DOI:
10.1039/C9RA02169H
(Paper)
RSC Adv., 2019,
9, 13243-13248
Design and synthesis of CoIIHMTAA-14/16 macrocycles and their nano-composites for oxygen reduction electrocatalysis†
Received
20th March 2019
, Accepted 16th April 2019
First published on 30th April 2019
Abstract
The major concerns in the design of macrocycle based-ORR catalysts are: (i) understanding the macrocyclic, π-conjugation, central metal and substituent effects on ORR electrocatalysis; and (ii) the use of macrocycles on the electrode surface for the retention of ORR activity because of their poor stability. In this work, we demonstrated the aromaticity/π-electron conjugation effect on ORR activity by using the same macrocycles [HMTAA-14 and 16 (hexamethyltetraaza [14] and [16] annulenes)] with a difference in their macrocyclic cavity/π-electron conjugation. The macrocycles CoIIHMTAA-14 and CoIIHMTAA-16 and their nanocomposites with highly conductive carbon black were prepared by a microwave-assisted method and characterized by using multiple spectroscopy techniques. Comparative redox and oxygen reduction activity studies of CoIIHMTAA-14 and CoIIHMTAA-16 were undertaken by using cyclic voltammetry and linear sweep voltammetry in an alkaline medium. The composite CoIIHMTAA-16@C showed good ORR activity compared to CoIIHMTAA-14@C in O2-saturated KOH electrolyte. Since the CoIIHMTAA-14 and CoIIHMTAA-16 systems have a similar central atom and substituents, the shift of the ORR peak position in the +ive potential region for HMTAA-16 can be attributed to the difference in the size of the macrocyclic cavity (macrocyclic effect) and the extra stability of HMTAA-16 annulene due to its aromaticity.
1. Introduction
Fuel cells have been developed to offer a more efficient way for energy conversion with the minimum CO2 emissions and a harmless by-product, namely water. But the fuel cell devices suffer from efficiency loss in cathode over-potential. Therefore, it has become necessary to develop high-performance cathode electro-catalysts that are capable of reducing efficiency loss of the cathode over-potential. Platinum derived catalysts were identified as the most promising catalysts in fuel cell devices.1 But Pt-based electro-catalysts have serious limitations including poor abundance, high-priced cost, and low viable accessibility, which have restricted the application of Pt-based electro-catalysts for marketable uses. Consequently, seeking a new potential alternative of Pt-based catalysts has become an apparent aim.2 Macrocyclic complexes have been largely considered as alternatives for Pt-based ORR catalysts.3 But the major challenges are (i) to understand the parameters of macrocycles like the macrocyclic, π-conjugation, central metal, and substituent effects and their effect on ORR electro-catalysis and (ii) the use of macrocycles on the electrode surface for the retention of ORR activity;4–9 principally because of the poor stability of these macrocycles in the electrolytic environment.10,11 We are mainly interested in N4-type tetraazaannulene (TAA) macrocycles. The TAA macrocyclic frameworks have a smaller macrocyclic cavity than those of other N4 type macrocycles such as porphyrins and phthalocyanines. The main advantage of these small macrocycles is that the electron density of these macrocycles can be increased on the electrode surface. Furthermore, the macrocyclic cavity size, π-conjugation, central metal, and substituents can have strong electronic effects on the active sites for ORR. This indicates that the ORR activity of TAA macrocycles might be enhanced by altering these macrocyclic parameters.12 The hexamethyl substituted TAA macrocycles like HMTAA-14 and HMTAA-16 macrocyclic ligands have been used as mimicking agents, electrocatalysts, and photocatalysts such as in naturally occurring systems of porphyrins and corrin rings.13 These macrocycles have the affinity to enable multiple 1 e− oxidation routes. Therefore, many tetraazamacrocyclic ligands and their metal complexes have been considered in the quest of finding their applications in