
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Experimental and computational investigation of a DNA-shielded 3D metal–organic framework for the prompt dual sensing of Ag+ and S2−†
Shao-Lan Cai‡
,
Zi-Chuan Yang‡,
Ke-Yang Wu,
Cheng Fan,
Ling-Yan Zhai,
Nai-Han Huang,
Rong-Tian Li,
Wen-Jun Duan* and
Jin-Xiang Chen *
*
Guangdong Provincial Key Laboratory of New Drug Screening, School of Pharmaceutical Sciences, Southern Medical University, Guangzhou 510515, P. R. China. E-mail: jxchen@smu.edu.cn; wjduan@smu.edu.cn; Fax: +86-20-61648533
First published on 17th May 2019
Abstract
We herein report an efficient Ag+ and S2− dual sensing scenario by a three-dimensional (3D) Cu-based metal–organic framework [Cu(Cdcbp)(bpea)]n (MOF 1, H3CdcbpBr = 3-carboxyl-(3,5-dicarboxybenzyl)-pyridinium bromide, bpea = 1,2-di(4-pyridinyl)ethane) shielded with a 5-carboxytetramethylrhodamine (TAMRA)-labeled C-rich single-stranded DNA (ss-probe DNA, P-DNA) as a fluorescent probe. The formed MOF-DNA probe, denoted as P-DNA@1, is able to sequentially detect Ag+ and S2− in one pot, with detection limits of 3.8 nM (for Ag+) and 5.5 nM (for S2−), which are much more lower than the allowable Ag+ (0.5 μM) and S2− (0.6 μM) concentration in drinking water as regulated by World Health Organization (WHO). The detection method has been successfully applied to sense Ag+ and S2− in domestic, lake, and mineral water with satisfactory recoveries ranging from 98.2 to 107.3%. The detection mechanism was further confirmed by molecular simulation studies.
Introduction
Silver ions (Ag+) are ubiquitous and widely applied in various fields including biomedicine, antibacterial manufacturing, etc.1 However, the biological accumulation of Ag+ raises serious concerns as a result of its relevance to multiple organ dysfunction syndrome, cytotoxicity, argyria and weakness of mitochondrial ability.2 Ag+ is accumulated via the water and food cycle, and thus a 0.5 μM concentration up-limit of Ag+ in drinking water is regulated by U.S. Environmental Protection Agency (US EPA) and World Health Organization (WHO).3On the other hand, sulfide (S2−) possesses an important biological function.4 S2− exhibits a strong affinity toward proton to give hydrogen sulfide (H2S), which is the third largest gas signal molecules. H2S takes part in the course of atherosclerosis, myocardial contraction, nerve transmission and regulation of insulin secretion.5 Moreover, high concentrations of H2S gives rise to diseases such as diabetes,6 hepatic sclerosis,7 Alzheimer's disease,8 and Down's syndrome.9
An equally important concern is the co-existence of Ag+ and S2− that would result in the formation of Ag2S precipitates due to its extremely low solubility product constant of Ag2S (Ksp = 6.3 × 10−50, r.t.). Ag2S particle formation is harmful to eyes, skin and respiratory system.10 It would, therefore, be essential to develop a sensor that can simultaneously detect the presence of Ag+ and S2−. Some hazardous ions can be monitored by fluorescence-based chemosensors as a result of their rapid detection speed, simple procedure, and high sensitivity.11 Among them, metal–organic frameworks (MOFs), consist of metal ions and various organic ligands, have shown great potential in sensing metal ions and small molecules due to their unique advantages such as diversity, porosity, stability, amenability toward further functionalization.12 However, sensors by integrating MOFs and bio-related species, such as DNA, remains relatively rare.13
Herein, we report the construction of a sensing system by hybridizing a three-dimensional (3D) Cu-based MOF [Cu(Cdcbp)(bpea)]n (MOF 1, H3CdcbpBr = 3-carboxyl-(3,5-dicarboxybenzyl)-pyridinium bromide, bpea = 1,2-di(4-pyridinyl)ethane, Fig. 1a) and a single-stranded, carboxytetramethylrhodamine (TAMRA)-labeled C-rich probe DNA (ss-DNA, P-DNA). The MOF and P-DNA are associated through π–π stacking, hydrogen bonding, and electrostatic interactions, and thereby quenches the TAMRA fluorescence of the latter (off-state) via a photo-induced electron transfer to eliminate the high background fluorescence (Fig. 1b).14 The formed MOF-DNA probe, denoted as P-DNA@1, is able to sense the presence of Ag+ through the formation of C–Ag+-C coordination bonds, yielding the double-stranded hairpin-like DNA, that is ds-DNA@Ag+ duplex.15 The much more rigid ds-DNA@Ag+ duplex formed is subsequently detached from the surface of MOF 1 with the recovery of the TAMRA fluorescence (on-state). When further adding S2− to the above formed 1 + ds-DNA@Ag+ mixture, Ag+ was extracted from ds-DNA@Ag+ to form Ag2S precipitates and the ds-DNA unwinded and converts back to P-DNA, and resorbed by MOF 1 to quench the fluorescence again (off-state). Thus, the “off-on-off” fluorescent sensing system was successfully constructed to monitor Ag+ and S2− in succession. The computational investigation revealed that P-DNA bounds to MOF 1 more tightly through multiple π–π stacking and hydrogen bonding interactions than ds-DNA@Ag+.
