained at 650 °C), the structure of g-C3N4 becomes excessively condensed into deformed multilayered crystals having reduced surface area, resulting in reduced density of active sites, and thus adversely affecting photocatalytic ability.40 Moreover, sp2 planar geometry also gets contorted because of buckling, thus could lead to trapping states of charge at nitrogen sites and therefore decreased activity.40 As revealed from BET analysis, the increased surface area for g-C3N4 prepared at 550 °C as a consequence of morphological evolution caused due to appropriate calcination temperature, which not only can lead to the formation of surface active sites for reaction but can also facilitate the separation of charge carrier because exfoliated and altered structures can reduce distance of migration for photogenerated charge carriers and thus can decrease the probability of charge recombination.32 From DRS analysis it was found that the g-C3N4 prepared at 550 °C also showed better ability for the absorption of light and thus may result in enhancement of photoactivity. The calculated value of reaction rate constant in case of g-C3N4 at 550 °C was found to be 2 to 1.5 times higher than g-C3N4 prepared at other temperature. Also, the factors like pH and catalyst loading resulted in the observable increment of nearly 6.6 times in the value of rate of reaction constant of g-C3N4 prepared at 550 °C which could be due to enhanced formation of reactive intermediates and increased active sites on catalyst surface respectively. In addition, value of reaction rate constant for MB dye degradation of g-C3N4 obtained at 550 °C (catalyst conc. of 0.1 g L−1) was found to be 1.35 times higher when compared with the value reported by Mo et al. for melamine derived g-C3N4 at 650 °C (the optimum temperature condition, catalyst conc. 0.5 g L−1).34 The comparison in reaction rate constant values suggest that urea might be considered as a better precursor option for preparing g-C3N4 over melamine and it can be further supported by the fact that BET specific surface area and calculated band gap was found to be better for optimally synthesised urea derived g-C3N4 that is 73.7 m2 g−1 and 2.72 eV where as for melamine derived g-C3N4 was 46.8 m2 g−1 and 2.74 eV respectively.34 The above discussion can lead to a conclusion that improved activity of g-C3N4 prepared at 550 °C or the observed trend between activities with calcination temperature has found to be clearly based on the evolutions related to optical, structural and morphological properties of g-C3N4 brought due to different temperature conditions, choice of precursor and other optimization factors like pH and catalyst concentration of MB dye solution.
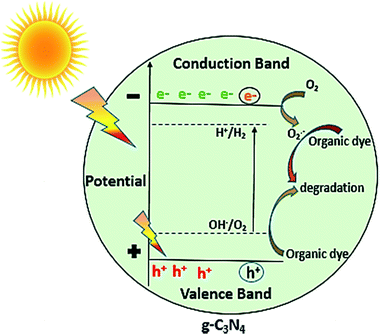 |
| | Fig. 11 Photocatalytic mechanism of g-C3N4. | |
3.10. Conclusion
In summary, the g-C3N4 was synthesised using urea at different calcination temperature. It was noted that the photocatalytic performance of g-C3N4 towards MB dye solution firstly got better with increasing calcination temperature up to 550 °C and further increment of temperature leads to decrease in the photoactivity of g-C3N4. XRD, FTIR, SEM, BET and DRS analysis revealed that the highest photoactivity performance of g-C3N4 prepared at 550 °C may be due to the appropriate degree of polymerisation and condensation, increased surface area resulting in enhanced light absorption ability and more number of active sites. Factors like pH of solution and catalyst loading also affected effectively the photodegradation process of MB dye solution. The g-C3N4 prepared at 550 °C also showed the good stability even after four cyclic runs. The study inferences that the optimally synthesised g-C3N4 along with various factors influencing photodegradation process can be used as a potential material for degrading MB dye and can be explored further for its photocatalytic performance towards various other organic pollutants.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
DRP thanks Deenbandhu Chhotu Ram University of Science and Technology for providing financial support under the University Doctoral Research Fellowship scheme. AS acknowledges Indo-German Science and Technology Centre and Department of Science and Technology, Govt. of India for providing Max Planck India Mobility Grant award and Women Scientists Scheme-A grant respectively. SPN thanks Indo-US Science and Technology Forum and Department of Science and Technology, Govt. of India for providing research opportunity under IUSSTF BASE Fellowships 2018/2.
References
- R. Andreozzi, Catal. Today, 1999, 53, 51–59 CrossRef CAS.
- M. A. Quiroz, E. R. Bandala and C. A. Martinez-Huitle, Advanced Oxidation Processes (AOPs) for Removal of Pesticides from Aqueous Media, Pesticides - Formulations, Effects, Fate, ed. M. Stoytcheva, 2011 Search PubMed.
- V. Binas, D. Venieri, D. Kotzias and G. Kiriakidis, J. Materiomics, 2017, 3, 3–16 CrossRef.
- A. Ibhadon and P. Fitzpatrick, Catalysts, 2013, 3, 189–218 CrossRef CAS.
- J. Liu, H. Wang and M. Antonietti, Chem. Soc. Rev., 2016, 45, 2308–2326 RSC.
- T. S. Miller, A. B. Jorge, T. M. Suter, A. Sella, F. Corà and P. F. McMillan, Phys. Chem. Chem. Phys., 2017, 19, 15613–15638 RSC.
- J. Zhu, P. Xiao, H. Li and S. A. C. Carabineiro, ACS Appl. Mater. Interfaces, 2014, 6, 16449–16465 CrossRef CAS PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893 RSC.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- M. Gaidi, A. Hajjaji, M. A. El Khakani, B. Chenevier, M. Labeau and B. Bessaïs, Jpn. J. Appl. Phys., 2009, 48, 072501 CrossRef.
- J. Ben Naceur, M. Gaidi, F. Bousbih, R. Mechiakh and R. Chtourou, Curr. Appl. Phys., 2012, 12, 422–428 CrossRef.
- L. Zhou, H. Zhang, H. Sun, S. Liu, M. O. Tade, S. Wang and W. Jin, Catal. Sci. Technol., 2016, 6, 7002–7023 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2008, 8, 76–80 CrossRef PubMed.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- J. Zhu, Y. Wei, W. Chen, Z. Zhao and A. Thomas, Chem. Commun., 2010, 46, 6965 RSC.
- R. Malik, V. K. Tomer, V. Chaudhary, M. S. Dahiya, A. Sharma, S. P. Nehra, S. Duhan and K. Kailasam, J. Mater. Chem. A, 2017, 5, 14134–14143 RSC.
- J. Cheng, Z. Hu, K. Lv, X. Wu, Q. Li, Y. Li, X. Li and J. Sun, Appl. Catal., B, 2018, 232, 330–339 CrossRef CAS.
- J. Cheng, Z. Hu, Q. Li, X. Li, S. Fang, X. Wu, M. Li, Y. Ding, B. Liu, C. Yang, L. Wen, Y. Liu and K. Lv, Appl. Catal., B, 2018, 245, 197–206 CrossRef.
- J. C. Wang, H. C. Yao, Z. Y. Fan, L. Zhang, J. S. Wang, S. Q. Zang and Z. J. Li, ACS Appl. Mater. Interfaces, 2016, 8, 3765–3775 CrossRef CAS PubMed.
- P. Niu, L. Zhang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- D. R. Paul, R. Sharma, P. Panchal, R. Malik, A. Sharma, V. K. Tomer and S. P. Nehra, J. Nanosci. Nanotechnol., 2019, 19, 1–8 CrossRef PubMed.
- H. Xu, Y. Song, Y. Song, J. Zhu, T. Zhu, C. Liu, D. Zhao, Q. Zhang and H. Li, RSC Adv., 2014, 4, 34539 RSC.
- M. Wu, J. M. Yan, X. W. Zhang, M. Zhao and Q. Jiang, J. Mater. Chem. A, 2015, 3, 15710–15714 RSC.
- N. Cheng, J. Tian, Q. Liu, C. Ge, A. H. Qusti, A. M. Asiri, A. O. Al-Youbi and X. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 6815–6819 CrossRef CAS PubMed.
- S. Le, T. Jiang, Q. Zhao, X. Liu, Y. Li, B. Fang and M. Gong, RSC Adv., 2016, 6, 38811–38819 RSC.
- B. Liu, L. Ye, R. Wang, J. Yang, Y. Zhang, R. Guan, L. Tian and X. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 4001–4009 CrossRef CAS PubMed.
- J. Thomas, K. S. Ambili and S. Radhika, Catal. Today, 2018, 310, 11–18 CrossRef CAS.
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X. Chen and S. Huang, ACS Nano, 2011, 6, 205–211 CrossRef CAS PubMed.
- W. J. Ong, L. L. Tan, S. P. Chai and S. T. Yong, Dalton Trans., 2015, 44, 1249–1257 RSC.
- J. Fang, H. Fan, M. Li and C. Long, J. Mater. Chem. A, 2015, 3, 13819–13826 RSC.
- S. Wang, C. Li, T. Wang, P. Zhang, A. Li and J. Gong, J. Mater. Chem. A, 2014, 2, 2885 RSC.
- H. B. Fang, X. H. Zhang, J. Wu, N. Li, Y. Z. Zheng and X. Tao, Appl. Catal., B, 2018, 225, 397–405 CrossRef CAS.
- G. Zhang, J. Zhang, M. Zhang and X. Wang, J. Mater. Chem., 2012, 22, 8083 RSC.
- Z. Mo, X. She, Y. Li, L. Liu, L. Huang, Z. Chen, Q. Zhang, H. Xu and H. Li, RSC Adv., 2015, 5, 101552–101562 RSC.
- Q. Gu, Z. Gao, H. Zhao, Z. Lou, Y. Liao and C. Xue, RSC Adv., 2015, 5, 49317–49325 RSC.
- H. B. Fang, Y. Luo, Y. Z. Zheng, W. Ma and X. Tao, Ind. Eng. Chem. Res., 2016, 55, 4506–4514 CrossRef CAS.
- Q. Su, J. Sun, J. Wang, Z. Yang, W. Cheng and S. Zhang, Catal. Sci. Technol., 2014, 4, 1556 RSC.
- J. Liu, T. Zhang, Z. Wang, G. Dawson and W. Chen, J. Mater. Chem., 2011, 21, 14398 RSC.
- Y. Zhang, J. Liu, G. Wu and W. Chen, Nanoscale, 2012, 5300 RSC.
- D. J. Martin, K. Qiu, S. A. Shevlin, A. D. Handoko, X. Chen, Z. Guo and J. Tang, Angew. Chem., Int. Ed., 2014, 12, 9394–9399 CrossRef.
- P. Niu, G. Liu and H. M. Cheng, J. Phys. Chem. C, 2012, 116, 11013–11018 CrossRef CAS.
- J. Xu, Y. Li, S. Peng, G. Lu and S. Li, Phys. Chem. Chem. Phys., 2013, 15, 7657 RSC.
- F. Dong, L. Wu, Y. Sun, M. Fu, Z. Wu and S. C. Lee, J. Mater. Chem., 2011, 21, 15171 RSC.
- Y. Yang, Q. Zhang, R. Zhang, T. Ran, W. Wan and Y. Zhou, Front. Chem., 2018, 6, 156 CrossRef PubMed.
- D. Das, D. Banerjee, B. Das, N. S. Das and K. K. Chattopadhyay, Mater. Res. Bull., 2017, 89, 170–179 CrossRef CAS.
- Y. Li, K. Lv, W. Ho, Z. Zhao and Y. Huang, Chinese J Catal., 2017, 38(2), 321–329 CrossRef CAS.
- J. L. Zimmerman, R. Williams, V. N. Khabashesku and J. L. Margrave, Nano Lett., 2001, 1, 731–734 CrossRef CAS.
- Q. Xiang, J. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CrossRef CAS.
- A. Bahuguna, A. Kumar, S. Kumar, T. Chhabra and V. Krishnan, ChemCatChem, 2018, 10, 3121–3132 CrossRef CAS.
- A. Bahuguna, P. Choudhary, T. Chhabra and V. Krishnan, ACS Omega, 2018, 3(9), 12163–12178 CrossRef CAS.
- R. S. Mane, S. J. Roh, O. S. Joo, C. D. Lokhande and S. H. Han, Electrochim. Acta, 2005, 50, 2453–2459 CrossRef CAS.
- A. F. Alkaim, A. M. Aljeboree, N. A. Alrazaq, S. J. Baqir, F. H. Hussein and A. J. Lilo, Asian J. Chem., 2014, 26, 8445–8448 CrossRef.
- L. Heymann, B. Schiller, H. Noei, A. Stierle and C. Klinke, ACS Omega, 2018, 3, 3892–3900 CrossRef CAS PubMed.
- S. Ahmed, M. G. Rasul, W. N. Martens, R. Brown and M. A. Hashib, Water, Air, Soil Pollut., 2010, 215, 3–29 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab and
Anshu Sharma*c
*ab and
Anshu Sharma*c