electrocatalysis. HMTAA-14, HMTAA-16, and porphyrin ligands all have four co-planar donor sites through the N-atom and form the corresponding dianions on deprotonation.14–16 The HMTAA-14 annulenes are Huckel antiaromatic (4nπ electrons) and flexible, and they can adopt various conformations with respect to planarity (known as saddle-shaped). Further, the dianion [HMTAA-14]2− does not show complete resonance through the entire annulene framework but the π-electron delocalization remains confined to the 1,3-diiminato linkages. In a similar fashion, HMTAA-16 and porphyrin type annulenes showed complete π-electron delocalization spread over the entire ligand framework. For HMTAA macrocycles, the stability was also controlled by the size of the macrocyclic ring; a larger macrocyclic ring leads to greater complex stabilization for small metal ions, while larger metal ions show progressively greater complex destabilization with the larger aromatic macrocyclic ring, displaying selectivity towards metal ions that fit best into it. The computational studies suggested that the TAA macrocyclic ligand in the process of activation and binding of molecular oxygen with a Mn metal ion, leading to the formation of MnN4–O2 adducts, shown to have good ORR activity.17 In addition, the CoIITAA macrocyclic complex is more active for ORR compared to cobalt phthalocyanine, catalyzing the reduction of O2 to H2O2 at potentials 400 mV more positive than that required using CoIITAA,18 with comparable performance to platinum in the ORR. These efforts inspired us to make an experimental assessment on the studies of ORR activities of CoHMTAA-14/16 macrocycles and the effect of macrocyclic cavity size on their ORR activity, which was not conducted to date. Further, we investigated the redox property and ORR activity of CoHMTAA-14/16 ligated by HMTAA under alkaline conditions and discussed the effect of the ligand structures on the electrochemical behavior and ORR electrocatalysis of CoHMTAA by emphasizing the macrocyclic parameters such as π-electron conjugation, size of the macrocyclic framework and aromaticity effect on their electrocatalytic ORR activity.
2. Results and discussion
2.1. Morphology and structural characterization of the composites
The macrocycles and their composites, CoIIHMTAA-14/16 and CoIIHMTAA-14/16@C, respectively, were synthesized via a microwave (MW) assisted method19,20 (see ESI for details†), as illustrated in Fig. 1.
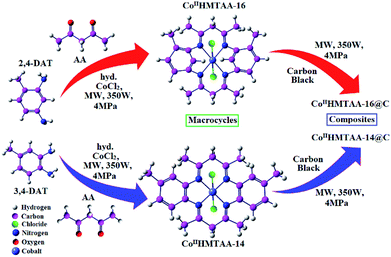 |
| | Fig. 1 A systematic schematic diagram of the synthesis of CoIIHMTAA-14/16 macrocycles and CoIIHMTAA-14/16@C composites. | |
2.1.1. Fourier transform infrared (FT-IR) spectroscopy studies. The FT-IR study was employed to confirm the structural features of the HMTAA macrocycles. The IR spectra of both HMTAA-14/16 macrocycles and their nanocomposites (Fig. S1 and S2†) showed the disappearance of the free keto group (near 1700 cm−1) and free amino group (near 3300 cm−1) and the emergence of new sharp absorption peaks near 1570–1595 cm−1, which verify the reaction between amine and ketone groups to form the “macrocyclic Schiff base”, as these peaks may be assigned to ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) stretching vibrations. Further, the absorption bands between 1450 and 1480 cm−1 can be assigned to ν(CC) stretching vibrations of the aromatic moiety. The medium intensity bands appearing from 2800 to 2950 cm−1 can be assigned to the ν(C–H) stretch of the methyl groups. In addition, the far infrared spectra displayed some bands near 450–525 cm−1 corresponding to ν(M–N) vibrations, and these bands originate from (M–N) –CN– vibrational modes, which indicates the coordination of the azomethine nitrogen. In the case of their nanocomposites, some minor shifting was observed towards higher wavenumbers, which may be due to some carbon black interaction.21,22
N) stretching vibrations. Further, the absorption bands between 1450 and 1480 cm−1 can be assigned to ν(CC) stretching vibrations of the aromatic moiety. The medium intensity bands appearing from 2800 to 2950 cm−1 can be assigned to the ν(C–H) stretch of the methyl groups. In addition, the far infrared spectra displayed some bands near 450–525 cm−1 corresponding to ν(M–N) vibrations, and these bands originate from (M–N) –CN– vibrational modes, which indicates the coordination of the azomethine nitrogen. In the case of their nanocomposites, some minor shifting was observed towards higher wavenumbers, which may be due to some carbon black interaction.21,22
2.1.2. Ultraviolet-visible (UV-vis) spectroscopic studies. The UV-vis studies were performed to confirm the electronic environment, the geometry of the macrocycle and the interaction between carbon black and the macrocycles. The UV-vis spectra (Fig. S3a and S4a†) of both macrocycles were recorded in methanol (10−3 M) in the range of 200–800 nm. The absorption bands at 253 and 270 nm may be allocated to the π → π* and n → π* electronic transitions, correspondingly. Both macrocycles CoIIHMTAA-14 and CoIIHMTAA-16 showed three bands in each case at (315 nm) 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 746 cm−1, (352 nm) 28409 cm−1 and (425 nm) 23529 cm−1, and (315 nm) 31746 cm−1, (370 nm) 27027 cm−1 and (460 nm) 21739 cm−1, respectively, and these bands can be attributed to 4T1g(F) → 4A2g(F), 4T1g(F) → 4T2g(F) and 4T1g(F) → 4T1g(P) electronic transitions. Thus, these electronic spectral data are in agreement with the molar conductance data, confirming the octahedral geometry of these macrocycles with axial chloride ions. In addition, the UV-vis spectra of CoIIHMTAA-14@C and CoIIHMTAA-16@C (Fig. S3b and S4b†) were quite different as they showed a bathochromic shift in the main absorption bands, which confirmed the adsorption of these macrocycles via π–π interactions. This clearly indicates the strong electronic coupling between CoIIHMTAA-14/16 and the carbon support.23,24
746 cm−1, (352 nm) 28409 cm−1 and (425 nm) 23529 cm−1, and (315 nm) 31746 cm−1, (370 nm) 27027 cm−1 and (460 nm) 21739 cm−1, respectively, and these bands can be attributed to 4T1g(F) → 4A2g(F), 4T1g(F) → 4T2g(F) and 4T1g(F) → 4T1g(P) electronic transitions. Thus, these electronic spectral data are in agreement with the molar conductance data, confirming the octahedral geometry of these macrocycles with axial chloride ions. In addition, the UV-vis spectra of CoIIHMTAA-14@C and CoIIHMTAA-16@C (Fig. S3b and S4b†) were quite different as they showed a bathochromic shift in the main absorption bands, which confirmed the adsorption of these macrocycles via π–π interactions. This clearly indicates the strong electronic coupling between CoIIHMTAA-14/16 and the carbon support.23,24
2.1.3. Mass spectra. The mass spectra, recorded in positive mode for both macrocycles, showed a more intense peak at m/z 502 that corresponds to the molecular ion peak (M)+, which is in good agreement with respect to the molecular formula (Fig. S4†). The other observed peaks at m/z 443, 175, 134 and 71 can be assigned to other different cleavage moieties.13,25
2.1.4. X-ray photoelectron spectroscopy (XPS) studies. The XPS studies were undertaken to characterize the chemical composition of the composites. The XPS spectra of N1s (Fig. 2a) of HMTAA-14@C showed two peaks at around 399.70 eV and 400.10 eV, which may be due to the presence of C–N and CN, respectively. From the literature, it is clear that Co(II) and Co(III) have comparative 2p binding energies however they can be separated by the Co2p1/2 and Co2p3/2 full width at half maximum for high-turn Co(II) and low-turn Co(III). A fine probe of Co2p (Fig. 2b) presented four peaks at 781.8, 787.1 (for Co2p3/2), 796.8, and 804.1 eV (Co2p1/2), indicating that Co2p demonstrates the 2+/3+ oxidation state of Co during the photo-reduction of HMTAA-14 by X-rays.26 Further, the XPS survey (Fig. 2c) for HMTAA-14 is in good agreement with the elemental composition, showing characteristics of Cl2p (∼199.0 eV), N1s (∼400.0 eV), O1s (∼530 eV) and Co2p (∼779 eV).27 Thus, the XPS studies further confirmed the composite compositions in accordance with the proposed structural features of the catalysts.
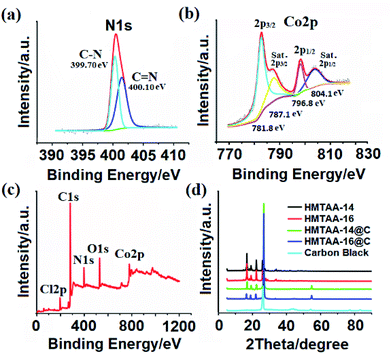 |
| | Fig. 2 XPS spectra of HMTAA-16: (a) N1s, (b) Co2p, and (c) survey; and (d) XRD patterns. | |
2.1.5. X-ray diffraction (XRD) studies. The structural features of the macrocycles were characterized using XRD. Fig. 2d shows the XRD patterns of carbon black and the composites HMTAA-14/16@C, and the inset shows the XRD patterns of the unsupported HMTAA-14/16 macrocycles. The unsupported HMTAA-14/16 showed sharp characteristic diffraction peaks at ca. 7–20 and the composites HMTAA-14/16@C also showed a similar pattern, which can be attributed to the aggregation of HMTAA-14/16 macrocycles on carbon black. The observed external intense peak at ca. 26 in each pattern of HMTAA-14/16@C can be assigned to the characteristic carbon black diffraction peak,28 indicating the graphitic structure of carbon black. In comparison to that of pure carbon black, the XRD peak of graphitic carbon in the composites HMTAA-14@C and HMTAA-16 shifted toward a higher degree, which can be attributed to the expansion of carbon black during the modification using microwave irradiation.29
2.1.6. Transmission electron microscopy (TEM) study. The morphological study of the synthesized composites was performed using TEM. The typical TEM images of the composite HMTAA-16@C showed the “particle aggregation morphology” of the carbon black (Fig. 3). However, no individual HMTAA-16 was observed in this composite. This indicates that the use of high surface area carbon black as a supporting material inhibits the pi–pi-stacking of the HMTAA-14 molecule; as a result, a core–shell structured composite where the HMTAA-14 molecule is adsorbed on the carbon black material is formed.30 Our results are in accordance with the previously reported similar work on an iron phthalocyanine composite, FePc@C.31
 |
| | Fig. 3 TEM images of HMTAA-16: (a) low magnification and (b) high magnification. | |
3. Electrochemical measurements
3.1. Redox studies of CoIIHMTAA-14 and CoIIHMTAA-16
The cyclic voltammograms (CVs) of both CoIIHMTAA-14 and CoIIHMTAA-16 were obtained in an inert atmosphere at a scan rate of 50 mV s−1. The macrocycles CoIIHMTAA-14 and CoIIHMTAA-16 showed similar redox behavior with a slight difference in the anodic peak potential (Ep,a) and cathodic peak potential (Ep,c) peak positions. The CVs of both CoIIHMTAA-14 and CoIIHMTAA-16 (Fig. 4a and b) showed first redox couples at +0.76 V and +0.81 V corresponding to Co3+/Co2+ and the second redox couple at +1.09 V and +1.13 V corresponding to L+/L0 (L = HMTAA), respectively.31,32 In addition, based on the peak separation (ΔEp = Ep,a − Ep,c), which was observed to be near to +60 mV, and peak current ratio (ip,a/ip,c) near to 0.81 (in both cases), a quasi-reversible process may be assigned to the Co3+/Co2+ redox couple. Further, the anodic and cathodic peak potentials were observed to be independent of scan rate (Fig. 4c) when the scan rate was varied from 25 mV s−1 to 500 mV s−1, and the plots of ip vs. v1/2 (inset Fig. 3c) were found to be linear and follow the “Randles–Sevcik” equation13,34,35
| i = 2.69 × 105n3/2AD1/2cv1/2 |
where n, number of electrons transferred; A, area of the electrode; D, diffusion coefficient; c, analyte concentration; v, scan rate.
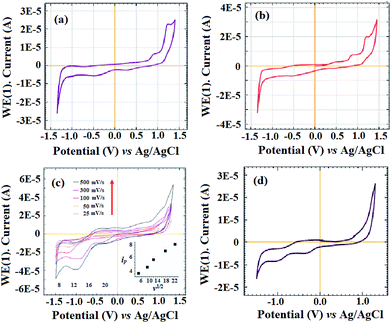 |
| | Fig. 4 CVs of (a) CoIIHMTAA-14 and (b) CoIIHMTAA-16, (c) a cyclic voltammogram of CoIIHMTAA-14 at varying scan rates and (d) a cyclic voltammogram of CoIIHMTAA-14 in the presence of pyridine. | |
Since both the CoIIHMTAA-14 and CoIIHMTAA-16 macrocycles have similar substituents, the difference in the shifting of peak position in the +ive potential region of the CoIIHMTAA-16 macrocyclic complex can be attributed to the difference in the macrocyclic cavity size/extent of unsaturation and extra stability as aromaticity of the CoIIHMTAA-16 complex.33 Next, we studied the axial ligation effect on the nature of electrochemistry of the planar N4 chelates of the macrocycles by using pyridine as a strong axial ligand donor.30,32–35 Fig. 4d shows the considerable effect of the addition of a small amount of pyridine on the voltammetry of CoIIHMTAA-14 by eliminating the CoII/CoIII redox couple. Our results are in good agreement with previous published similar analogous CoIIDPTAA systems.33 The observed redox parameters for both macrocycles are given in Table 1.