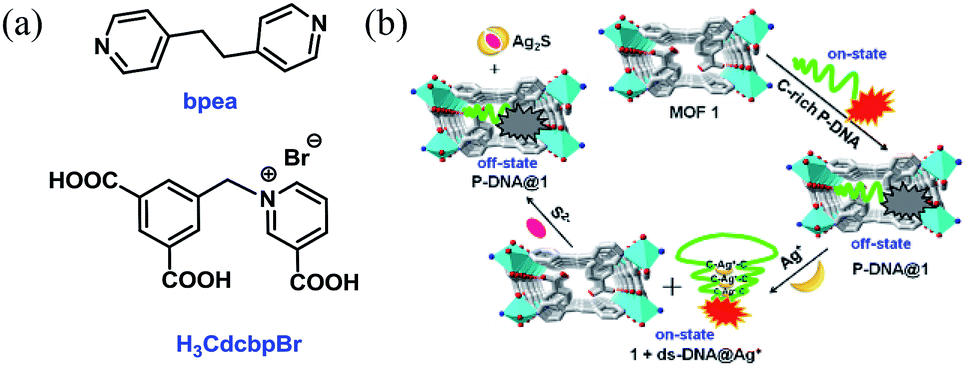 | ||
| Fig. 1 (a) The structures of bpea and H3CdcbpBr. (b) The detection mechanism of Ag+ and S2− based on the hybrid of MOF 1 and C-rich P-DNA. | ||
Experimental
General
IR spectra were recorded on a Nicolet MagNa-IR 550 infrared spectrometer. Elemental analyses for C, H, and N were carried out with an EA1110 CHNS elemental analyzer. Powder X-ray diffraction (PXRD) spectra were obtained with a Rigaku D/max2200/PC. The X-ray generated from a sealed Cu tube was monochromated by a graphite crystal and collimated by a 0.5 mm MONOCAP (λ Cu-Kα = 1.54178 Å). The tube voltage and current were 40 kV and 40 mA, respectively. Fluorescence spectra were measured on a PE LS-55 fluorescence spectrophotometer.The synthesis of amphoteric tricarboxylic acid ligand-H3CdcbpBr was based on our reported procedure.14c The TAMRA-labeled cytimidine-rich probe DNA sequence (P-DNA: 5′-TAMRA-ACCTCTTCTCTTCATTTTTCAACACAACACCG-3′) was purchased from Sangon Inc. and stored at −20 °C for use, and at −80 °C for long-term preservation. All the other reagents and solvents were obtained from commercial sources and used without further purification.
Synthesis of [Cu(Cdcbp)(bpea)]n (1)
Powder of H3CdcbpBr (764 mg, 2 mmol) was dispersed in distilled H2O (70 mL) by sonication and the pH adjusted to 7.0 with 0.1 M NaOH to give a clear solution. This is followed by slow dropwise addition of CuSO4·5H2O (391 mg, 2 mmol) dissolved in H2O (20 mL). The formed mixture was stirred for 30 min to give a clear blue solution. Subsequently, bpea (380 mg, 2 mmol) dissolved in DMF (5 mL) was slowly added and the mixture was shaken for a while to give a blue solution containing a small amount of blue precipitate. Upon filtration, the filtrate was sub-packaged in thick-walled pressure bottle and transferred to a programmed oven. The temperature of the oven was smoothly increased from r.t. to 100 °C in 1 h, maintained at 100 °C for 72 h, before finally cooled to r.t. within 48 h to give blue block crystals. The crystals obtained were collected by filtration and washed with diethyl ether and dried in vacuo (910 mg, 83%). Anal. calcd for C27H21N3O6Cu·H2O: C 57.35%, H 4.07%, N 7.43%; found: C 57.16%, H 3.98%, N 7.37%. IR (KBr disc, cm−1) ν 3324 (s), 3123 (m), 1635 (s), 1489 (m), 1378 (s), 1220 (m), 1154 (m), 1018 (w), 832 (m), 815 (m), 727 (m), 614 (m), 478 (m).X-ray crystal structure determination
Crystallographic measurements were made on a Bruker APEX II diffractometer by using graphite-monochromated Mo Kα (λ = 0.71073 Å) irradiation for MOF 1. The data were corrected for Lorentz and polarization effects with the SMART suite of programs and for absorption effects with SADABS.16 All crystal structures were solved by direct methods and refined on F2 by full-matrix least-squares techniques with SHELXTL-97 program.17 The location of the two hydrogen atoms on the coordinated water was suggested by Calc-OH program in the WinGX suite,18 the water molecules were then refined as a rigid model with their thermal parameters constrained to Uiso(H) = 1.2Ueq(O). Spatially delocalized electron density in the lattice was found but acceptable refinement results could not be obtained for this electron density. The solvent contribution was then modeled using SQUEEZE in Platon.19 A summary of the key crystallographic data for 1 is listed in Table S1.†Results and discussion
Characterization of MOF 1
Single crystal X-ray diffraction analysis revealed that MOF 1 crystallizes in the monoclinic space group C2/c and the asymmetric unit consists one [Cu(Cdcbp)(bpea)] molecule. As shown in Fig. 2a, each two Cu(II) ions are linked by a couple of μ–COO− (η1:η1) groups from two Cdcbp ligands, and each Cu(II) ion further coordinated one chelating carboxylate to form a [Cu2(Cdcbp)4] unit (Fig. 2a). Such unit extends to six equivalents to form a two-dimensional (2D) structure within the bc plane as shown in Fig. 2b. These adjacent 2D layers have their associated benzoate oriented face-to-face and approximate 13.4 Å apart, an ideal distance to further accommodate one bpea ligand (13.4 Å approximates the distance of one bpea ligand of ca. 9.4 Å and a pair of Cu–N bonds with ca. 2.0 Å for each) (Table S2†). The Cu–bpea association is aligned in the a direction, completes the octahedral coordination geometry of Cu2+ (Fig. 2a), and result in a 3D network of MOF 1 as shown in Fig. 2c.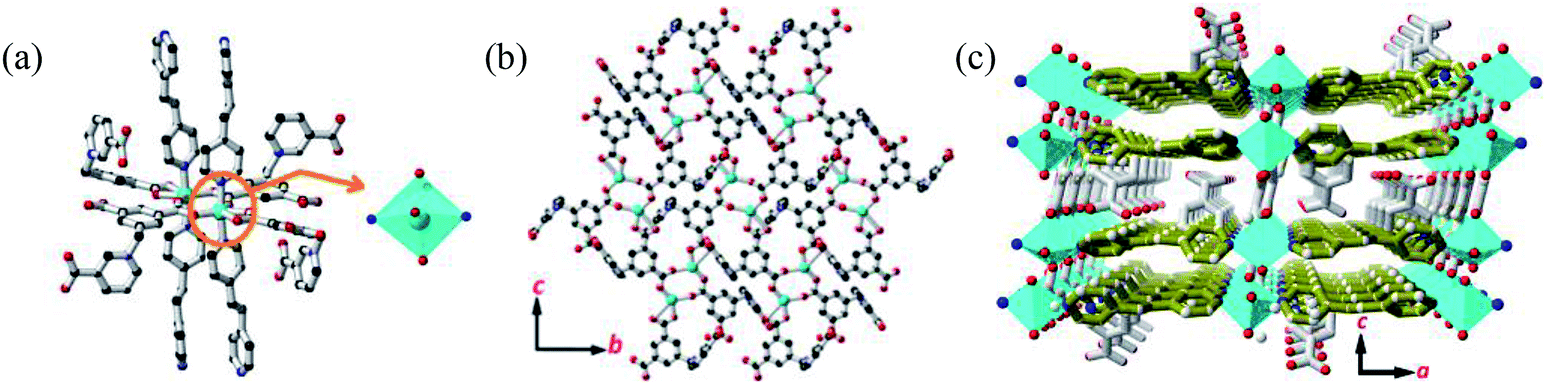 | ||
