DOI:
10.1039/C9RA02043H
(Paper)
RSC Adv., 2019,
9, 26238-26247
Anti-inflammatory effects of hederagenin on diabetic cardiomyopathy via inhibiting NF-κB and Smads signaling pathways in a type-2 diabetic mice model
Received
17th March 2019
, Accepted 15th August 2019
First published on 21st August 2019
Abstract
Hederagenin (HED) is a bioactive natural compound of pentacyclic triterpenes extracted from many medicinal plants. It has a wide range of antitumor cytotoxic effects and significant anti-inflammation effects. However, at present, it is unclear whether HED can inhibit cardiac remodelling caused by diabetic cardiomyopathy. In this study, we evaluated the effects of HED on pathological abnormalities in cardiac structures and cardiac insufficiency caused by diabetic cardiomyopathy and focused on the inflammatory signalling pathways of the diabetic heart. Treatment with HED reduced pro-inflammatory cytokines, the heart and body mass of diabetic db/db mice but had no effect on fasting plasma glucose (FPG). Moreover, after HED treatment, the cardiac dysfunction of diabetic mice was relieved, and myocardial hypertrophy and fibrosis decreased. Furthermore, HED inhibited the nuclear translocation of nuclear factor-κB (NF-κB) and Smads and decreased the transcriptional activity of NF-κB and Smads. Additionally, the expression levels of transforming growth factor (TGF)-β1 and collagen I, which are target downstream molecules of the NF-κB and Smads signalling pathways, were also decreased in diabetic hearts. Taken together, our findings suggest that the cardioprotective effect of HED may be achieved by reducing the activation of inflammation-associated NF-κB and Smads signalling. We suggest that the protective effect of HED on the diabetic heart, as revealed in this study, should be further explored in-depth to elucidate its cell biology and molecular mechanisms.
1. Introduction
Diabetes is a disease characterized by a series of carbohydrate metabolism disorders.1 The risk of compilations caused by diabetes, such as cardiomyopathy, stroke, retinopathy, chronic kidney disease, and diabetic foot caused by peripheral neuropathy, is extremely high.2 In fact, cardiomyopathy is a common cardiovascular disease that affects the function and structure of the heart. When cardiomyopathy occurs as an outcome of the progression of diabetes and in the absence of coronary heart disease or hypertension, we refer to it as diabetic cardiomyopathy (DCM).3 The Framingham Heart Study revealed that compared with the age-matched control group, the risk of heart failure in patients with diabetes increased fivefold.4 The main features of DCM are a decreased diastolic function of heart, concurrent significant structural abnormalities and left ventricular hypertrophy.3 DCM has some additional histological and cytological features, such as a large amount of pathological collagen deposition between cardiomyocytes and cardiomyocyte hypertrophy.5 Studies have found that the DCM-caused remodelling in diabetic heart is closely related to the activation of inflammatory signalling molecules and fibrosis-related signals.6 The activation of inflammatory signalling pathway often results in structural and functional abnormalities in diabetic hearts, such as the hypertrophy of cardiomyocytes and a significant increase in the myocardial collagen content, eventually triggering a reduction in the systolic and (or) diastolic function.6
Pro-inflammatory cytokines and adhesion molecules are often regulated by NF-κB.7 In general, hyperglycaemia often induces NF-κB-mediated systemic inflammation.8 However, studies have found that some bioactive natural compounds extracted from plants can directly inhibit inflammation in diabetic hearts without lowering blood glucose levels.9,10 Moreover, NF-κB, which plays a key role in the inflammatory signalling pathway, is also involved in the diabetes-induced fibrosis of the heart, liver and kidney.11,12 Diabetes-induced expression of TNF-α, IL-1β and IL-6 further promotes myocardial inflammation, which leads to cardiac hypertrophy and fibrosis during the progression of cardiac remodelling.13 Members of the Smads family are signalling molecules that are closely associated with increased expression of collagen and increased content of fibrosis.14 In vitro studies have found that chronic hyperglycaemia stimulates fibroblasts, causing them to transdifferentiate from fibroblasts into cardiac fibroblasts, and promotes increased expression levels of extracellular matrix and collagen.14 Many studies have found that regulation of Smad2/3 can attenuate fibroblast proliferation, inhibit activation of TGF-β signalling pathway and inhibit collagen production.14,15 Moreover, NF-κB is also involved in the activation of TGF-β signalling in diabetic cardiomyopathy.7
Hederagenin (HED; Fig. 1A) is a natural extract of pentacyclic triterpenes isolated from the leaves of ivy widely grown in subtropical zone, from the leaves of Cyclocarya paliurus, which only exists in China, and from the leaves of other common plants in China.16 Many studies have demonstrated that HED has a broad spectrum of biological effects that kill many types of tumour cells.17 Other studies have also found that HED has pharmacological effects, such as reducing lipids and lipid oxidation and inhibiting platelet aggregation.18 Kim et al.19 found that HED can inhibit the activation of mitogen-activated protein kinase (MAPK) signalling in an alcohol-induced liver injury rat model. Moreover, in the vascular injury model generated in Wistar rats fed with a high-fat diet, HED reduces the expression levels of pro-inflammatory cytokines by inhibiting the NF-κB signalling pathway, thereby correcting the endothelial dysfunction stimulated by free fatty acids.20 Lee et al.21 shown that HED can inhibit lipopolysaccharide (LPS)-induced activation of the NF-κB signalling pathway and reduce the secretion of pro-inflammatory cytokines, thereby exhibiting anti-inflammatory activity.
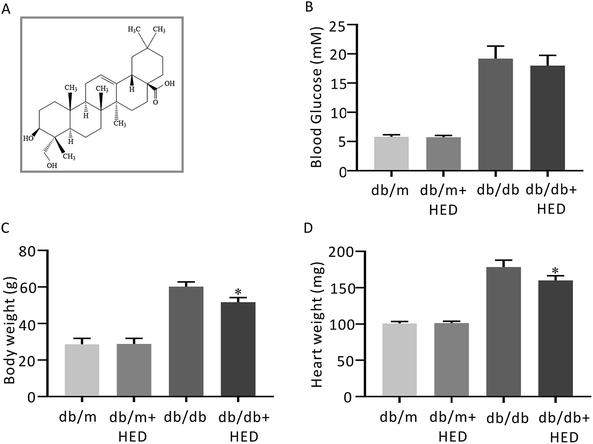 |
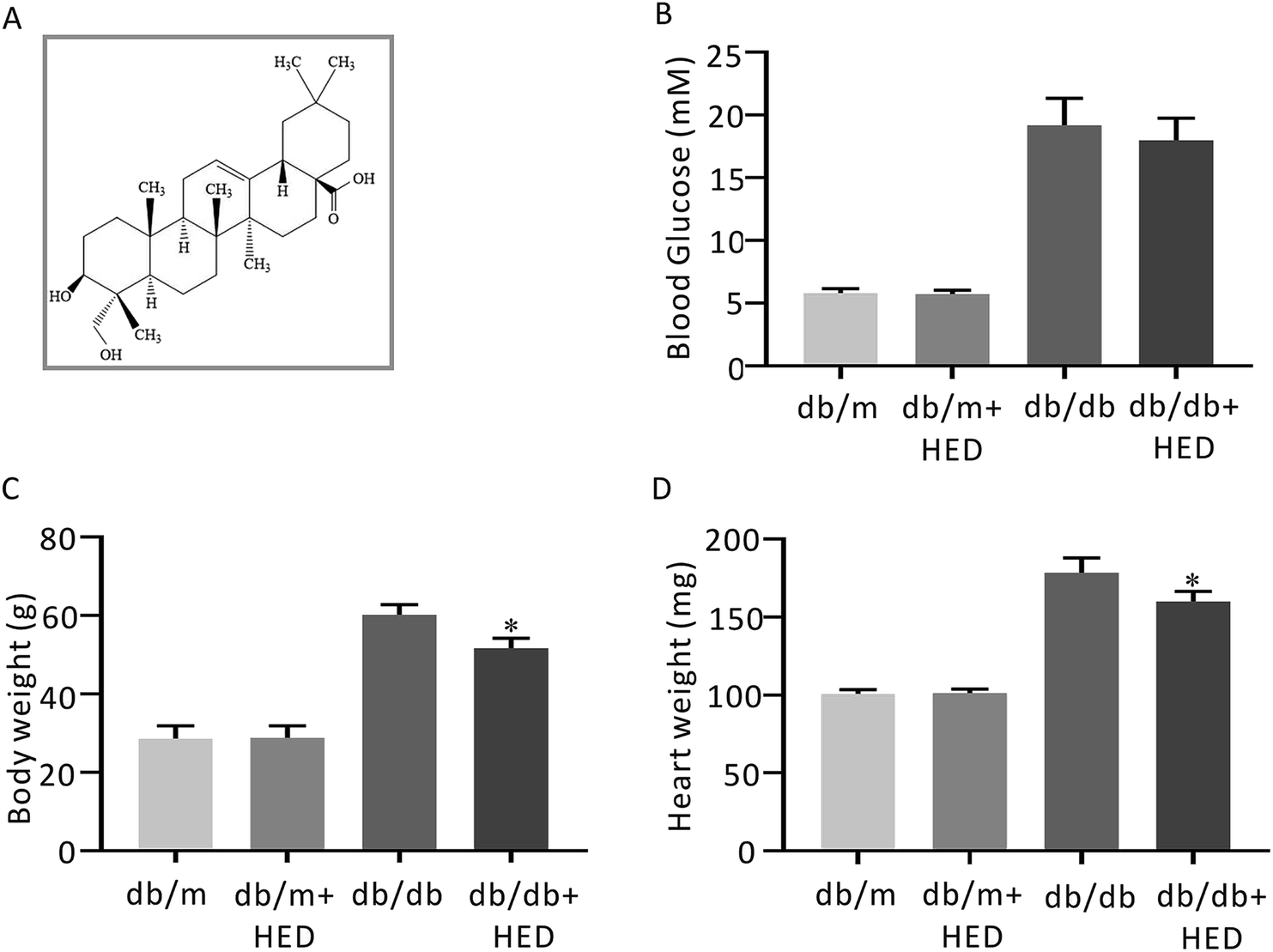
| | Fig. 1 The structure of HED and the effect of HED on the fasting plasma glucose (FPG), heart mass and body mass of mice. (A) Schematic diagram of the structure of HED. (B) FPG of each group of mice. (C) Comparison of the body mass of each group of mice. (D) Comparison of the heart mass of each group of mice. db/db + HED group vs. db/db group, *P < 0.05. | |
The db/db mouse is an obese diabetic mouse model caused by defects in the leptin receptor gene on chromosome 4. Bulimia and obesity appear in four-week old db/db mice.22 The mice exhibit obvious characteristics, such as hyperglycaemia, hyperlipidaemia, and insulin resistance with increasing age, and the pathogenic process is very similar to that of patients with type 2 diabetes.22 Therefore, our study was based on the following: (1) can HED inhibit cardiac remodelling and improve cardiac function in diabetic db/db mice? (2) What effect does HED have on the activation of NF-κB and Smads signalings in the diabetic heart?
2. Materials and methods
2.1 Animal experiment design
All animal experiments were approved by the Experimental Animal Centre of Xiangya Medical College of Central South University. The breeding and welfare of experimental animals were carried out strictly following the regulations issued by the Hunan Provincial Experimental Animal Centre and the National Institutes of Health (NIH) of the United States. The db/db mice and db/m mice involved in the animal experiments were purchased from SJA Experimental Animal Co., Ltd. (Changsha, China). All mice were maintained on a 12 hour light/dark cycle and given adequate water and food. Prior to the treatment, all animals were allowed to adapt to the SPF environment for one week after purchase. Twelve db/db mice were randomly divided into the db/db group (n = 6) and db/db + HED group (n = 6); twelve db/m mice were randomly divided into the db/m group (n = 6) and db/m + HED group (n = 6). HED (Desite, Chengdu, China) was administered orally at a dose of 50 mg kg−1 (ref. 19) for a period of 6 weeks. Before the end point of the observation, three mice were randomly selected from each group for hemodynamics determination. After the tests were completed, all animals were weighed and anaesthetized. The whole blood was collected using a heparin-containing BD Vacutainer® blood collection tubes (BD, shanghai, China) and the plasma was separated for future use. Next, the hearts of the mice were removed and weighed. The left heart was separated; one part of it was dried and stored in liquid nitrogen for future use, and the other part was fixed in 4% paraformaldehyde/phosphate buffered-saline (PBS) fixative (Servicebio, Wuhan, China).
2.2 FPG detection
FPG testing was performed according to the method in an FPG detection kit (Jincheng Biotechnology Institute, Nanjing, China).
2.3 Measurement of inflammation-related cytokines
The levels of TNF-α, IL-1β and IL-6 in plasma of all mice were performed for determination of proinflammatory cytokines. Elisa kits of TNF-α, IL-1β and IL-6 were purchase from Boster Bio (Wuhan, China).
2.4 Hemodynamics determination
Hemodynamics testing was performed according to the instructions of the Powerlab system (ADInstruments, New Zealand) and the Millar catheter system (SPR-407) (Millar, Inc., Houston, USA). All mice were anaesthetized by intraperitoneal injection of 20% urethane (5 ml kg−1) at the end of the observation. Depilation and skin preparation were performed. An incision was made at the middle of the neck. Blunt dissection of various layers of tissue was performed to expose the right common carotid sheath. 6 to 10 mm of cervical artery was freed. The distal segment was ligated, and the proximal end was clamped with a small artery clamp. Ophthalmic scissors were used to transversely cut open approximately 1 mm of carotid artery, and a catheter with a pressure electrode was passed through. At this time, the changes in the pressure channel connected with catheter were observed. If a better pressure–volume loop appeared, then the steady-state hemodynamics data, such as pressure and volume changes, were recorded.
2.5 Histological staining evaluation
After blood collection from the mouse, the heart was immediately removed and immersed in 4% paraformaldehyde PBS fixative for 48 hours. Stepwise dehydration was performed until the heart became transparent. The heart was embedded in formaldehyde-containing paraffin. An inverted microscope (Zeiss, Germany) was used for observation and imaging. The wheat germ agglutinin (WGA) was obtained from Thermo Fisher Scientific (Shanghai, China), and WGA staining was performed to determine the cross-sectional area of the heart for evaluating the hypertrophy of cardiomyocytes. Masson's trichrome staining kit was purchased from Boster Bio (Wuhan, China) and used to assess the extent of fibrosis in the cardiac tissue. The Masson's trichrome staining procedure was performed by strictly following the method described in the instructions. Collagen fibres were stained blue, while the myocardium was labelled red. The amount of fibrosis in the heart tissue was calculated using the collagen/myocardium ratio in each group. The primary antibodies for performing immunohistochemical analysis were anti-collagen I (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 dilution; 22734-1-AP, Proteintech, Rosemont, USA) and anti-TGF-β1 (1:50; 21988-1-AP, Proteintech, Rosemont USA). The sections were exposed to methanol-hydrogen peroxide for 10 minutes and then washed with PBS. Antigen retrieval was performed. The sections were blocked with serum for 20 minutes and then incubated with the primary antibody against collagen I or TGF-β1 at 4 °C overnight. After completion of the above performance, the sections were washed three times with PBS and incubated with biotinylated secondary antibody at room temperature for 20 minutes. The sections, which were washed with PBS, were incubated with streptomycin–biotin–peroxidase complex for 10 minutes. After the sections were washed again with PBS, diaminobenzidine was added as the coloration reagent. The nuclei were counterstained with haematoxylin. The positive staining with primary antibodies against collagen I or TGF-β1 was brown. The expression of the protein of interest was compared between the groups by counting the number of positive cells. For each section, the number of positively stained cells was counted in random 10 fields under a light microscope. The above analysis and calculations were performed independently by two independent researchers using Image-Pro Plus 6.2 software (Media Cybernetics, Inc., Rockville, USA).
:50 dilution; 22734-1-AP, Proteintech, Rosemont, USA) and anti-TGF-β1 (1:50; 21988-1-AP, Proteintech, Rosemont USA). The sections were exposed to methanol-hydrogen peroxide for 10 minutes and then washed with PBS. Antigen retrieval was performed. The sections were blocked with serum for 20 minutes and then incubated with the primary antibody against collagen I or TGF-β1 at 4 °C overnight. After completion of the above performance, the sections were washed three times with PBS and incubated with biotinylated secondary antibody at room temperature for 20 minutes. The sections, which were washed with PBS, were incubated with streptomycin–biotin–peroxidase complex for 10 minutes. After the sections were washed again with PBS, diaminobenzidine was added as the coloration reagent. The nuclei were counterstained with haematoxylin. The positive staining with primary antibodies against collagen I or TGF-β1 was brown. The expression of the protein of interest was compared between the groups by counting the number of positive cells. For each section, the number of positively stained cells was counted in random 10 fields under a light microscope. The above analysis and calculations were performed independently by two independent researchers using Image-Pro Plus 6.2 software (Media Cybernetics, Inc., Rockville, USA).
2.6 Protein extraction
Total protein, cytoplasmic protein and nuclear protein were extracted from the heart tissue using radioimmunoprecipitation assay (RIPA) lysis buffer (Abcam, Shanghai, China), a cytoplasmic and nuclear protein isolation kit (Thermo Scientific, Shanghai, China), respectively, along with protease/phosphorylase inhibitors (Transgene Bio, Beijing, China), all according to the manufacturer instructions. All extracted proteins were stored at −80 °C until use. The protein concentration was determined using the bicinchoninic acid (BCA) assay (Boster Bio, Wuhan, China).
2.7 Western blot analysis
Nuclear proteins and cytoplasmic proteins of the heart tissue from the db/m group, db/m + HED group, db/db group, and db/db + HED group were collected, and the BCA assay was used for protein quantification. Approximately 20 to 25 μg of protein was loaded into each lane and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The transfer apparatus was placed in a refrigerator at 4 °C. The voltage was set at 25 V, and the transfer was performed for 12 hours. The proteins were transferred to a polyvinylidene fluoride (PVDF) membrane, which was blocked with 5% bovine serum albumin (BSA) at room temperature for 2–3 hours. The PVDF membrane was incubated with Smad2/3 (1:1000, Abcam, Shanghai, China), Smad4 (1:1000, Abcam, Shanghai, China), NF-κB p65 (1:1000, Abcam, Shanghai, China), β-actin (1:2000, Santacruz Biotechnology, USA) and proliferating cell nuclear antigen (PCNA) (1:1000, Santacruz Biotechnology, USA) overnight. The membrane was then washed with PBST three times and incubated with horseradish peroxidase (HRP)-conjugated rabbit, mouse and goat secondary antibodies (1:5000, Millipore, USA) at room temperature for 2 hours. A chemiluminescent imager (SmarChemi™, SAGECREATION, Beijing, China) was used for the exposure, development and imaging. The ratio between the absorbance of the protein band and that of the corresponding internal control was calculated and represented by the mean ± standard deviation (S.D.).
2.8 Electrophoretic mobility shift assay (EMSA)
First, the EMSA probe was labelled with biotin. The NF-κB oligonucleotide probe was labelled according to the instructions of the 3′ biotin probe labelling kit from Thermo Fisher Scientific (Shanghai, China). The incubated complex of the double-stranded DNA probe and the nuclear protein from heart tissue was electrophoresed on a 6% non-denaturing polyacrylamide gel. In the competition experiment, a 100-fold concentration of unlabelled probe was first incubated with the nucleoprotein on ice for 20 minutes before the labelled probe was added. The sequence information of the NF-κB and Smad3/4 oligonucleotide probe is as follows, respectively:
| 5′-AGTTGAGGG GACTTTCCCAGG C-3′, 3′-TCAACTCCCCTGAAAGGGTCCG-5′; |
| 5′-AGTATGTCTAGACTGA-3′, 3′-TCATACAGATCTGACT-5′. |
2.9 Statistical analysis
The statistical software SPSS 16.0 and GraphPad Prism 5.0 were used. The data were analysed by one-way analysis of variance (ANOVA), the chi-squared test and the t-test. Values were expressed as the mean ± S.D.; P < 0.05 was considered statistically significant.
3. Results
3.1 Effects of HED on the blood glucose, body mass and heart mass of db/m and db/db mice
After 6 weeks of oral administration of HED, there was no significant change in FPG in the db/db + HED group compared with the db/db group (P > 0.05; Fig. 1B). The body mass and heart mass in the db/db + HED group showed a significant reduction compared with the db/db group (P < 0.05; Fig. 1C and D). These results suggest that the increase in heart mass and body weight induced by long-term diabetes was inhibited by HED, and this inhibition was not achieved by lowering the blood glucose in type 2 diabetic db/db mice.
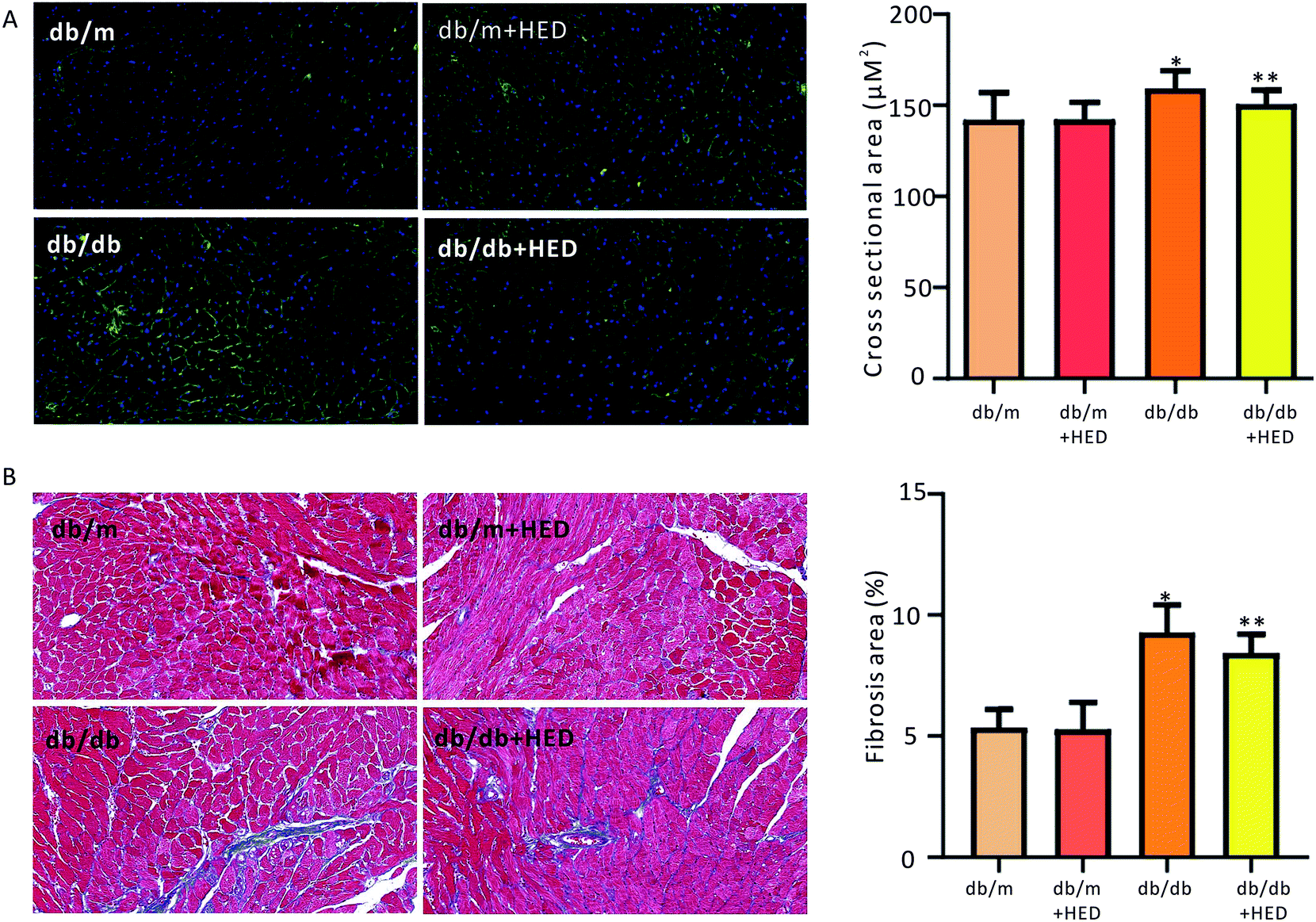
3.2 HED inhibits myocardial hypertrophy and fibrosis in db/db mice
To investigate the effect of HED on the histomorphological changes in diabetic heart in db/db mice, we used both of WGA staining and Masson's trichrome staining to assess the structural changes in the diabetic heart. As seen in Fig. 2A, after 6 weeks of HED treatment, the db/m mice, which served as the healthy control group, showed no significant difference in the cross-sectional area of the left ventricular (reflecting the size of cardiomyocytes) compared with the db/m mice that did not receive HED treatment (P > 0.05). However, after the diabetic db/db mice received 6 weeks of HED oral administration, the size of their left ventricular cardiomyocytes were significantly reduced compared with that of the db/db control group mice (P < 0.05, Fig. 2A). Similar to the above results, compared with the db/db mice, oral administration of HED could significantly reduce the fibrosis content in the db/db group mice (P < 0.05, Fig. 2B). Taken together, the long-term HED application can significantly ameliorate myocardial hypertrophy and fibrosis in the db/db mouse model.
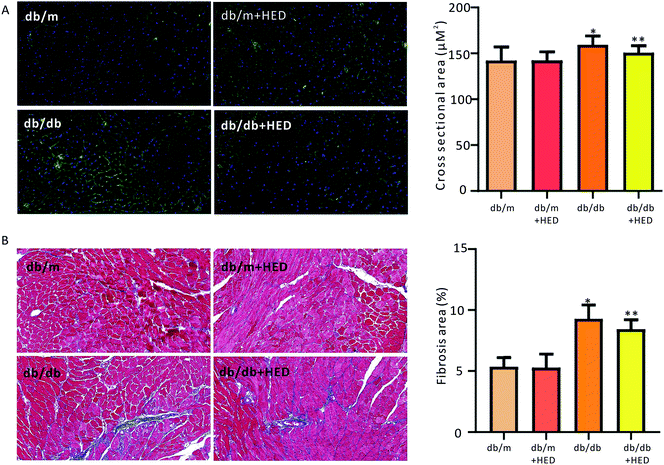 |
| | Fig. 2 Effect of HED on the myocardial structure of mice. The microscope magnification is 20 times. (A) Comparison of the cross-sectional area of hearts in each group when the hearts of mice were stained with WGA. Green fluorescence represents the myocardial cell membrane, and blue represents the nucleus. (B) Comparison of the cardiac collagen area ratio in mice upon Masson staining. Red indicates cardiomyocytes, and blue indicates collagen. db/db group vs. db/m group, *P < 0.05; db/db + HED groups vs. db/db group, **P < 0.05. | |
3.3 HED improves the cardiac insufficiency induced by diabetes in db/db mice
Previous results have shown that the effect of HED on the structures of diabetic hearts. We performed a catheter system of the pressure–volume loop (PV loop) to examine whether HED can also relieve chronic cardiac dysfunction in mice caused by diabetes. We found that for all healthy db/m control mice, regardless of whether they received 6 weeks of HED treatment, there were no significant differences in the parameters of hemodynamic, including the heart rate (HR), left ventricular end-systolic pressure (ESP), left ventricular end-diastolic pressure (EDP), maximal rate of rise of left ventricular pressure (dp/dtmax) and maximal rate of decline of left ventricular pressure (dp/dtmin) (P > 0.05, Table 1). Further, we found that HED significantly improved the cardiac dysfunction of db/db mice, including the left ventricular ESP, left ventricular EDP, dp/dtmax and dp/dtmin (Table 1). Meanwhile, we observed the displacement of the PV loop. The PV loops of all db/db mice were displaced to the left on the horizontal axis, indicating that db/db mice had significant cardiac dysfunction compared with the db/m mice (P < 0.05, Fig. 3). However, after 6 weeks of HED treatment, the PV loop of db/db mice were significantly shifted to the right, indicating that their cardiac function was significantly enhanced, although the PV loop of db/db + HED mice was still to the left of the PV loop of the db/m control mice (Fig. 3).
Table 1 Comparison of hemodynamic parameters of the mice groupsa
| |
db/m |
db/m + HED |
db/db |
db/db + HED |
| HR: heart rate, Pes: LV end systolic pressure, Ped: LV end diastolic pressure, dp/dtmax: maximal rates of rise of ventricular pressure, dp/dtmin: maximal rates of decline of ventricular pressure. Values are presented as mean ± S.D., *P < 0.01 vs. db/m, ΔP < 0.01 vs. db/db + HED. |
| HR (bpm) |
551 ± 2.9 |
553 ± 5.1 |
555 ± 6.6 |
553 ± 6.2 |
| ESP (mmHg) |
82.6 ± 5.92 |
81.3 ± 1.3 |
79.5 ± 7.9* |
78.8 ± 6.1Δ |
| EDP (mmHg) |
3.5 ± 1.1 |
4.9 ± 1.9 |
2.3 ± 1.1* |
2.8 ± 1.5Δ |
| dp/dtmax (mmHg s−1) |
7035 ± 268 |
6998 ± 367 |
6723 ± 158* |
6861 ± 256Δ |
| dp/dtmin (mmHg s−1) |
−6992 ± 323 |
−6916 ± 239 |
−6578 ± 420* |
−6727 ± 394Δ |
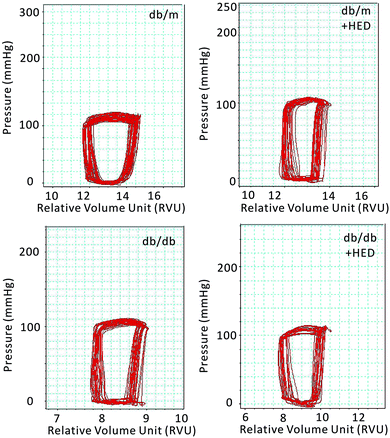 |
| | Fig. 3 Pressure–volume loop of the cardiac function of mice in each group. The horizontal axis represents the relative volume of the left heart (RVU); the vertical axis represents the left ventricular intraventricular pressure (mmHg). The red ring represents the change in PV during the cardiac cycle. | |
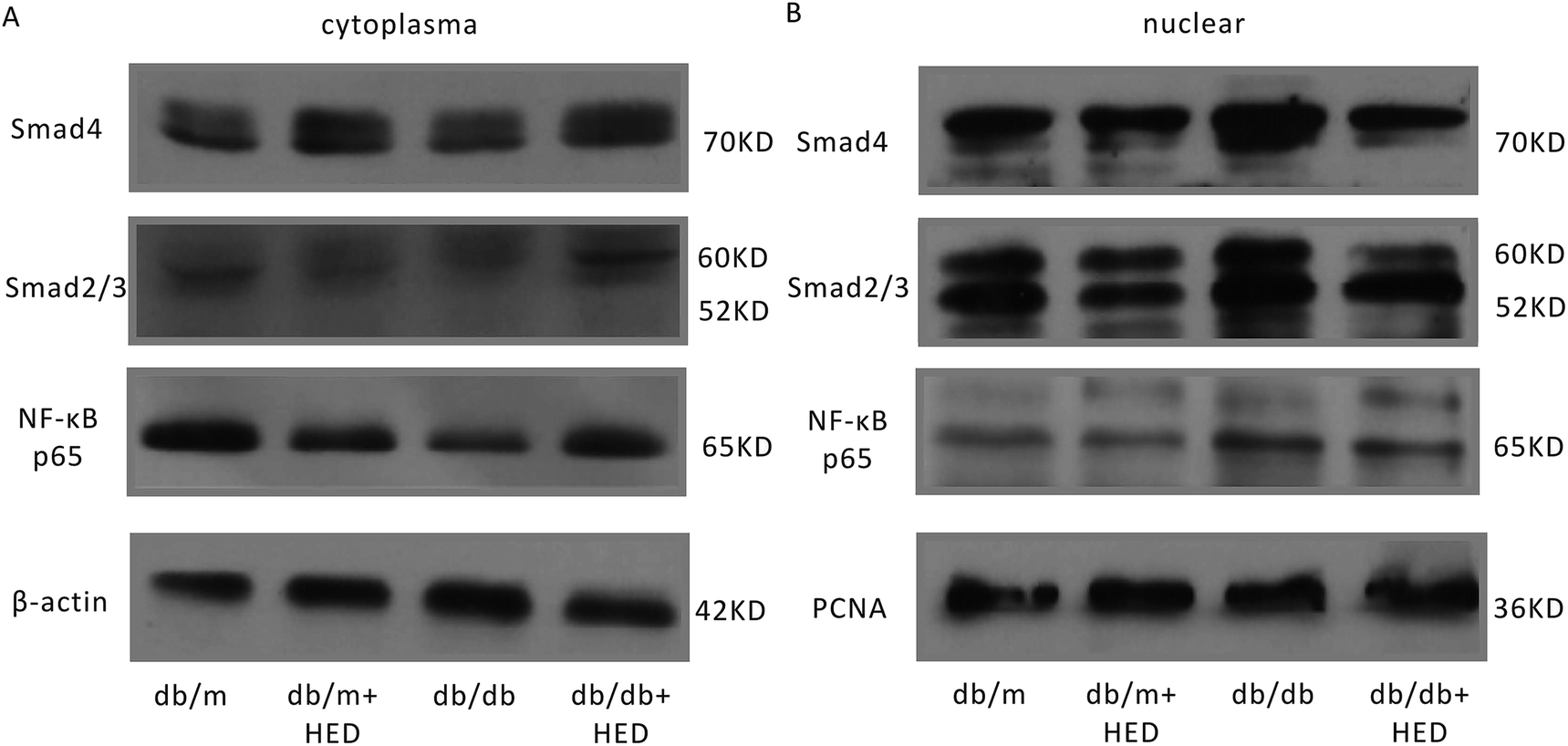
3.4 HED attenuates the nuclear translocation of NF-κB and Smads in the hearts of db/db mice
It is well known that the NF-κB and Smads signalling pathways are closely associated with the cardiac remodelling induced by DCM. To evaluate the effect of HED on the activation of the aforementioned signalling pathways in cardiac remodelling caused by DCM, we used Western blotting and EMSA to determine the nuclear translocation of NF-κB and Smads in the hearts of db/db and db/m mice. The Western blot results suggested that HED had no effect on the nuclear translocation of NF-κB, Smad4 and Smad2/3 in the hearts of the db/m mice (Fig. 4A and B). We further analysed the nuclear translocation of these proteins in the hearts of db/db mice without HED treatment and in the hearts of db/m mice and found that the NF-κB, Smad4 and Smad2/3 protein levels in the cardiomyocyte nuclei of db/db mice were significantly increased (Fig. 4A and B). However, HED could significantly inhibit the nuclear translocation of NF-κB, Smad4 and Smad2/3 in the hearts of db/db mice (Fig. 4A and B).
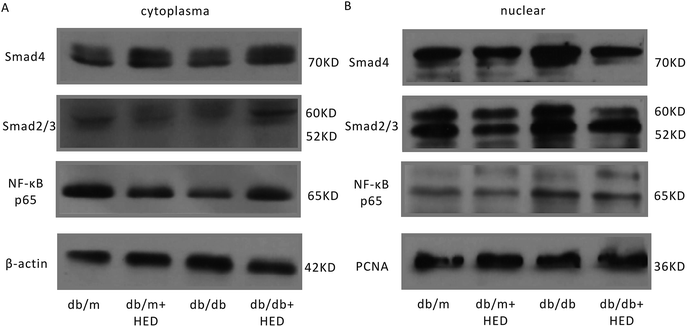 |
| | Fig. 4 HED inhibits the nuclear translocation of NF-κB and Smads. (A) The protein expression levels of NF-κB, Smad2/3 and Smad4 in the cytoplasm of the myocardial tissue of db/m mice and db/db mice. (B) The protein expression levels of NF-κB, Smad2/3 and Smad4 in the cardiomyocyte nuclei of db/m mice and db/db mice. | |
Moreover, our findings showed that the changes in EMSA were consistent with those in Western blotting analysis. The binding bands of NF-κB and Smad3/4 in both the db/m group and the db/m + HED group did not show a significant migration retardation (Fig. 5A and B), whereas the shifted NF-κB and Smad3/4 bands in the db/db group showed a highly significant migration retardation (Fig. 5A and B). This result indicates that the levels of NF-κB and Smad3/4 were significantly increased in the nuclear protein extracted from the diabetic hearts. There were changed after the HED treatment: the binding activities of the NF-κB and Smad3/4 probes to nuclear proteins were significantly diminished (Fig. 5A and B). Taken together, HED can significantly attenuate the nuclear translocation of NF-κB and Smads in diabetic hearts.
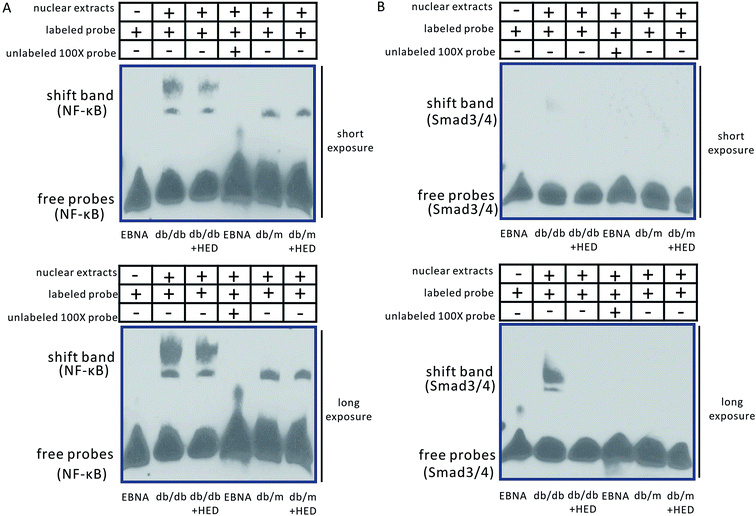 |
| | Fig. 5 EMSA assay of NF-κB and Smad3/4. (A) Changes in NF-κB transcriptional activity. (B) Changes in Smad3/4 transcriptional activity. The Epstein–Barr virus nuclear antigen (EBNA) is the nuclear antigen of the Epstein–Barr virus and serves as a positive control for EMSA. The nucleic acid probes of the db/db, db/db + HED, db/m and db/m + HED groups are oligonucleotide probes that bind to NF-κB and Smad3/4; the oligonucleotide probe that binds to EBNA is the detection probe that comes with the kit. | |
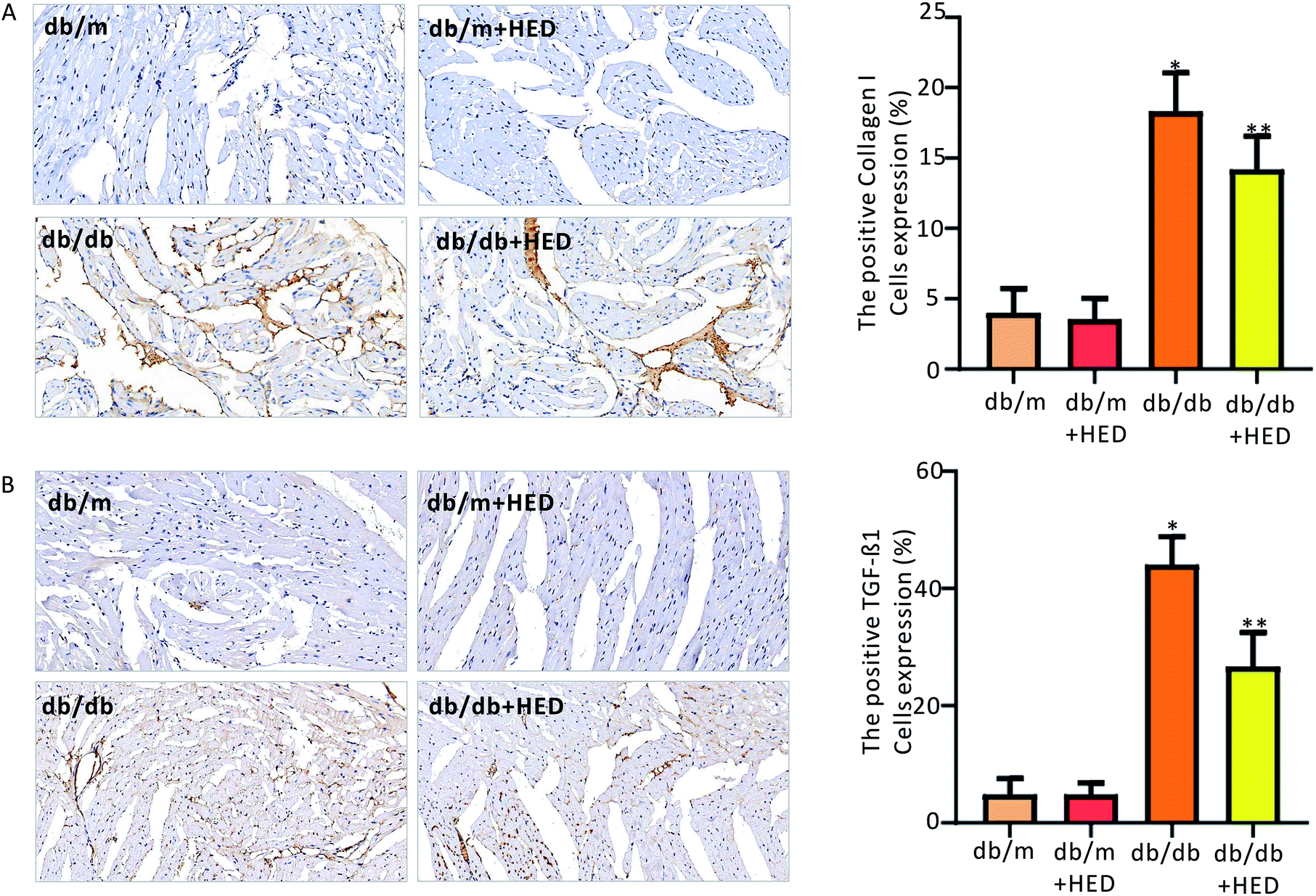
3.5 HED inhibits the synthesis of TGF-β1 and collagen I in the myocardium of db/db mice
To clarify the effect of HED on the downstream proteins of cardiac fibrosis, we used an immunohistochemical assay to evaluate changes in TGF-β1 and collagen I synthesis. We found that the number of positive cells expressing TGF-β1 and collagen I proteins in the hearts of the db/m control mice was much less than those in the diabetic db/db mice and the db/db + HED mice (P < 0.05, Fig. 6A and B). Meanwhile, HED had no significant effect on the expression levels of TGF-β1 and collagen I in the cardiomyocytes of db/m mice (Fig. 6A and B). Further analysis showed that after the db/db mice received 6 weeks of HED treatment, the production of synthesized TGF-β1 and collagen I in their cardiomyocytes was significantly reduced compared with the db/db mice without the HED administration (Fig. 6A and B), which were consistent with the decreased content of fibrosis.
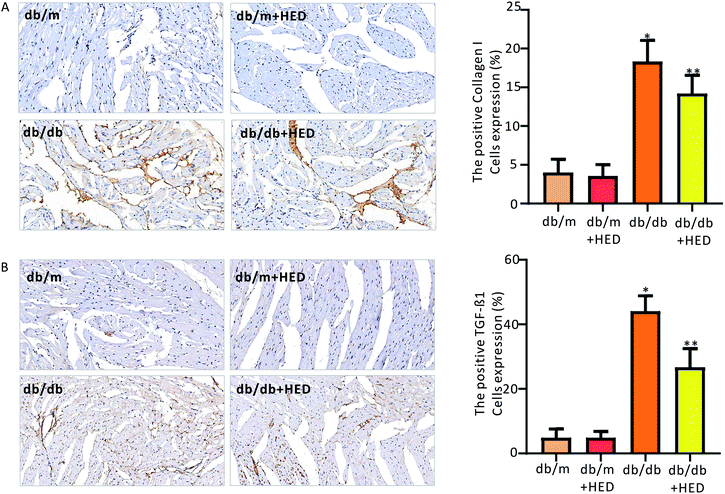 |
| | Fig. 6 Effect of HED on the expression levels of TGF-β1 and collagen I in the hearts of mice in each group. (A) Changes in TGF-β1 expression under immunohistochemical staining. A brown colour indicates positive expression of TGF-β1. (B) Immunohistochemical staining of collagen I. A brown colour indicates positive expression of collagen I. db/db group vs. db/m group, *P < 0.05; db/db + HED groups vs. db/db group, **P < 0.05. | |
3.6 Effect of HED on plasma levels of TNF-α, IL-1β and IL-6
The plasma levels of TNF-α, IL-1β and IL-6 were significantly up-regulated by long-term stimulation of diabetes without HED administration (Table 2). However, HED significantly down-regulated increasing plasma pro-inflammatory cytokines (TNF-α, IL-1β and IL-6) secretion (Table 2).
Table 2 Comparison of mean levels of inflammation-related cytokines in plasma of all micea
| Group |
db/m |
db/m + HED |
db/db |
db/db + HED |
| Values are presented as mean ± S.D., *P < 0.01 vs. db/db. |
| TNF-α (pg ml−1) |
19.47 ± 3.31 |
19.37 ± 2.12 |
69.42 ± 3.93 |
57.82 ± 5.85* |
| IL-6 (pg ml−1) |
11.07 ± 1.42 |
11.14 ± 0.74 |
41.96 ± 2.64 |
34.78 ± 2.52* |
| IL-1β (pg ml−1) |
18.92 ± 1.85 |
19.02 ± 1.46 |
51.02 ± 2.03 |
41.75 ± 1.31* |
4. Discussion
HED, a pentacyclic triterpenoid isolated from a variety of medicinal plants, such as Dipsacaceae, Caprifoliaceae, Ranunculaceae, and Araliaceae, and has natural biological activity.23 A growing body of research confirms that HED has anti-inflammatory, anti-fungal, anti-protozoal and antibacterial activities as well as cytotoxic effects on various tumour cell lines.24 Moreover, HED also improves lipid metabolism and inhibits lipid deposition to protect blood vessels.20 Hence, it was considered that HED might have other pharmacological effects. Kim et al.19 shown that HED treatment ameliorated alcohol-induced liver injury in Wistar rats, and simultaneously, the activities of molecules in inflammatory signalling pathways, such as extracellular signal-regulated kinase (ERK) and p38 mitogen-activated protein kinase (p38MAPK), were significantly inhibited, indicating that HED may protect the liver through an anti-inflammatory pathway.19 For the treatment of diabetic complications, especially DCM, hypoglycaemic drugs cannot wholly reverse the cardiac remodelling that has already occurred.25 However, many studies have found that some natural bioactive extracts have the pharmacological effect of treating diabetes-induced myocardial injury.9 Our results showed that after 6 weeks of gavage treatment with HED, there was no significant difference in FPG levels between the db/db + HED mice and the db/db control mice. These results suggest that HED, which plays an anti-inflammatory role, cannot be used alone as a hypoglycaemic drug. Meanwhile, chronic inflammation may contribute to myocardial remodelling by inducing increased levels of proinflammatory cytokines during the progression of DCM.6 In the present study, HED has an obvious effect on down-regulating secretion of pro-inflammatory cytokines.
The diabetic heart is characterized by marked hypertrophy of the left ventricle, dilated dysfunction and increased extracellular collagen deposition, all of which affect cardiac output and ultimately lead to cardiac insufficiency.26 In this study, we found that db/db mice as the control had significantly reduced cardiac function, with substantial changes in the structure of the heart, such as significantly increased cardiac mass, marked hypertrophy of cardiomyocytes (as indicated by WGA staining) and massive collagen deposition or pathological fibrosis (as indicated by Masson's trichrome staining); all of these abnormalities were induced by diabetes. The diabetic heart secretes large amounts of pro-inflammatory cytokines and chemokines.27 The continuous increase in pro-inflammatory cytokines and chemokines is closely related to the inflammation status of the heart.6 It is generally considered that the expression levels of pro-inflammation cytokines and chemokines are regulated by the transcription factor NF-κB.3,28 In particular, the activation of NF-κB is even more significant during myocarditis, congestive heart failure and ventricular hypertrophy. Certain endogenous stimuli can significantly induce the transcriptional activity of NF-κB p65/p55 heterodimers, such as hyperglycaemia, hyperlipidaemia and disorders of lipoprotein metabolism.28 As a central molecule in the inflammatory signalling pathway, NF-κB is often used as a target molecule for the treatment of cardiac remodelling caused by DCM.7 Some studies using natural compounds to treat DCM have found that inhibiting the level of NF-κB activation can play a role in improving diabetic heart injury.12,28 Our results showed that HED could significantly improve the chronic cardiac dysfunction; meanwhile, it significantly attenuated the myocardial hypertrophy and the content of fibrosis. Furthermore, at the molecular level, we found that the level of nuclear translocation of the NF-κB p65 subunit was significantly inhibited. The EMSA results also furtherly confirmed that the level of nuclear translocation was downregulated. Therefore, it can be argued that HED may inhibit cardiac remodelling in db/db mice by inhibiting the activation of NF-κB.
Studies have shown that high glucose levels stimulate the transcription of the TGF-β1 gene, resulting in a significant increase in TGF-β1 levels.14 The activation of TGF-β in diabetic hearts is regulated by the ERK-MAPK signalling pathway.29 The ERK-MAPK signalling pathway is closely associated with multiple damages in diabetes, and at the same time, NF-κB is in the most critical position in the entire MAPK signalling pathway.30,31 The db/db mice were orally administered HED for up to 6 weeks. This natural extract inhibited the translocation of Smad2/3 and Smad4 to the nucleus. The EMSA results also revealed that nuclear protein binding to Smad3/4 was attenuated in the heart of db/db mice treated with HED, which also reflected the inhibition of nuclear translocation of the Smads family proteins. Moreover, we also confirmed by immunohistochemical staining that the protein expression levels of TGF-β1 and collagen I, which are target downstream molecules of the inflammatory signalling pathway, were also significantly decreased. All of these findings indicate that HED acts pharmacologically to protect the diabetic heart by inhibiting the key molecules of the inflammatory and fibrotic signalling pathways.
5. Conclusion
In summary, we clearly revealed that HED relieved diabetes-induced cardiac insufficiency, cardiac hypertrophy and myocardial fibrosis (Fig. 7). It was also found that HED may reduce the transcriptional activity of NF-κB and Smads by inhibiting their nuclear translocation, thereby downregulating the synthesis of their target molecules TGF-β1 and collagen I. However, this study provides only a basic clarification of the phenomenon that HED protects the heart of diabetic mice, and it is necessary to conduct further explorations to elucidate the cellular and molecular mechanisms.
 |
| | Fig. 7 Schematic diagram of HED treatment for diabetic cardiomyopathy via inhibition of the inflammatory signalling pathway. | |
Conflicts of interest
The authors declare that they have no conflict of interest.
Abbreviations
| HED | Hederagenin |
| DM | Diabetes mellitus |
| DCM | Diabetic cardiomyopathy |
| NF-κB | Nuclear factor-κB |
| TGF-β | Transforming growth factor |
Acknowledgements
This study was supported by National Natural Science Foundation of China (No. 81301507) and Scientific Research Guidance Project of Qinghai Provincial Health and Family Planning Commission (2018-wjzdx-17). We express our sincere appreciation to Yuanyuan Wu for the graphic design.
References
- F. B. Hu, Diabetes Care, 2011, 34, 1249–1257 CrossRef PubMed.
- W. D. Strain and P. M. Paldanius, Cardiovasc. Diabetol., 2018, 17, 57 CrossRef CAS PubMed.
- G. Jia, M. A. Hill and J. R. Sowers, Circ. Res., 2018, 122, 624–638 CrossRef CAS PubMed.
- S. S. Mahmood, D. Levy, R. S. Vasan and T. J. Wang, Lancet, 2014, 383, 999–1008 CrossRef.
- T. H. Marwick, R. Ritchie, J. E. Shaw and D. Kaye, J. Am. Coll. Cardiol., 2018, 71, 339–351 CrossRef PubMed.
- G. Frati, L. Schirone, I. Chimenti, D. Yee, G. Biondi-Zoccai, M. Volpe and S. Sciarretta, Cardiovasc. Res., 2017, 113, 378–388 CrossRef CAS PubMed.
- A. Oeckinghaus, M. S. Hayden and S. Ghosh, Nat. Immunol., 2011, 12, 695–708 CrossRef CAS PubMed.
- G. Jia, A. Whaley-Connell and J. R. Sowers, Diabetologia, 2018, 61, 21–28 CrossRef CAS PubMed.
- J. Tian, Y. Zhao, Y. Liu, Y. Liu, K. Chen and S. Lyu, Oxid. Med. Cell. Longevity, 2017, 2017, 8214541 Search PubMed.
- X. Palomer, L. Salvado, E. Barroso and M. Vazquez-Carrera, Int. J. Cardiol., 2013, 168, 3160–3172 CrossRef PubMed.
- M. Cameron, J. J. Gagnier and S. Chrubasik, Cochrane Database Syst. Rev., 2011, CD002948, DOI:10.1002/14651858.CD002948.pub2.
- V. Jha, Nephrology, 2010, 15(suppl. 2), 10–17 CrossRef PubMed.
- R. A. Frieler and R. M. Mortensen, Circulation, 2015, 131, 1019–1030 CrossRef PubMed.
- H. H. Hu, D. Q. Chen, Y. N. Wang, Y. L. Feng, G. Cao, N. D. Vaziri and Y. Y. Zhao, Chem.-Biol. Interact., 2018, 292, 76–83 CrossRef CAS PubMed.
- Y. Yue, K. Meng, Y. Pu and X. Zhang, Diabetes Res. Clin. Pract., 2017, 133, 124–130 CrossRef CAS PubMed.
- B. X. Liu, J. Y. Zhou, Y. Li, X. Zou, J. Wu, J. F. Gu, J. R. Yuan, B. J. Zhao, L. Feng, X. B. Jia and R. P. Wang, BMC Complementary Altern. Med., 2014, 14, 412 CrossRef PubMed.
- D. Rodriguez-Hernandez, A. J. Demuner, L. C. Barbosa, L. Heller and R. Csuk, Eur. J. Med. Chem., 2016, 115, 257–267 CrossRef CAS PubMed.
- L. Choi, Y. N. Han, K. T. Lee, K. Y. Park, T. S. Kwak, S. H. Kwon and H. L. Park, Arch. Pharmacal Res., 2001, 24, 536–540 CrossRef CAS.
- G. J. Kim, D. H. Song, H. S. Yoo, K. H. Chung, K. J. Lee and J. H. An, Nutrients, 2017, 9, 41 CrossRef PubMed.
- C. Jiang, Q. Wang, Y. Wei, N. Yao, Z. Wu, Y. Ma, Z. Lin, M. Zhao, C. Che, X. Yao, J. Zhang and Z. Yin, J. Ethnopharmacol., 2015, 176, 17–26 CrossRef CAS PubMed.
- C. W. Lee, S. M. Park, R. Zhao, C. Lee, W. Chun, Y. Son, S. H. Kim, J. Y. Jung, K. H. Jegal, I. J. Cho, S. K. Ku, Y. W. Kim, S. A. Ju, S. C. Kim and W. G. An, Int. Immunopharmacol., 2015, 29, 528–537 CrossRef CAS PubMed.
- B. Wang, P. C. Chandrasekera and J. J. Pippin, Curr. Diabetes Rev., 2014, 10, 131–145 CrossRef CAS PubMed.
- Z. Lin, Z. F. Wu, C. H. Jiang, Q. W. Zhang, S. Ouyang, C. T. Che, J. Zhang and Z. Q. Yin, Phytomedicine, 2016, 23, 1475–1483 CrossRef CAS PubMed.
- S. H. Lu, J. H. Guan, Y. L. Huang, Y. W. Pan, W. Yang, H. Lan, S. Huang, J. Hu and G. P. Zhao, J. Evidence-Based Complementary Altern. Med., 2015, 2015, 456354 Search PubMed.
- A. A. Tahrani, A. H. Barnett and C. J. Bailey, Nat. Rev. Endocrinol., 2016, 12, 566–592 CrossRef CAS PubMed.
- S. Gray and J. K. Kim, Trends Endocrinol. Metab., 2011, 22, 394–403 CrossRef CAS PubMed.
- I. Russo and N. G. Frangogiannis, J. Mol. Cell. Cardiol., 2016, 90, 84–93 CrossRef CAS PubMed.
- S. V. Suryavanshi and Y. A. Kulkarni, Front. Pharmacol., 2017, 8, 798 CrossRef PubMed.
- A. K. Ghosh, R. Rai, P. Flevaris and D. E. Vaughan, J. Cell. Physiol., 2017, 232, 1941–1956 CrossRef CAS PubMed.
- Z. Xu, J. Sun, Q. Tong, Q. Lin, L. Qian, Y. Park and Y. Zheng, Int. J. Mol. Sci., 2016, 17, 2001 CrossRef PubMed.
- M. S. Shah and M. Brownlee, Circ. Res., 2016, 118, 1808–1829 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *e
*e