Table 1 Redox data for CoIIHMTAA-14 and CoIIHMTAA-16 macrocycles observed in DMF containing 0.1 M TEAP and 10−3 M concentrations of each complex at a scan rate 25 mV s−1
| Macrocycle |
Co2+/3+ |
HMTAA+/0 |
| CoIIHMTAA-14 |
+0.76 V |
+1.09 V |
| CoIIHMTAA-16 |
+0.81 V |
+1.13 V |
| CoIIDPTAA29 |
+0.24 V |
+0.56 V |
| CoIIHMTAA-14 + excess pyridine |
+0.60 V |
+0.86 V |
| CoIIDPTAA-14 + excess pyridine29 |
+0.02 V |
+0.64 V |
3.2. ORR activity of the composites CoIIHMTAA-14@C and CoIIHMTAA-16@C
The CVs of CoIIHMTAA-14@C, CoIIHMTAA-16@C and 20% Pt/C (Fig. 5a) were recorded in O2-saturated 0.1 M KOH electrolyte at a scan rate of 50 mV s−1 and with 0.25 μg cm−2 loading amounts of each catalyst onto the glassy carbon electrode. The onset potential of CoIIHMTAA-14@C, CoIIHMTAA-16@C and 20% Pt/C was found to be approximately +0.85 V, +0.93 V and +0.95 V while the formal potential (E1/2) was observed in the order of CoIIHMTAA@C (E1/2 = +0.70 V) < CoIIHMTAA-16@C (E1/2 = +0.82 V) < 20% Pt/C (E1/2 = +0.85 V).36 These results are in good agreement with the corresponding LSV curves (Fig. 5b). Thus, CoIIHMTAA-16@C showed a more positive formal potential than CoIIHMTAA-14@C, indicating the enhanced ORR performance of CoIIHMTAA-16@C as compared to CoIIHMTAA-14@C.
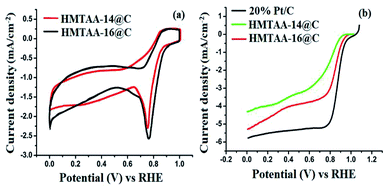 |
| | Fig. 5 (a) CVs collected of CoIIHMTAA-14@C and CoIIHMTAA-16@C, and (b) LSV curves collected of CoIIHMTAA-14@C, CoIIHMTAA-16@C and 20% Pt/C in O2-saturated 0.1 M KOH electrolyte at a scan rate of 50 mV s−1 and 5 mV s−1, respectively, at room temperature. | |
Both composites, CoIIHMTAA-14@C and CoIIHMTAA-16@C, have a similar chemical composition and carbon support and only differ in the electronic structure inside of the macrocycles. The electrochemistry of the unsupported CoIIHMTAA-14 and CoIIHMTAA-16 macrocycles suggested that the difference in redox peak position can be attributed to the different electronic environment.
Similarly, we observed that the ORR catalytic activity varied with the size of the macrocyclic cavity and extent of π-electron conjugation. CoIIHMTAA-14 and CoIIHMTAA-16 macrocyclic frameworks have coplanar N4-donor atom arrangements that can form corresponding dianions on deprotonation. Thus, the protonated HMTAA-14 showed an anti-aromatic nature (4nπ electron rule) followed by incomplete resonance through the entire framework; as a result, it showed poor activity and stability towards ORR. Besides, HMTAA-16 is a member of the porphyrin and phthalocyanine family, which have complete π-electron delocalization over the entire macrocyclic framework followed by an aromatic nature (4n + 2π electron rule).7–10,13 Thus, this special difference between the antiaromatic and aromatic nature of HMTAA-14 and HMTAA-16, respectively, and macrocyclic effect can explain the higher ORR activity of the CoIIHMTAA-16 as compared to CoIIHMTAA-14 macrocycle containing composites.37 The ORR activity of macrocycles is associated with e− transfer processes that involve the π-electron conjugation in the macrocycle, d-electrons of the metal center and charge transfer transitions between them. Although these macrocycles show comparable ORR activity with that of 20% Pt/C, it seems that their ORR activity can be further improved by tailoring the electronic structure using appropriate substitution and carbon support. Usually, for the CoII–N4 macrocycles, CoIII is probably not formed upon interaction with the oxygen molecule since the CoIII/CoII formal potential is much more positive, so the CoII/CoI redox process can be considered as the ORR mediator by providing the CoII active site for ORR.38 The cyclic voltammograms of both CoIIHMTAA-14 and CoIIHMTAA-16 showed distinct cathodic peaks near −0.50 V, and maybe this redox couple played an important role as mediator to catalyze ORR. So, our future intention is to design several excellent macrocycle-based ORR electrocatalysts by using different macrocyclic framework sizes, metal ions, electron donating or withdrawing groups and carbon supports to alter their redox potential and electronic structure.
4. Conclusions
In the present studies, two macrocycles, CoIIHMTAA-14 and CoIIHMTAA-16, were synthesized and characterized by using multiple spectroscopic techniques that confirmed an octahedral geometry for both macrocycles. The cyclic voltammograms of CoIIHMTAA-14 and CoIIHMTAA-16 showed a considerable difference in the values of Ep,a and Ep,c. The cyclic voltammogram of CoIIHMTAA-14 shows two distinct redox couples at +0.76 V and +1.09 V, whereas that of CoIIHMTAA-16 shows the corresponding redox couples at +0.81 V and +1.13 V, respectively. Since both the CoIIHMTAA-14 and CoIIHMTAA-16 complexes have similar substituents, the difference in the shifting of peak position in the +ive potential region of the CoIIHMTAA-16 macrocyclic complex can be attributed to the difference in the size of the macrocyclic cavity or the extent of unsaturation and extra stability in terms of the aromaticity of HMTAA-16 annulene. Similarly, the ORR activities of their composites could be correlated and it was concluded that the external stability of HMTAA-16 annulene can play a key role in enhancing the ORR activity of CoIIHMTAA-16 macrocycle based electrocatalysts as compared to antiaromatic or nonaromatic systems. Further, the present work can be further explored to design macrocycle-based electrocatalysts for various electrocatalytic processes.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are thankful to GLA University, Mathura and IIT Roorkee, Roorkee for infrastructural support, and SAIF Panjab University Chandigarh and Beijing University of Chemical Technology, China for aiding in completion of the studies.
References
- S. Walch, A. Dhanda, M. Aryanpour and H. Pitsch, J. Phys. Chem. C, 2008, 112, 831 CrossRef.
- C. H. Tsai, C. J. Shih, W. S. Wang, W. F. Chi, W. C. Huang, Y. C. Hu and Y. H. Yu, Appl. Surf. Sci., 2018, 434, 414 CrossRef.
- Y. Liu, Y. Y. Wu, G. J. Lv, T. Pu, X. Q. He and L. L. Cui, Electrochim. Acta, 2013, 112, 269 CrossRef CAS.
- Y. Zhao, H. Tang, N. Yang and D. Wang, Adv. Sci., 2018, 5, 1800959 CrossRef PubMed.
- Y. Zhao, J. Wan, H. Yao, L. Zhang, K. Lin, L. Wang, N. Yang, D. Liu, L. Song, J. Zhu, L. Gu, L. Liu, H. Zhao, Y. Li and D. Wang, Nat. Chem., 2018, 10, 924 CrossRef CAS PubMed.
- Y. Zhao, L. Zhang, J. Qi, Q. Jin, K. Lin and D. Wang, Acta Phys.-Chim. Sin., 2018, 34, 1048 Search PubMed.
- N. Parvin, Q. Jin, Y. Wei, R. Yu, B. Zheng, L. Huang, Y. Zhang, L. Wang, H. Zhang, M. Gao, H. Zhao, W. Hu, Y. Li and D. Wang, Adv. Mater., 2017, 29(18), 1606755 CrossRef PubMed.
- H. Tang, J. Wang, H. Yin, H. Zhao, D. Wang and Z. Tang, Adv. Mater., 2015, 27, 1117 CrossRef CAS PubMed.
- H. Tang, C. M. Hessel, J. Wang, N. Yang, R. Yu, H. Zhaod and D. Wang, Chem. Soc. Rev., 2014, 43, 4281 RSC.
- K. Wiesener, D. Ohms, V. Neumann and R. Franke, Mater. Chem. Phys., 1989, 22, 457 CrossRef CAS.
- J. H. Zagala, F. J. Recio, C. A. Gutierrez, C. Zuñiga, M. A. Páez and C. A. Caro, Electrochem. Commun., 2014, 41, 24 CrossRef.
- P. Gouérec, A. Biloul, O. Contamin, G. Scarbeck, M. Savy, J. Riga, L. T. Wengand and P. Bertrand, J. Electroanal. Chem., 1997, 422, 61 CrossRef.
- Á. R. Escudero, G. L. Estiú, J. Costamagna and G. I. C. Jirón, J. Coord. Chem., 2003, 56, 1257 CrossRef.
- J. F. M. Oth, H. Baumann, J. M. Giles and G. Schroeder, J. Am. Chem. Soc., 1972, 94, 3498 CrossRef CAS.
- J. F. M. Oth, G. Anthoine and J. M. Gilles, Tetrahedron Lett., 1968, 9, 6265 CrossRef.
- F. L. Hirshfeld and D. Rabinovich, Acta Crystallogr., 2010, 19, 235 Search PubMed.
- A. Costa, A. Luís, P. Silva, R. B. Viana, A. A. Tanaka, J. de Jesus and G. Varela Jr, J. Mol. Model., 2016, 22, 217 CrossRef PubMed.
- A. Putten, A. Elzing, W. Visscher and E. Barendrecht, J. Electroanal. Chem. Interfacial Electrochem., 1987, 221, 95 CrossRef.
- A. R. Cutler and D. Dolphin, J. Coord. Chem., 1976, 6, 59 CrossRef CAS.
- K. Sakata, Y. Saitoh, K. Kawakami, N. Nakamura, T. Hori, Y. Nakano and M. Hashimoto, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2006, 26, 1267 CrossRef.
- R. Sustmann, H. G. Korth, D. Kobus, J. Baute, K. H. Seiffert, E. Verheggen, E. Bill, M. Kirsch and H. Groot, Inorg. Chem., 2007, 46, 11416 CrossRef CAS PubMed.
- M. A. Ali, A. H. Mirza, A. M. S. Hossain and M. Nazimuddin, Polyhedron, 2001, 20, 1045 CrossRef CAS.
- K. Sakata, H. Shibata, M. Hashimoto and A. Tsuge, J. Coord. Chem., 2002, 55, 15 Search PubMed.
- K. Sakata and T. Hori, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 1990, 20, 263 CrossRef CAS.
- D. P. Singh, R. Kumar, M. Kamboj and K. Jain, J. Coord. Chem., 2009, 62, 2995 CrossRef CAS.
- J. Stoch and A. Capecki, Surf. Interface Anal., 1990, 15, 206 CrossRef CAS.
- L. Xuguang, X. Wei, L. Tianhong, J. Yiping, L. Hongying and S. Yun, Chem. Res. Chin. Univ., 2003, 24, 1246 Search PubMed.
- K. Sakata, Z. Wang, M. Hashimoto, A. Tsuge and Y. Tanoue, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 1999, 29, 265 CrossRef CAS.
- H. Shang, Y. Lu, F. Zhao, C. Chao, B. Zhang and H. Zhang, RSC Adv., 2015, 5, 75728 RSC.
- Y. Yuan, J. Ahmed and S. Kim, J. Power Sources, 2011, 196, 1103 CrossRef CAS.
- S. Zhang, H. Zhang, X. Hua and S. Chen, J. Mater. Chem. A, 2015, 3, 10013 RSC.
- V. B. Rana, P. Singh, D. P. Singh and M. P. Teotia, Transition Met. Chem., 1982, 7, 174 CrossRef CAS.
- B. D. Alexander, J. A. Crayston and T. J. Dines, J. Electroanal. Chem., 2007, 605, 109 CrossRef CAS.
- A. Deronzier and M. J. Marques, Electrochim. Acta, 1994, 39, 1377 CrossRef CAS.
- S. Karunakaran and M. Kandaswamy, Dalton Trans., 1994, 10, 1595 RSC.
- K. Liu, Y. Lei, R. Chen and G. Wang, Oxygen Electroreduction on M-N4 Macrocyclic Complexes, Electrochemistry of N4 Macrocyclic Metal Complexes, 2nd edn, 2016 Search PubMed.
- J. H. Cameron and S. Graham, Dalton Trans., 1992, 3, 385 RSC.
- J. H. Zagal, S. Griveau, J. F. Silva, T. Nyokong and F. Bedioui, Coord. Chem. Rev., 2010, 254, 2755 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02169h |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b