| Fig. 2 The structure of MOF 1. (a) The coordination environment of Cu2+ in MOF 1. (b) The 2D plane sheet structure constructed by the [Cu2(Cdcbp)4] units. (c) The 3D structure of MOF 1. Color codes: Cu (turquoise), O (red), N (blue), C (black) and bpea (olive green). | ||
Ag+ detection with P-DNA@1 hybrid and S2− detection with 1 + ds-DNA@Ag+ system
MOF 1 is water stable and its powder X-ray diffraction (PXRD) pattern indicated that the as-synthesized MOF 1 and its fresh powder immersed in Hepes buffer (pH = 6.5, 7.0, 7.4) for 24 h matched very well with the simulated one, suggesting its phase purity and buffer stability (Fig. S1†). The scanning electron microscopic (SEM) images of the fresh MOF 1 gave the size of 831.25 ± 80 nm, further indicating its phase homogeneity (Fig. S2†).In MOF 1, the Cdcbp and bpea ligands contain conjugated π-electron system, Cu2+, and quaternary ammonium groups, all facilitate its binding with ss-P-DNA molecules through a variety of weak intermolecular interactions, such as π–π interactions and hydrogen bonding, and ultimately lead to fluorescence quenching.20 In order to verify our hypothesis, we first studied the stability of P-DNA in Hepes buffer at three different pH conditions (6.5, 7.0, 7.4) that approximate the physiological conditions. As shown in Fig. S3,† the emission profile is retained in these condition in 4 h. Then we studied the interaction of MOF 1 with P-DNA at these pH conditions to make the Ag+ sensor. As shown in Fig. 3a, S4a and S5a,† in all three conditions, the fluorescence intensity of P-DNA gradually decreased with the increasing concentration of MOF 1 up to 9.0 μM with a quenching efficiency (QE, %) of 89.3% due to the formation of P-DNA@1 (QE = (F0 − FM)/F0 × 100%, wherein FM and F0 are the fluorescent intensities at 582 nm in the presence and absence of MOF 1, respectively). Our second step involves the sensing of Ag+ using P-DNA@1 also at three different pH conditions (6.5, 7.0, 7.4). As shown in Fig. 3b, S4b and S5b,† in all three conditions, when Ag+ was added into the P-DNA@1 sensing system with gradually increasing concentrations, the fluorescence intensity recovered to saturation with a concentration of Ag+ from 0 to 6.0 μM. The recovery efficiency (RE) was 4.9 on the basis of RE = (FT − FM)/FM (FT and FM are the fluorescence intensities at 582 nm with and without Ag+, respectively). The fluorescence recovery spectra exhibits a linear relationship with the concentration of Ag+ (Fig. 4a), and a linear equation of Y = 212.559X + 91.532 with a related coefficient R2 = 0.9995 can be derived. The detecting limit (LOD) of Ag+ was 3.8 nM calculated from 3σ/slope (σ = standard deviation for 10 blank samples), which is much lower than the reported materials, such as tetraphenyl ethylene (874 nM),21 carbon dots (320 nM),22 imidazole derivatives (101 nM),23 and comparable to gold nanoparticle (7.3 nM)24 and g-C3N4 nanosheets (4.2 nM) (Table S3†).25 Our third experiment concerns the sensing of S2− using the above formed 1 + ds-DNA@Ag+ system at three different pH conditions (6.5, 7.0, 7.4). As shown in Fig. 3c, 4c and S5c,† in all three conditions, when various concentrations of S2− were added to the 1 + ds-DNA@Ag+ system, the fluorescence intensity gradually decreased to stabilization with a maximum S2− consumption of 6.0 μM (QE value being 86.9%). The fluorescence quenching spectra of 1 + ds-DNA@Ag+, as depicted in Fig. 4b, also showed good linearity between fluorescence intensity and S2− concentration with the linear equation of Y = −89.855X + 544.153 (R2 = 0.9978), giving an LOD value of 5.5 nM for S2−. Such a value is much lower than some nanocomposite (138 nM),26 gold nanoparticles (80 nM),27 DNA@copper nanoparticles (80 nM),28 and nano Ag–carbon (27 nM),29 and comparable to g-C3N4 nanosheets (3.5 nM) (Table S4†).25 All the results indicate that the detection process was not disturbed by the variation of the pH values.
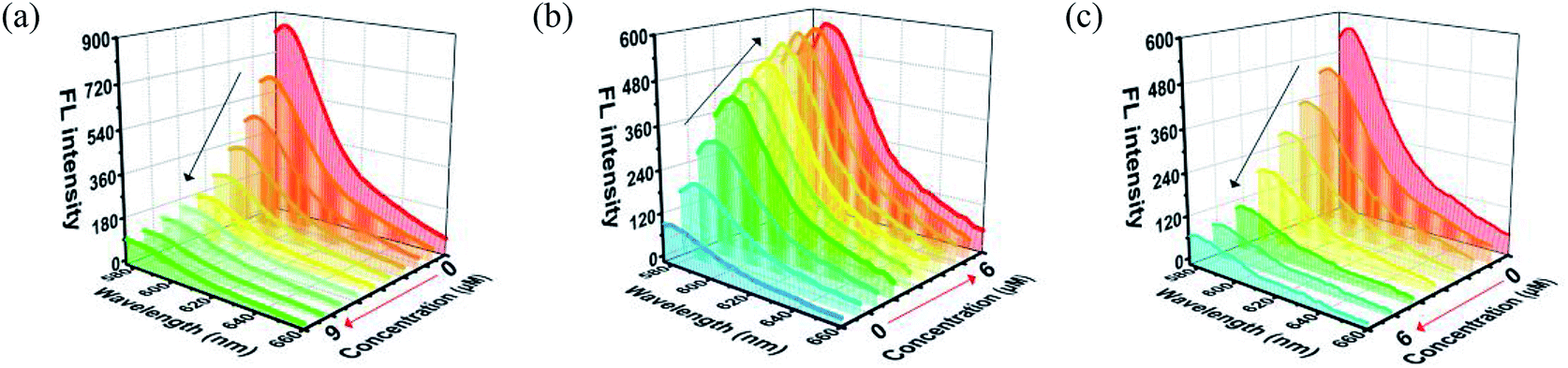 | ||
| Fig. 3 (a) The fluorescence quenching of the P-DNA (50 nM) incubated with different concentrations of MOF 1. (b) The fluorescence recovery of P-DNA@1 (50 nM/9.0 μM) sensing system towards different concentrations of Ag+. (c) The fluorescence quenching of 1 + ds-DNA@Ag+ (9.0 μM/50 nM/6.0 μM) sensing system towards various concentrations of S2−. | ||
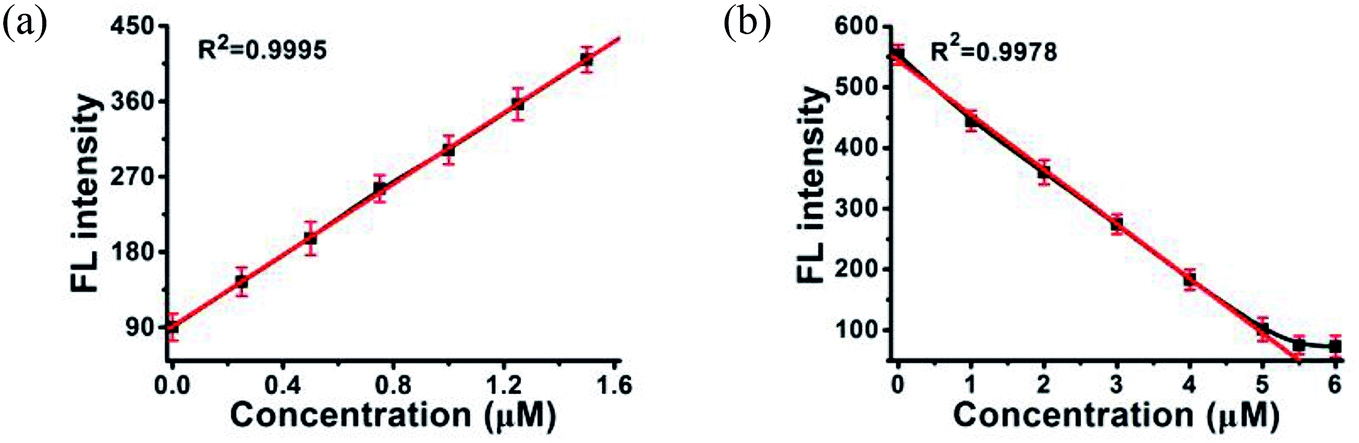 | ||
| Fig. 4 A linear relationship between the fluorescence intensity of (a) P-DNA@1 at 582 nm and the concentrations of Ag+, (b) 1 + ds-DNA@Ag+ at 582 nm and the concentrations S2−. Error bars represent the standard deviation for three measurements. | ||
The selectivity of the sensor
The anti-interference ability is another critical characteristic for the biosensors to ensure their practical application. Herein, the P-DNA was designed with specific bases that can pair with silver exclusively to form “C–Ag+–C”.15 To further confirm our hypothesis, we carried the following experiments. First, different kinds of metal ions were used as interferences, including Hg2+, Mn2+, Cd2+, Ca2+, Pb2+, Mg2+, Cu2+, Ba2+, Ni2+, K+, Na+, Zn2+, Cr3+, Co2+ and Fe2+ with a concentration of 30 μM. As depicted in Fig. 5a, the biosensor of P-DNA@1 showed no remarkable response to these interfering metal ions. Subsequently, we added Ag+ (6.0 μM) to each of the above metal ion solutions, and the fluorescence intensities increased dramatically in contrast with other interfering metal ions with 5-fold concentrations higher than that of Ag+. It is thus conclusive that the system had a high selectivity towards Ag+. Likewise, the specificity of the S2− sensor was also evaluated by comparing its response to S2− and other small anions (HSO4−, SO42−, OH−, H2PO4−, CO32−, NO3−, F−, Cl−, Br− and I−) with a concentration of 30 μM (5-fold higher than S2−). As illustrated in Fig. 5b, there was no obvious fluorescence intensity changes induced by other anions while the fluorescence intensity declined significantly when S2− (6.0 μM) was presented. Thus, the interference of the other small anions to the biosensor 1 + ds-DNA@Ag+ was also negligible.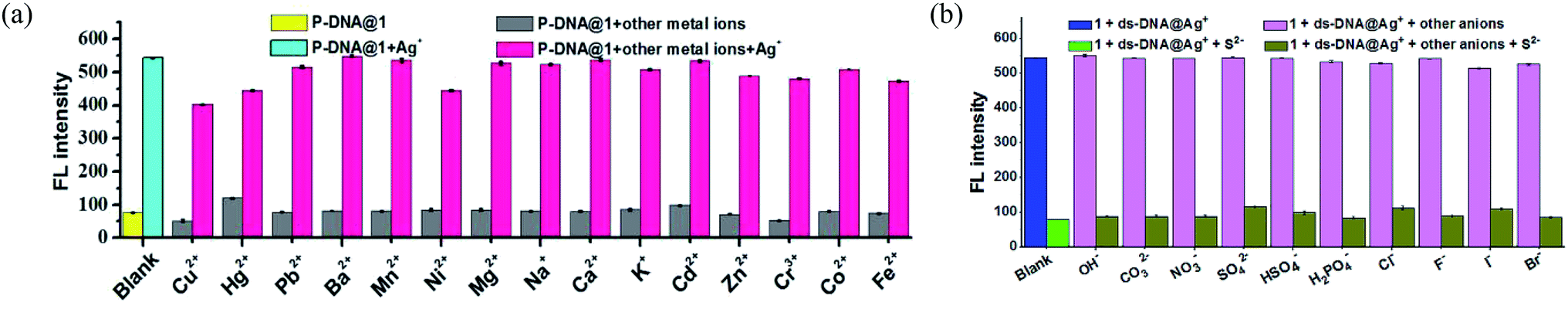 | ||
| Fig. 5 (a) The detection selectivity of Ag+ sensor (blank: P-DNA@1 (50 nM/9.0 μM); Ag+: 6.0 μM, other metal ions: 30 μM). (b) The detection selectivity of S2− sensor (blank: 1 + ds-DNA@Ag+ (9.0 μM/50 nM/6.0 μM); S2−: 6.0 μM, other anions: 30 μM). Error bars represent the standard deviation for three measurements. | ||
In order to further clarify the detection process, the conformational changes of the P-DNA induced by Ag+ and S2− were investigated by circular dichroism (CD) spectroscopy. Single-stranded P-DNA with a concentration of 5 μM exhibited a strong positive CD peak at 278 nm. As shown in Fig. 6a, the intensity of the peak gradually decreased with increased concentrations of MOF 1. MOF 1 fails to absorb all the P-DNA (5 μM) at its highest concentration of 150 μM, and only lead to partial disappearance of the CD peak of P-DNA. With an increased concentration of Ag+ in the P-DNA@1 sensing system, the intensity of positive peak weakened and ultimately disappeared, accompanied by the continuous intensity increase of the negative peak (Fig. 6b), attributable to the formation of a large amount of C–Ag+–C ds-DNA@Ag+ complex.30 Finally, with the addition of S2− to the 1 + ds-DNA@Ag+ system, the negative peak disappeared and the positive peak appeared again, indicating that the ds-DNA@Ag+ unwind to release P-DNA (Fig. 6c).
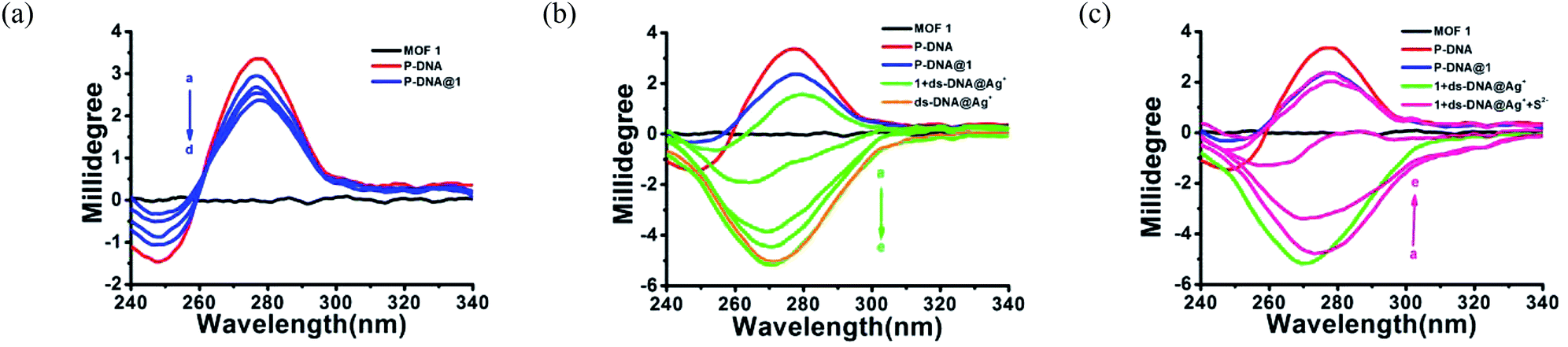 | ||
| Fig. 6 (a) The CD spectra of MOF 1 (150 μM) and P-DNA (5 μM) with different concentrations of MOF 1 (50, 100, 125, 150 μM, a–d). (b) The CD spectra of P-DNA@1 (5 μM/150 μM) with different concentrations of Ag+ (12, 24, 36, 48, 60 μM, a–e) and ds-DNA@Ag+ (5 μM/60 μM). (c) The CD spectra of 1 + ds-DNA@Ag+ (150 μM/5 μM/60 μM) with different concentrations of S2− (20, 40, 60, 80, 100 μM, a–e). | ||
The detection mechanism
In order to elucidate the detection mechanism, the binding free energy difference (ΔΔG) between reactions of MOF 1 with P-DNA (ΔGP-DNA@MOF) and MOF 1 with ds-DNA@Ag+ (ΔGMOF+ds-DNA@Ag+) is calculated. The Gibbs free energy calculations were simplified by calculating single point energies. The result (Table S5†) showed that ΔΔG = −180.24 kcal mol−1 < 0, suggesting that P-DNA bounds to MOF 1 more tightly than ds-DNA@Ag+, corroborating the experimental observation.The electrostatic surface showed that the surface of MOF 1 was largely positive and P-DNA covered by a negative and neutral surface with the scattered positive area, which led to good electronic complement between MOF 1 and P-DNA. Single chain P-DNA exhibits a higher contact area with MOF 1 due to its structural flexibility (Fig. 7A), while double chain ds-DNA@Ag+ only has a small interface (Fig. 7F). MOF 1 bound to P-DNA mainly through 19 π–π stacking (Fig. 7B–E) between the aromatic rings in MOF 1 and nucleobases in P-DNA with the ring centroid distances in the range of 3.8–5.9 Å. This is in addition to 18 hydrogen bonding (Fig. 7B–E) between the nucleobases in P-DNA and carboxylate groups in MOF 1, with the donor–acceptor distances in the range of 2.9–3.4 Å. In sharp contrast, there are only 9 π–π stacking between the aromatic rings in MOF 1 and the nucleobases in ds-DNA@Ag+, and 5 hydrogen-bondings between oxygen/nitrogen atoms of the phosphate group, or the nucleobases in ds-DNA@Ag+ and carboxylate groups in MOF 1 (Fig. 7G–J), with the ring centroid distances in the range of 4.2–5.8 Å and donor–acceptor distances in the range of 2.8–3.5 Å.
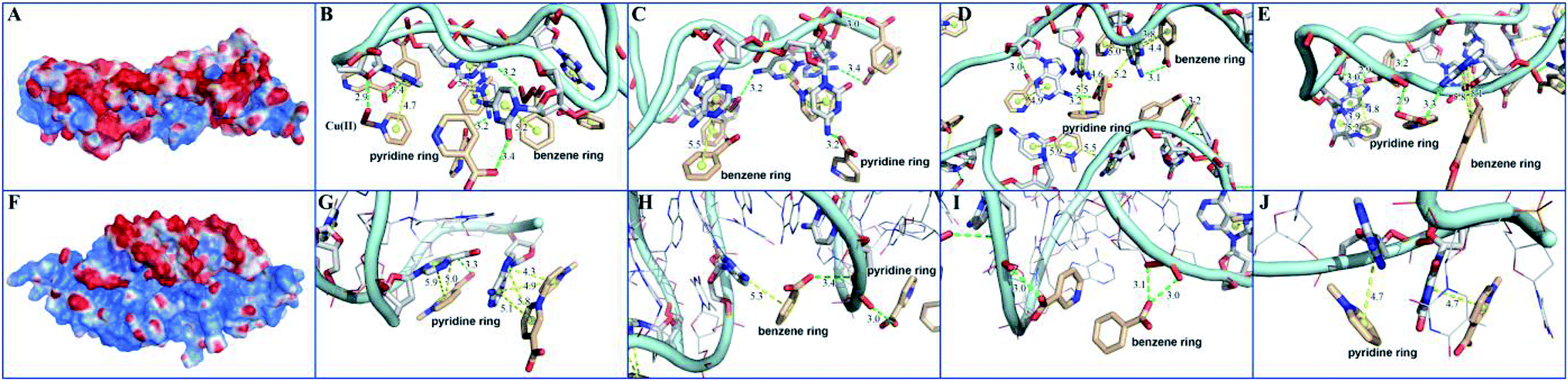 | ||
| Fig. 7 The interactions between MOF 1 and P-DNA and ds-DNA@Ag+: (A) and (F) showed the charge distributions where red area represented negative charges, blue positive, and white neutral; (B–E) and (G–J) showed the local binding modes for MOF 1 with P-DNA (B–E) and MOF 1 with ds-DNA@Ag+ (G–J), where MOF 1 was displayed by smudge lines and sticks, while P-DNA and ds-DNA@Ag+ by light blue lines and sticks. Aromatic ring centers were denoted by yellow spheres, π–π stacking, and hydrogen bonding interactions by yellow dashes and green dashes. | ||
Detect Ag+ and S2− in environmental water
To further demonstrate the applications of these two biosensors, the recovery efficiencies of Ag+ and S2− were studied in three kinds of environmental water samples, including domestic water, lake water, and mineral water. The environmental water samples of 0.6 μM Ag+ and S2− were prepared from a condensed stock solution of silver ion (10 mM) and sulfide ion (10 mM), respectively. Both Ag+ and S2− in environmental water samples were measured as mentioned in the previous section. Each measurement was carried out three times repeatedly. As shown in Tables S6 and S7,† the recovery efficiencies of the Ag+ and S2− were calculated to be in the range of 98.2–101.8% with standard deviations (RSD) ≤ 1.0% for Ag+, and from 99.0% to 107.3% with RSD ≤ 4% for S2−, respectively. These results verified the reliability and practicability of the proposed fluorescence sensor for the successive detection of Ag+ and S2−.Conclusions
In summary, a highly sensitive and selective fluorescence sensor was created based on the Cu-MOF shielded with fluorescence-labeled C-rich ss-P-DNA for the specific and successive detection of Ag+ and S2− with a low detection limit of 3.8 nM and 5.5 nM, respectively, which were two orders of magnitudes lower than those allowed in drinking water. The uniqueness of the present probe arises from the use of C–C mismatched P-DNA sequences 5′-TAMRA-ACCTCTTCTCTTCATTTTTCAACACAACACCG-3′ that can exclusively recognize Ag+ to form C–Ag(I)–C duplex.15 The use of Cu-based MOF 1 as fluorescence quencher serves as an additional advantage as the Ag+ sensing process is against a dark background. The sensing events are stable at three different pH conditions (6.5, 7.0, 7.4) that mimicking physiological conditions, and are further transferrable to domestic, lake, and mineral waters, indicating the good adaptability of the present sensor. This discovery further inspired us to develop relevant sensors for the pairwise detection of other biologically relevant cations and anions, such as Hg2+ and S2−, Pb2+ and Br−, Fe3+ and ascorbic acid, and etc., using a shared mechanism and beyond.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (21874064), the Natural Science Foundation from Guangdong Provincial Department of Science and Technology of China (2018A030313456) and One Belt One Road Incubation Project from Southern Medical University (YD2018N003). We also thank Dr Ziying Zhang from Guangzhou Yinfo Information Technology Co., Ltd. for doing the molecular simulation studies.Notes and references
- T. W. Purcell and J. J. Peters, Environ. Toxicol. Chem., 2010, 17, 539 CrossRef.
- (a) S. Kim, J. E. Choi, J. Choi, K. H. Chung, K. Park, J. Yi and D. Y. Ryu, Toxicol. in Vitro, 2009, 23, 1076 CrossRef CAS PubMed; (b) C. Greulich, D. Braun, A. Peetsch, J. Diendorf, B. Siebers, M. Epple and M. Koller, RSC Adv., 2012, 2, 6981 RSC; (c) B. Z. Zhang and C. Y. Wei, ChemistrySelect, 2017, 2, 6844 CrossRef CAS.
- (a) M. M. Hu, J. L. Fan, J. F. Cao, K. D. Song, H. Zhang, S. G. Sun and X. J. Peng, Analyst, 2012, 137, 2107 RSC; (b) Z. Y. Xiao, A. J. Tang, H. S. Huang and Z. Wang, Can. J. Chem., 2017, 95, 1267 CrossRef CAS.
- (a) S. A. Patwardhan and S. M. Abhyankar, Colourage, 1988, 35, 15 CAS; (b) R. Huang, X. Zheng and Y. Qu, Anal. Chim. Acta, 2007, 582, 267 CrossRef CAS PubMed.
- (a) E. Lowicka and J. Beltowski, Pharmacol. Rep., 2007, 59, 4 CAS; (b) J. W. Calvert, S. Jha, S. Gundewar, J. W. Elrod, A. Ramachandran, C. B. Pattillo, C. G. Kevil and D. J. Lefer, Circ. Res., 2009, 105, 365 CrossRef CAS PubMed; (c) C. A. Wagner, J. Nephrol., 2009, 22, 173 CAS; (d) G. D. Yang, L. Y. Wu, B. Jiang, W. Yang, J. S. Qi, K. Cao, Q. H. Meng, A. K. Mustafa, W. T. Mu and S. M. Zhang, Science, 2009, 322, 587 CrossRef PubMed.
- W. Yang, G. Yang, X. Jia, L. Wu and R. Wang, J. Physiol., 2005, 569, 519 CrossRef CAS PubMed.
- S. Fiorucci, E. Antonelli, A. Mencarelli, S. Orlandi, B. Renga, G. Rizzo, E. Distrutti, V. Shah and A. Morelli, Hepatology, 2005, 42, 539 CrossRef CAS PubMed.
- K. Eto, T. Asada, K. Arima, T. Makifuchi and H. Kimura, Biochem. Biophys. Res. Commun., 2002, 293, 1485 CrossRef CAS PubMed.
- P. Kamoun, M. C. Belardinelli, A. Chabli, K. Lallouchi and B. Chadefaux-Vekemans, Am. J. Med. Genet., Part A, 2003, 116, 310 CrossRef PubMed.
- Y. Yin, W. Xu, Z. Tan, Y. Li, W. Wang, X. Guo, S. Yu, J. Liu and G. Jiang, Environ. Pollut., 2017, 220, 955 CrossRef CAS PubMed.
- (a) G. B. Demirel, B. Daglar and M. Bayindir, Chem. Commun., 2013, 49, 6140 RSC; (b) B. Gole, A. K. Bar and P. S. Mukherjee, Chem.–Eur. J., 2014, 20, 2276 CrossRef CAS PubMed; (c) X. Y. Guo, F. Zhao, J. J. Liu, Z. L. Liu and Y. Q. Wang, J. Med. Chem., 2017, 5, 20035 CAS.
- (a) L. Li, Q. Chen, Z. G. Niu, X. H. Zhou, T. Yang and W. Huang, J. Mater. Chem. C, 2016, 4, 1900 RSC; (b) A. M. Spokoyny, D. Kim, A. Sumrein and C. A. Mirkin, Chem. Soc. Rev., 2009, 40, 1218 RSC; (c) G. F. Ji, J. J. Liu, X. C. Gao, W. Sun, J. Z. Wang, S. L. Zhao and Z. L. Lin, J. Mater. Chem. A, 2017, 5, 10200 RSC; (d) D. Liu, J. P. Lang and B. F. Abrahams, J. Am. Chem. Soc., 2011, 133, 11042 CrossRef CAS PubMed; (e) X. Y. Tang, J. X. Chen, G. F. Liu, Z. G. Ren, Y. Zhang and J. P. Lang, Eur. J. Inorg. Chem., 2008, 16, 2593 CrossRef; (f) F. L. Yuan, Y. Q. Yuan, M. Y. Chao, D. J. Young, W. H. Zhang and J. P. Lang, Inorg. Chem., 2017, 56, 6522 CrossRef CAS PubMed; (g) Z. X. Zhang, N. N. Ding, W. H. Zhang, J. X. Chen, D. J. Young and T. S. A. Hor, Angew. Chem., Int. Ed., 2014, 53, 4628 CrossRef CAS PubMed.
- (a) H. Weng and B. Yan, Anal. Chim. Acta, 2017, 988, 89 CrossRef CAS PubMed; (b) L. L. Wu, Z. Wang, S. N. Zhao, X. Meng, X. Z. Song, J. Feng, S. Y. Song and H. J. Zhang, Chem.–Eur. J., 2016, 22, 477 CrossRef CAS PubMed; (c) P. P. Hu, N. Liu, K. Y. Wu, L. Y. Zhai, B. P. Xie, B. Sun, W. J. Duan, W. H. Zhang and J. X. Chen, Inorg. Chem., 2018, 57, 8382 CrossRef CAS PubMed; (d) B. P. Xie, G. H. Qiu, B. Sun, Z. F. Yang, W. H. Zhang, J. X. Chen and Z. H. Jiang, Inorg. Chem. Front., 2019, 6, 148 RSC; (e) B. P. Xie, Q. H. Qiu, P. P. Hu, Z. Liang, Y. M. Liang, B. Sun, L. P. Bai, Z. H. Jiang and J. X. Chen, Sens. Actuators, B, 2018, 254C, 1133 CrossRef; (f) G. H. Qiu, Z. H. Weng, P. P. Hu, W. J. Duan, B. P. Xie, B. Sun, X. Y. Tang and J. X. Chen, Talanta, 2018, 180, 396 CrossRef CAS PubMed.
- (a) S. P. Yang, W. Zhao, P. P. Hu, K. Y. Wu, Z. H. Jiang, L. P. Bai, M. M. Li and J. X. Chen, Inorg. Chem., 2017, 56, 14880 CrossRef CAS PubMed; (b) H. Q. Zhao, G. H. Qiu, Z. Liang, M. M. Li, B. Sun, L. Qin, S. P. Yang, W. H. Chen and J. X. Chen, Anal. Chim. Acta, 2016, 922, 55 CrossRef CAS PubMed; (c) H. Q. Zhao, S. P. Yang, N. N. Ding, L. Qin, G. H. Qiu, J. X. Chen, W. H. Zhang, W. H. Chen and T. S. A. Hor, Dalton Trans., 2016, 45, 5092 RSC.
- A. Ono, S. Cao, H. Togashi, M. Tashiro, T. Fujimoto, T. Machinami, S. Oda, Y. Miyake, I. Okamoto and Y. Tanaka, Chem. Commun., 2008, 39, 4825 RSC.
- G. M. Sheldrick, SADABS, Program for empirical absorption correction of area detector data, University of Göttingen: Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- (a) Z. W. Tang, H. Wu, J. R. Cort, G. W. Buchko, Y. Y. Zhang, Y. Y. Shao, I. A. Aksay, J. Liu and Y. H. Lin, Small, 2010, 6, 1205 CrossRef CAS PubMed; (b) L. H. Tang, H. X. Chang, Y. Liu and J. H. Li, Adv. Funct. Mater., 2012, 22, 3083 CrossRef CAS.
- Y. Li, H. Yu, G. Shao and F. J. Gan, J. Photochem. Photobiol., A, 2015, 301, 14 CrossRef CAS.
- X. H. Gao, Y. Z. Lu, R. Z. Zhang, S. J. He, J. Ju, M. M. Liu, L. Li and W. Chen, J. Mater. Chem. C, 2015, 3, 2302 RSC.
- B. Zhao, Y. Xu, Y. Fang, L. Y. Wang and Q. G. Deng, Tetrahedron Lett., 2015, 56, 2460 CrossRef CAS.
- J. J. Du, H. Du and H. Y. Ge, Sens. Actuators, B, 2017, 4005, 31456 Search PubMed.
- S. Wang, D. Du, M. Yang, Q. Lu, R. Ye and X. Yan, Talanta, 2017, 168, 168 CrossRef CAS PubMed.
- T. Zhou, N. Wang, C. Li, H. Yuan and D. Xiao, Anal. Chem., 2010, 82, 1705 CrossRef CAS PubMed.
- H. H. Deng, S. H. Weng, S. L. Huang, L. N. Zhang, A. L. Liu and X. H. Lin, Anal. Chim. Acta, 2014, 852, 218 CrossRef CAS PubMed.
- J. Liu, J. H. Chen and Z. Y. Fang, Analyst, 2012, 137, 5502 RSC.
- Z. X. Wang, C. L. Zheng, Q. L. Li and S. N. Ding, Analyst, 2014, 139, 1751 RSC.
- S. Zhan, Y. Wu, L. Wang, X. Zhan and Z. Pei, Biosens. Bioelectron., 2016, 86, 353 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General, X-ray crystal structure determinations, Ag+ and S2− detection experiments, computational molecular simulation studies, crystallographic data, selected bond distances (Å) and angles (°) for 1, the analytical performance of various Ag+ sensors, comparison of different sensing platforms for S2− detection, the single point energy results of P-DNA, P-DNA@1, ds-DNA@Ag+ and 1 + ds-DNA@Ag+, detection of Ag+ and S2− in environmental water samples, PXRD patterns of MOF 1 showing agreement among the simulated, as-synthesized and fresh powder of MOF 1 immerse in Hepes buffer (20 mM, pH = 6.5, 7.0, 7.4) for 24 h respectively, the SEM image of MOF 1, comparison of the intensity of the emission peak (582 nm) of the P-DNA in 20 mM Hepes buffer (pH = 6.5, 7.0, 7.4) for 4 h, the fluorescence quenching of the P-DNA (50 nM) incubated with MOF 1 with increasing concentrations in Hepes buffer (pH 7.4, 20 mM), the fluorescence recovery of P-DNA@1 (50 nM/9 μM) sensor towards Ag+ with different concentrations in Hepes buffer (pH 7.4, 20 mM), the fluorescence quenching of 1 + ds-DNA@Ag+ (9 μM/50 nM/6 μM) sensing system towards S2− with different concentrations in Hepes buffer (pH 7.4, 20 mM), the fluorescence quenching of the P-DNA (50 nM) incubated with different concentrations of MOF 1 in different pH Hepes buffer solutions (pH = 6.5, 7.0, 7.4), the fluorescence recovery of P-DNA@1 (50 nM/9.0 μM) sensing system towards different concentrations of Ag+ in different pH buffer solutions (pH = 6.5, 7.0, 7.4), the fluorescence quenching of 1 + ds-DNA@Ag+ (9.0 μM/50 nM/6.0 μM) sensing system towards various concentrations of S2− in different pH buffer solutions (pH = 6.5, 7.0, 7.4), reference. CCDC 1892132. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra02028d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |