
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLabel-free cell phenotypic study of FFA4 and FFA1 and discovery of novel agonists of FFA4 from natural products
Fangfang Xuab,
Han Zhoua,
Xiumei Liua,
Xiuli Zhang *ac,
Zhiwei Wangd,
Tao Houa,
Jixia Wanga,
Lala Quab,
Pengyu Zhange,
Hailong Piaoa and
Xinmiao Liang*ac
*ac,
Zhiwei Wangd,
Tao Houa,
Jixia Wanga,
Lala Quab,
Pengyu Zhange,
Hailong Piaoa and
Xinmiao Liang*ac
aKey Lab of Separation Science for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China. E-mail: zhangxiuli@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China
cCo-innovation Center of Neuroregeneration, Nantong University, Nantong 226019, China
dDICP-CMC Innovation Institute of Medicine, Taizhou 225300, China
eLiaoning Provincial Core Lab of Medical Molecular Biology, Dalian Medical University, Dalian 116044, China
First published on 15th May 2019
Abstract
In this article, pharmacological studies of the free fatty acid receptor (FFA) 4 and FFA1 were conducted in transfected CHO cells (FFA4&FFA1) and HT29 cells with application of a label-free dynamic mass redistribution (DMR) assay. Commercially available compounds including α-linolenic acid (ALA), GW9508, TUG891, GSK137647A, TAK875, MEDICA16, AH7614 and GW1100, were used to validate the assay; real-time tracing of ligand-induced cell responses elucidated pharmacological properties of ligand–receptor interactions. A pool of 140 natural compounds was screened using the CHO-FFA4 cells. Three new FFA4 agonists with novel skeletons were discovered and they were dihydrotanshinone, emodin and acetylshikonin (EC50 values were 32.88, 38.18 and 10.17 μM, respectively). Ligand selectivity was compared between FFA4 and FFA1; dihydrotanshinone and emodin displayed FFA4 selectivity, while acetylshikonin shared FFA1 and FFA4 agonist activities with EC50 values comparable to the endogenous ligand ALA. The three novel FFA4 agonists provide a promising chemical starting point for identification and optimization of drugs used for treating metabolic and inflammatory diseases. Besides, this work will help to explain the mechanism of actions of natural products.
1. Introduction
The free fatty acid receptor (FFA) 4 (previously known as GPR120) is a member of the G-protein coupled receptors (GPCRs).1,2 The receptor plays important roles in various physiological processes and is relevant in a number of diseases. Hirasawa et al. found that dietary free fatty acids regulated gut incretin glucagon-like peptide-1 secretion through FFA4,2 indicating the potential of FFA4 ligands in diabetics treatment. Based on the genotyping data in 6942 unrelated obese individuals and 7654 control subjects, it was shown that people whose FFA4 function was impaired by a deleterious non-synonymous mutation (p.R270H) were significantly more likely to be obese.3 In mouse models, it was also found that with dysfunctional FFA4 receptors, mice fed with a high-fat diet tended to develop severer fatty liver, peripheral inflammatory infiltration and insulin intolerance. Nakamoto et al. reported docosahexenoic acid (DHA) via FFA4 signaling prevented progression of nonalcoholic steatohepatitis.4 Increasing evidence suggested FFA4 could be a potential therapeutic target for treatment of a variety of disorders. Hence, pharmacological study and development of new FFA4 ligands will be beneficial for treatment of related diseases. The free fatty acid receptor (FFA) 1 (previously known as GPR40) is another subtype of the FFA receptors, and it shares the same endogenous ligands with FFA4.1 Ligand selectivity between FFA1 and FFA4 is a challenge for research.1,5 Therefore, discovery of FFA4-selective ligands with selectivity is highly desired.Different functional assays have been performed on FFA4, most of which were Ca2+ mobilization assay and β-arrestin assay.6–8 These assays often rely on the measurement of a loaded fluorescent dye or fluorescently tagged targets, and the cellular physiology of the target protein might be altered in the tests as a consequence.9 In this study, a label-free dynamic mass redistribution (DMR) technique was applied as a noninvasive assay that offers real-time and integrated measurement of the receptor–ligand pharmacology in whole cells.10–14 It can be carried out in various formats and supports intervention with probes, allowing mechanistic elucidation of drug pharmacology.
Natural products (NPs)-derived compounds provide inherent large-scale structural diversity and have become important sources for drug discovery.15 Diet-derived bioactive compounds, for example polyphenols and some fatty acids, are known to cure inflammation and related to the obesity associated metabolic disorders.16 Reviews also describe that NPs-derived compounds had a major impact on the treatment of metabolic diseases.17,18 Therefore, NPs can be a source for discovering FFA4 ligands. By literature retrieval, two kinds of natural FFA4 agonists were found, grifolic acid (EC50 > 30 μM) and its methyl ether19 and phytosphingosine (EC50 = 33.4 μM) and its derivatives.20,21 Structural diversity of FFA4 ligands need to be expanded urgently as it will not only serve as a source of new drug candidates, but also benefit investigation of the pharmacological and physiological functions of FFA4.
In the study, we specifically investigated pharmacology of endogenously or heterologously expressed FFA4 and FFA1 receptors, using multiple commercial available ligands with DMR assay. This technique was established as an efficient method for FFA4 ligand screening. Importantly, we identified three novel FFA4 agonists from a pool of 140 natural compounds, and they are dihydrotanshinone, emodin and acetylshikonin with EC50 values of 32.88 μM, 38.18 μM and 10.17 μM, respectively. It is worth noting that the potency of acetylshikonin is comparable to the natural ligand α-linolenic acid (ALA), while dihydrotanshinone and emodin are FFA4 selective.
2. Experimental
2.1. Materials
 | ||
| Fig. 1 Chemical structure of ligand molecules and three NPs-derived FFA4 agonists. | ||
Hank's balanced salt solution (HBSS), HEPES, fetal bovine serum (FBS) were obtained from Gibco, McCoy's 5A and Ham's F12K media were obtained from Sigma (St Louis, MO, USA). Epic® 384-well biosensor cell culture compatible microplates were obtained from Corning Incorporated (Corning, NY, USA).
GW9508, TUG891, GSK137647A, MEDICA16, AH7614 and ALA were stocked at 100 mM and TAK875, GW1100 were stocked at 10 mM. The stock solution were stored at −20 °C. ALA was prepared in 95% ethanol, and others were dissolved in 100% dimethyl sulfoxide (DMSO). All compounds were freshly diluted with the assay buffer (1× Hank's balanced salt solution (HBSS) buffer, 10 mM HEPES, pH = 7.2) containing 0.1% bovine serum albumin (BSA).
2.2. Cell culture
HT29 cells (Human colorectal carcinoma cells) and CHO-K1 cells (Chinese hamster ovary cells) were obtained from the Type Culture Collection of the Chinese Academy of Science (Shanghai, China). HT29 cells were cultured in McCoy's 5A medium supplemented with 10% FBS, 50 μg mL−1 penicillin and 100 μg mL−1 streptomycin at 37 °C under air/5% CO2. CHO-K1 cells were cultured in Ham's F12K medium supplemented with 10% FBS.2.3. Transfection of FFA4 and FFA1 cell line
CHO-K1 cells stably transfected with human FFA4 and FFA1 were cultured in F12K medium with 10% FBS. The plasmids was kindly provided by Professor Olivier Civelli (University of California, Irvine, USA). CHO-K1 cells were transfected with 8 μg of pcDNA3.1-FFAR4 or pcDNA3.1/v5-His-topo-FFAR1 plasmids mixed with 24 μL of lipofectamine 2000 reagent (Invitrogen). After 24 hour post-transfection, clones were selected using complete medium containing 600 μg mL−1 zeocin (Invitrogen) for FFA4, and 400 μg mL−1 G418 (TransGen Biotech Co., Ltd, Beijing, China) for FFA1, respectively. Stable clones were selected with zeocin or G418 treatment after 3–4 weeks for the FFA4- and FFA1-tranfected cells.2.4. Dynamic mass redistribution assay
Cells were seeded on 384-well biosensor microplate and cultured overnight to form a confluent monolayer. The cells were then manually washed twice, and maintained with the assay buffer for 1–2 h before measurement. The DMR assays were operated on Epic® BT system (Corning, NY, USA).For profiling agonism, a 2 min baseline was established, followed by adding compounds and monitoring the agonists-induced DMR signals for 1 h. Then a 2 min baseline was re-established for desensitization profiling followed by adding the agonists at a fixed concentration (typically at its EC80–EC100 concentration) and monitoring the cellular responses induced by the agonists for 1 h.
Similarly, for DMR antagonism assays, cells were initially treated with an antagonist for 1 h. Afterwards, the baseline was re-established, followed by adding agonists at a fixed concentration (typically at its EC80–EC100). All compounds were dissolved in the assay buffer with 0.1% BSA.
2.5. RT-PCR analysis
Total cellular RNAs were extracted from HT29 cells using the Takara RNAiso Plus (Takara Biomedical Technology Co., Ltd., Beijing, China) according to the instruction. Then extracted RNAs were reverse-transcribed to cDNA using PrimeScript RT reagent Kit with gDNA Eraser (Takara). cDNA templates were amplified by polymerase chain reaction (PCR) with the primers for human FFA4 (forward 5′-CGATTTGCACACTGATTTGGC-3′; reverse 5′-AAGAGCCGGAAGTCCTGCTG-3′), human FFA1 (forward 5′-GTCTGGTCTTTGGGTTGGAGG-3′; reverse 5′-AGAGCAGGAGAGAGAGGCTG-3′). The amplification program was set as following, first denature at 95 °C for 10 min, then repeat 40 cycles at 95 °C for 5 s and 61 °C for 30 s, finally detect melt curve dissociation. Quantitative real-time PCR was performed using BIO-RAD CFX96 Real-Time System. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a control housekeeping gene with the following primers (forward 5′-GACAGTCAGCCGCATCTTCT-3′; reverse 5′-TTAAAAGCAGCCCTGGTGAC-3′). For data analysis, the normalized expression level of individual gene was normalized to GAPDH and calculated by ΔΔCt method.2.6. FLIPR assay
CHO-FFA4 cells seeded on black-walled 96-well plates (Greiner, USA) at a density of 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well were loaded for 1 h at 37 °C with Calcium-6 dye (Molecular Devices, USA) in Hanks' balanced salt solution containing 20 mM HEPES (pH 7.4). The level of [Ca2+] was then monitored using a FLIPRTetra system (Molecular Devices, USA).
000 cells per well were loaded for 1 h at 37 °C with Calcium-6 dye (Molecular Devices, USA) in Hanks' balanced salt solution containing 20 mM HEPES (pH 7.4). The level of [Ca2+] was then monitored using a FLIPRTetra system (Molecular Devices, USA).
For antagonists to work, AH7614 was first incubated with the cells for 10 min, before the addition of the compounds (i.e. the agonists). Data were expressed as fluorescence (arbitrary units) versus time.
2.7. Data analysis
All DMR data were acquired by Epic Imager software (Corning, NY, USA) and processed by Imager Beta 3.7 (Corning), Microsoft Excel 2010, and GraphPad Prism 6.02 (GraphPad Software Inc., San Diego, CA, USA).3. Results
3.1. DMR characterization of ligands in CHO-FFA4 and CHO-FFA1 cells
Commercially available ligands were tested to evaluate the FFA1 and FFA4 stable cell lines. These compounds could be classified into five types. The first was the endogenous agonist ALA, which could non-selectively bind to the two receptors.22 The second kind was FFA4 selective agonists, including TUG891 and GSK137647A. Both were reported to be two orders of magnitude more potent on FFA4 than on FFA1.5,23 The third was a FFA1 selective agonist, TAK875 that almost had no activity on FFA4.24 The fourth was a non-selective synthetic agonist, GW9508. It was considered as an FFA1 agonist despite the fact that it had similar activity at FFA4.25 The last was two selective antagonists, AH7614 for FFA4 and GW1100 for FFA1.23,25Two types of DMR assays were carried out for ligand characterization. First was agonist assay to monitor the DMR response induced by ligands. The second was desensitization assay and antagonism assay, to examine if the DMR responses were specifically representing the ligand-stimulated FFA4 or FFA1.
In CHO-FFA4 cells, the endogenous agonist ALA induced a strong DMR response (Fig. 2A), indicating stimulation of ALA to the cells. Furthermore, the responses of ALA were reduced by different concentrations of FFA4 antagonist AH7614 (Fig. 2B). Interestingly, there was a residual response induced by ALA even in the presence of 25 μM AH7614 (Fig. 2B), indicating that ALA might stimulate other targets in the cells than FFA4 to generate responses that participated in the general DMR. So we examined the performance of ALA in CHO-K1 the parental cell line. Fig. 2C showed a moderate DMR response induced by ALA, suggesting that the residual DMR in the presence of AH7614 could be caused by interactions with something endogenous in the CHO-K1 cells. Secondly, GSK137647A and TUG891 also stimulated the cells similarly as ALA did (Fig. 2A). AH7614 completely suppressed the DMR responses of GSK137647A and TUG891 (Fig. 2D and E, respectively). Furthermore, on the parental CHO-K1 cells, neither GSK137647A (Fig. 2F) nor TUG891 (data not shown) produced detectable DMR events in the cells. These results suggested that GSK137647A and TUG891 had receptor specificity with FFA4 in this cell line. Lastly, the FFA1 selective agonist TAK875 did not induce any response in the FFA4-expressing cells, while the non-discriminating GW9508 induced a minor response (Fig. 2A). AH7614 reduced DMR response of GW9508 (Fig. 2G). These results suggested GW9508 specifically targeting FFA4. On the parental cell line, TAK875 and GW9508 generated negligible DMR response on the cells (data not shown).
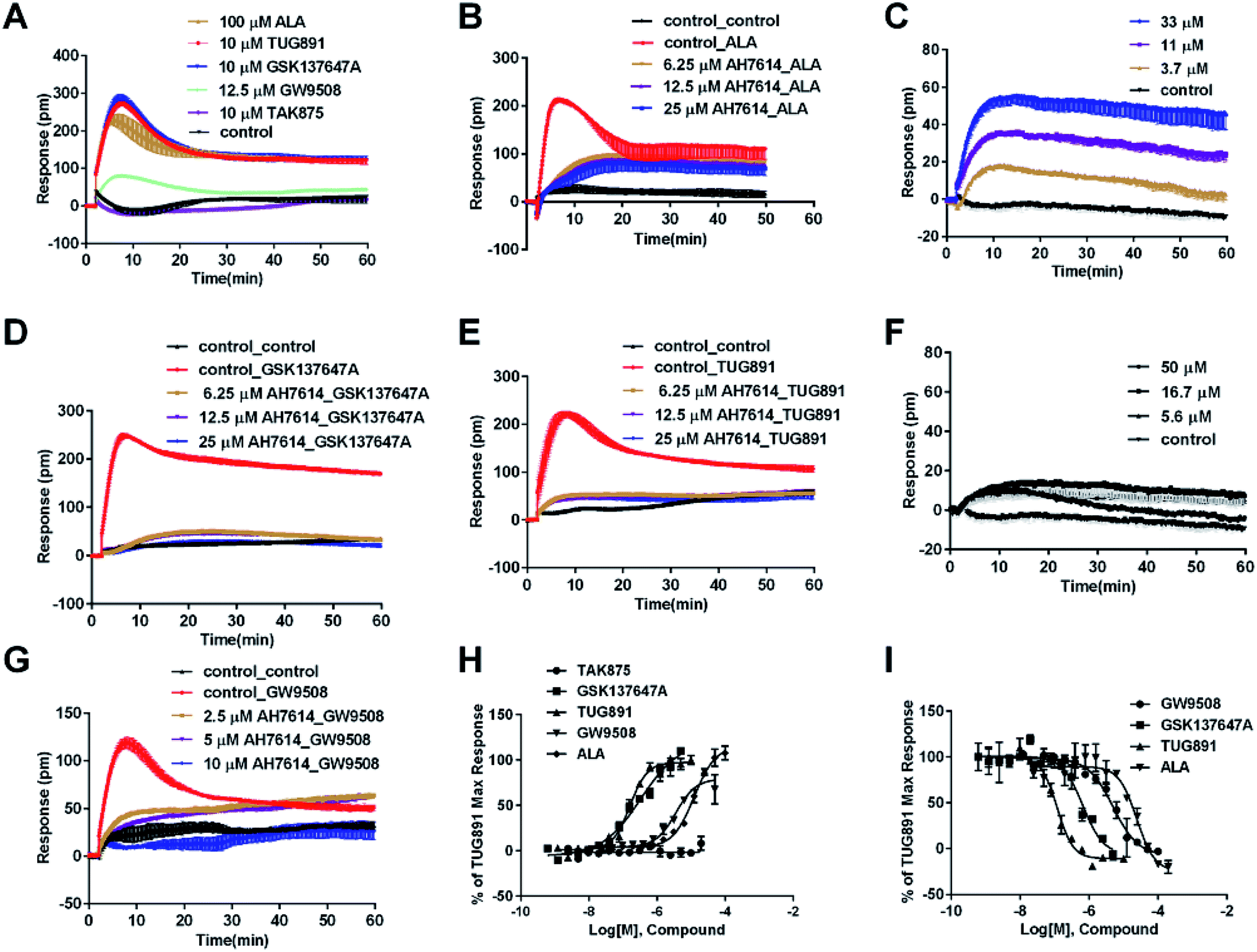 | ||
| Fig. 2 DMR response of ligands on CHO-FFA4 cells. (A) Real-time DMR response of 100 μM ALA, 10 μM TUG891, 10 μM GSK137647A, 12.5 μM GW9508 and 10 μM TAK875. (B) Real-time DMR of 25 μM ALA after CHO-FFA4 cells pretreated with AH7614 at indicated concentrations. (C) Real-time DMR of ALA at indicated concentrations on the parental cell line CHO-K1. (D) Real-time DMR response of 0.5 μM GSK137647A after CHO-FFA4 cells pretreated with AH7614 at indicated concentrations. (E) Real-time DMR response of 0.5 μM TUG891 after CHO-FFA4 cells pretreated with AH7614 at indicated concentrations. (F) Real-time DMR of GSK137647A at indicated concentrations on CHO-K1 cells. (G) Real-time DMR of 25 μM GW9508 on CHO-FFA4 cells after cells pretreated with AH7614 at indicated concentrations. (H) Amplitudes as a function of each agonist doses. (I) The concentration-dependent inhibition of the 0.5 μM TUG891 by GW9508, GSK137647A, TUG891 and ALA. All data represent mean ± s.d. from three independent measurements, each in duplicate (n = 6). | ||
DMR amplitudes of each compound at 5 min were collected and fitted in non-linear regression curves (Fig. 2H) to calculate EC50 values (Table 1). The rank order of potency was TUG891 (0.16 ± 0.01 μM) > GSK137647A (0.34 ± 0.07 μM) > GW9508 (3.62 ± 0.50 μM) > ALA (12.54 ± 0.95 μM) > TAK875 (≫20 μM). TUG891 was therefore chosen as probe in desensitization assay, which examined the ability of each compound to reduce the cellular response induced by TUG891 to further confirm the receptor specificity of the compounds to FFA4 in this cell line. DMR amplitudes of TUG891 at 5 min were used (Fig. 2I) to calculate IC50 values (Table 1). All results confirmed that the ligands specifically interacted with FFA4 in a concentration-dependent manner.
| Class | Compound | CHO-FFA4 cell | CHO-FFA1 cell | HT 29 cell | |||
|---|---|---|---|---|---|---|---|
| EC50 (μM) | Desensitizationa IC50 (μM) | EC50 (μM) | Desensitizationb IC50 (μM) | EC50 (μM) | Desensitizationc IC50 (μM) | ||
| a IC50 to desensitize the DMR of 0.5 μM TUG891 in CHO-FFA4 cells.b IC50 to desensitize the DMR of 1 μM GW9508 in CHO-FFA1 cells.c IC50 to desensitize the DMR of 5 μM TUG891 in HT29 cells. Slash (/) indicates that the EC50 is greater than the highest concentration tested of 100 μM (GSK137647A) or 20 μM (TAK875). All data represent mean ± s.d. from three independent measurements, each in duplicate (n = 6). | |||||||
| Class 1, endogenous ligand | ALA | 12.54 ± 0.95 | 24.58 ± 3.55 | 28.66 ± 0.94 | 67.60 ± 23.48 | 50.88 ± 10.01 | ∼153 |
| Class 2, FFA4 selective ligand | TUG891 | 0.16 ± 0.01 | 0.12 ± 0.01 | ∼32 | 4.98 ± 1.29 | 0.29 ± 0.03 | 0.48 ± 0.12 |
| GSK137647A | 0.34 ± 0.07 | 0.67 ± 0.12 | / | / | 2.98 ± 0.64 | 18.38 ± 6.54 | |
| Class 3, FFA1 selective ligand | TAK875 | / | / | 0.031 ± 0.0045 | 0.0016 ± 0.00041 | / | / |
| Class 4, non-selective ligand | GW9508 | 3.62 ± 0.50 | 5.04 ± 0.65 | 0.40 ± 0.03 | 0.14 ± 0.01 | 5.33 ± 1.42 | 12.02 ± 2.12 |
In CHO-FFA1 cells, firstly, ALA induced a DMR response (Fig. 3A). In the antagonism assay, the DMR responses of ALA were reduced gradually by GW1100 (Fig. 3B), implying that ALA interacted with the FFA1 receptor. Similarly, 20 μM of GW1100 did not completely inhibit ALA. As mentioned above, ALA induced a moderate DMR response (∼60 pm) in CHO-K1 cells (Fig. 2C). Therefore, the residual response herein was caused by stimulation of the host cells. Secondly, TAK875 and GW9508 also induced strong DMR responses (Fig. 3A), but displayed partial agonism compared with ALA. GW1100 at 20 μM was able to completely inhibit TAK875 and GW9508 (Fig. 3C and D, respectively). These results indicated that TAK875 and GW9508 stimulated the cells through interaction with FFA1. Lastly, FFA4-selective agonists were tested in this cell line. GSK137647A did not induce detectable DMR even at 100 μM, but TUG891 produced a moderate DMR in the CHO-FFA1 cells (Fig. 3A). GW1100 could reduce the response stimulated by TUG891 (Fig. 3E). These results suggested that TUG891 also functioned as a FFA1 agonist.
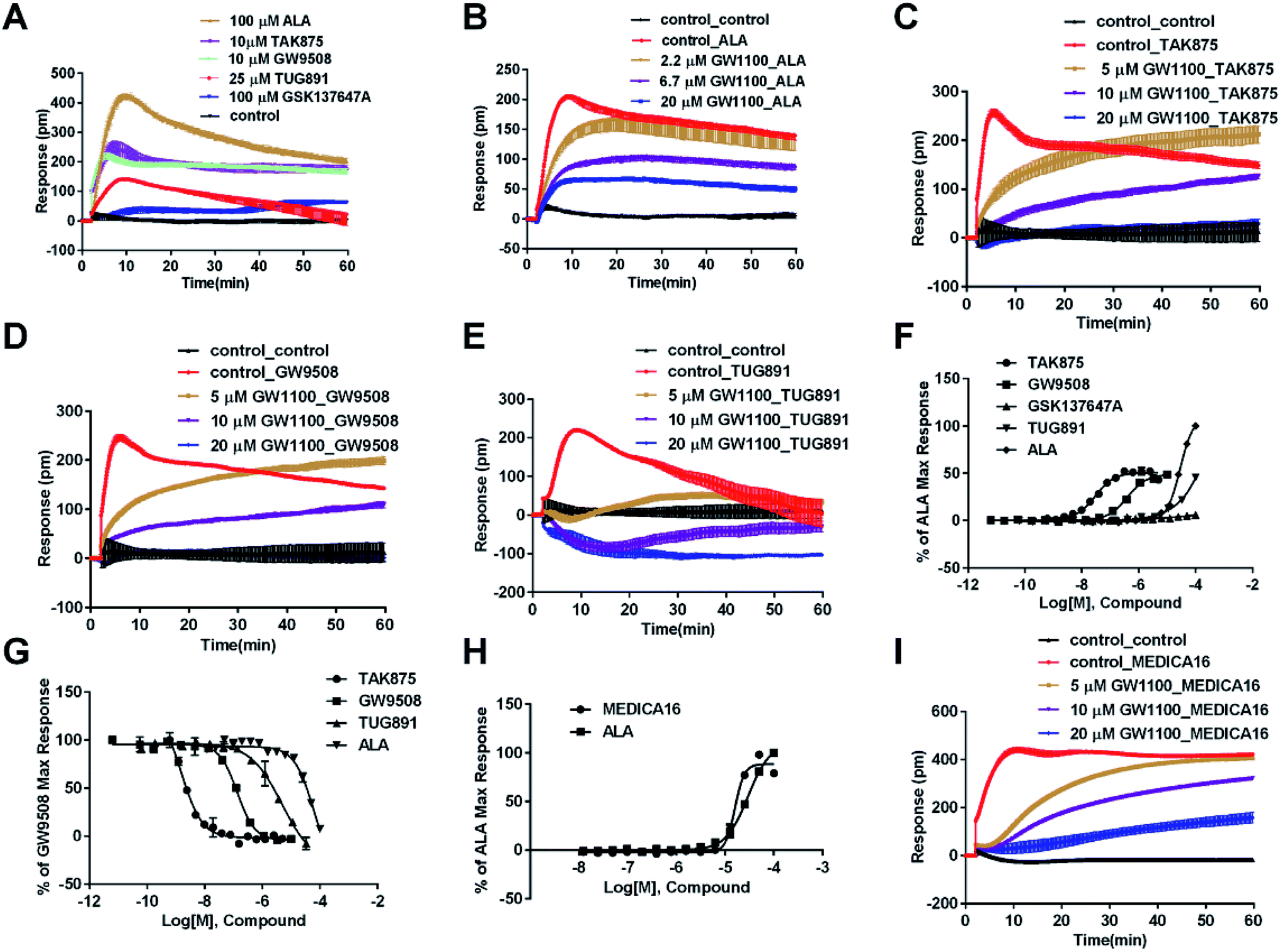 | ||
| Fig. 3 DMR response of ligands in CHO-FFA1 cells. (A) Real-time DMR response of 100 μM ALA, 10 μM TAK875, 10 μM GW9508, 25 μM TUG891 and 100 μM GSK137647A. (B) Real-time DMR of 30 μM ALA after CHO-FFA1 cells pretreated with GW1100 at indicated concentrations. (C–E) Real-time DMR response of agonists (0.5 μM TAK875, 1 μM GW9508 and 30 μM TUG891) after cells pretreated with GW1100 at indicated concentrations. (F) Amplitudes as a function of each agonist doses. (G) The concentration-dependent inhibition of the 1 μM GW9508 DMR by TAK875, GW9508, TUG891 and ALA. (H) Amplitudes as a function of MEDICA16 and ALA doses. (I) Real-time DMR response of 25 μM MEDICA16 after cells pretreated with GW1100 at indicated concentrations. All data represent mean ± s.d. from three independent measurements, each in duplicate (n = 6). | ||
DMR amplitudes of each compound at 5 min were collected and fitted in nonlinear regression curves (Fig. 3F) to calculate EC50 values (Table 1). The rank order of potency was TAK875 (0.031 ± 0.0045 μM) > GW9508 (0.40 ± 0.03 μM) > ALA (28.66 ± 0.94 μM) > TUG891 (∼32 μM) > GSK137647A (>100 μM). ALA was a non-selective agonist for FFA4 and FFA1, TUG891 exhibited clear selectivity to FFA4 for being 200-fold more potent than to FFA1. GW9508 was slightly selective to FFA1. GSK137647A showed great selectivity to FFA4, while TAK875 had good selectivity to FFA1, both bearing two orders of magnitude higher potency on corresponding receptor. GW9508 was chosen as probe in desensitization assay to confirm that these compounds provoked responses through FFA1. DMR amplitudes of GW9508 at 5 min were used (Fig. 3G) to calculate IC50 values (Table 1).
3.2. DMR characterization of ligands in HT29 cells
The above ligands were tested in non-transfected cells to study the pharmacology of endogenously expressed FFA receptors. HT29 cells (human colorectal carcinoma cells) were used for this purpose because FFA1 and FFA4 receptors were found in intestine cells.26–28In HT29 cells, ALA-induced DMR response (Fig. 4A) was partially inhibited by either AH7614 or GW1100 (Fig. 4B and C, respectively). These results demonstrated both FFA1 and FFA4 might be involved in the ALA-induced DMR response. FFA4 selective agonists TUG891 and GSK137647A induced DMR responses (Fig. 4A) that were gradually suppressed by the FFA4 selective antagonist AH7614 (Fig. 4D and E, respectively). GW1100 the FFA1-selective antagonist almost had no effect on blocking TUG891 (Fig. 4F), or GSK147647A (data not shown). These results showed that FFA4 receptors existed in the HT29 cell line and they were functional to specific probe molecules and detectable by the DMR technique.
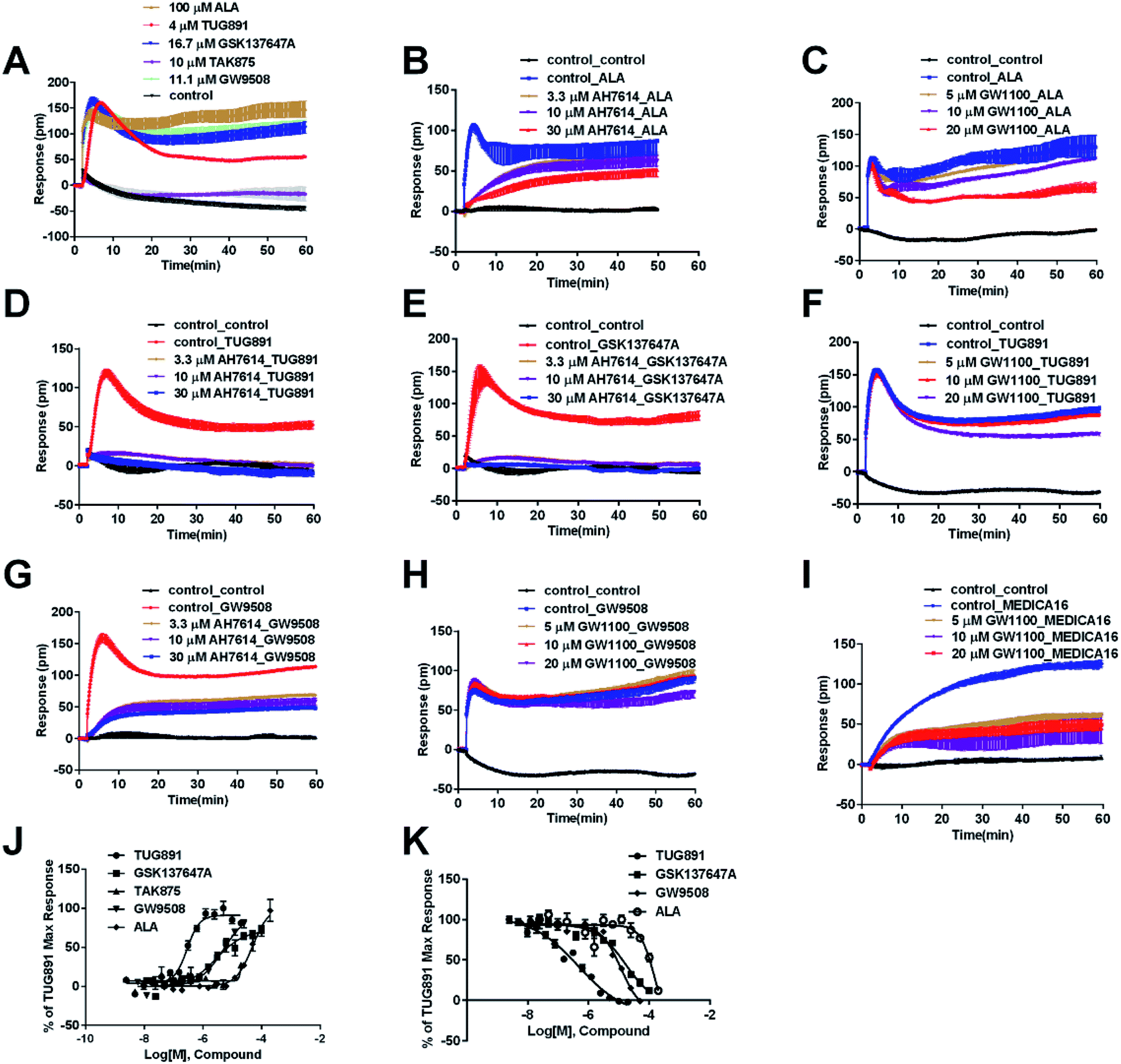 | ||
| Fig. 4 DMR response of ligands in HT29 cells. (A) Real-time DMR response of 100 μM ALA, 4 μM TUG891, 16.7 μM GSK137647A, 10 μM TAK875 and 11.1 μM GW9508. (B) Real-time DMR of 60 μM ALA after HT29 cells pretreated with AH7614 at indicated concentrations. (C) Real-time DMR of 60 μM ALA after HT29 cells pretreated with GW1100 at indicated concentrations. (D) Real-time DMR of 2 μM TUG891 after HT29 cells pretreated with AH7614 at indicated concentrations. (E) Real-time DMR of 16 μM GSK137647A after HT29 cells pretreated with AH7614 at indicated concentrations. (F) Real-time DMR of 2 μM TUG891 after HT29 cells pretreated with GW1100 at indicated concentrations. (G) Real-time DMR of 16 μM GW9508 after HT29 cells pretreated with AH7614 at indicated concentrations. (H) Real-time DMR of 16 μM GW9508 after HT29 cells pretreated with GW1100 at indicated concentrations. (I) Real-time DMR of 50 μM MEDICA16 after HT29 cells pretreated with GW1100 at indicated concentrations. (J) Amplitudes as a function of each agonist doses. (K) The concentration-dependent inhibition of the 5 μM TUG891 DMR by TUG891, GSK137647A, GW9508 and ALA. All data represent mean ± s.d. from three independent measurements, each in duplicate (n = 6). | ||
Interestingly, the FFA1-selective TAK875 did not induce an observable signal in the HT29 cells, even at a concentration of 10 μM (Fig. 4A). Another non-selective agonist GW9508 (11.1 μM) can induce a DMR event (Fig. 4A) that was suppressed by the FFA4-selective AH7614 (Fig. 4G), but not by the FFA1-selective GW1100 (Fig. 4H), suggesting that the DMR response of GW9508 was through FFA4 and/or other targets in the cells. Although neither GW9508 nor TAK875 produced detectable FFA1 DMR events in HT29 cells. It was premature to conduct that HT29 does not express FFA1. Both compounds function as partial agonists to FFA1 and the efficacies of partial agonists depend on the receptor expression levels.29 Yabuki et al. showed that the maximal responses of TAK875 and GW9508 decreased more quickly compared with that of ALA when the amount of expression plasmids decreased.30
Therefore another FFA1 full agonist, MEDICA16, was selected to confirm our hypothesis that the expression level of FFA1 might affect the ligand performance.19,31 First in the CHO-FFA1 cell line, MEDICA16 displayed the same efficiency as ALA did (Fig. 3H) and its response was inhibited by GW1100 (Fig. 3I), proving itself as a full agonist. MEDICA16 produced a DMR response in HT29 cells and it was also inhibited by GW1100 (Fig. 4I). These results revealed that MEDICA16 activated FFA1. To summarize, ALA and MEDICA16 were able to activate FFA1 on HT29 cells, while TAK875 and GW9508 seemed to fail; such results confirmed our hypothesis that a low expression level of receptors indeed affected the performance of partial agonists.
DMR amplitudes of each compound at 6 min were used (Fig. 4J) to calculate EC50 values (Table 1). The rank order of potency was TUG891 (0.29 ± 0.03 μM) > GSK137647A (2.98 ± 0.64 μM) > GW9508 (5.33 ± 1.42 μM) > ALA (50.88 ± 10.01 μM) > TAK875 (≫20 μM) (Table 1). TUG891 was chosen as probe in DMR desensitization assay. DMR amplitudes of TUG891 at 6 min were used (Fig. 4K) to calculate IC50 values (Table 1).
RT-PCR assay was conducted to assess the relative expression levels of FFA4 and FFA1 receptors in HT29 cells. Primers were designed as described in Materials and methods (2.5). GAPDH was the reference gene. The RT-PCR results indicated a slightly lower expression level of FFA1 compared to FFA4 (Fig. 5). Combining these results, HT29 was a suitable cell line for the DMR assay in the study of endogenous FFA1 and FFA4 receptors.
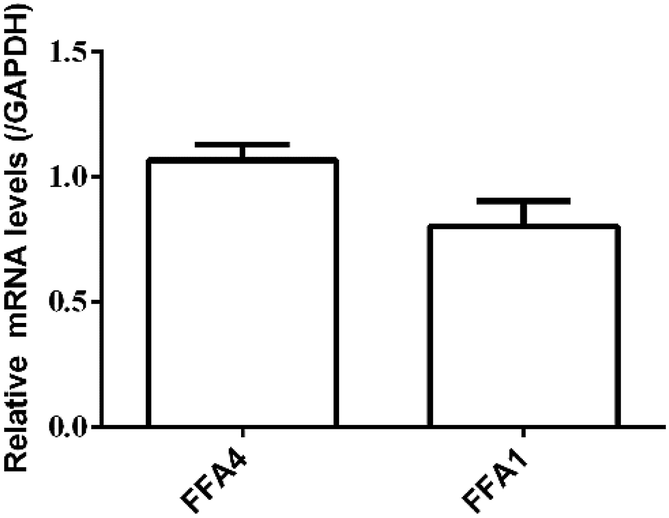 | ||
| Fig. 5 Relative expression levels of FFA4 and FFA1 mRNA in HT29 cells by RT-PCR. Results are shown as mean ± s.d. from two independent measurements, each in triplicate (n = 6). | ||
Since the functional test of the FFA1 receptor in HT29 cells had some complicated phenomena, brief summary is made here. First, based on the results of PCR, the expression of FFA1 mRNA was confirmed. Then, in the DMR experiment, partial agonist (TAK875 and GW9508) did not cause a FFA1-related DMR response, whereas a full agonist ALA was able to stimulate a FFA1-related DMR response. According to the literature, the efficiency of partial agonists is related to the level of receptor expression. So we speculated that the expression level of the endogenous FFA1 in HT29 cells was low. As a result the two FFA1 partial agonists did not induce detectable FFA1-related DMR signals. We then tested another full agonist, MEDICA16. Results showed that MEDICA16 could induce DMR responses which could be attenuated by the antagonist GW1100. Thus, the results supported our speculation.
3.3. Screening of natural compounds for FFA4 activation
To identify agonists from natural products, we used CHO-FFA4 cells that grew fast and were easy to be cultured for screening. A pool of 140 natural compounds from various natural products was screened against the CHO-FFA4 cells. DMR responses, induced by these compounds, were displayed in Fig. 6. Compounds emodin (no. 93), dihydrotanshinone (no. 95) and acetylshikonin (no. 137) generated responses greater than 30% of the TUG891-induced response (∼80 pm), indicating that they were potential agonists for FFA4. Therefore, they were considered as hit compounds for further verification.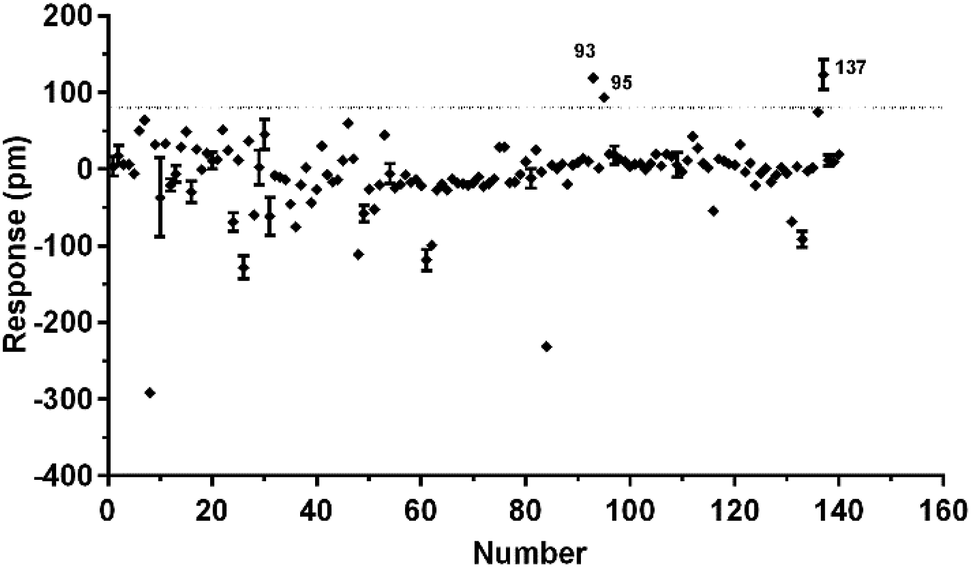 | ||
| Fig. 6 High throughput screening of natural compounds against FFA4. The DMR amplitudes of 140 natural compounds as a function of their concentrations in CHO-FFA4 cells. All data represent mean ± s.d. from two independent measurements, each in duplicate (n = 4). | ||
3.4. Validation of the activity of hit compounds on FFA4
Agonistic activity of the three natural compounds on FFA4 were further investigated by DMR agonist, desensitization and antagonist assays. Emodin triggered a dose-dependent DMR event (Fig. 7A), indicating possible FFA4 agonist activity. Dihydrotanshinone and acetylshikonin also induced concentration-dependent DMR responses (data not shown). DMR amplitudes of individual compounds were collected (Fig. 7B) to calculate EC50 values (Table 2). In the desensitization assay, the compounds gradually desensitized FFA4 to TUG891 (Fig. 7C). In the antagonism assay, AH7614 blocked the three molecules in a concentration-dependent manner (Fig. 7D). These results confirmed the direct interaction between the compounds and FFA4. It is worth noting that the EC50 value of acetylshikonin (10.17 ± 2.95 μM) is comparable to that of ALA (12.54 ± 0.95 μM), although this compound is structurally different from any reported FFA4 agonists. The potency order was acetylshikonin (10.17 ± 2.95 μM) > dihydrotanshinone (32.88 ± 2.28 μM) > emodin (38.18 ± 5.02 μM) (Table 2). The compounds were also assayed in CHO-K1 cells to confirm that responses they induced were not from the host cells. None of them triggered detectable responses in CHO-K1 cells (Fig. 7E), suggesting that these compounds specifically activated FFA4 in the CHO-FFA4 cells.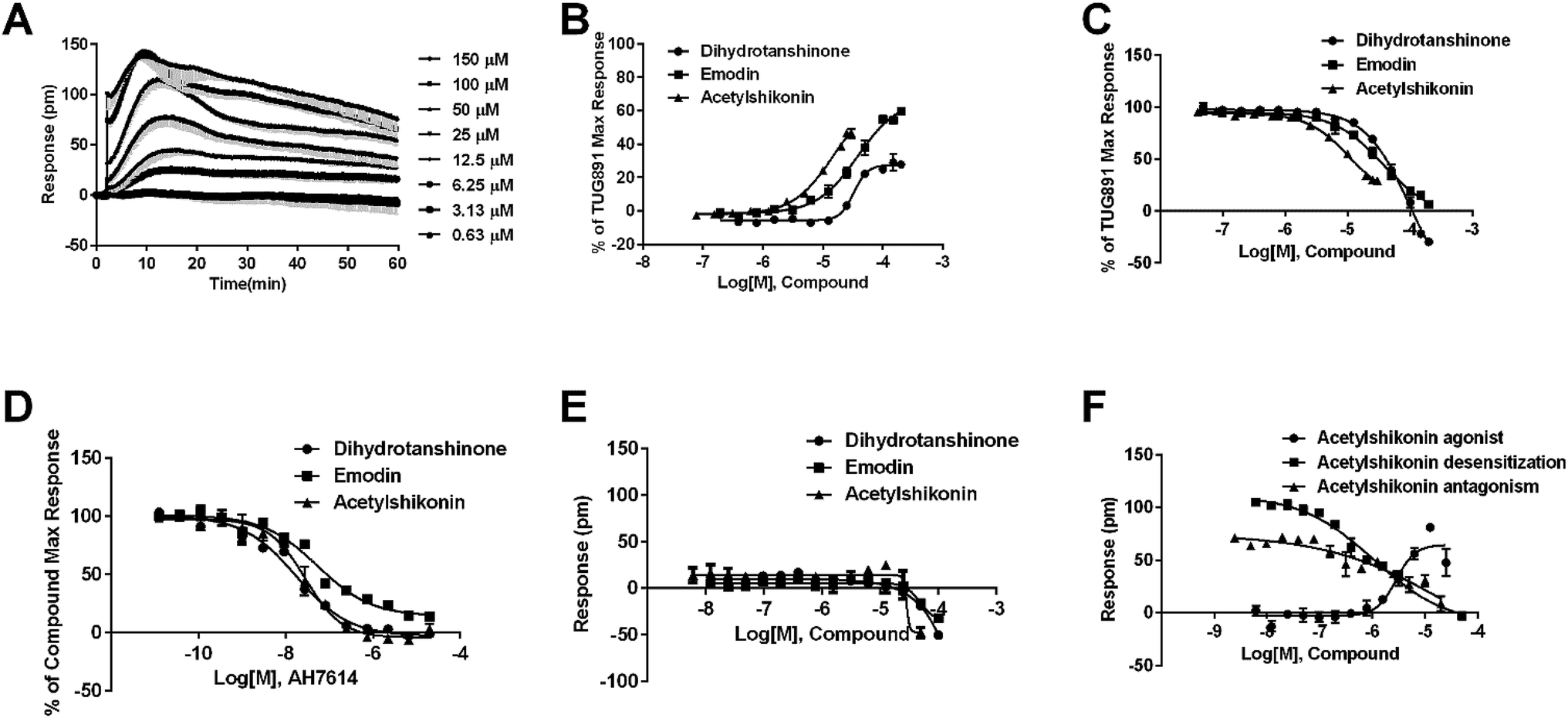 | ||
| Fig. 7 Validation of the activity of hit compounds on FFA4. (A) Real-time DMR of emodin at indicated concentrations on the CHO-FFA4 cells. (B) Amplitudes as a function of each agonist doses. (C) The concentration-dependent inhibition of the 1 μM TUG891 DMR by compounds. (D) The DMR amplitudes of compounds as a function of AH7614 doses in CHO-FFA4 cells after being pre-treated with the antagonist for 1 h. (E) The DMR amplitudes as a function of compounds dose in CHO-K1 cells. (F) Concentration-response data of agonist, desensitization and antagonism for acetylshikonin in HT29 cells. All data represent mean ± s.d. from three independent measurements, each in duplicate (n = 6). | ||
| Compound | CHO-FFA4 cell, EC50 (μM) | CHO-FFA1 cell, EC50 (μM) | HT29 cell, EC50 (μM) |
|---|---|---|---|
| Dihydrotanshinone | 32.88 ± 2.28 | >100 | 21.35 ± 2.20 |
| Emodin | 38.18 ± 5.02 | >100 | ∼39 |
| Acetylshikonin | 10.17 ± 2.95 | 6.17 ± 2.81 | 2.82 ± 0.39 |
The three compounds were tested in HT29 cells. The result of acetylshikonin was presented as an example (Fig. 7F), that it activated the FFA4 receptors in the cells. Dihydrotanshinone and emodin also induced agonistic DMR responses in HT29 (data not shown). The EC50 values of the compounds were shown in Table 2. The potency order was similar to the order generated in the CHO-FFA4 cells, acetylshikonin (2.82 ± 0.39 μM) > dihydrotanshinone (21.35 ± 2.20 μM) > emodin (∼39 μM). Acetylshikonin, dihydrotanshinone and emodin were confirmed to be FFA4 agonists in the HT29 cells, and acetylshikonin was more potent than ALA (EC50 = 50.88 μM) in this cell line.
We further examined the activity of the most active compound (acetylshikonin) on FFA4 by calcium flux assay (FLIPR) in CHO-FFA4 cells. As shown in Fig. 8, acetylshikonin produced a Ca2+ response in the cells and the response was inhibited by the FFA4 antagonist AH7614. The known agonist TUG891 of FFA4 also produced a Ca2+ signal, which was attenuated by AH7614. The two agonists therefore showed similar active pattern in the CHO-FFA4 cell line. In summary, the FLIPR assay confirmed activity of acetylshikonin on FFA4.
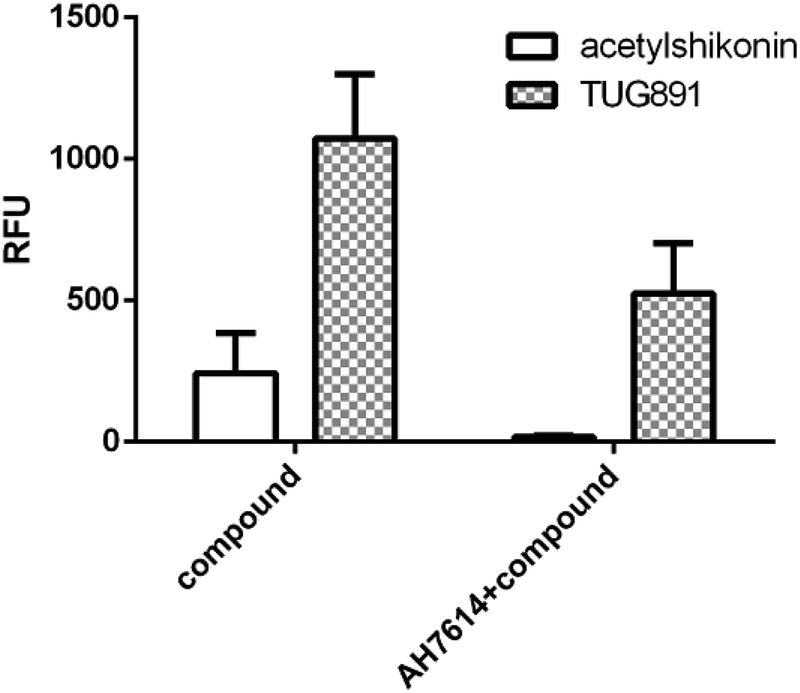 | ||
| Fig. 8 (Left bars) Acetylshikonin (100 μM) and TUG891 (15 μM) induced Ca2+ responses on CHO-FFA4 cells and (right bars) Ca2+ signals of the two compounds after antagonist AH7614 (2 μM) pretreatment. | ||
3.5. Selectivity of novel agonists between FFA1 and FFA4
Since FFA1 and FFA4 share endogenous ligands, subtype selectivity was investigated in this study. The three natural compounds were evaluated in CHO-FFA1 cells and their activities were compared between subtypes for selectivity. Emodin (Fig. 9A) and dihydrotanshinone (Fig. 9B) did not induce detectable agonistic responses in CHO-FFA1 as they did in CHO-FFA4, thus indicating selectivity on FFA4. However, acetylshikonin turned out to be non-selective. It induced dose-dependent agonistic responses in CHO-FFA1 cells (Fig. 9C), which were gradually reduced by the FFA1 antagonist GW1100 (Fig. 9D). The EC50 value of acetylshikonin in CHO-FFA1 was 6.17 ± 2.81 μM (Table 2), comparable to the EC50 value tested in CHO-FFA4 (10.17 ± 2.95 μM).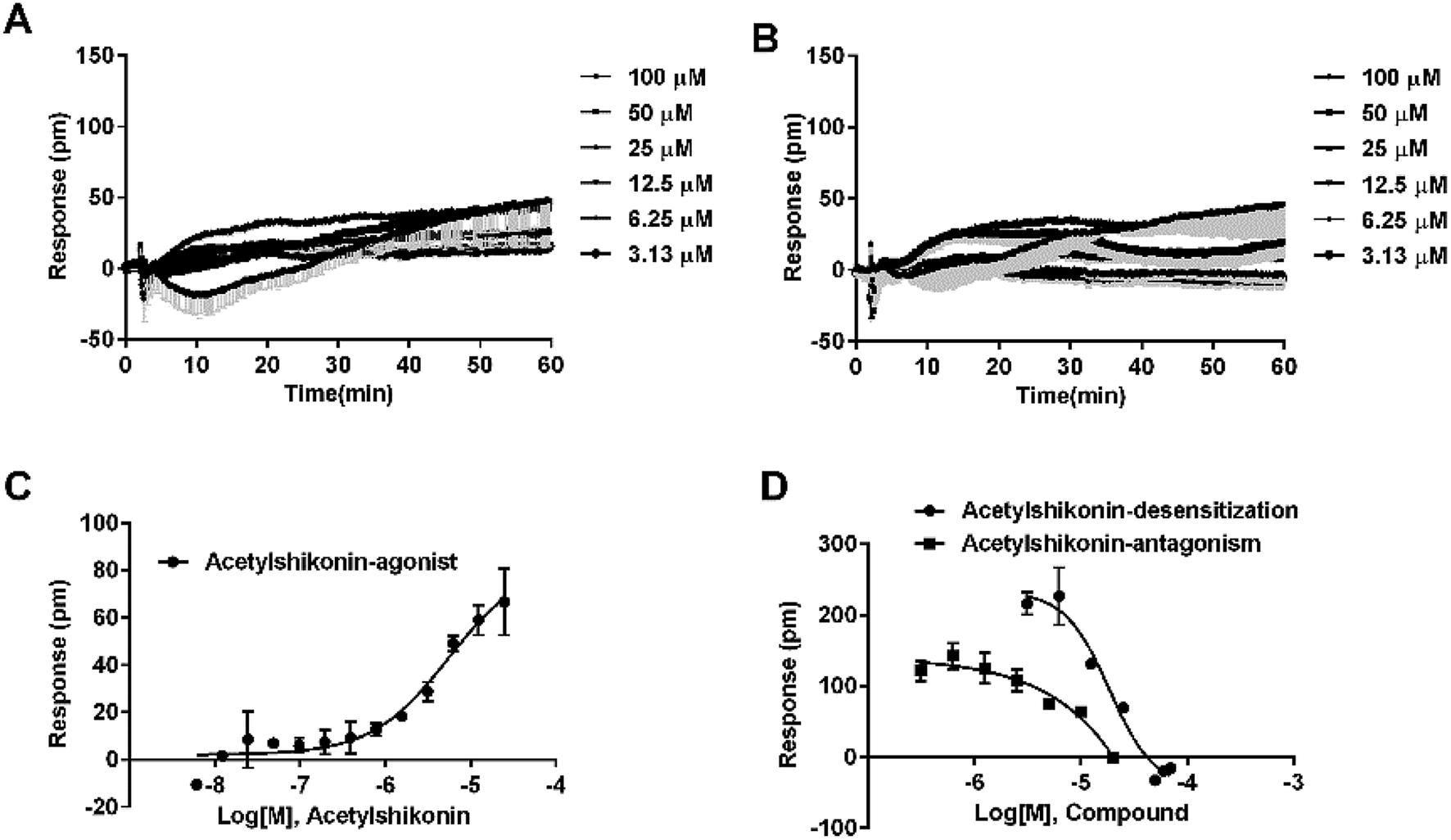 | ||
| Fig. 9 Receptor selectivity of novel compounds. Real-time DMR of emodin (A) and dihydrotanshinone (B) at indicated concentrations on the CHO-FFA1 cells. (C) The DMR amplitudes as a function of acetylshikonin dose in CHO-FFA1 cells. (D) Concentration-response of desensitization and antagonism for acetylshikonin in CHO-FFA1 cells. All data represent mean ± s.d. from three independent measurements, each in duplicate (n = 6). | ||
4. Discussion
In this study, five types of commonly-used probe ligands were characterized with the label-free DMR assay in FFA4 or FFA1 transfected CHO-K1 cells, and a relatively comprehensive investigation was conducted in terms of selectivity and potency of the ligands on the receptors. The EC50 values were consistent with those reported in the literature.6,24 There is no Ki value of these commercially available ligands, except TAK875. Most studies focused on one or two types of ligands,32–34 and only few reports mentioned systematic characterization of these types by the same method in the same lab. Though Hudson et al. used BRET assays to test selectivity of four ligands (ALA, GW9508, NCG21 and TUG891) in transiently transfected HEK293T-FFA1 and HEK293T-FFA4 cells, no commercial FFA1-selective ligands tested in the same work.6 Data generated by various detection methods and different sources make it difficult and inconvenient at times to choose proper molecules. This study provided potency data of five types of probe molecules tested by the same technique, which would aid further studies in this area.We noticed some phenomena during data analysis. First, ALA is probably not a good standard compound in pharmacological studies where selectivity is a major consideration. For tool molecules, potency, selectivity, chemical stability, solubility, in vivo pharmacokinetics and availability are key factors.35 In the case of ALA, apart from its non-discriminating activation between subtypes, this molecule also stimulates the CHO-K1 cells through some targets that are unclear. Second, among the FFA4 agonists, TUG891 was a more suitable tool compound than GSK137647A in this work, because TUG891 had higher potency in the CHO-FFA4 and HT29 cells (Table 1), and the EC50 values in both cell lines were very close. The potencies of GSK137647A showed an approximately ten fold difference in the two lines, indicating that GSK137647A might have certain special properties in endogenous cells not reported yet, such as pathway bias. This finding suggests that certain FFA4 agonists should be evaluated on both transfected and endogenous cell lines. However, Hudson et al. noted that TUG891 hardly distinguish mouse FFA4 and FFA1 and this point should be considered in related studies. Lastly, the EC50 value of TAK875 appeared to be one order of magnitude larger than the IC50 value (Table 1), and thus we modified the experiment protocol with a prolonged incubation time of 3 h. Nevertheless, the results remained the same (IC50 = 0.0018 ± 0.00019 μM). There was a chance that this ligand had a relatively slow koff, and further investigation would be needed on kon and koff of TAK875. This property may be the main factor affecting the clinical dosing frequency of TAK875 (once per day).24,36,37
The probe molecules were also tested in HT29 cells with DMR assays. It was reported that HT29 cells expressed FFA4, but no FFA1, by RT-PCR.6,8 However, we detected FFA1 mRNA and the DMR technique managed to detect FFA1 in the HT29 cells, albeit expression level of which was relatively low. This finding reflected differences between detection methods. By detecting real-time cellular responses in living cells, the DMR assay in HT29 found that GW1100 and AH7614 affected the time kinetic DMR profiles of ALA in different manners (Fig. 4B and C). DMR responses of ALA were mainly affected by AH7614 before 10 min, while by GW1100, after 10 min. What caused this difference needs to be studied in future.
FFA4 ligands have a potential benefit of treating diabetes, obesity, inflammatory and nonalcoholic steatohepatitis.38,39 There is a great interest in identifying novel agonists of FFA4 with structural diversity and receptor selectivity,40 and thus efficient screening methods become important. On the established screening GPCR models, three new NP-derived FFA4 agonists (dihydrotanshinone, emodin and acetylshikonin) were identified, which provided a good starting point to design new FFA4 ligands on those templates. As quinone derivatives, the three compounds are structurally different from any FFA4 agonists reported by far, indicating different ligand–receptor binding modes. Isolated from the plant lithospermum, acetylshikonin exhibited potency higher than ALA in this work. This compound has been reported to play roles in the treatment of obesity and preventing hepatic steatosis,41,42 but mechanisms of action are unclear. Its interaction with FFA4, an important target of obesity-related diseases, may contribute to the clinical effects. Lithospermum contains a number of compounds structurally similar to acetylshikonin,43 and they could be potential FFA4 agonists, too. So it is meaningful to carry out in-depth research on this plant. The anthraquinone derivative, emodin, showed selectivity to the FFA4 receptor, providing some guidance for structure–activity relationship on this kind of agonists.
5. Conclusions
In summary, we systematically characterized activities of ligands of FFA1 and FFA4 receptors by DMR assay, and three novel agonists from natural products were discovered to be selective to FFA4 by this assay. The study benefits the pharmacological investigation of FFA4 and FFA1, and helps to explain the mechanism of action of natural products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the State Key Program of National Natural Science of China (grant no. U1508221); Project of National Science Foundation of China (81403100); and the innovation program of science and research from DICP, CAS (DICP TMSR201601 and ZZBS201803).References
- G. Milligan, B. ShimpukadE, T. Ulven and B. D. Hudson, Chem. Rev., 2017, 117, 67–110 CrossRef CAS PubMed.
- A. Hirasawa, K. Tsumaya, T. Awaji, S. Katsuma, T. Adachi, M. Yamada, Y. Sugimoto, S. Miyazaki and G. Tsujimoto, Nat. Med., 2005, 11, 90–94 CrossRef CAS PubMed.
- A. Ichimura, A. Hirasawa, O. Poulain-Godefroy, A. Bonnefond, T. Hara, L. Yengo, I. Kimura, A. Leloire, N. Liu, K. Iida, H. Choquet, P. Besnard, C. Lecoeur, S. Vivequin, K. Ayukawa, M. Takeuchi, K. Ozawa, M. Tauber, C. Maffeis, A. Morandi, R. Buzzetti, P. Elliott, A. Pouta, M. R. Jarvelin, A. Korner, W. Kiess, M. Pigeyre, R. Caiazzo, W. Van Hul, L. Van Gaal, F. Horber, B. Balkau, C. Levy-Marchal, K. Rouskas, A. Kouvatsi, J. Hebebrand, A. Hinney, A. Scherag, F. Pattou, D. Meyre, T. A. Koshimizu, I. Wolowczuk, G. Tsujimoto and P. Froguel, Nature, 2012, 483, 350–354 CrossRef CAS PubMed.
- K. Nakamoto, K. Shimada, S. Harada, Y. Morimoto, A. Hirasawa and S. Tokuyama, Eur. J. Pharmacol., 2018, 820, 31–38 CrossRef CAS PubMed.
- B. Shimpukade, B. D. Hudson, C. K. Hovgaard, G. Milligan and T. Ulven, J. Med. Chem., 2012, 55, 4511–4515 CrossRef CAS PubMed.
- B. D. Hudson, B. Shimpukade, A. E. Mackenzie, A. J. Butcher, J. D. Pediani, E. Christiansen, H. Heathcote, A. B. Tobin, T. Ulven and G. Milligan, Mol. Pharmacol., 2013, 84, 710–725 CrossRef CAS PubMed.
- R. Formicola, P. Pevarello, C. Kuhn, C. Liberati, F. Piscitelli and M. Sodano, Pharm. Pat. Anal., 2015, 4, 443–451 CrossRef CAS PubMed.
- J. M. Kim, K. P. Lee, S. J. Park, S. Kang, J. Huang, J. M. Lee, K. Sato, H. Y. Chung, F. Okajima and D. S. Im, Acta Pharmacol. Sin., 2015, 36, 813–820 CrossRef CAS PubMed.
- P. H. Lee, J. Recept. Signal Transduction Res., 2009, 29, 146–153 CrossRef CAS PubMed.
- Y. Fang, Drug Discovery Today: Technol., 2010, 7, e5–e11 CrossRef CAS PubMed.
- R. Zhang and X. Xie, Acta Pharmacol. Sin., 2012, 33, 372–384 CrossRef CAS PubMed.
- R. Schroder, J. Schmidt, S. Blattermann, L. Peters, N. Janssen, M. Grundmann, W. Seemann, D. Kaufel, N. Merten, C. Drewke, J. Gomeza, G. Milligan, K. Mohr and E. Kostenis, Nat. Protoc., 2011, 6, 1748–1760 CrossRef PubMed.
- M. F. Peters, F. Vaillancourt, M. Heroux, M. Valiquette and C. W. Scott, Assay Drug Dev. Technol., 2010, 8, 219–227 CrossRef CAS PubMed.
- E. E. Codd, J. R. Mabus, B. S. Murray, S. P. Zhang and C. M. Flores, Assay Drug Dev. Technol., 2011, 9, 362–372 CrossRef CAS PubMed.
- Q. Liu, L. Chen, L. Hu, Y. Guo and X. Shen, Biochim. Biophys. Acta, 2010, 1799, 854–865 CrossRef CAS PubMed.
- N. Siriwardhana, N. S. Kalupahana, M. Cekanova, M. LeMieux, B. Greer and N. Moustaid-Moussa, J. Nutr. Biochem., 2013, 24, 613–623 CrossRef CAS PubMed.
- M. S. Butler, Nat. Prod. Rep., 2008, 25, 475–516 RSC.
- M. S. Butler, A. A. Robertson and M. A. Cooper, Nat. Prod. Rep., 2014, 31, 1612–1661 RSC.
- T. Hara, A. Hirasawa, Q. Sun, K. Sadakane, C. Itsubo, T. Iga, T. Adachi, T. A. Koshimizu, T. Hashimoto, Y. Asakawa and G. Tsujimoto, Naunyn-Schmiedeberg's Arch. Pharmacol., 2009, 380, 247–255 CrossRef CAS PubMed.
- T. Nagasawa, H. Nakamichi, Y. Hama, S. Higashiyama, Y. Igarashi and S. Mitsutake, J. Biochem., 2018, 164, 27–32 CrossRef CAS PubMed.
- S. Esaki, T. Nagasawa, H. Tanaka, A. Tominaga, D. Mikami, S. Usuki, H. Hamajima, H. Hanamatsu, S. Sakai, Y. Hama, Y. Igarashi, H. Kitagaki and S. Mitsutake, J. Food Biochem., 2018, e12624, DOI:10.1111/jfbc.12624.
- T. Ulven and E. Christiansen, in Annual Review of Nutrition, ed. B. A. Bowman and P. J. Stover, 2015, vol. 35, pp. 239–263 Search PubMed.
- S. M. Sparks, G. Chen, J. L. Collins, D. Danger, S. T. Dock, C. Jayawickreme, S. Jenkinson, C. Laudeman, M. A. Leesnitzer, X. Liang, P. Maloney, D. C. McCoy, D. Moncol, V. Rash, T. Rimele, P. Vulimiri, J. M. Way and S. Ross, Bioorg. Med. Chem. Lett., 2014, 24, 3100–3103 CrossRef CAS PubMed.
- N. Negoro, S. Sasaki, S. Mikami, M. Ito, M. Suzuki, Y. Tsujihata, R. Ito, A. Harada, K. Takeuchi, N. Suzuki, J. Miyazaki, T. Santou, T. Odani, N. Kanzaki, M. Funami, T. Tanaka, A. Kogame, S. Matsunaga, T. Yasuma and Y. Momose, ACS Med. Chem. Lett., 2010, 1, 290–294 CrossRef CAS PubMed.
- C. P. Briscoe, A. J. Peat, S. C. McKeown, D. F. Corbett, A. S. Goetz, T. R. Littleton, D. C. McCoy, T. P. Kenakin, J. L. Andrews, C. Ammala, J. A. Fornwald, D. M. Ignar and S. Jenkinson, Br. J. Pharmacol., 2006, 148, 619–628 CrossRef CAS PubMed.
- J. Hidalgo, S. Teuber, F. J. Morera, C. Ojeda, C. A. Flores, M. A. Hidalgo, L. Nunez, C. Villalobos and R. A. Burgos, Int. J. Mol. Sci., 2017, 18(4), 750 CrossRef PubMed.
- K. Takahashi, K. Fukushima, Y. Onishi, K. Minami, S. Otagaki, K. Ishimoto, N. Fukushima, K. Honoki and T. Tsujiuchi, Exp. Cell Res., 2018, 369, 54–60 CrossRef CAS PubMed.
- S. Miyauchi, A. Hirasawa, T. Iga, N. Liu, C. Itsubo, K. Sadakane, T. Hara and G. Tsujimoto, Naunyn-Schmiedeberg's Arch. Pharmacol., 2009, 379, 427–434 CrossRef CAS PubMed.
- D. Hoyer and H. W. G. M. Boddeke, Trends Pharmacol. Sci., 1993, 14, 270–275 CrossRef CAS PubMed.
- C. Yabuki, H. Komatsu, Y. Tsujihata, R. Maeda, R. Ito, K. Matsuda-Nagasumi, K. Sakuma, K. Miyawaki, N. Kikuchi, K. Takeuchi, Y. Habata and M. Mori, PLoS One, 2013, 8, e76280 CrossRef CAS PubMed.
- K. Kotarsky, N. E. Nilsson, E. Flodgren, C. Owman and B. Olde, Biochem. Biophys. Res. Commun., 2003, 301, 406–410 CrossRef CAS PubMed.
- E. Christiansen, K. R. Watterson, C. J. Stocker, E. Sokol, L. Jenkins, K. Simon, M. Grundmann, R. K. Petersen, E. T. Wargent, B. D. Hudson, E. Kostenis, C. S. Ejsing, M. A. Cawthorne, G. Milligan and T. Ulven, Br. J. Nutr., 2015, 113, 1677–1688 CrossRef CAS PubMed.
- A. Li, Y. Li and L. Du, Future Med. Chem., 2015, 7, 1457–1468 CrossRef CAS PubMed.
- A. Li, Y. Li and L. Du, Future Med. Chem., 2015, 7, 1457–1468 CrossRef CAS PubMed.
- S. V. F. Hansen and T. Ulven, Handb. Exp. Pharmacol., 2017, 236, 33–56 CAS.
- C. F. Burant, P. Viswanathan, J. Marcinak, C. Cao, M. Vakilynejad, B. Xie and E. Leifke, Lancet, 2012, 379, 1403–1411 CrossRef CAS.
- T. Araki, M. Hirayama, S. Hiroi and K. Kaku, Diabetes, Obes. Metab., 2012, 14, 271–278 CrossRef CAS PubMed.
- S. Kang, J. Huang, B. K. Lee, Y. S. Jung, E. Im, J. M. Koh and D. S. Im, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2018, 1863, 105–116 CrossRef CAS PubMed.
- D. Y. Oh, S. Talukdar, E. J. Bae, T. Imamura, H. Morinaga, W. Fan, P. Li, W. J. Lu, S. M. Watkins and J. M. Olefsky, Cell, 2010, 142, 687–698 CrossRef CAS PubMed.
- C. M. Azevedo, K. R. Watterson, E. T. Wargent, S. V. Hansen, B. D. Hudson, M. A. Kepczynska, J. Dunlop, B. Shimpukade, E. Christiansen, G. Milligan, C. J. Stocker and T. Ulven, J. Med. Chem., 2016, 59, 8868–8878 CrossRef CAS PubMed.
- M. Su, W. Huang and B. Zhu, PLoS One, 2016, 11, e0146884 CrossRef PubMed.
- M.-L. Su, Y. He, Q.-S. Li and B.-H. Zhu, Molecules, 2016, 21, 976 CrossRef PubMed.
- M. Liao, A. Li, C. Chen, H. Ouyang, Y. Zhang, Y. Xu, Y. Feng and H. Jiang, J. Chromatogr. A, 2015, 1425, 158–172 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |