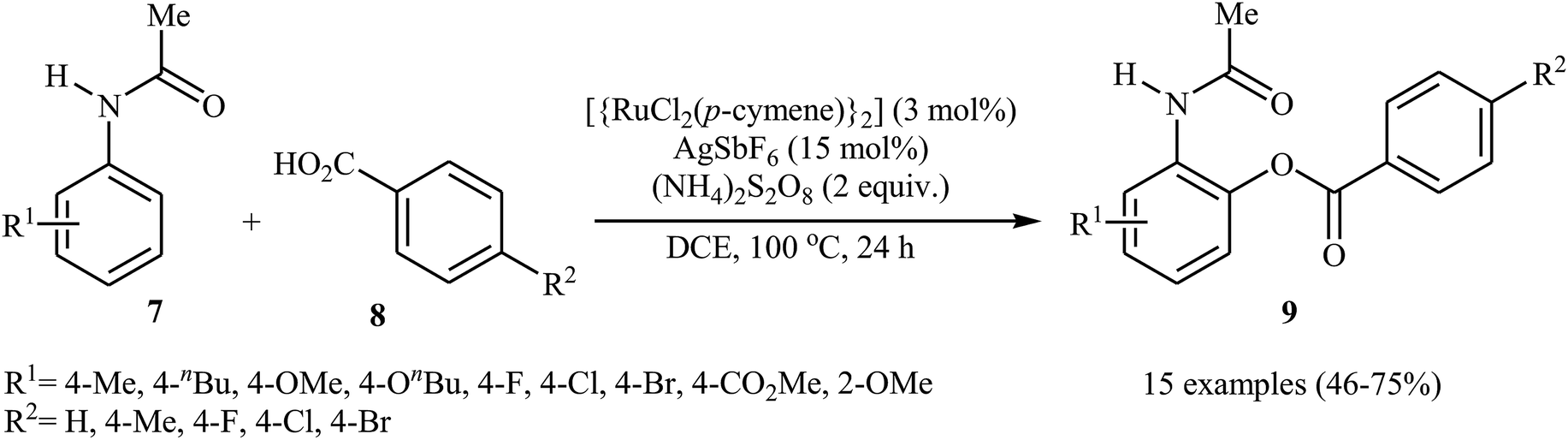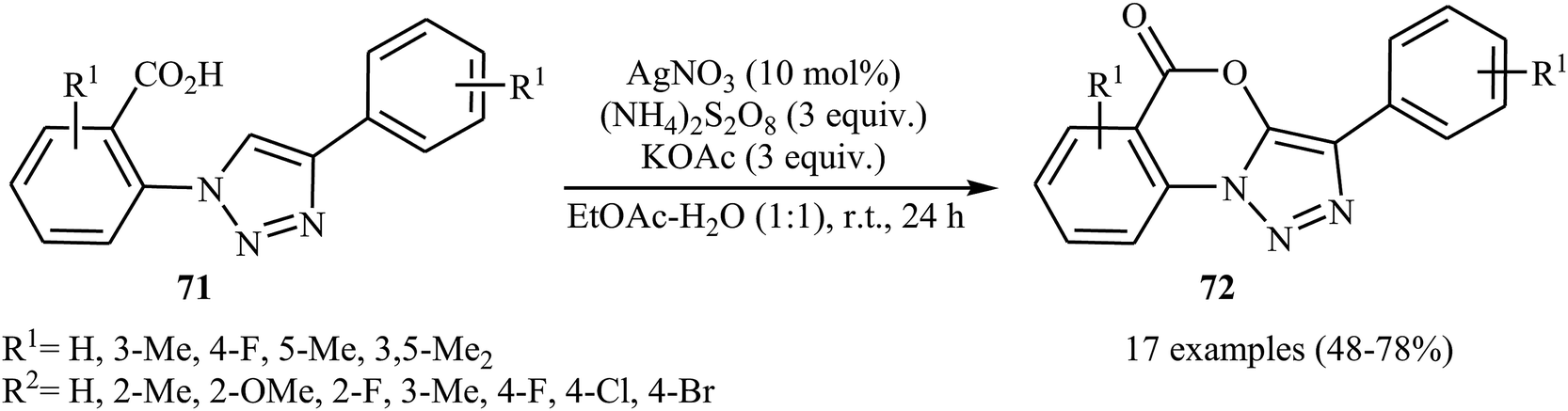DOI:
10.1039/C9RA01941C
(Review Article)
RSC Adv., 2019,
9, 17101-17118
Cross-dehydrogenative coupling reactions between arenes (C–H) and carboxylic acids (O–H): a straightforward and environmentally benign access to O-aryl esters
Received
13th March 2019
, Accepted 17th May 2019
First published on 31st May 2019
Abstract
Transition-metal catalyzed cross-dehydrogenative-coupling reactions encompass highly versatile and atom economical methods for the construction of various carbon–carbon and carbon–heteroatom bonds by combining two C(X)–H (X = heteroatom) bonds. Along this line, direct acyloxylation of C–H bonds with carboxylic acids has emerged as a powerful and green approach for the synthesis of structurally diverse esters. In this focus-review we will describe recent progress in direct esterification of aromatic C–H bonds with special emphasis on the mechanistic features of the reactions. Literature has been surveyed until the end of February 2019.
 Sattar Arshadi | Sattar Arshadi was born in Miandoab, west Azerbaijan province, Iran, in 1973. He received his B.Sc. degree in chemistry in the University of Kermanshah (1997) and his M.Sc. (2000) under supervision of Professor Issa Yavari and PhD (2004) in organic chemistry under supervision of Professor M. Z. Kassaee both in Tarbiat Modarres University, Tehran, Iran. Then, he went to the University of Payame Noor as an assistant professor (2005). His main research interest is computational chemistry (especially on rearrangements and interactions in nano systems), organic synthesis and spectral studies of organic compounds. |
 Alireza Banaei | Alireza Banaei was born in Ardabil, Iran, in 1966. He received his B.S. degree in Pure Chemistry from University of Shahid Beheshti Tehran, Iran, and his M.S. degree in organic chemistry from Tabriz University, Tabriz, Iran, in 1994 under the supervision of Prof. A. Shahrisa. He completed his PhD degree in 2000 under the supervision of Prof. A. Shahrisa. Now he is working at the University of Payame Noor as Associate Professor. His research interests include inorganic and organic synthesis, new methodologies in nano material synthesis, carbon dots. |
 Aazam Monfared | Aazam Monfared was born in Tehran, Iran, in 1965. She received her B.S. degree in Pure Chemistry from University of Shahid Beheshti, Tehran, Iran, and her M.S. degree in Organic chemistry from Shahid Beheshti University, Tehran, Iran, in 1991 under the supervision of Prof. A. Rustaiyan. She received her PhD degree in 1999 under the supervision of Prof. A. Rustaiyan in Shahid Beheshti University, Tehran, Iran. Now, she is working at Payame Noor University of Tehran as Associate Professor. Her research interests include organic synthesis, phytochemistry, drug synthesis, nano chemistry, methodologies and theoretical chemistry. |
 Saeideh Ebrahimiasl | Saeideh Ebrahimiasl was born in Tabriz, Iran, in 1973. She received her PhD degree in nanomaterials and nanotechnology from Institute of Advanced Materials, University Putra Malaysia in 2010. Professor Majid Monajjemi, Professor Wan Zin Wan Yunus, Professor Zulkarnain Zainal, Professor Mohd Zobir in physical chemistry, nanotechnology and electrochemistry were her most famous supervisors and examiners. Now she is head of Industrial nanotechnology Research Center in Tabriz and academic member of Islamic Azad University of Ahar. Her research interests include synthesis and application of nanomaterials in different sciences. |
 Akram Hosseinian | Akram Hosseinian was born in Ahar, Iran, in 1973. She received her B.S. degree in Pure Chemistry from University of Tehran, Iran, and her M.S. degree in inorganic chemistry from Tarbiat Modares University, Tehran, Iran, in 2000 under the supervision of Prof. A. R. Mahjoub. She completed her PhD degree in 2007 under the supervision of Prof. A. R. Mahjoub. Now she is working at University of Tehran as Associate Professor. Her research interests include inorganic and organic synthesis, new methodologies in nano material synthesis. |
1. Introduction
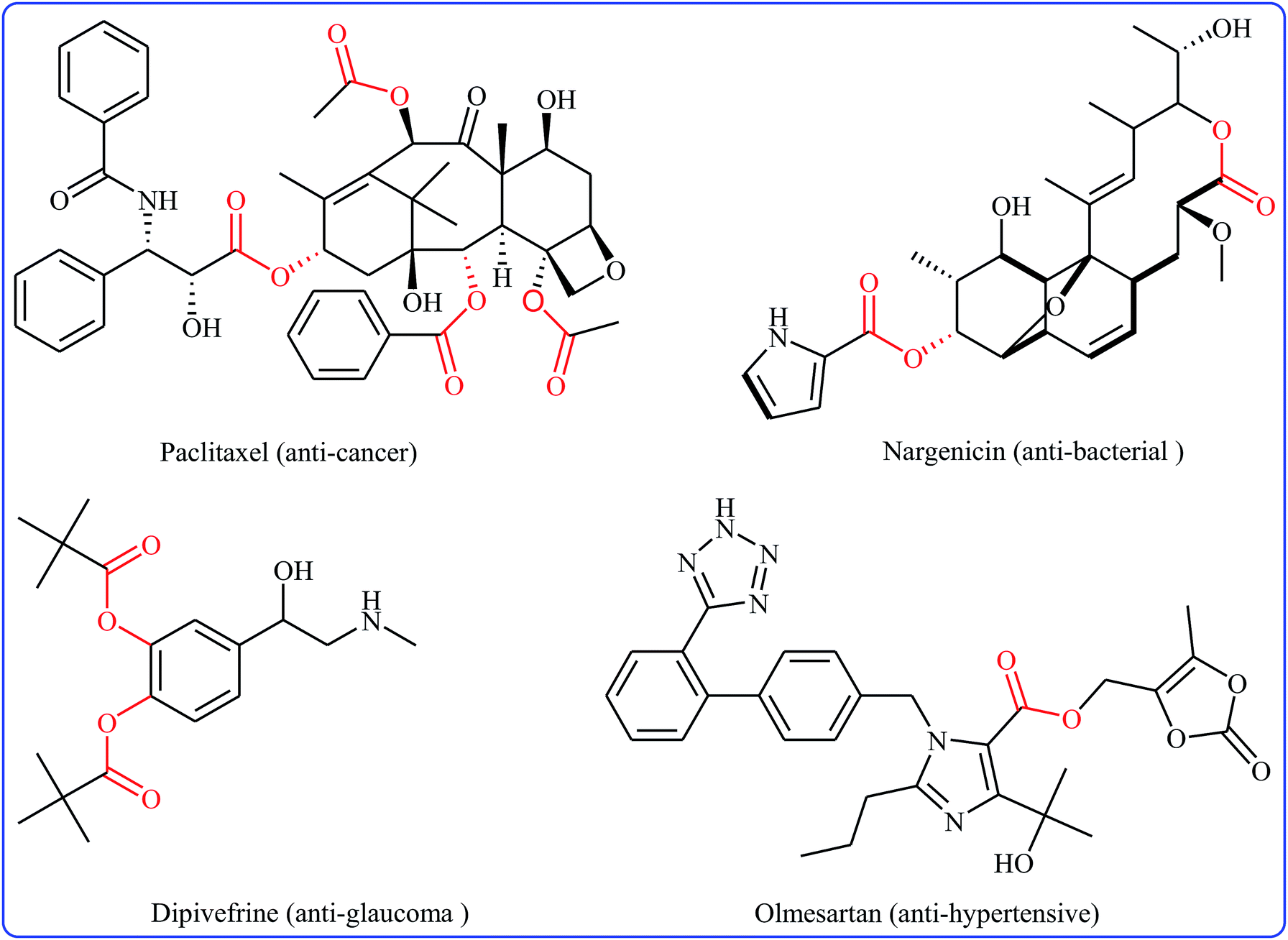
Without the slightest doubt, esters are one the most significant classes of carbonyl compounds, featuring in a large number of natural products,1 synthetically useful intermediates,2 and medicinally relevant compounds (Fig. 1).3 Molecules containing this prominent medical moiety have showed widespread biological activities such as anti-cancer,4 anti-HIV,5 anti-asthma,6 anti-glaucoma,7 anticonvulsant,8 and antibiotic9 activities. Furthermore, this motif is widely distributed in agrochemicals like malathion, permethrin, clodinafop propargyl, and many more.10 In light of the above-mentioned benefits, the development of novel, practical and economical approaches for the high yielding synthesis of the titled compounds, from simple, easily accessible and inexpensive starting materials is always interesting.
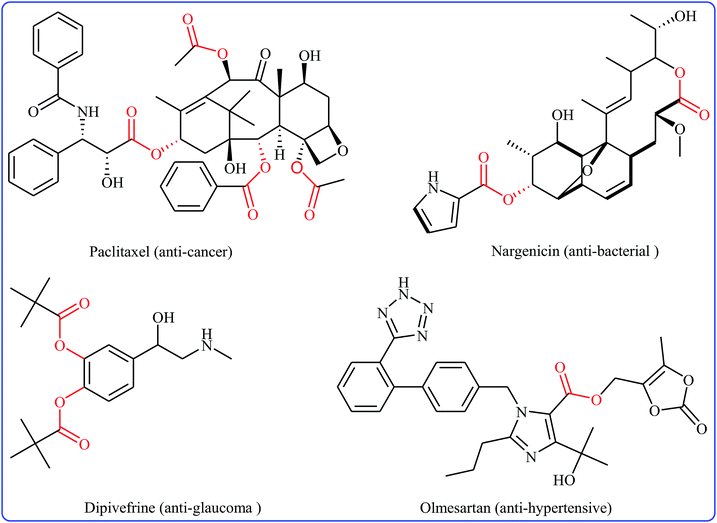 |
| | Fig. 1 Some ester-containing commercialized drugs. | |
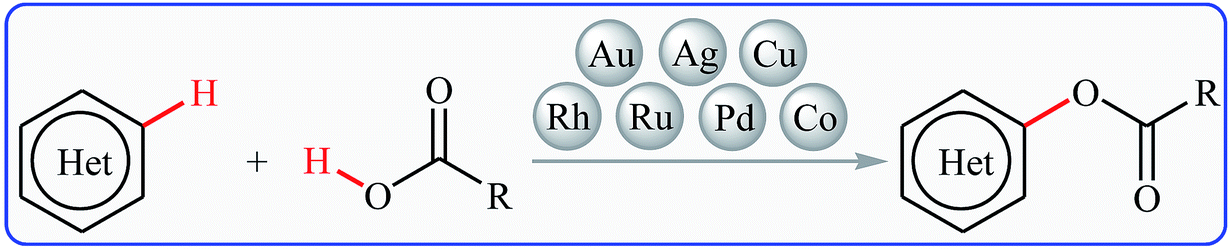
Cross-dehydrogenative-coupling reactions, which combine two unmodified C(X)–H (X = heteroatom) bonds for the construction of new C(X)–C(X) bonds, represent one of the most versatile and atom-economical methods in organic synthesis that can avoid the use of pre-functionalized substrates to make synthetic schemes shorter and cleaner than traditional cross-coupling reactions.11–13 In this regard, direct esterification of C–H bonds with carboxylic acids have got ever-increasing attention because of the highly atom-economical and environmentally friendly characters. In connection with our continuing reviews on the cross-coupling reactions14 and new methodologies in organic synthesis,15 herein, we will describe the recent advances in the cross-dehydrogenative coupling reactions of C(aryl)–H bonds with carboxylic acids (Fig. 2), with special emphasis on the mechanistic features of the reactions. It should be noted that we have not discussed cross-dehydrogenative coupling reactions of sp3-hybridized C–H bonds with carboxylic acids16 and direct acetoxylation of aromatic C–H bonds,17 since they have recently been described in another publications.
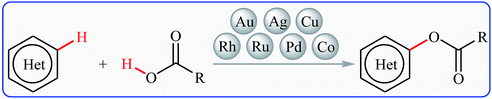 |
| | Fig. 2 Direct esterification of aromatic C–H bonds with carboxylic acids. | |
2. Rhodium-catalyzed reactions
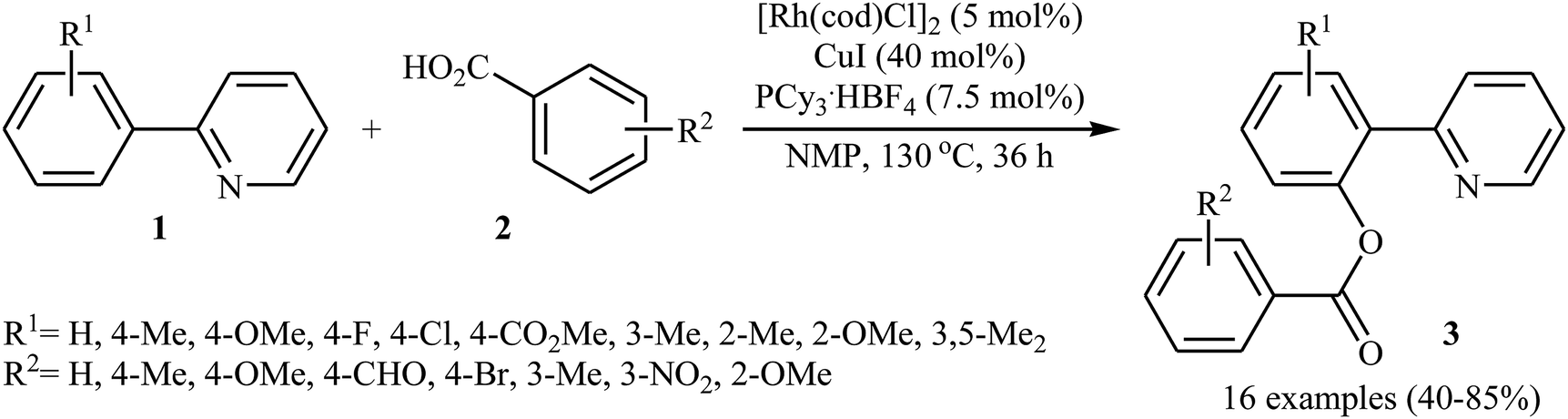
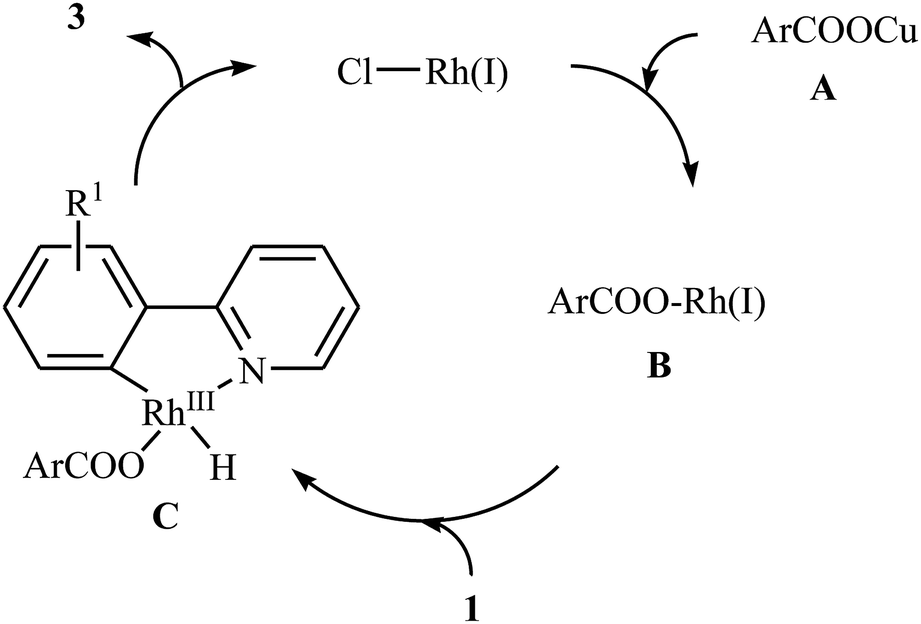
In 2009, Cheng and co-workers reported the first Rh-catalyzed direct acyloxylation of aromatic C–H bonds using pyridyl as the ortho-selective directing group.18 Screening of various rhodium catalysts such as Rh(CO)2(C5H7O2), Rh(PPh3)3Cl, [Rh(cod)Cl]2; ligands like binap, PPh3, P(1-Nap)3, PCy3 + HBF4, iPr2N-PPh2; and Cu source namely CuCl, CuBr, CuI, Cu2O, and Cu(OAc)2 led to [Rh(cod)Cl]2/PCy3 + HBF4/CuI combination as the most suitable catalytic system for this transformation. Solvent has a dramatic impact on this process and among the common solvents (e.g., toluene, xylene, 1,4-dioxane, NMP, DMF); N-methyl-2-pyrrolidone (NMP) was the most effective solvent. Under optimized conditions, various 2-arylpyridines 1 underwent ortho-selective mono-acyloxylation with aromatic carboxylic acids 2 to give the corresponding esters 3 in moderate to good yields (Scheme 1). Of note, alkyl and alkenyl carboxylic acids are also suitable substrates leading to the expected esters in almost moderate yields. It should be mentioned that the little intramolecular kinetic isotope effect of the reaction (kH/kD = 51/49) indicated that the break of the C–H(D) bond is not the rate-determining step in this transformation. The authors illustrated a preliminary mechanism for this reaction as shown in Scheme 2. Initially, the reaction of rhodium(I) species with the copper(I) carboxylate A (generated from carboxylic acid 2 and CuI) afforded intermediate B. Afterward, the ortho-C–H bond of 2-arylpyridine 1 undergo oxidative addition with rhodium(I) species B to form Rh(III) intermediate C, which produce the desired product 3 and regenerate the rhodium(I) catalyst by reductive elimination.
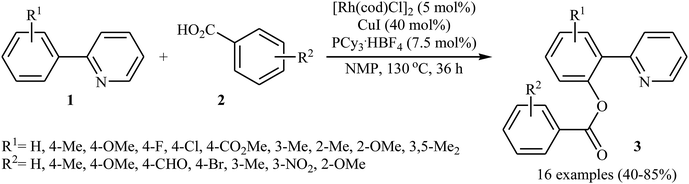 |
| | Scheme 1 Rh-catalyzed direct benzoxylation of 2-arylpyridines 1 with benzoic acids 2. | |
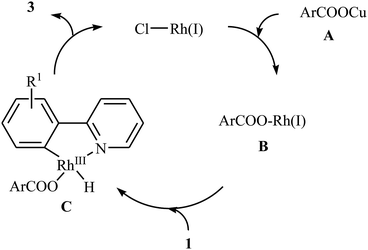 |
| | Scheme 2 Mechanistic proposal for the formation of ortho-acyloxylated 2-arylpyridines 3. | |
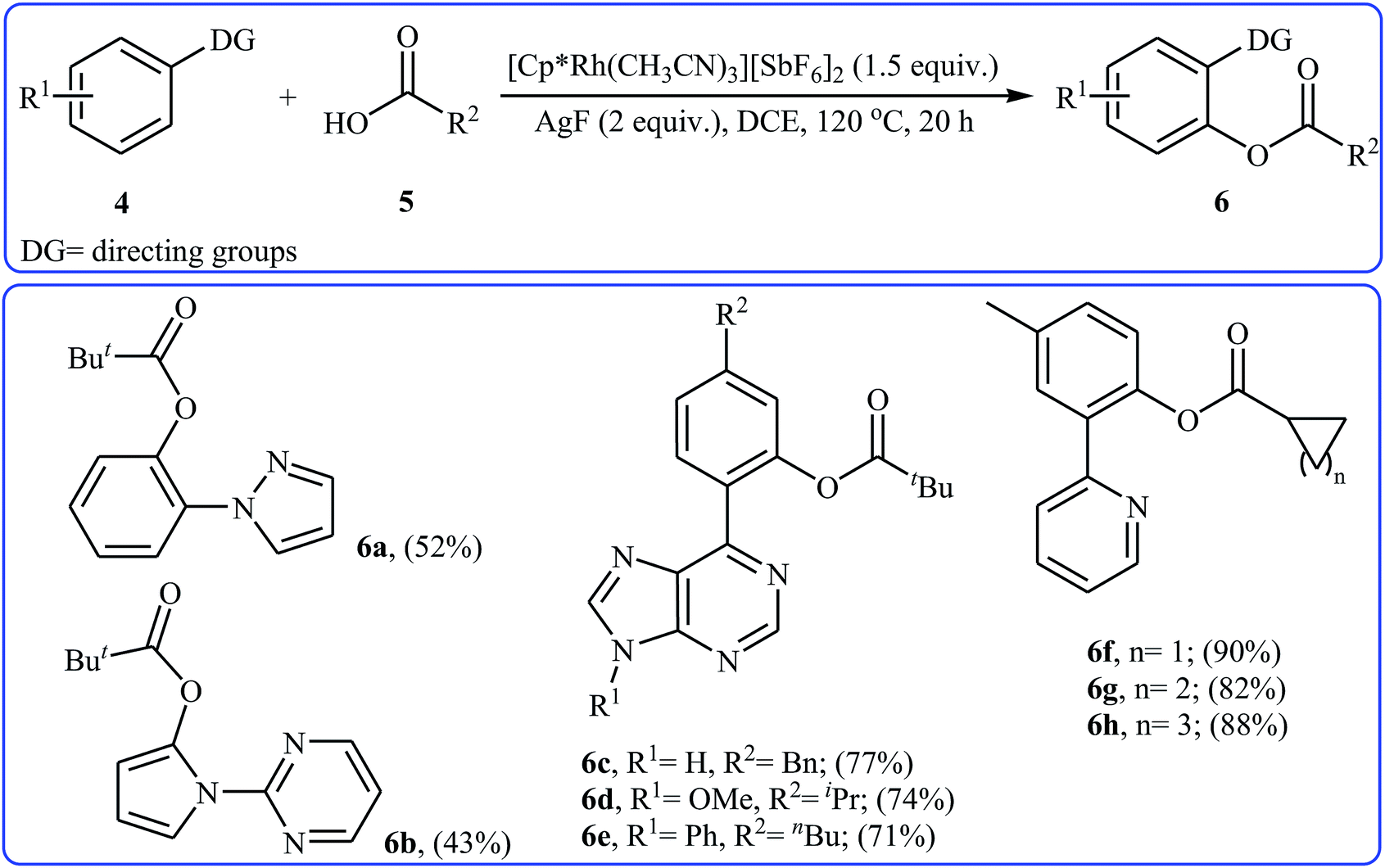
Eight years later, Xu and Li along with their co-workers illustrated a similar dehydrogenative cross-coupling of arene substrates 4 bearing coordinating directing groups (pyridyl, pyrimidinyl, pyrazolyl, oxazolyl, purinyl and etc.) with a range of (hetero)aromatic and aliphatic carboxylic acids 5 for the synthesis O-aromatic esters 6 using [Cp*Rh(CH3CN)3][SbF6]2 as a commercially available catalyst and AgF as an oxidant (Scheme 3).19 Noteworthy, the presence of both catalyst and oxidant were crucial to the success of the reaction and so no product was observed in the lack of any of them. This Rh-catalyzed system gave moderate to excellent yields (43–92% for 83 examples) and was tolerant to various important functional groups (OMe, F, Cl, Br, I, CF3, CHO, CO2Et, COMe, CN, NO2). The reaction was even successfully conducted on a gram scale. The synthetic utility of this transformation was further demonstrated by the esterification of bioactive carboxylic acids such as fenbufen, dehydrocholic acid, and glycyrrhetinic acid.
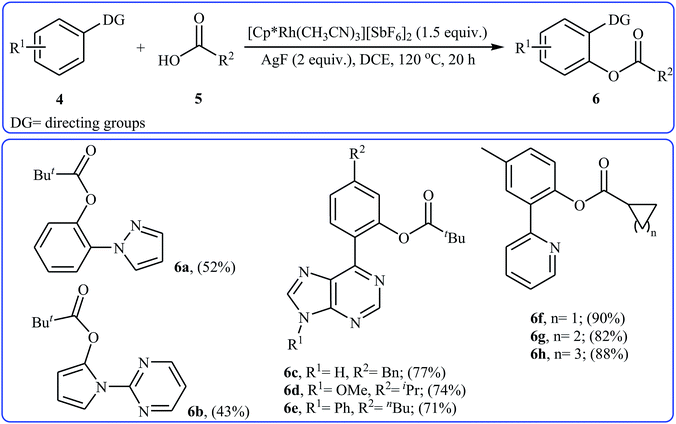 |
| | Scheme 3 Some reported examples on the Rh-catalyzed ortho-selective acyloxylation of arene substrates 4 bearing coordinating directing groups with aliphatic carboxylic acids 5 reported by Xu and Li. | |
3. Ruthenium-catalyzed reactions
In 2013, Padala and Jeganmohan revealed for the first time the usefulness of ruthenium catalysts for the direct acyloxylation of arene C–H bonds with carboxylic acids.20 Thus, in the presence of [{RuCl2(p-cymene)}2]/AgSbF6/(NH4)2S2O8 combination as a catalytic system in DCE at 100 °C, dehydrogenative cross-coupling of substituted acetanilides 7 with aromatic carboxylic acids 8 furnished the corresponding ortho-benzoxylated acetanilides 9 in moderate to good yields (Scheme 4). Both electron-rich and electron-deficient acetanilide derivatives participate effectively in this highly monoselective transformation. In the case of carboxylic acids, substrates bearing both moderate electron-donating and electron-withdrawing groups worked well in the reaction. However, strong electron-donating as well as electron-withdrawing substituents on the aromatic acids were incompatible in the reaction.
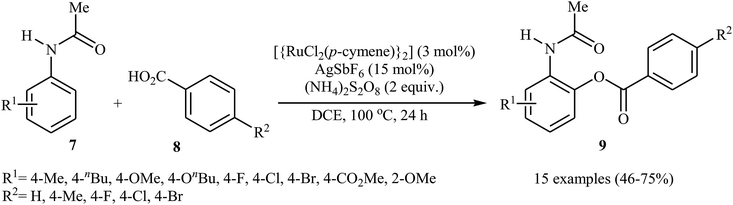 |
| | Scheme 4 Ru-catalyzed synthesis of ortho-benzoxylated acetanilides 9 from acetanilides 7 and benzoic acids 8. | |
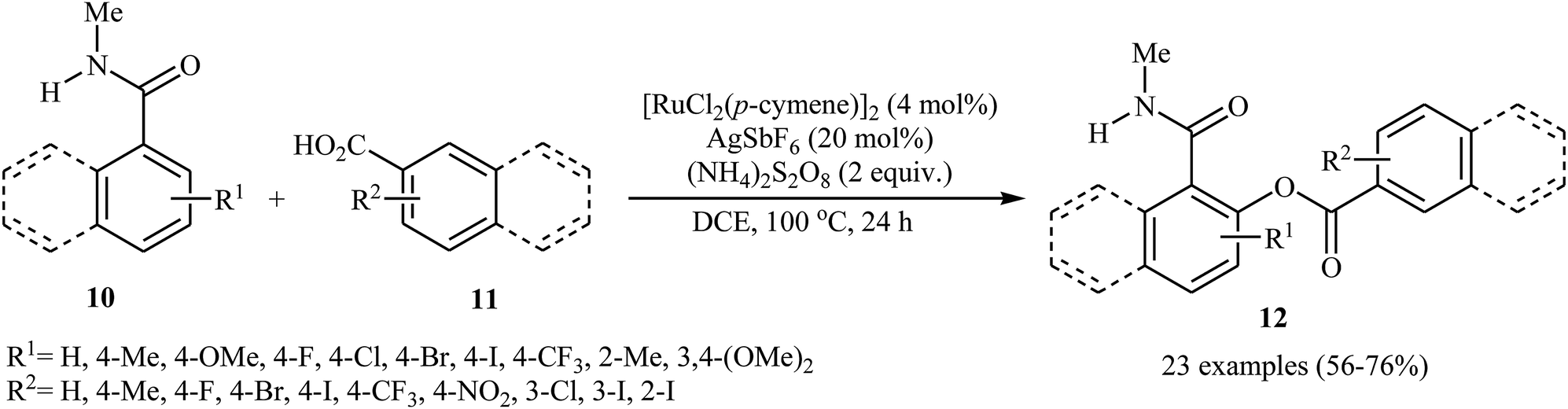
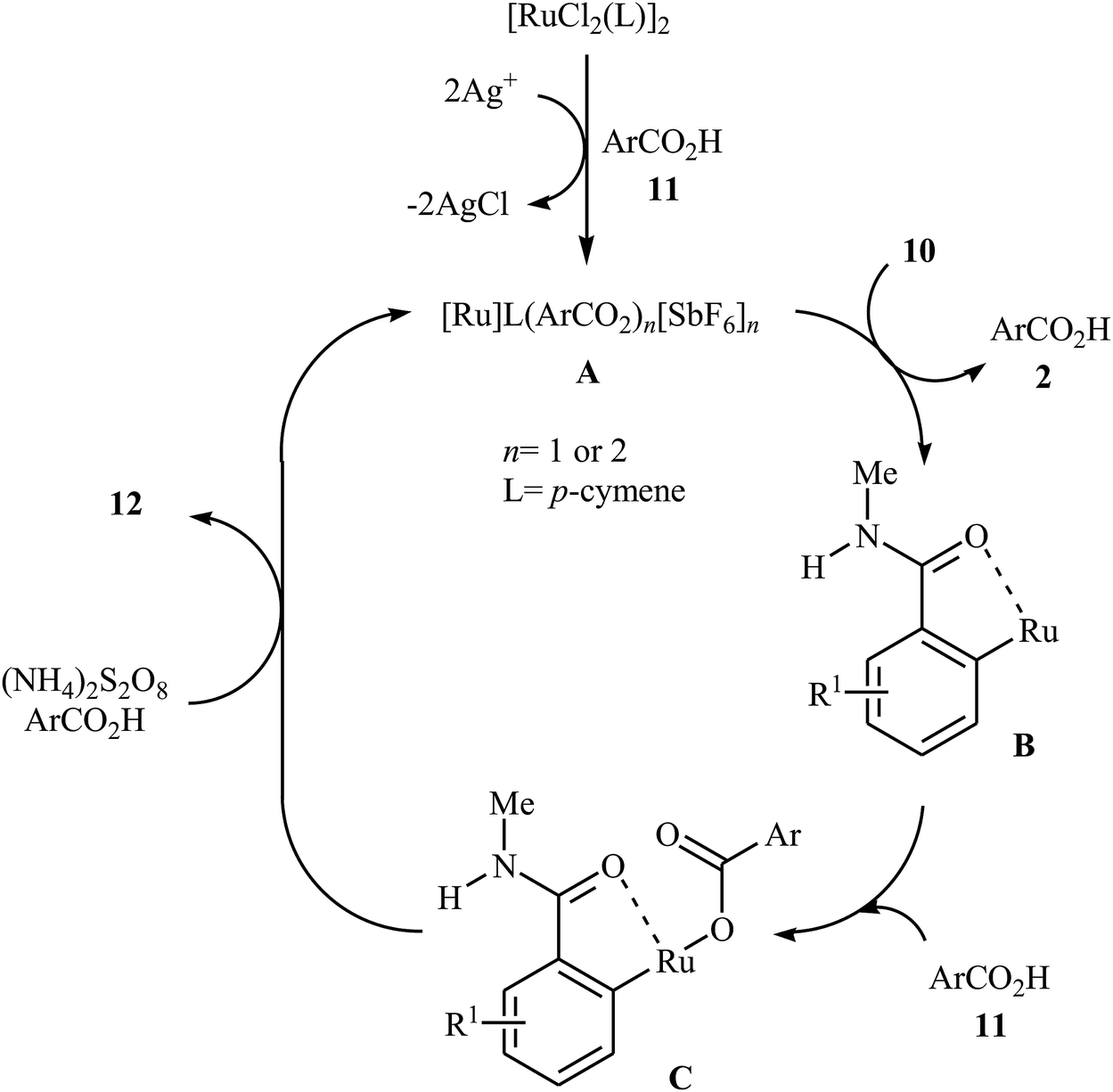
Later on, an extension to N-alkyl benzamides as coupling partners was reported by the same group also relying on the use of Ru(II) catalyst.21 Upon treatment with 4 mol% of [{RuCl2(p-cymene)}2], 20 mol% of AgSbF6, and 2.0 equiv. of (NH4)2S2O8, various N-methyl benzamide derivatives 10 underwent regioselective ortho-benzoxylation with aromatic carboxylic acids 11 to give ortho-benzoxylated N-alkyl benzamides 12 in moderate to good yields (Scheme 5). The mechanism proposed for this transformation consists of the following key steps (Scheme 6): (i) initial formation of cationic ruthenium carboxylate complex A via removing of the chloride ligand from the [{RuCl2(p-cymene)}2] complex by silver salt; (ii) coordination of the carbonyl oxygen of the benzamide 10 to the cationic species A followed by ortho-metalation to afford the five-membered metalacycle intermediate B; (iii) coupling of acid 11 into the ruthenacycle B to give the intermediate C; and (iv) reductive elimination of intermediate C to form the final product 12. Besides N-alkyl benzamides, N,N-dialkyl benzamides proved also applicable in this reaction. Interestingly, the products were successfully converted into ortho-benzoxylated benzaldehydes by employing Schwartz reagent (Cp2ZrHCl) at room temperature.22 Subsequently, the group of Ackermann extended this approach to the ortho-C–H-benzoxylation of phenols masked by removable pyridyl and pyrimidyl directing groups using Jeganmohan's standard condition giving good yields in the 55–78% range.23
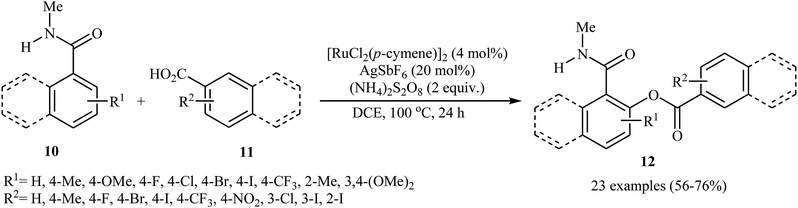 |
| | Scheme 5 Cross-dehydrogenative coupling of N-methyl benzamides 10 with aromatic carboxylic acids 11 catalyzed by [{RuCl2(p-cymene)}2]. | |
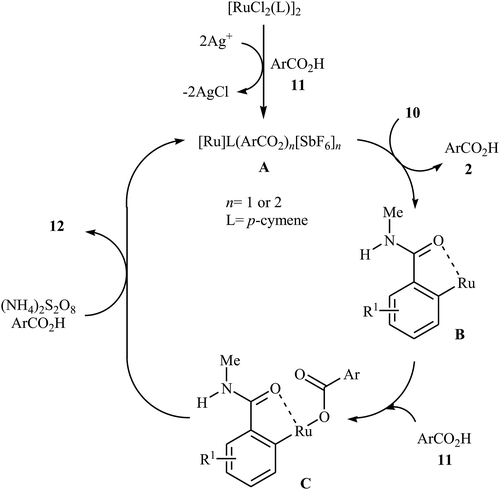 |
| | Scheme 6 Suggested mechanism for the formation of ortho-benzoxylated N-alkyl benzamides 12. | |
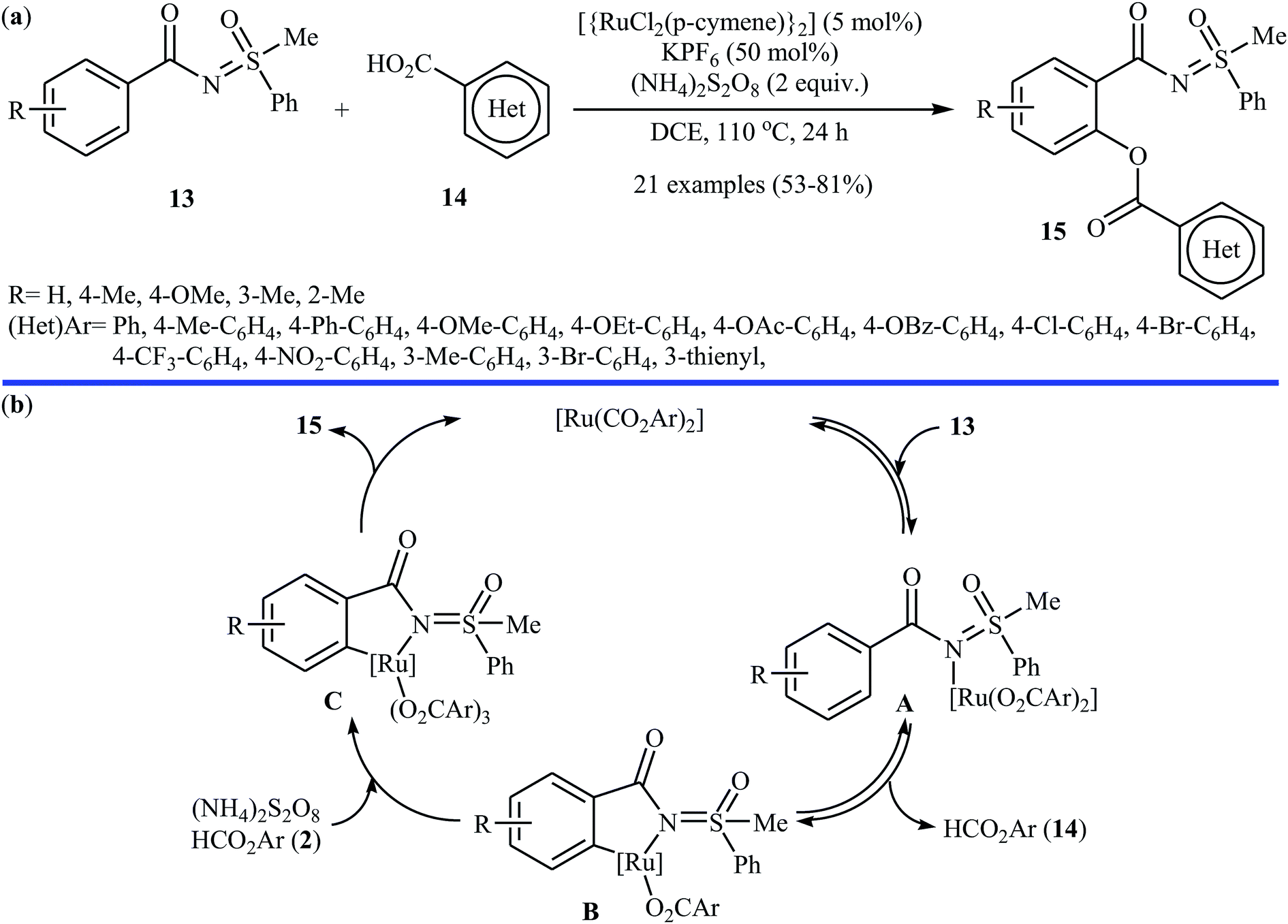
Following these works, the Ackermann group further expanded the Ru-catalyzed C–H benzoxylation to sulfoximines.24 Under optimized conditions ([{RuCl2(p-cymene)}2], KPF6, (NH4)2S2O8, DCE, 110 °C), various functionalized sulfoximines 13 coupled with diverse (hetero)aromatic carboxylic acids 14 to produce their corresponding ortho-benzoxylated sulfoximines 15 in moderate to good yields (Scheme 7a). Competition experiments between an electron-poor sulfoximine and electron-rich sulfoximine indicated that the latter reacted preferentially. Likewise, electron-rich aromatic carboxylic acids proved to be inherently more reactive than their electron-deficient counterparts. Of note, the benzoxylated sulfoximines can be easily converted into the corresponding salicylic acids under acidic conditions. Based on a series of control experiments, the authors suggested that this dehydrogenative cross-coupling reaction proceeds through a coordination/C–H metalation/oxidation/reductive elimination sequential process (Scheme 7b). Miura and colleague also used the commercially available [{RuCl2(p-cymene)}2] as a catalyst along with Ag2CO3 as the oxidant for the regioselective C–H acetoxylation of carbazole and indole frameworks with acetic acid in chlorobenzene at 140 °C.25
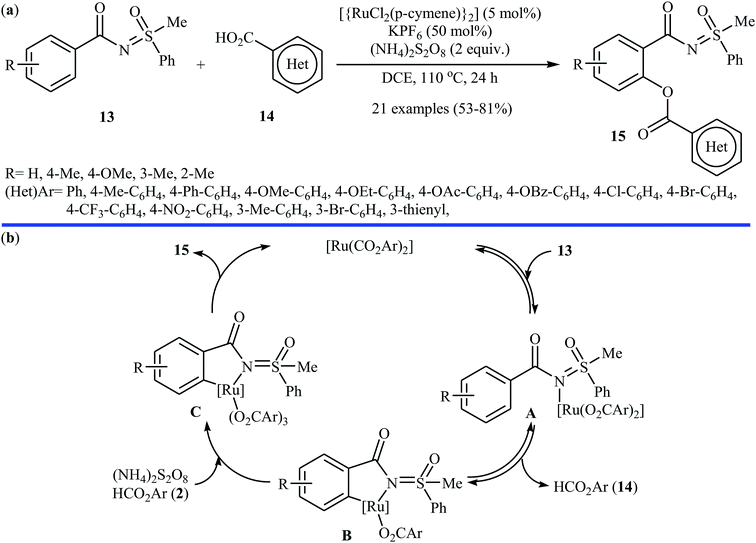 |
| | Scheme 7 (a) Ackermann's synthesis of ortho-benzoxylated sulfoximines 15; (b) plausible mechanism for the formation of ortho-benzoxylated sulfoximines 15. | |
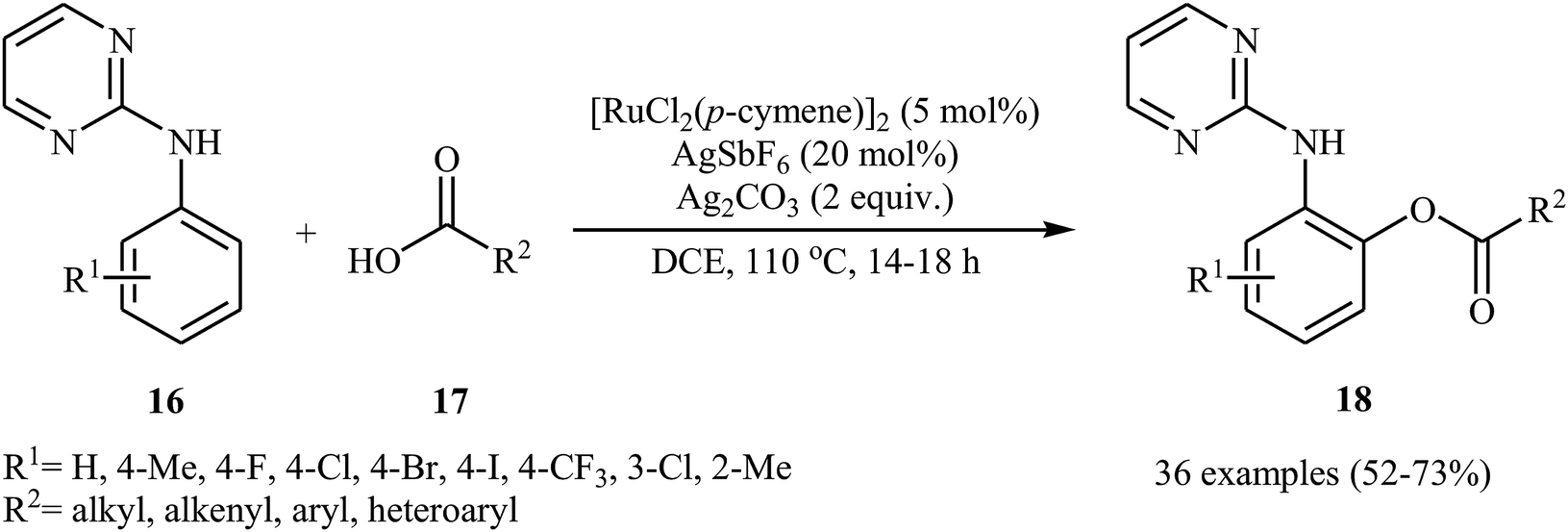
In a related investigation, Punniyamurthy's research team reported on ruthenium-catalyzed C–H oxygenation of N-aryl-2-pyrimidines 16 with carboxylic acids 17 in the presence of AgSbF6 as an additive and Ag2CO3 as an oxidant (Scheme 8).26 Various alkyl, aryl, heteroaryl, and α,β-unsaturated carboxylic acids, and a library of functionalized N-aryl-2-pyrimidines were used to establish the general applicability of the method. The results indicated that the efficiency of this reaction strongly depended on the electronic character of the substituents on the aromatic ring of N-aryl-2-pyrimidines, clearly in favour of electron-donating ones. The reaction can also be scaled up (3 mmol) to produce the desired ester in good yield. The authors showed that the ester products 18 were converted into the corresponding 2-aminophenols upon hydrolysis with aqueous HCl under microwave irradiation.
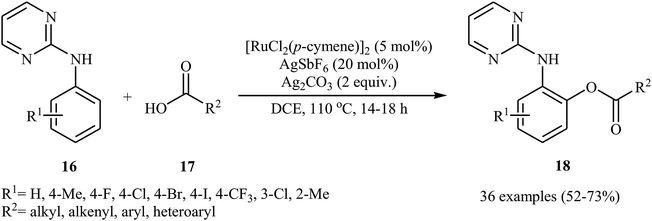 |
| | Scheme 8 Ru-catalyzed ortho-directed aromatic amines 16 with carboxylic acids 17. | |
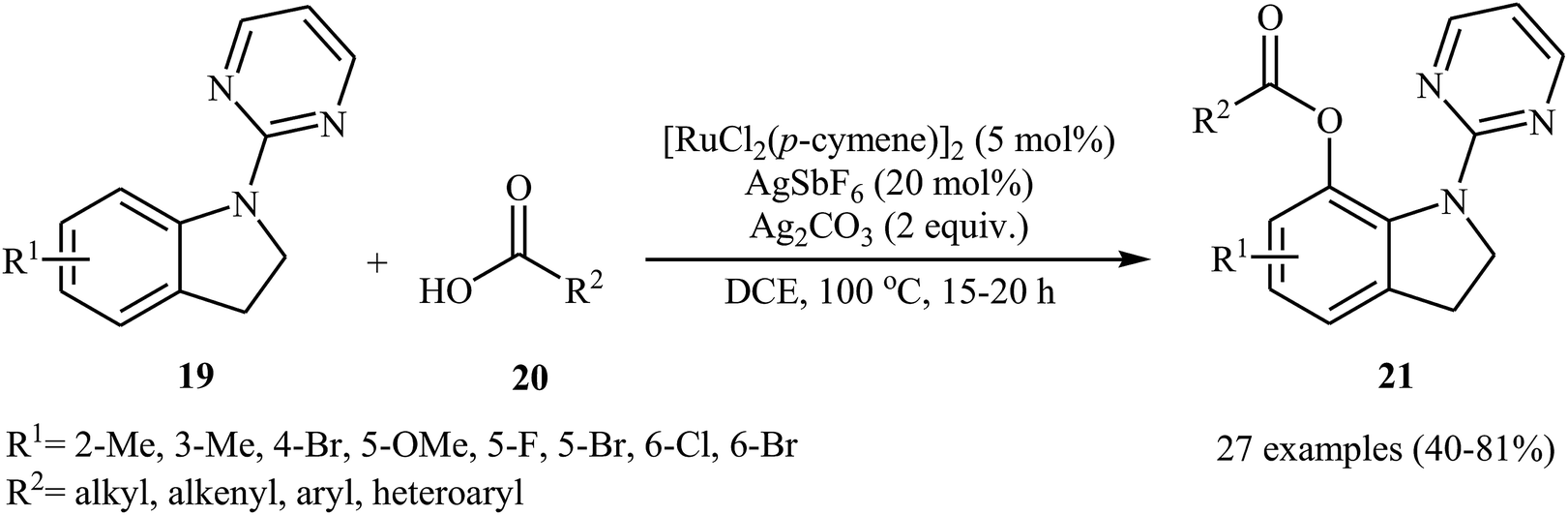
Recently, the same group reported that the system could also be applied to site-selective acyloxylation of indoline derivatives.27 Thus, a variety of N-pyrimidyl indolines 19 were coupled with carboxylic acids 20 to give the desired C7-acyloxylated indoline products 21 in modestly to high yields (Scheme 9). Various of aliphatic, aromatic, heteroaromatic and α,β-unsaturated carboxylic acids were found compatible with this mono-selective C–H transformation. However, picolinic and isonicotinic acids failed to participate in this reaction. The authors showed the synthetic application of synthesized 7-acyloxylated indolines for the high yielding preparation of 7-hydroxy indole scaffolds by heating in the basic medium. They also found that oxidation of indoline products in the presence of DDQ afforded the corresponding indoles in high yields.
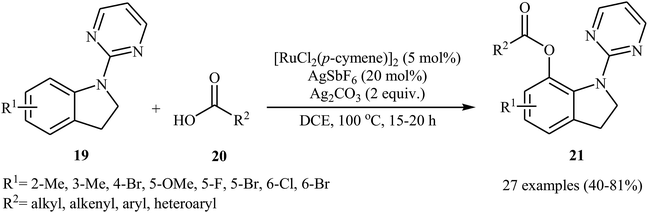 |
| | Scheme 9 Ru-catalyzed site-selective acyloxylation of indolines 19 with carboxylic acids 20. | |
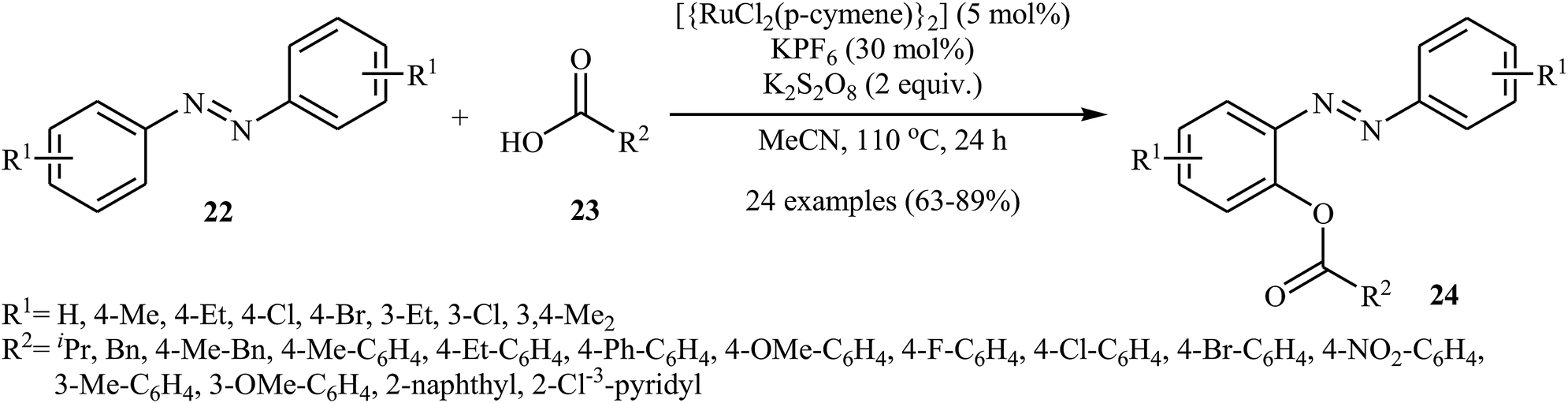
Very recently, Kianmehr and Bahrami-Nasab employed standard [{RuCl2(p-cymene)}2] catalyst, in combination with K2S2O8 and KPF6 for regioselective ortho-acyloxylation of azoarenes 22 with carboxylic acids 23.28 These reactions were performed in MeCN at 110 °C and afforded the corresponding ortho-acyloxylated azoarene derivatives 24 in moderate to high yields (Scheme 10). Notably, other ruthenium catalysts such as RuCl2(PPh3)2 and RuCl3 + H2O were also found to catalyze this dehydrogenative cross-coupling reaction, albeit at considerably lower efficiencies. The proposed mechanism for this transformation is similar to that one depicted in Scheme 7b.
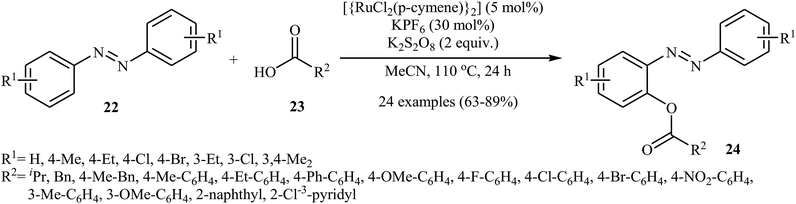 |
| | Scheme 10 ortho-Selective acyloxylation of azoarenes 22 with carboxylic acids 23 reported by Kianmehr. | |
4. Palladium-catalyzed reactions
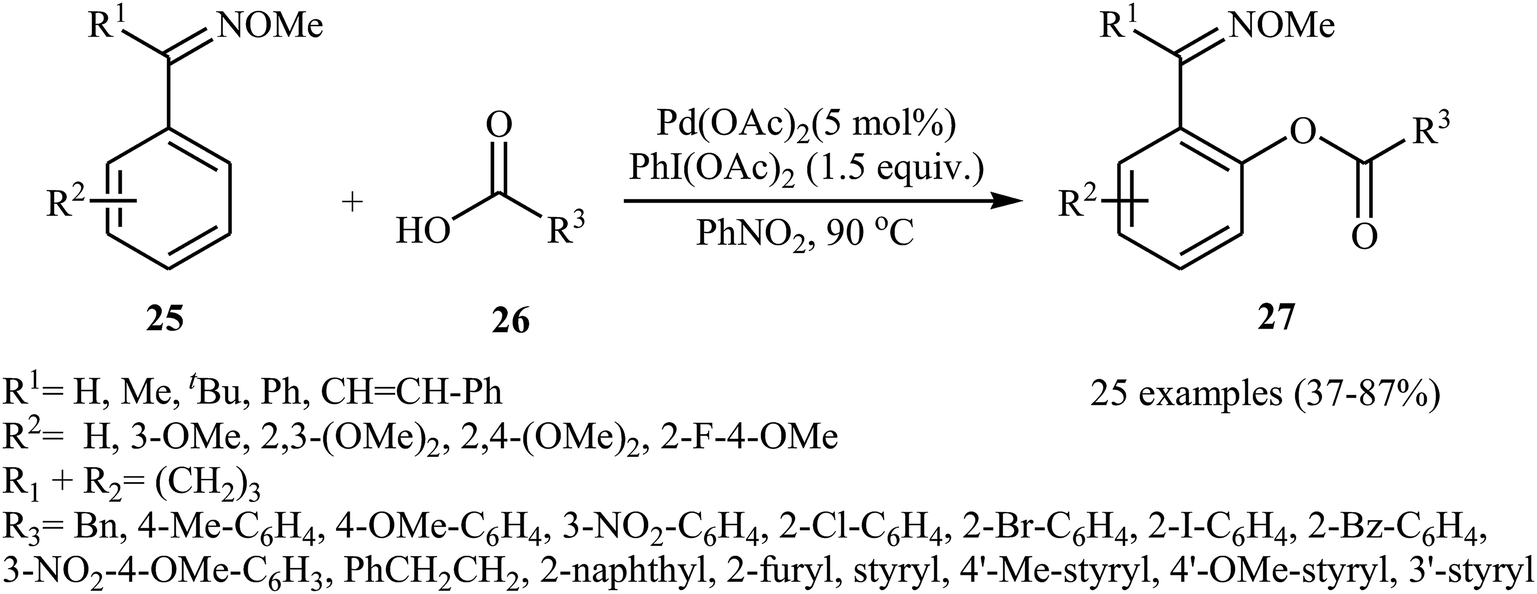
In recent years, palladium salts have been extensively applied as effective catalysts for the cross-dehydrogenative coupling reactions between (hetero)arenes and carboxylic acids. One of the first palladium-catalyzed direct acyloxylation of aromatic C–H bonds with carboxylic acids was published by Shi and collaborates in 2012.29 Here, various O-methyl aryloximes 25 were selectively acyloxylated at the 2-position with carboxylic acids 26 using 10 mol% of Pd(OAc)2 as a catalyst and 1.5 equiv. of PhI(OAc)2 an oxidant in PhNO2. The target esters 27 were obtained with yield ranging from 37% to 90%. As shown in Scheme 11, the process had a broad tolerance toward a wide range of important functional groups and both aliphatic and aromatic carboxylic acids were compatible with the reaction conditions. In addition, the reaction was also successfully extended to the synthesis of biologically important 6H-benzo[c]chromen-6-one derivatives via a sequential Pd-catalyzed double activations of adjacent sp2 C–H bond.
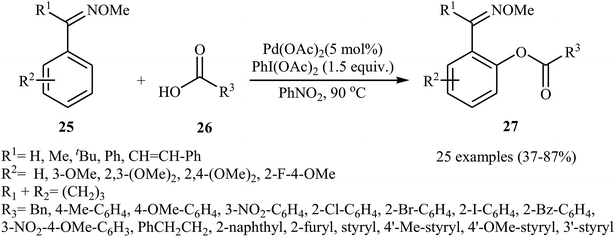 |
| | Scheme 11 Pd-catalyzed cross-dehydrogenative coupling of O-methyl aryloximes 25 with carboxylic acids 26. | |
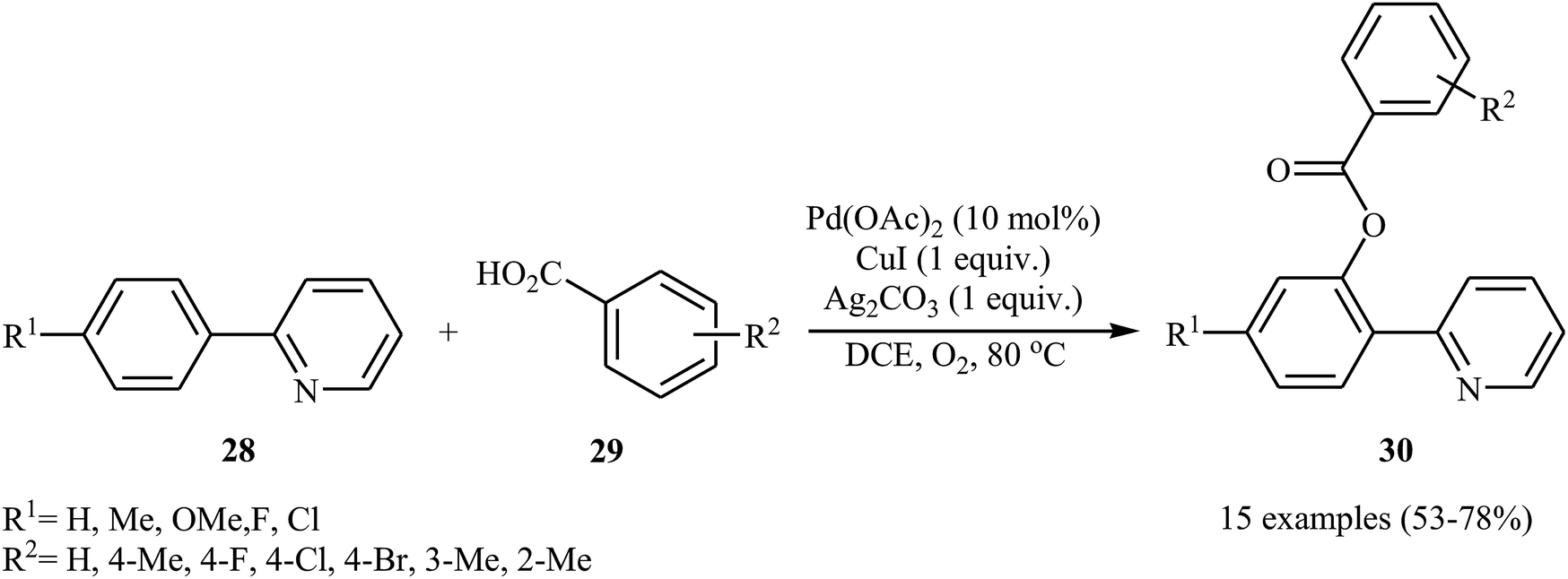
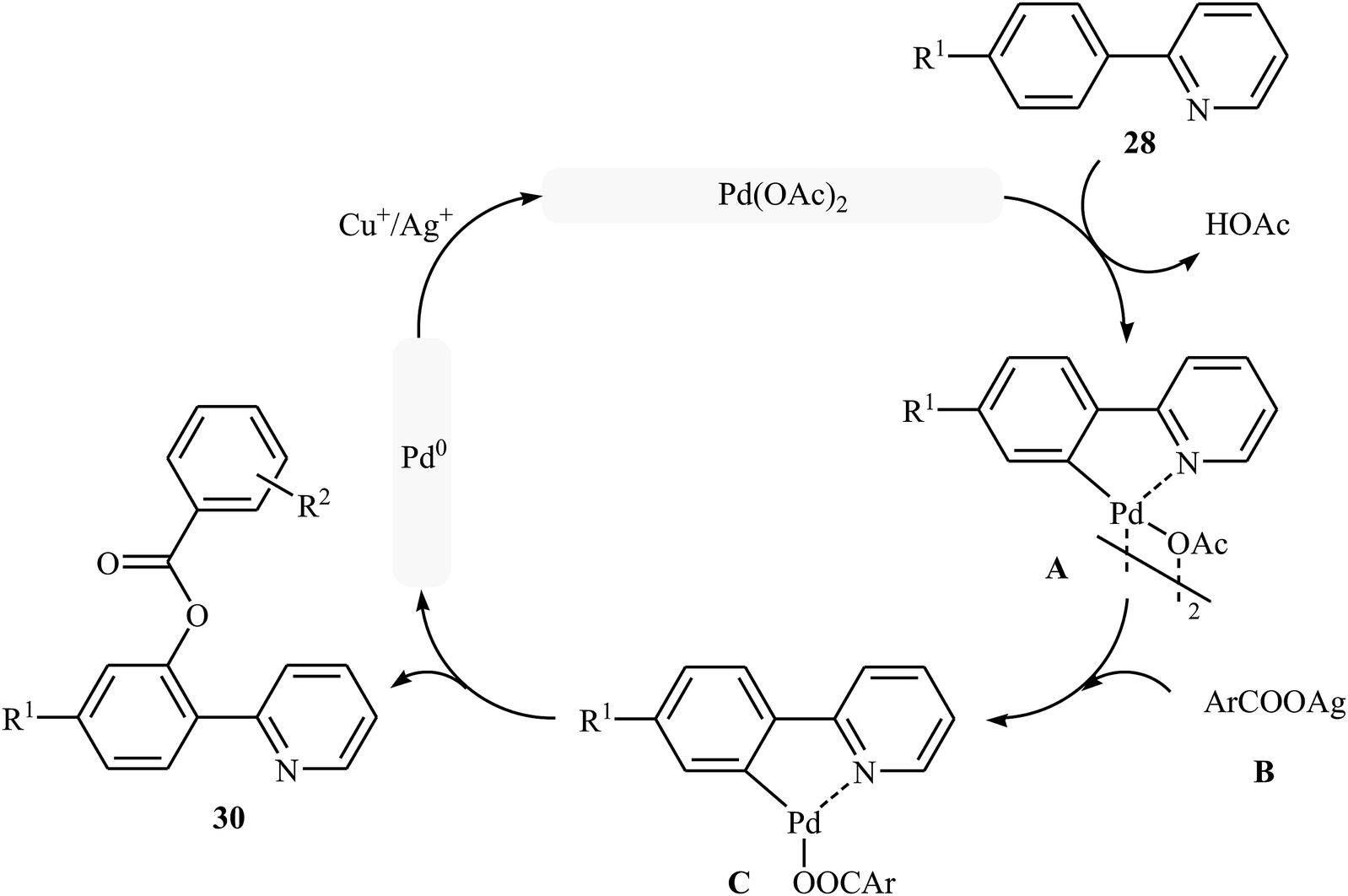
Shortly afterwards, Zhong and co-workers showed that the reaction of 2-arylpyridines 28 with aromatic carboxylic acids 29 in the presence of Pd(OAc)2/CuI/Ag2CO3 combination as a catalytic system in DCE under the O2 atmosphere afforded the corresponding mono-benzoxylated arylpyridines 30 in moderate to good yields (Scheme 12).30 Other palladium catalysts such as PdCl2 and Pd2(dba)3 were also tested in this reaction. However, only poor yield of isolated product was observed. Both electron-rich and electron-poor 2-arylpyridines were good coupling partners in this regioselective protocol, although carboxylic acids containing strongly electron-withdrawing substituents (e.g., NO2) were not tolerated. The following mechanism was proposed by the authors for this transformation (Scheme 13): initially, Pd(OAc)2 reacts with 2-arylpyridine 28 by removing HOAc to form a cyclopalladated intermediate A. Subsequently, the reaction of this intermediate with in situ generated silver carboxylate B (from the reaction of carboxylic acid 29 with Ag2CO3) gives the ligand exchanged product C. Finally, the reductive elimination of intermediate C affords the benzoxylated product 30 and Pd(0) species that is reoxidized to the Pd(II) catalyst by Ag(I) and Cu(I) salts.
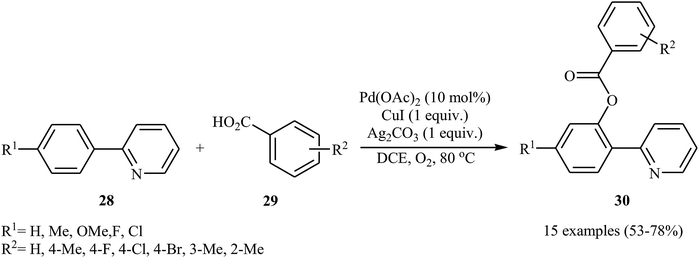 |
| | Scheme 12 Coupling of 2-arylpyridines 28 with carboxylic acids 29 catalyzed by Pd(OAc)2. | |
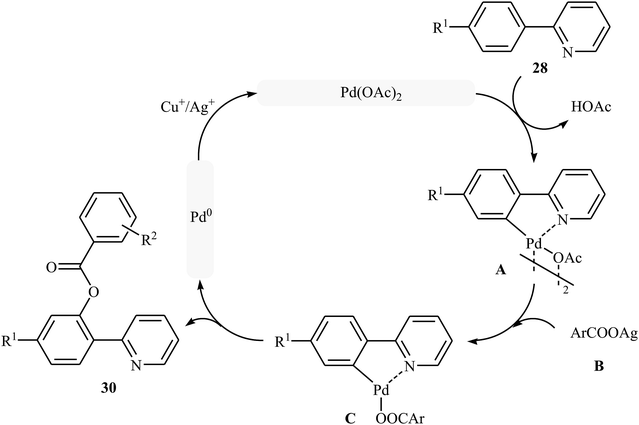 |
| | Scheme 13 Plausible mechanism for the Pd-catalyzed acyloxylation of 2-arylpyridines 28 with carboxylic acids 29. | |
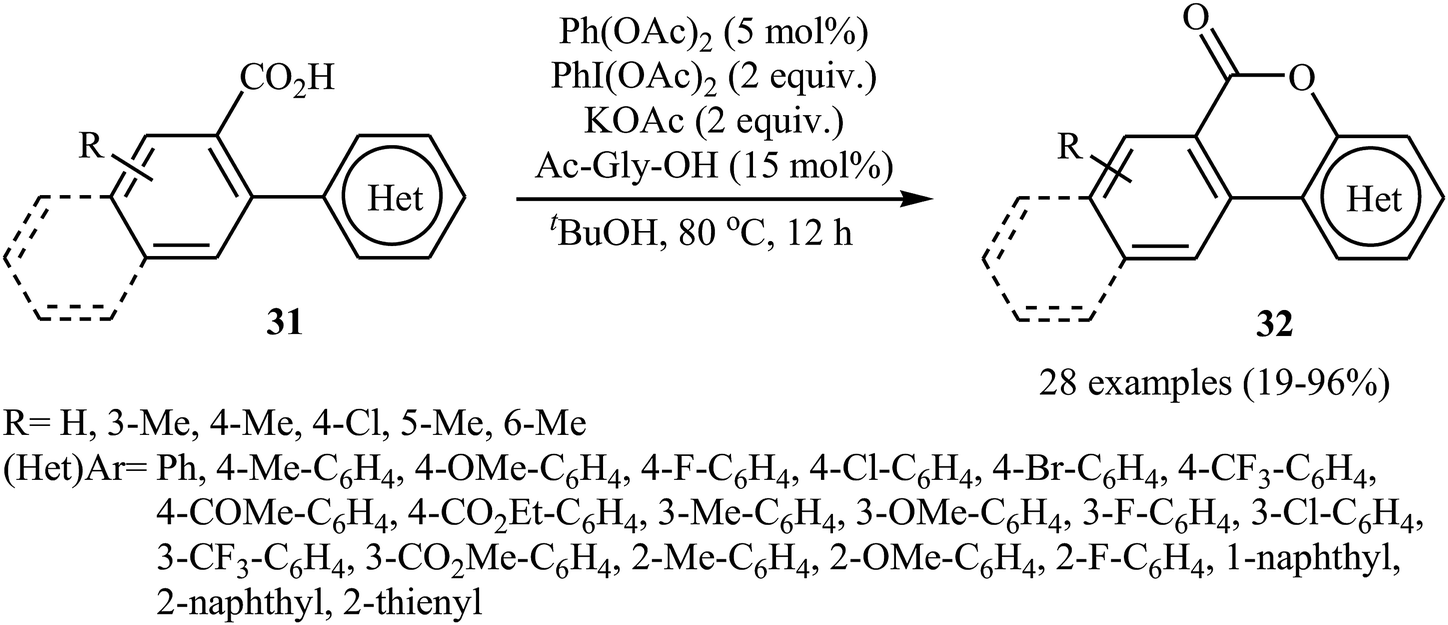
The Pd-catalyzed cross-dehydrogenative coupling of aryl C–H bonds with carboxylic acids in an intramolecular version was reported by Wang and co-workers in 2013.31 The combination of Pd(OAc)2/PhI(OAc)2/KOAc/Ac-Gly-OH was used as a catalytic system to prepare a series of biaryl lactones 32 from the corresponding 2-aryl carboxylic acids 31, according to Scheme 14.
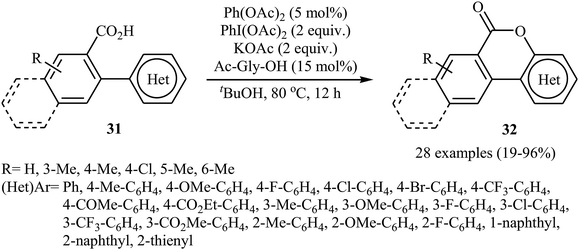 |
| | Scheme 14 Synthesis of lactones 32 from the corresponding 2-aryl carboxylic acids 31. | |
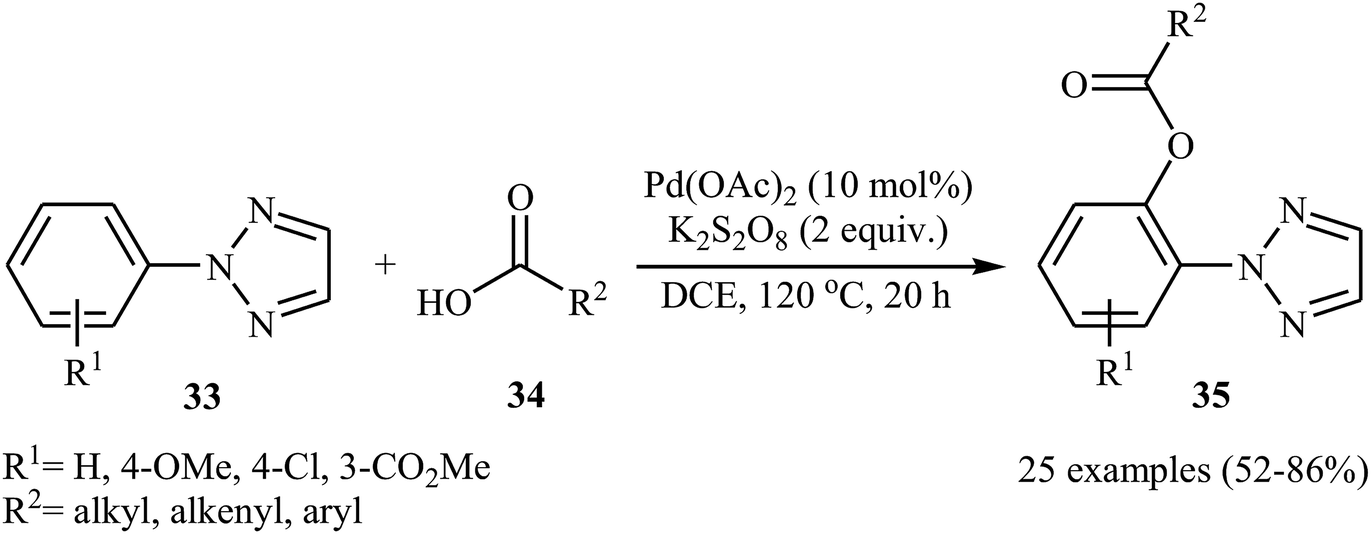
Later, Wang and Kuang presented the Pd-catalyzed direct acyloxylation of 2-substituted 1,2,3-triazoles 33 with various alkyl, alkenyl, and aryl carboxylic acids 34.32 The optimized condition for this C–H activation reaction is the use of 10 mol% of Pd(OAc)2 as a catalyst and 2.0 equiv. of K2S2O8 as an oxidant in DCE at 120 °C for 20 h. With the optimized protocol a number of examples were reported in fair to good yield range (Scheme 15). A significant kinetic isotope effect (kH/kD = 3.0) was observed with deuterium-labelled compounds, signifying that the cleavage of C–H bond was involved in the rate-determining step of this transformation. Similarly, 2-arylbenzothiazoles were also regioselectively acyloxylated at the ortho-position using Pd(OAc)2 as a catalyst in conjunction with ceric ammonium nitrate (CAN) as an oxidant.33
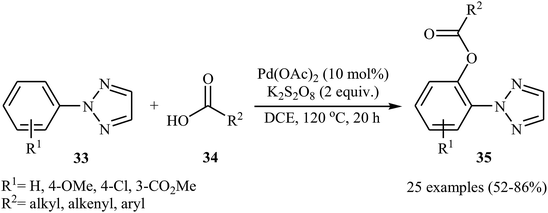 |
| | Scheme 15 Pd-catalyzed ortho-acyloxylation of 2-substituted 1,2,3-triazoles 33 with different carboxylic acids 34. | |
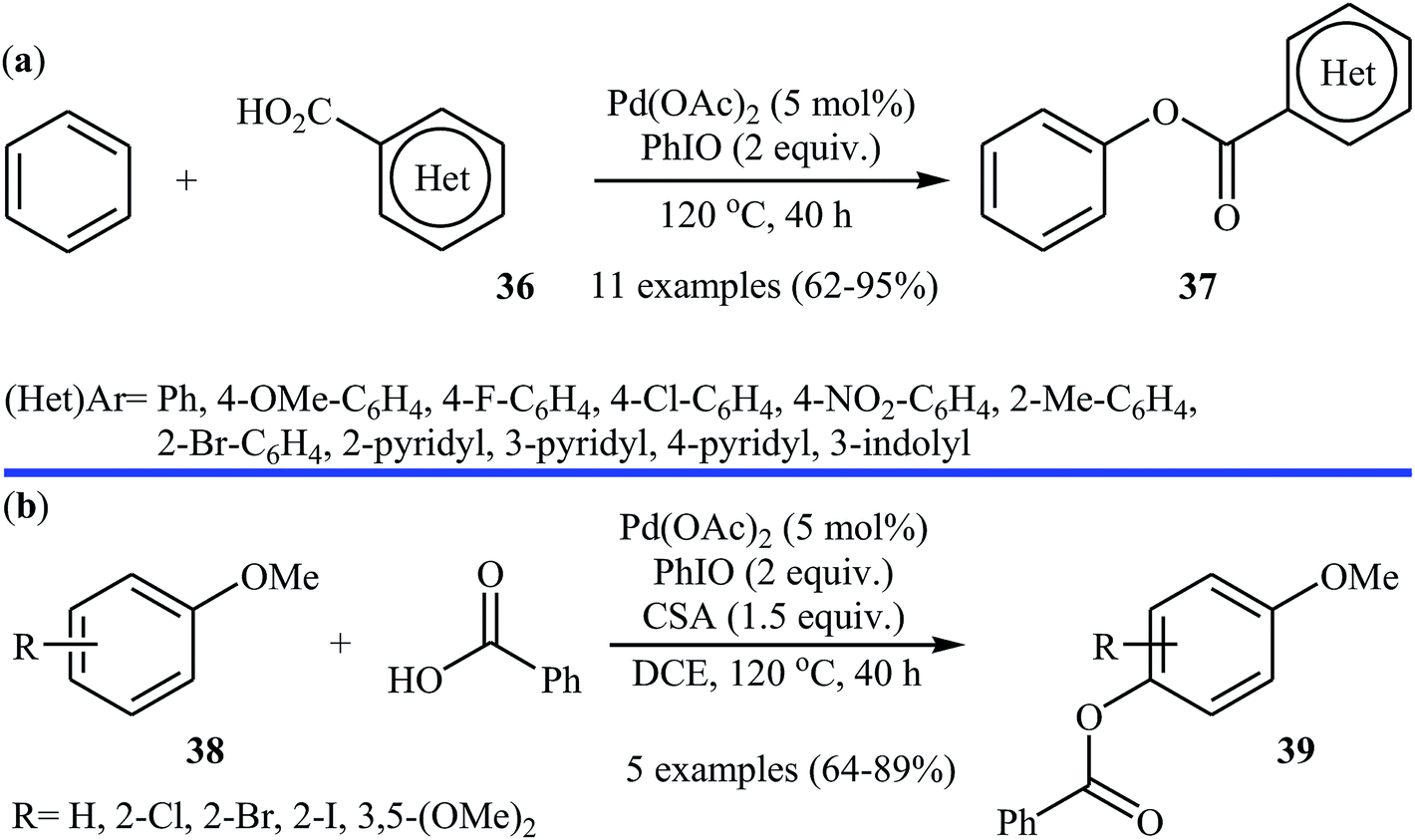
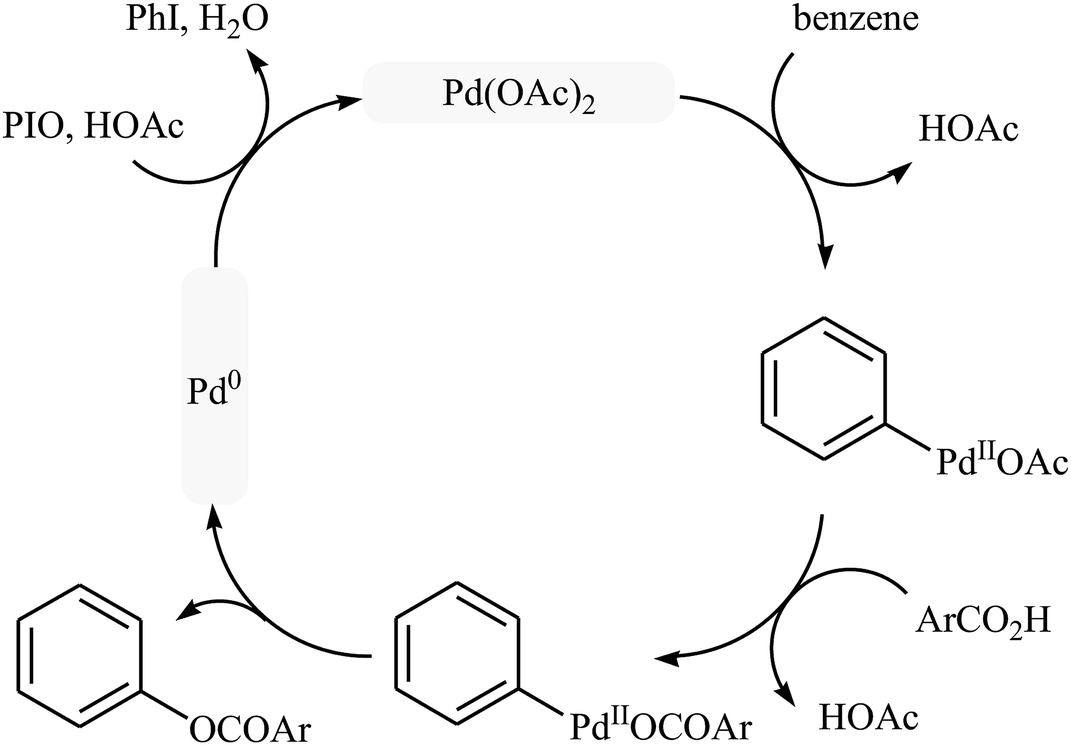
Following these works, Liu and collaborators informed an interesting Pd-catalyzed dehydrogenative cross-coupling of simple benzene with carboxylic acids under solvent-free conditions.34 To evaluate the activity of different catalysts, benzoic acid was chosen as the model substrate. Like previous works Pd(OAc)2 was found to be more effective catalyst, which gave a better yield of benzoxylated product. The oxidants such as Na2S2O8, oxone, Ag2O, Cu(OAc)2, PhI(OAc)2, PhIO, PhIO/CSA, PhIO/TFA, and PhIO/TfOH were examined and a good yield of product was obtained when using 2.0 equiv. of iodosobenzene as an oxidant. Under standard conditions, various (hetero)aromatic carboxylic acids 36 reacted efficiently with benzene to give the corresponding esters 37 in good to high yields (Scheme 16a). It was proven that the reaction conditions could also be applied to the benzoxylation of anisole derivatives 38 with simple benzoic acid (Scheme 16b). In this case, over-stoichiometric amounts of camphorsulfonic acid (CSA) as an additive and DCE as the solvent was required to promote the transformation. The mechanism proposed by the authors to explain this C–O coupling reaction involves the initial formation of an Ar–Pd(II) intermediate A via Pd(II)-catalyzed C–H cleavage of the C–H bond of benzene. Next, a ligand exchange process of this intermediate A with carboxylic acid leads to the intermediate B, which after C–O reductive elimination affords the final product (Scheme 17).
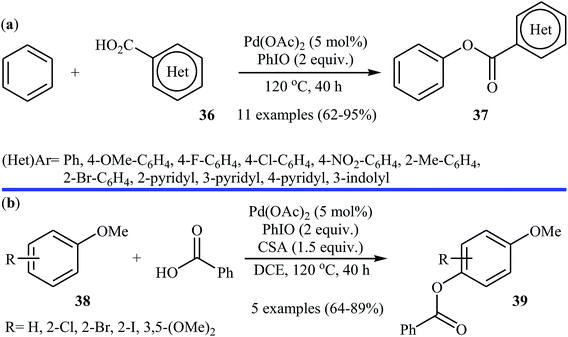 |
| | Scheme 16 (a) Pd-catalyzed dehydrogenative cross-coupling of simple benzene with carboxylic acids 36; (b) Pd-catalyzed para-benzoxylation of anisole derivatives 38 with simple benzoic acid. | |
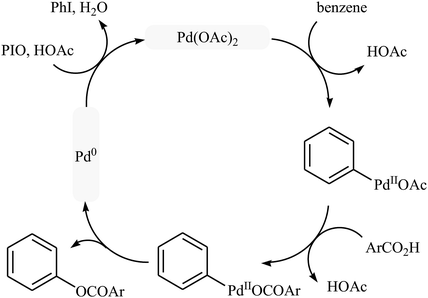 |
| | Scheme 17 Proposed mechanism for the reaction in Scheme 16a. | |
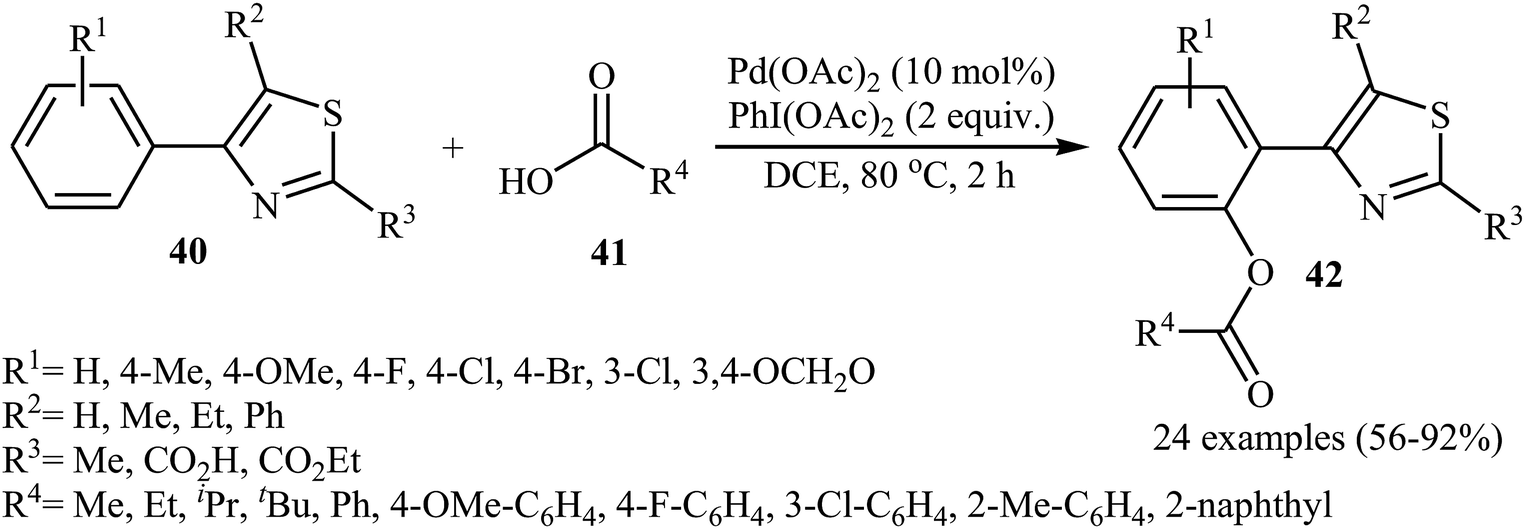
In 2016, Liao and Ji along with their co-workers broadened the applicability of Pd-catalyzed acyloxylation by developing a catalytic system [Pd(OAc)2, PhI(OAc)2] which allowed for the site-selective mono-acyloxylation of highly substituted 4-aryl-thiazoles 40 with diverse carboxylic acids 41 (Scheme 18).35 Importantly, the reaction was equally efficient for both aromatic and aliphatic carboxylic acids. In a related investigation, Zhang and colleagues reported that benzothiadiazole-arene derivatives can undergo mild mono-selective acetoxylation with acetic acid in the presence of 5 mol% of Pd(OAc)2 and 1.0 equiv. of PhI(OAc)2 at room temperature.36
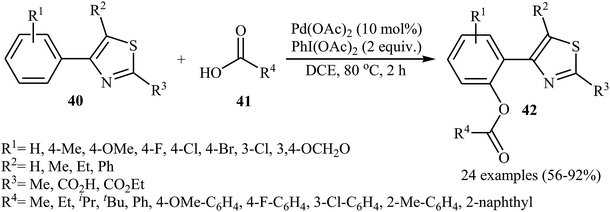 |
| | Scheme 18 Liao-Ji's synthesis of mono-acyloxylated 4-aryl-thiazoles 42. | |
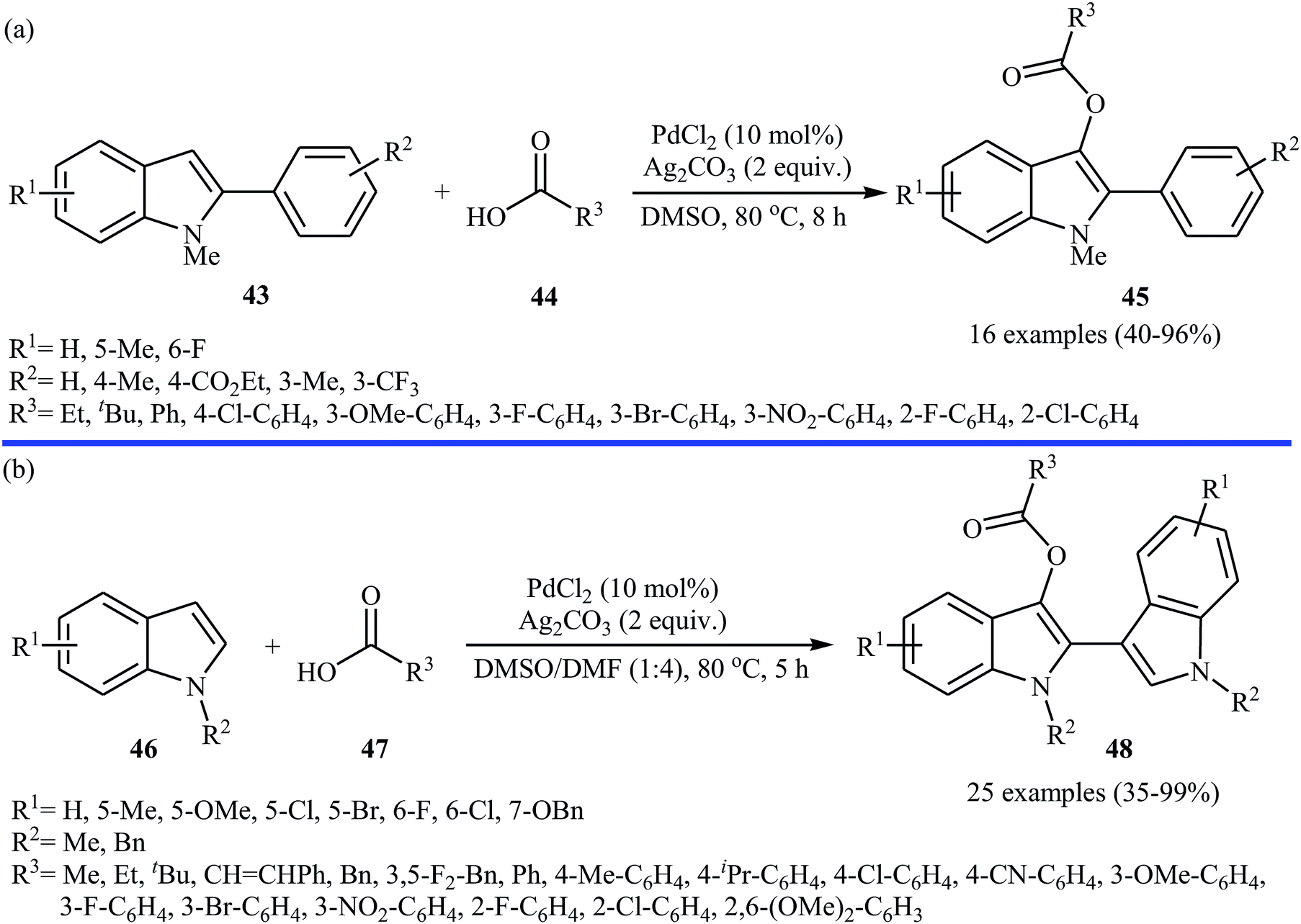
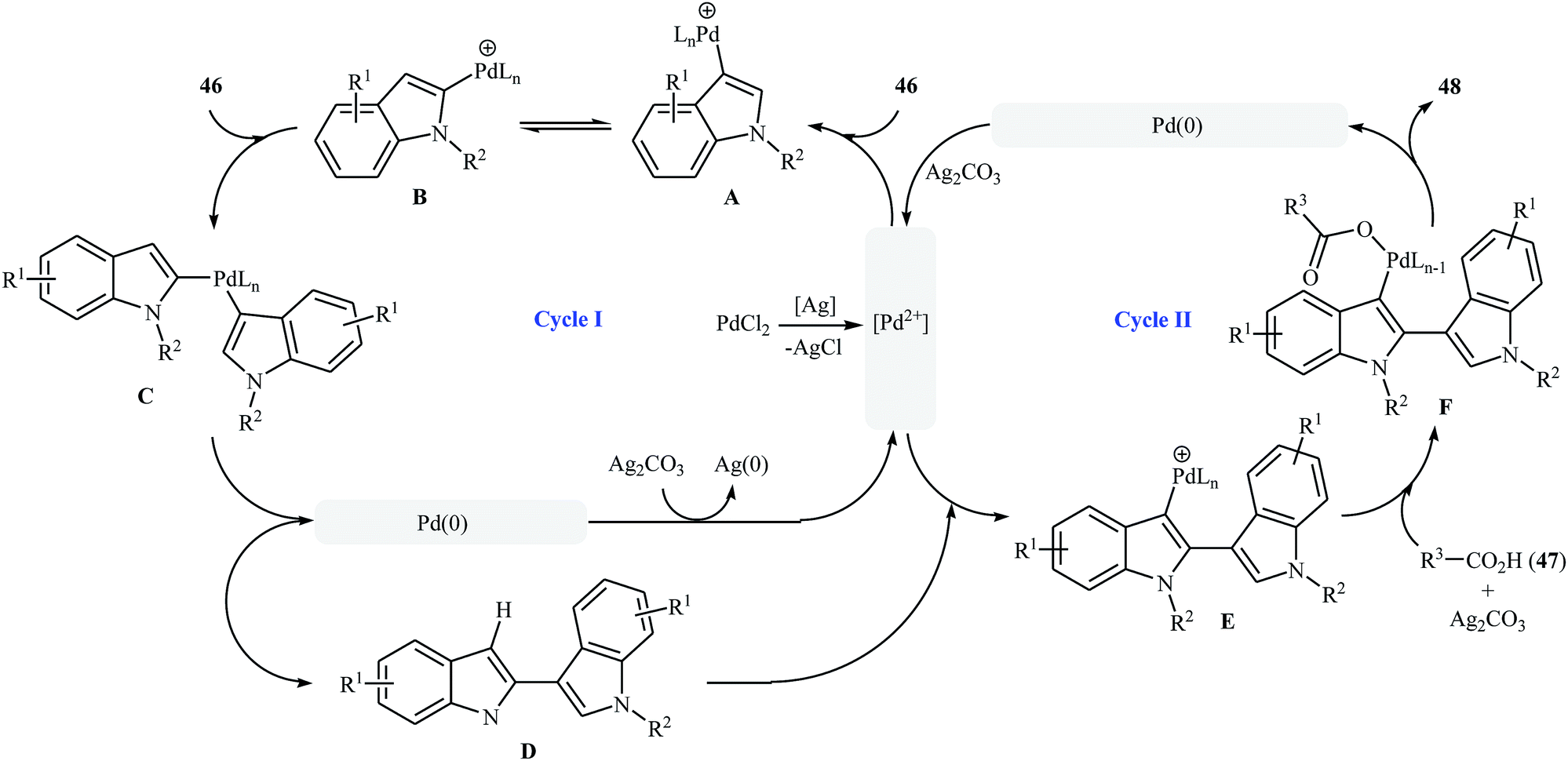
Very recently, a beautiful and efficient Pd-catalyzed direct esterification of 2-substituted indoles 43 with carboxylic acids 44 using PdCl2/Ag2CO3 combination as a catalytic system was developed by Liu and co-workers.37 The reactions were performed in DMSO at 80 °C for 8 h, tolerated a wide range of functional groups (e.g., F, Cl, Br, NO2, CF3, CO2Et, OMe) and afforded C3-acyloxylated indoles 45 in moderate to excellent yields with outstanding regioselectivity (Scheme 19a). Interestingly, under the standard conditions, C2-unsubstituted indoles 46 underwent homocoupling with high C2/C3′ selectivity and C3-acyloxylation with various alkyl/alkenyl/aryl carboxylic acids 47 in one step and provided the corresponding 3-(3-acyloxylated-2-indolyl)-indoles 48 in fair to almost quantitative yields (Scheme 19b). The proposed mechanism for this transformation is a little bit complex involving two catalytic cycles (Scheme 20). Initially, cationic palladium(II) generates from PdCl2 in the presence of Ag2CO3. In cycle I, electrophilic palladation of C3 position of the indole 46 by palladium(II) affords the palladium complex A, which converts into complex B through migration. Next, the electrophilic palladation with the second indole 46 forms intermediate C that after reductive elimination produces the homocoupling product 2,3′-dimer D and regenerates the cationic palladium(II) in the presence of Ag2CO3. In cycle II, the electrophilic palladation of the 2,3′-dimer D yields intermediate E, which then undergoes ligand exchange with carboxylic acid 47 in the presence of Ag2CO3 to form the palladium complex F. Finally, the reductive elimination of this complex F affords the expected acyloxylated indole product 48.
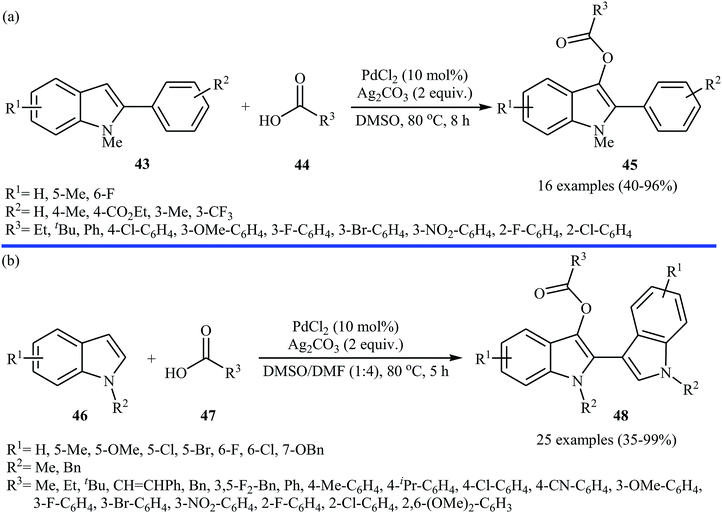 |
| | Scheme 19 (a) Pd-catalyzed direct esterification of 2-substituted indoles 43 with carboxylic acids 44; (b) Pd-catalyzed direct esterification of 2,3-unsubstituted indoles 46 with carboxylic acids 47. | |
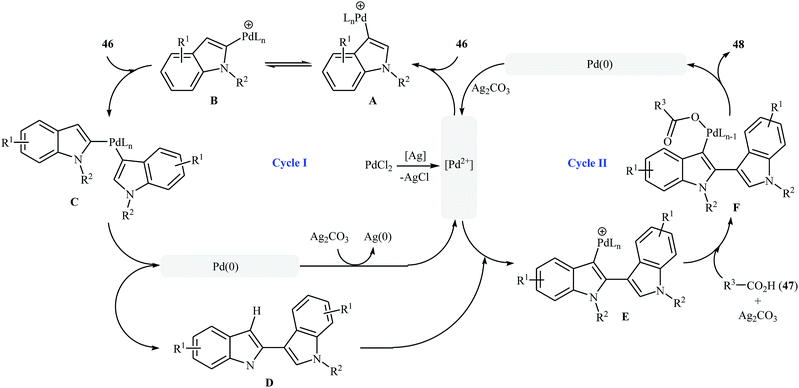 |
| | Scheme 20 Mechanistic explanation for the synthesis of acyloxylated indoles 48. | |
5. Cobalt-catalyzed reactions
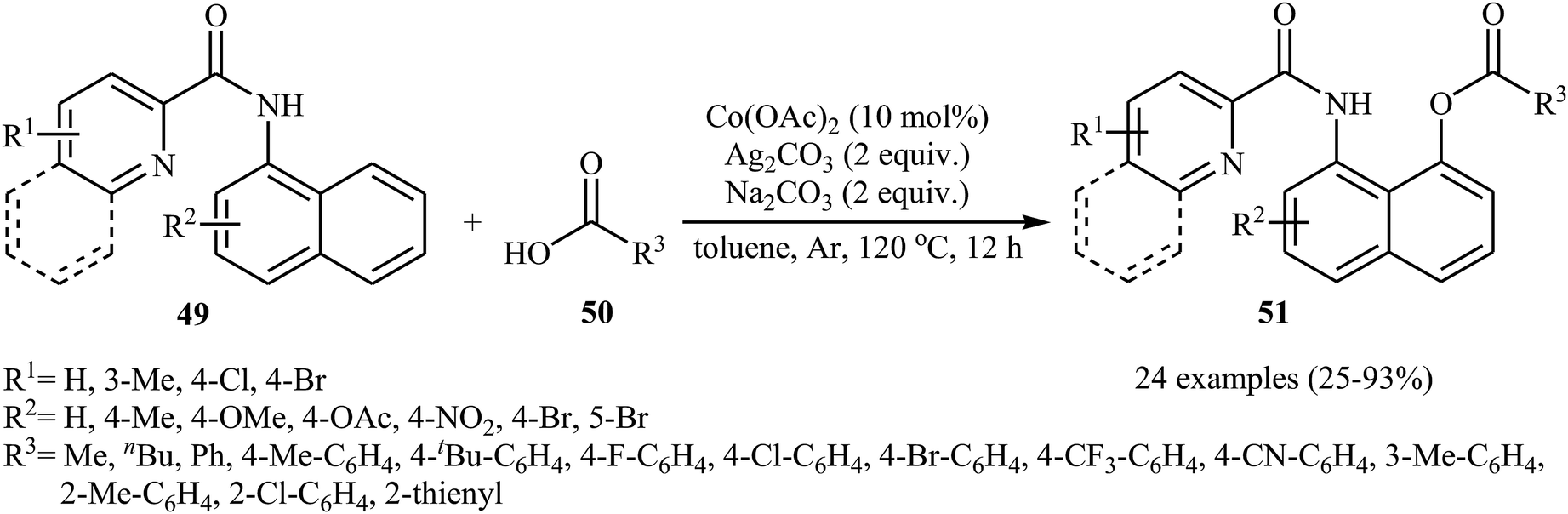
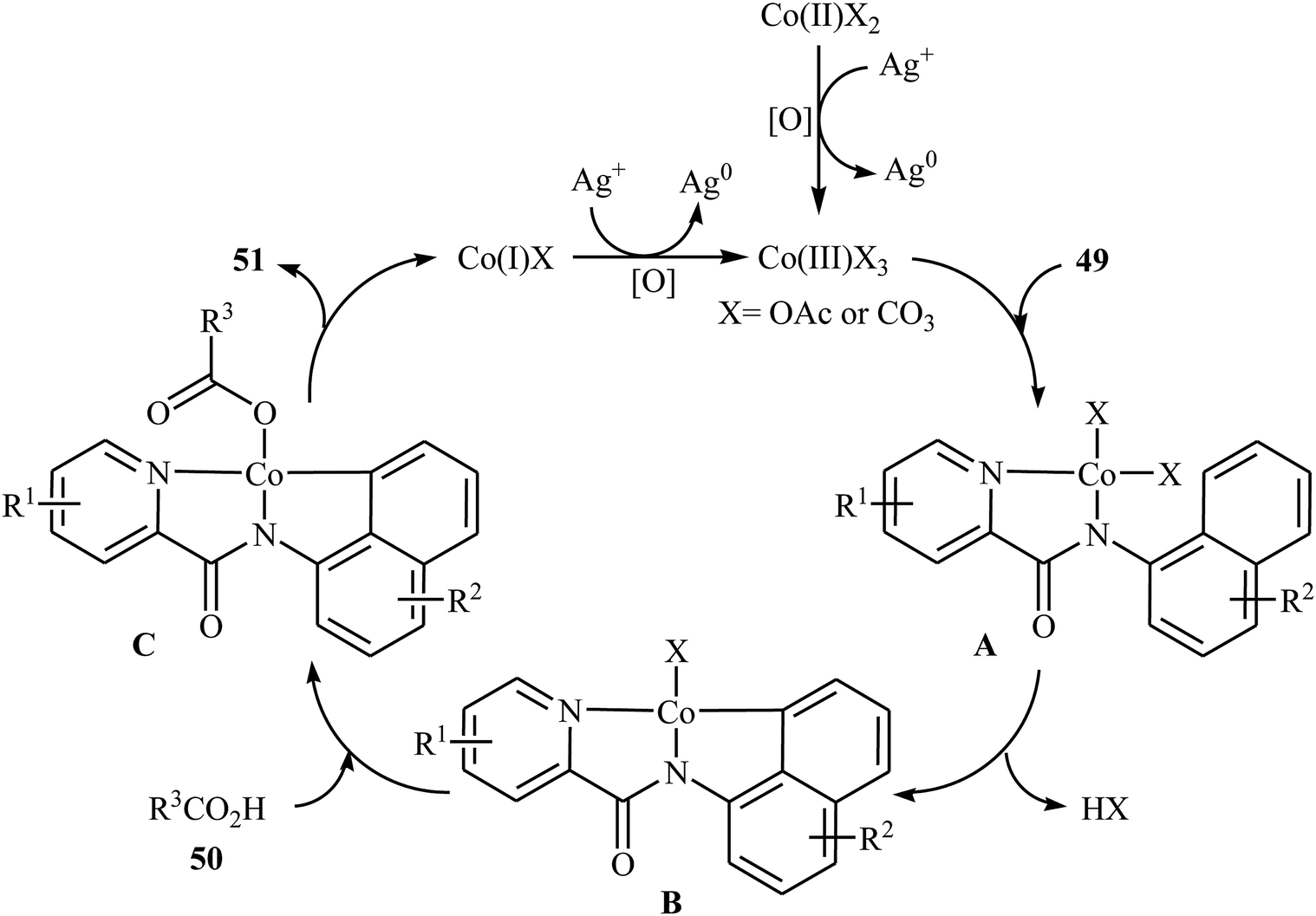
In 2017, the group of Zeng reported the first cobalt-catalyzed cross-dehydrogenative coupling of aryl C–H bonds with carboxylic acids.38 In this study, various cobalt catalysts (e.g., CoCl2, Co(acac)2, Co(OAc)2, CoSO4 + 7H2O), oxidants (e.g., O2, MnO2, Cu(OAc)2, AgOAc, Ag2CO3, K2S2O8), and bases (e.g., NaOAc, K2CO3, Na2CO3, NaHCO3) were examined and the Co(OAc)2/Ag2CO3/Na2CO3 system in toluene was found to be optimal for this heterocoupling reaction. Various N-aryl-2-pyridylcarboxamides 49 and alkyl/aryl/heteroaryl carboxylic acids 50 were reacted well under the optimized reaction conditions to produce the corresponding mono-acyloxylated products 51 with yield range from 25 to 93% (Scheme 21). However, 4-nitro-2-pyridyl carboxamide and 4-alkoxybenzoic acid could not tolerate this reaction system. Mechanistically, this esterification reaction is believed to proceed through a metalation/deprotonation/ligand-exchange/reductive elimination sequential process (Scheme 22). A related acyloxylation of arene C–H bonds under cobalt catalysis was also informed by Zhang and co-workers in 2018.39 Here, N-(quinolin-8-yl)benzamide scaffolds were ortho-selective mono-acyloxylated in the presence of Co(OAc)2 as the catalyst and Ag2SO4 as the oxidant in DCE medium under an inert atmosphere.
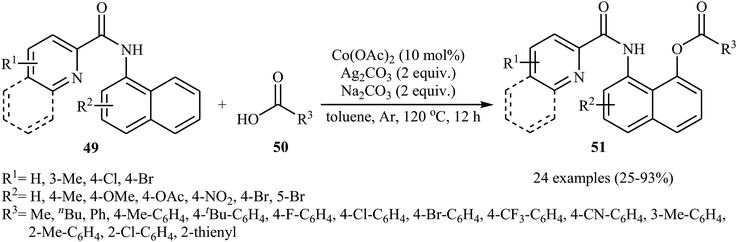 |
| | Scheme 21 Co-catalyzed cross-dehydrogenative coupling of N-aryl-2-pyridylcarboxamides 49 with carboxylic acids 50. | |
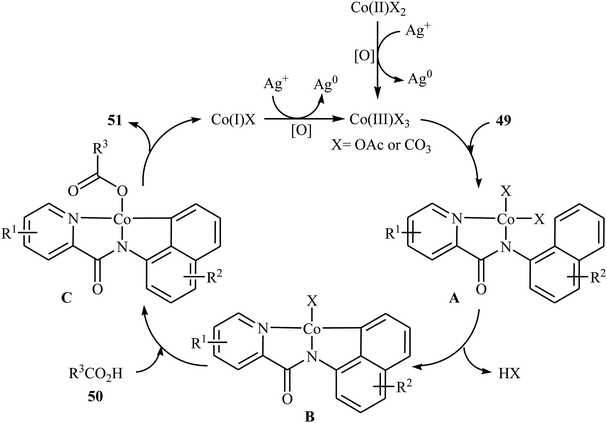 |
| | Scheme 22 Possible mechanism for the Co-catalyzed oxidative coupling between an arene 49 and carboxylic acid 50. | |
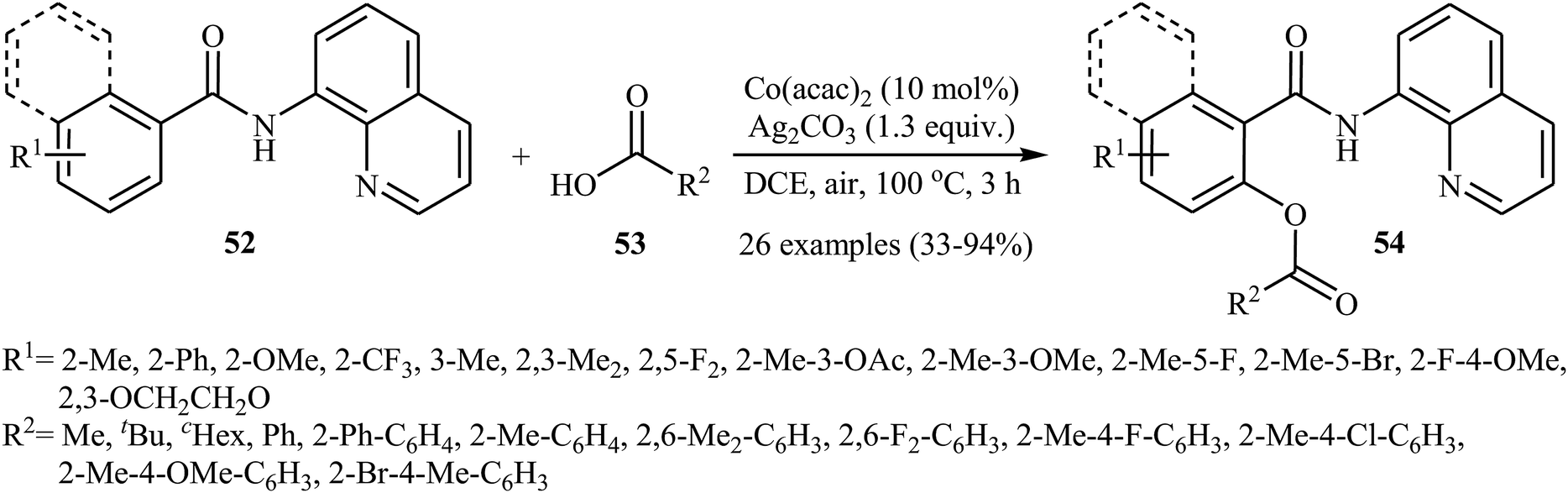
In a closely related investigation, Ueno, Natsui, and Chatani revealed that the treatment of N-(quinolin-8-yl)benzamides 52 with aromatic and aliphatic carboxylic acids 53 in the presence of 10 mol% of Co(acac)2 and 1.3 equiv. of Ag2CO3 in DCE under the open air afforded the corresponding mono-acyloxylated N-(quinolin-8-yl)benzamides 54 in relatively low to excellent yields (Scheme 23).40 This reaction tolerated many functional groups, including fluoro, bromo, trifluoromethyl, ether, and ester functionalities that offers scope for further transformations on the benzamide ring.
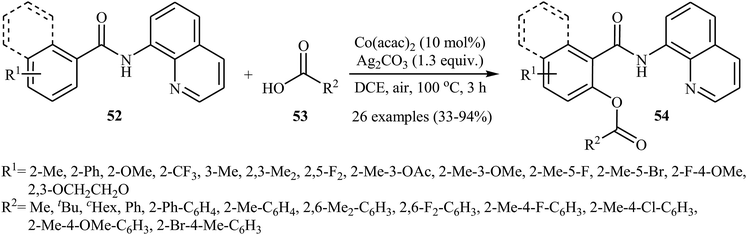 |
| | Scheme 23 Coupling of N-(quinolin-8-yl)benzamides 52 with carboxylic acids 53 catalyzed by Co(acac)2. | |
6. Copper-catalyzed reactions
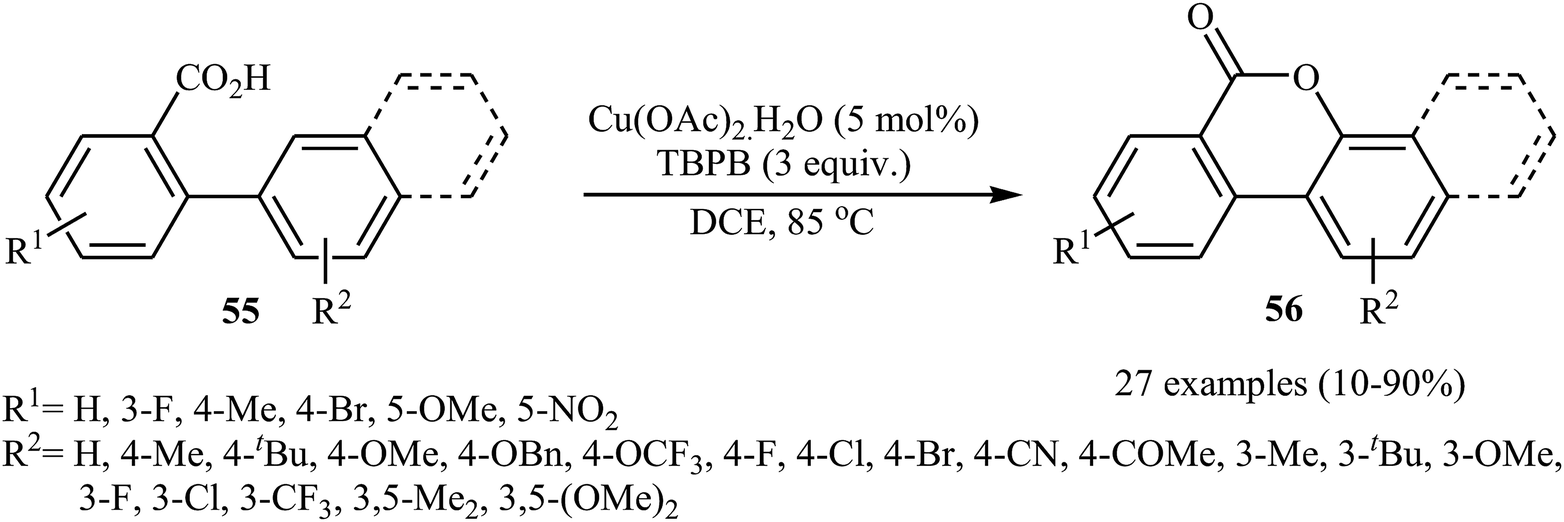
Copper salts, which are incredibly diverse, low-cost, versatile, and possess low toxicity, have proven to be very efficient catalysts for a wide variety of organic transformations.41 In 2013, Wang, Gulevich, and Gevorgyan reported on the first Cu-catalyzed cross-dehydrogenative coupling of arenes with carboxylic acids.42 They discovered that the intramolecular aromatic C–H oxygenation reaction of 2-arylbenzoic acid derivatives 55 in the presence of Cu(OAc)2 + H2O and tert-butyl peroxybenzoate (TBPB) afforded 3,4-benzocoumarins 56 (Scheme 24). The scope was rather narrow and limited for electron-neutral and electron-rich substrates. In particular, meta-substituted substrates gave a mixture of both possible isomers. The same lactones were also obtained in reactions performed with K2S2O8 as the oxidant in the presence of an small amount of AgNO3 as an additive. The results demonstrated that the later method was more general and practical, which is applicable to various electron-rich and electron-deficient 2-arylbenzoic acids.
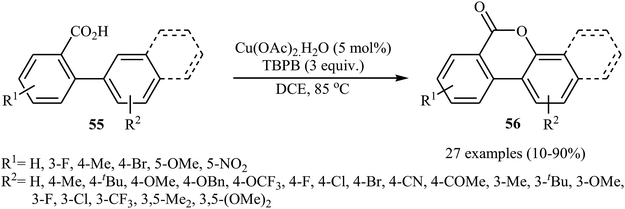 |
| | Scheme 24 Cu-catalyzed intramolecular C–H oxygenation reaction of 2-arylbenzoic acid derivatives 55. | |
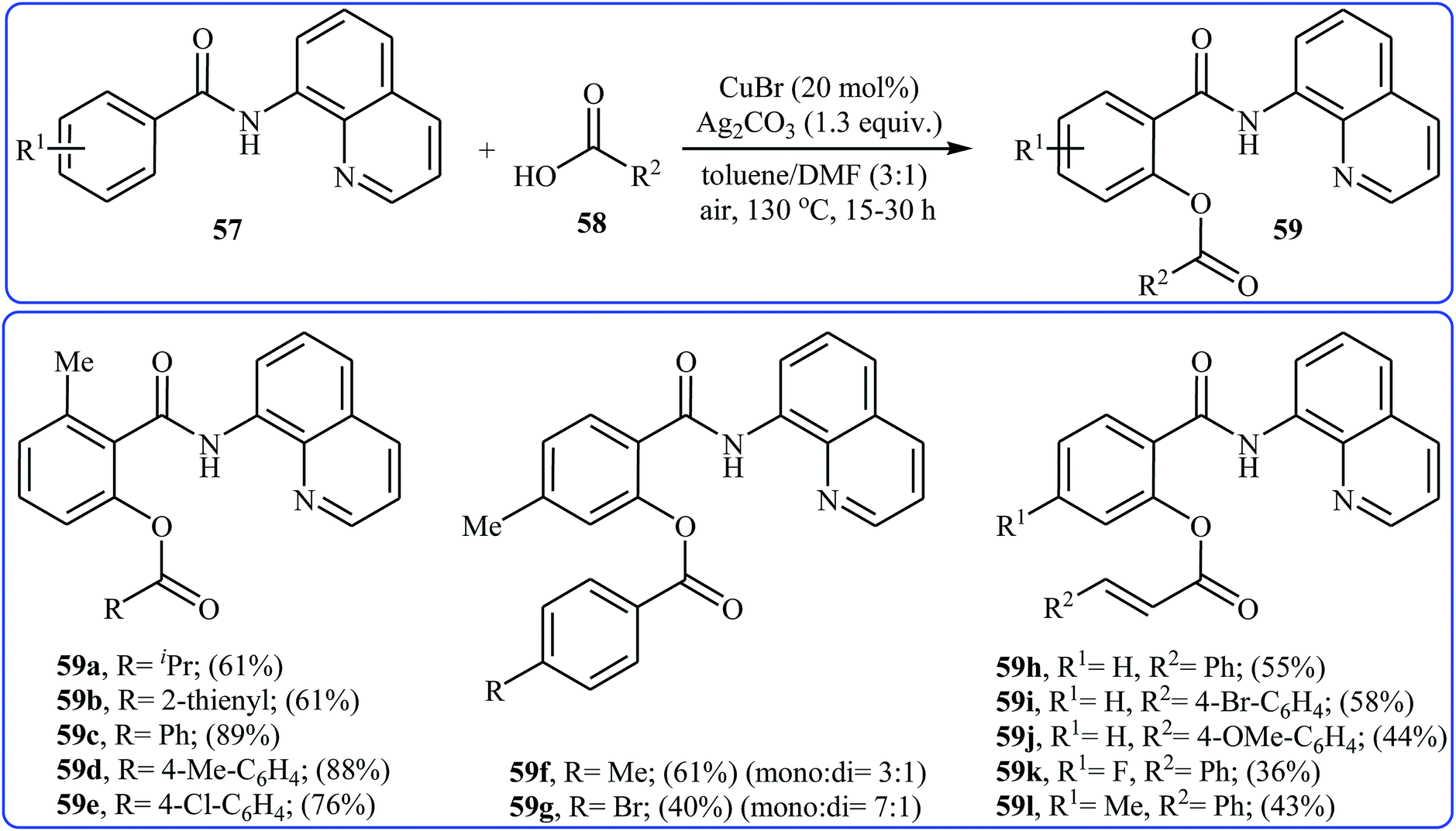
Four years later, Zhang and co-workers investigated the intermolecular version of this reaction.43 The optimization study revealed that efficient C–O coupling occurs when using commercially available CuBr catalyst in combination with Ag2CO3 as oxidant in the binary solvent toluene/DMF with ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 under the open air and at 130 °C. The optimized protocol allows direct mono-acyloxylation of N-(quinolin-8-yl)benzamides 57 with various alkyl, alkenyl, aryl, and heteroaryl carboxylic acids 58 furnishing the desired products 59 in rather moderate to high yields; albeit the reaction of unsubstituted and meta- and para-substituted benzamides with aromatic carboxylic acids offered a mixture of mono- and di-functionalized products. Furthermore, meta-methoxy-substituted benzamide gave a mixture of ortho, para, and di-functionalized products with poor selectivity. The authors explained this fact by the coordination effect of the methoxy substituent that stabilized the organo-copper intermediate. Some reported examples are shown in Scheme 25. According to the author proposed mechanism, this esterification reaction proceeds along the similar mechanistic pathway that depicted in Scheme 22.
:1 under the open air and at 130 °C. The optimized protocol allows direct mono-acyloxylation of N-(quinolin-8-yl)benzamides 57 with various alkyl, alkenyl, aryl, and heteroaryl carboxylic acids 58 furnishing the desired products 59 in rather moderate to high yields; albeit the reaction of unsubstituted and meta- and para-substituted benzamides with aromatic carboxylic acids offered a mixture of mono- and di-functionalized products. Furthermore, meta-methoxy-substituted benzamide gave a mixture of ortho, para, and di-functionalized products with poor selectivity. The authors explained this fact by the coordination effect of the methoxy substituent that stabilized the organo-copper intermediate. Some reported examples are shown in Scheme 25. According to the author proposed mechanism, this esterification reaction proceeds along the similar mechanistic pathway that depicted in Scheme 22.
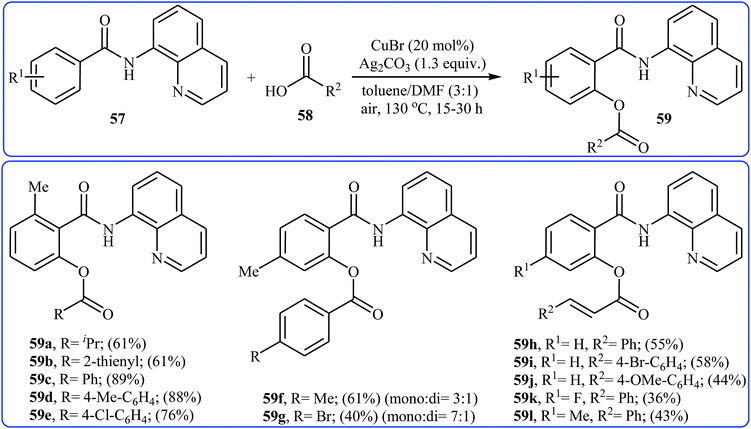 |
| | Scheme 25 Zhang's synthesis of mono-acyloxylated of N-(quinolin-8-yl)benzamides 59. | |
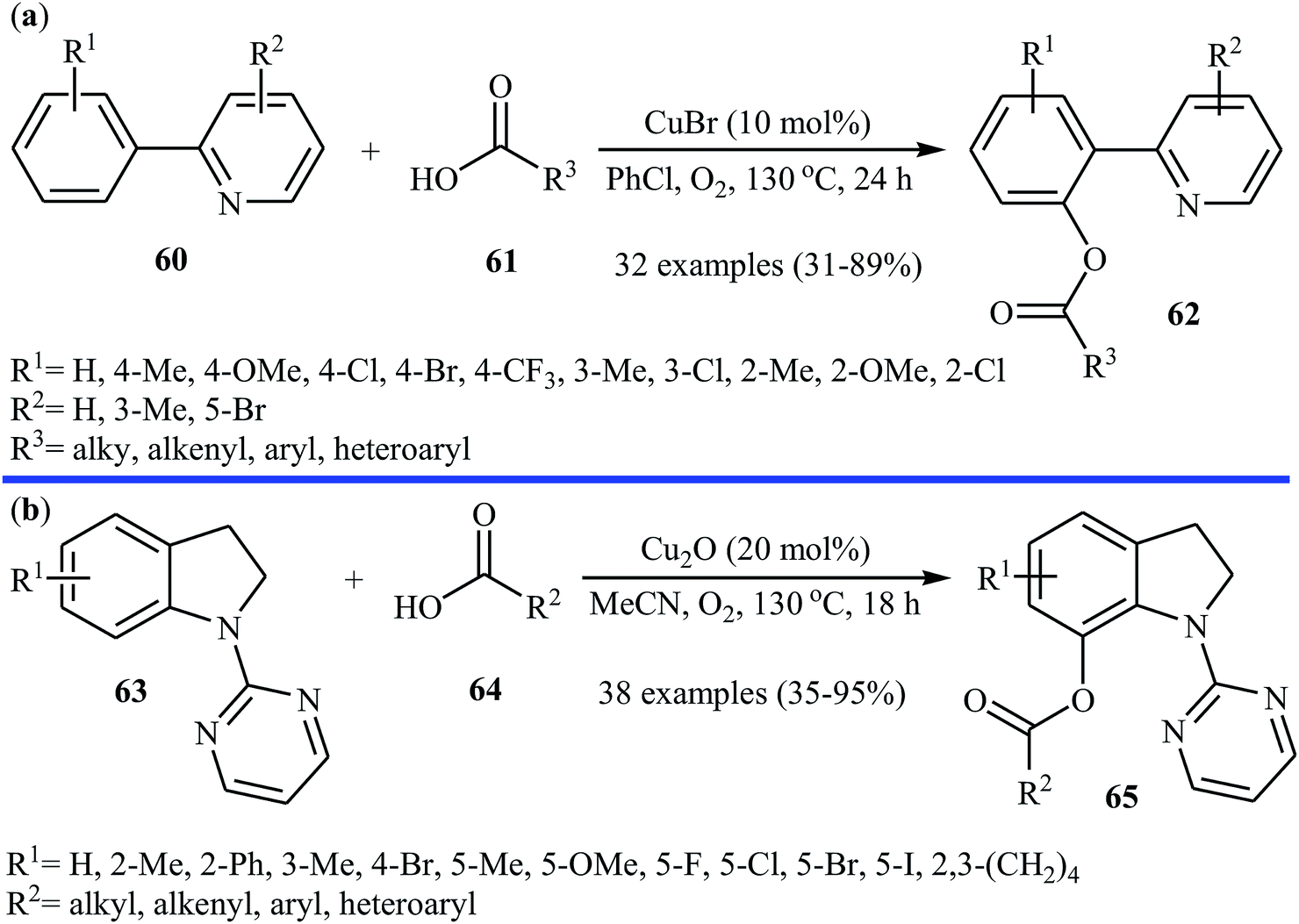
Subsequently, the same research team explored the copper-catalyzed C–H acyloxylation of 2-arylpyridines 60 with carboxylic acids 61 using oxygen as the oxidant.44 The investigation showed that like previous work the use of CuBr as a catalyst gave the best yield, while other copper salts such as CuCl, CuI, CuBr2, Cu(OAc)2, CuSO4 + 5H2O, CuCl2 + 2H2O, and Cu(OAc)2 + H2O promoted the reaction with reduced efficiency. To identify the solvent potentially suitable for this esterification reaction, the authors first chose DMF, DMSO, THF, PhCl, 1,2-DCB, and toluene. For this reaction, PhCl was the most effective solvent, giving the desired o-acyloxylated of 2-arylpyridines 62 with yield range from 31 to 89% (Scheme 26a). The results proved that 2-arylpyridines possessing electron-donating groups afforded relatively higher yields compared to the electron-poor 2-arylpyridines and the relative reaction rates of carboxylic acids followed the order: benzoic acids > cinnamic acids > aliphatic acids. Along this line, Koley and colleagues showed that Cu2O could effectively catalyze the cross-dehydrogenative coupling reactions between indolines 63 and a variety of carboxylic acids 64 in MeCN under oxygen atmosphere.45 The corresponding mono-acyloxylated indolines 65 were obtained in relatively low to good yields (Scheme 26b). Noteworthy, CuBr failed to promote this reaction.
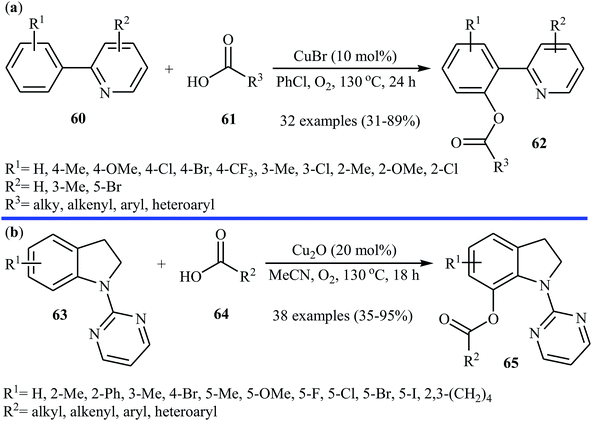 |
| | Scheme 26 (a) Cu-catalyzed C–H acyloxylation of 2-arylpyridines 60 with carboxylic acids 61; (b) Cu-catalyzed C–H acyloxylation of indolines 63 with carboxylic acids 64. | |
7. Gold-catalyzed reactions
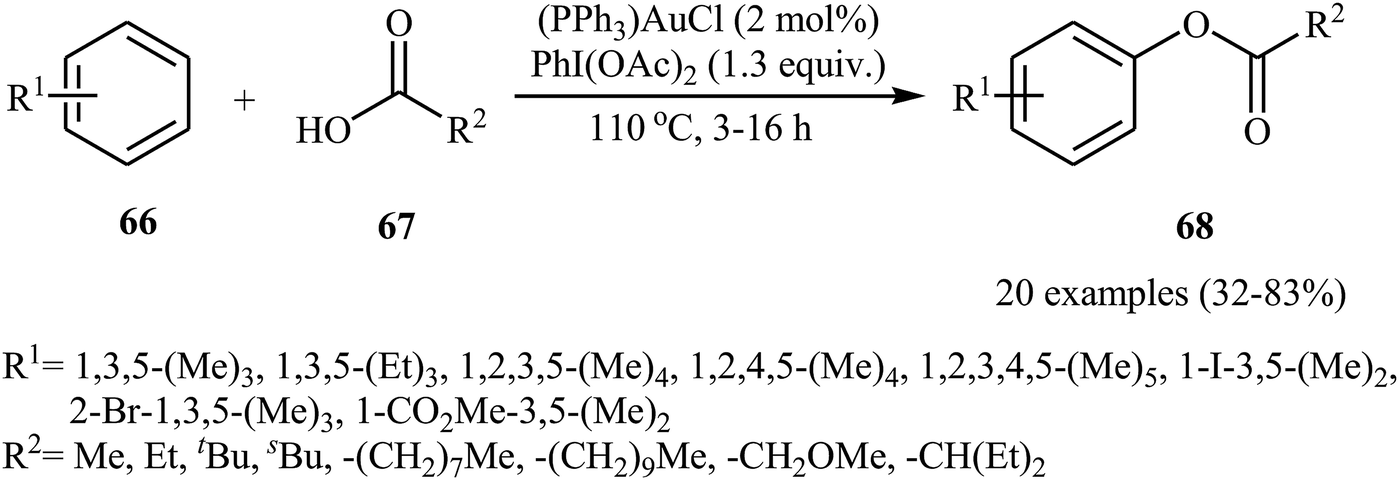
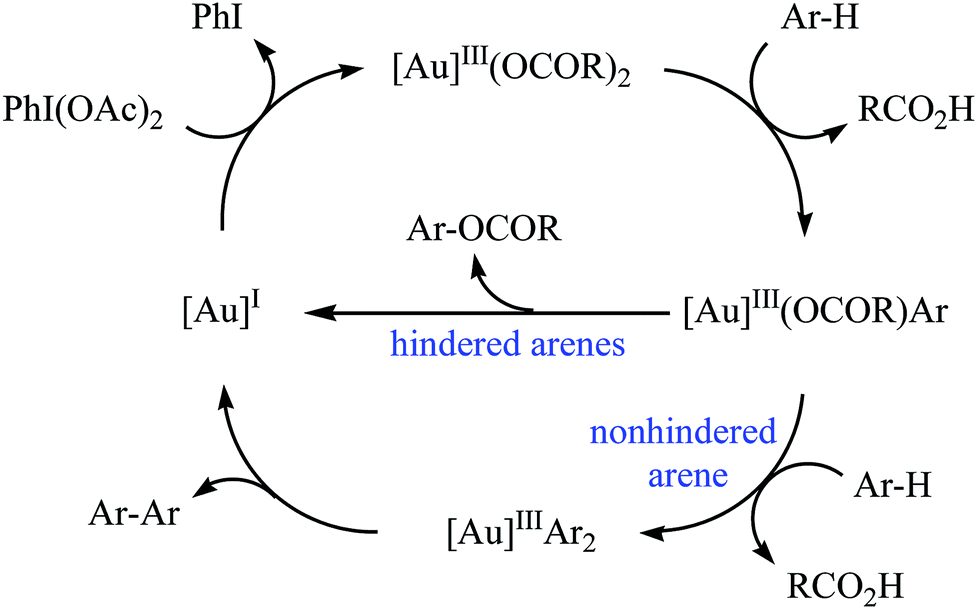
Gold-catalyzed direct acyloxylation of arene C–H bonds has been rarely studied; indeed, only one example of such a reaction was reported in the literature till date. In 2011, Pradal, Toullec, and Michelet revealed that the treatment of simple arenes 66 with aliphatic carboxylic acids 67 in the presence of only 2 mol% of (PPh3)AuCl as a catalyst and 1.3 equiv. of PhI(OAc)2 as an oxidant under solvent-free conditions afforded the corresponding mono-acyloxylated arenes 68 with yield range from 32 to 83% (Scheme 27).46 Indole did not work well in the reaction and therefore no other heteroaromatic compounds were examined in the protocol. It should be noted that other gold catalysts (e.g., AuCl, AuCl3, HAuCl4, Au2O3, and (PPh3)AuNTf2) were also found to promote the reaction; albeit in lower yields. Of note, the protocol was only applicable for hindered arenes, since nonhindered arene substrates afforded the corresponding biphenyl products under this condition. The authors suggested mechanism of this transformation is shown in Scheme 28.
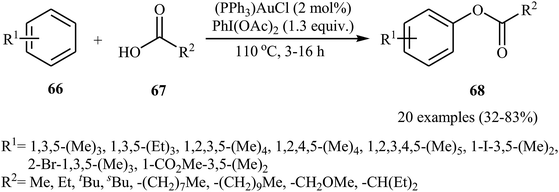 |
| | Scheme 27 Au-catalyzed oxidative acyloxylation of simple arenes 66. | |
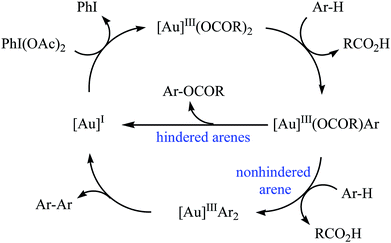 |
| | Scheme 28 Proposed mechanism for the reaction in Scheme 27. | |
8. Silver-catalyzed reactions
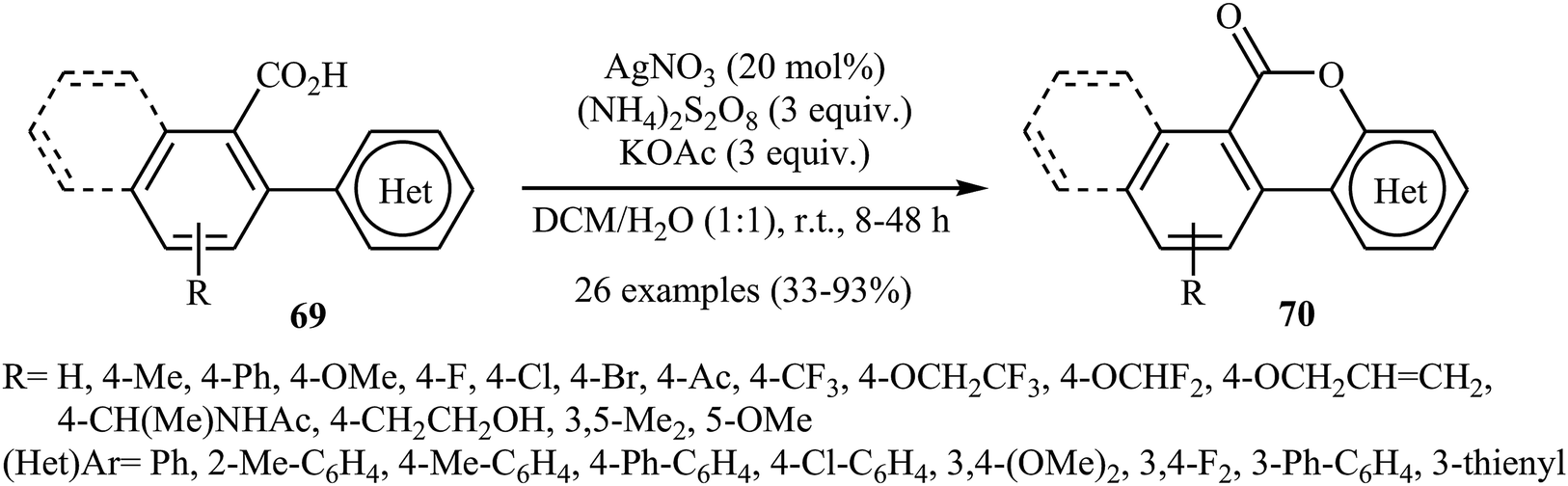
In 2015, in an innovative investigation, the group of Xu introduced the use of silver salts as effective catalysts for intramolecular C–O cross-coupling of biaryl-2-carboxylic acids through C–H functionalization/C–O cyclization.47 They studied the effects of reaction variables such as catalyst, oxidant, additive, and solvent. It was found that using 20 mol% of AgNO3 and 3 equiv. of (NH4)2S2O8 in a 1:1 mixture of DCM and H2O resulted in the best yields. Among the various additives such as HOAc, NaOAc, KOAc, K2HPO4; KOAc was the most efficient for this interesting reaction. Various 2 biaryl-2-carboxylic acids 69 bearing different electron-donating and electron-withdrawing substituents on the arene rings were compatible with the reaction conditions and provided the corresponding lactones 70 in moderate to excellent yields (Scheme 29). The procedures could also be adapted to larger-scale synthesis.
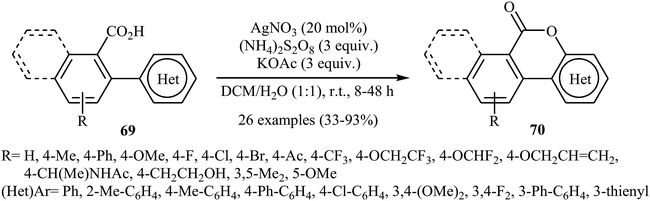 |
| | Scheme 29 Xu's synthesis of lactones 70. | |
The same silver-catalyzed method was applied for the intramolecular C(5)–H acyloxylation of a variety of 2-(4-aryl-1H-1,2,3-triazol-1-yl) benzoic acids 71 (Scheme 30).48 In this transformation, Jiang's group reported that the use of a binary solvent EtOAc/H2O with ratio 1:1 afforded the best results.
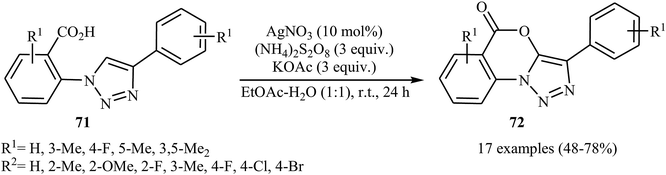 |
| | Scheme 30 Ag-catalyzed intramolecular C–O cross-coupling of biaryl-2-carboxylic acids 71 through C–H acyloxylation. | |
9. Conclusion
This review summarized the available literature on cross-dehydrogenative coupling reactions between arenes and carboxylic acids. As illustrated, a variety of O-aryl esters could be easily achieved by this simple and atom economical procedure indicating the versatility and practicality of this page of ester synthesis. Interestingly, all of the reactions covered in this review were catalyzed by commercially available metal catalysts. Despite remarkable progresses in this attractive research arena, several challenges still need to be addresses: (i) most of the reactions covered in this review were carried out at high temperatures. Therefore there is still a need for the discovery of new catalytic systems, which can allow the efficient coupling under milder conditions; (ii) the current strategies mostly limit to the use of expensive metal catalysts such as palladium and ruthenium. Thus the exploration of low-cost metal catalysts (e.g., iron and copper) for this transformation is highly desirable; (iii) other reactions such as coupling of arenes with alkynyl acids should be explored.
Conflicts of interest
There are no conflicts to declare.
References
- M. Tsakos, E. S. Schaffert, L. L. Clement, N. L. Villadsen and T. B. Poulsen, Nat. Prod. Rep., 2015, 32, 605–632 RSC.
-
(a) P. Wipf, T. Tsuchimoto and H. Takahashi, Pure Appl. Chem., 1999, 71, 415–421 Search PubMed;
(b) B. Gnanaprakasam and D. Milstein, J. Am. Chem. Soc., 2011, 133, 1682–1685 CrossRef CAS PubMed;
(c) R. Takise, K. Muto and J. Yamaguchi, Chem. Soc. Rev., 2017, 46, 5864–5888 RSC;
(d) E. Vessally, A. Hosseinian, M. Babazadeh, L. Edjlali and R. Hosseinzadeh-Khanmiri, Curr. Org. Chem., 2018, 22, 315–322 CrossRef CAS.
-
(a) M. V. Blagosklonny and T. Fojo, Int. J. Cancer, 1999, 83, 151–156 CrossRef CAS PubMed;
(b) J. K. Sohng, T. Yamaguchi, C. N. Seong, K. S. Baik, S. C. Park, H. J. Lee, S. Y. Jang, J. R. Simkhada and J. C. Yoo, Arch. Pharmacal Res., 2008, 31, 1339–1345 CrossRef CAS PubMed;
(c) D. A. Lee and E. J. Higginbotham, Am. J. Health-Syst. Pharm., 2005, 62, 691–699 CrossRef PubMed;
(d) L. J. Scott and P. L. McCormack, Drugs, 2008, 68, 1239–1272 CrossRef CAS PubMed.
- G. M. Keating, Drugs, 2015, 75, 1131–1140 CrossRef CAS.
- H. B. Fung, H. L. Kirschenbaum and R. Hameed, Clin. Ther., 2000, 22, 549–572 CrossRef CAS.
- C. J. Dunn and K. L. Goa, Drugs, 2001, 61, 285–315 CrossRef CAS PubMed.
- M. Digiuni, P. Fogagnolo and L. Rossetti, Expert Opin. Pharmacother., 2012, 13, 723–745 CrossRef CAS PubMed.
- A. Wauquier, Anaesthesia, 1983, 38, 26–33 CrossRef CAS.
- B. A. Sobin and F. W. Tanner Jr, J. Am. Chem. Soc., 1954, 76, 4053 CrossRef CAS.
-
(a) M. Gershater, K. Sharples and R. Edwards, Phytochemistry, 2006, 67, 2561–2567 CrossRef CAS;
(b) S. Giri, S. Prasad, A. Giri and G. Sharma, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2002, 514, 223–231 CrossRef CAS.
-
(a) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed;
(b) B. V. Varun, J. Dhineshkumar, K. R. Bettadapur, Y. Siddaraju, K. Alagiri and K. R. Prabhu, Tetrahedron Lett., 2017, 58, 803–824 CrossRef CAS.
-
(a) J. Yuan, C. Liu and A. Lei, Chem. Commun., 2015, 51, 1394–1409 RSC;
(b) T. Chen, J.-S. Zhang and L.-B. Han, Dalton Trans., 2016, 45, 1843–1849 RSC.
-
(a) A. Hosseinian, S. Farshbaf, L. Z. Fekri, M. Nikpassand and E. Vessally, Top. Curr. Chem., 2018, 376, 23 CrossRef PubMed;
(b) A. Hosseinian, S. Ahmadi, F. A. H. Nasab, R. Mohammadi and E. Vessally, Top. Curr. Chem., 2018, 376, 39 CrossRef PubMed.
-
(a) K. Nejati, S. Ahmadi, M. Nikpassand, P. D. K. Nezhad and E. Vessally, RSC Adv., 2018, 8, 19125–19143 RSC;
(b) A. Hosseinian, R. Mohammadi, S. Ahmadi, A. Monfared and Z. Rahmani, RSC Adv., 2018, 8, 33828–33844 RSC;
(c) A. Hosseinian, F. A. H. Nasab, S. Ahmadi, Z. Rahmani and E. Vessally, RSC Adv., 2018, 8, 26383–26398 RSC;
(d) A. Hosseinian, P. D. K. Nezhad, S. Ahmadi, Z. Rahmani and A. Monfared, J. Sulfur Chem., 2019, 40, 88–112 CrossRef CAS;
(e) A. Monfared, S. Ebrahimiasl, M. Babazadeh, S. Arshadi and E. Vessally, J. Fluorine Chem., 2019, 220, 24–34 CrossRef CAS;
(f) S. Sarhandi, M. Daghagheleh, M. Vali, R. Moghadami and E. Vessally, Chem. Rev. Lett., 2018, 1, 9–15 Search PubMed;
(g) S. Mohammadi, M. Musavi, F. Abdollahzadeh, S. Babadoust and A. Hosseinian, Chem. Rev. Lett., 2018, 1, 49–55 CrossRef.
-
(a) S. Arshadi, E. Vessally, M. Sobati, A. Hosseinian and A. Bekhradnia, J. CO2 Util., 2017, 19, 120–129 CrossRef CAS;
(b) M. Babazadeh, S. Soleimani-Amiri, E. Vessally, A. Hosseinian and L. Edjlali, RSC Adv., 2017, 7, 43716–43736 RSC;
(c) E. Vessally, M. Babazadeh, A. Hosseinian, L. Edjlali and L. Sreerama, Curr. Org. Chem., 2018, 22, 199–205 CrossRef CAS;
(d) E. Vessally, M. Babazadeh, K. Didehban, A. Hosseinian and L. Edjlali, Curr. Org. Chem., 2018, 22, 286–297 CrossRef CAS;
(e) M. Daghagheleh, M. Vali, Z. Rahmani, S. Sarhandi and E. Vessally, Chem. Rev. Lett., 2018, 1, 23–30 Search PubMed;
(f) A. Hosseinian, S. Farshbaf, R. Mohammadi, A. Monfared and E. Vessally, RSC Adv., 2018, 8, 17976–17988 RSC;
(g) S. Shahidi, P. Farajzadeh, P. Ojaghloo, A. Karbakhshzadeh and A. Hosseinian, Chem. Rev. Lett., 2018, 1, 37–44 Search PubMed;
(h) S. Farshbaf, L. Zare Fekri, M. Nikpassand, R. Mohammadi and E. Vessally, J. CO2 Util., 2018, 25, 194–204 CrossRef CAS;
(i) S. Farshbaf, L. Sreerama, T. Khodayari and E. Vessally, Chem. Rev. Lett., 2018, 1, 56–67 Search PubMed;
(j) E. Vessally, A. Hosseinian, L. Edjlali, M. Babazadeh and R. Hosseinzadeh-Khanmiri, Iran. J. Chem. Chem. Eng., 2017, 36, 1–13 CAS.
- G. Majji, S. K. Rout, S. Rajamanickam, S. Guin and B. K. Patel, Org. Biomol. Chem., 2016, 14, 8178–8211 RSC.
-
(a) S. Moghimi, M. Mahdavi, A. Shafiee and A. Fouroumadi, Eur. J. Org. Chem., 2016, 3282–3299 CrossRef CAS;
(b) I. B. Krylov, V. A. Vil' and A. O. Terent'ev, Beilstein J. Org. Chem., 2015, 11, 92–146 CrossRef PubMed.
- Z. Ye, W. Wang, F. Luo, S. Zhang and J. Cheng, Org. Lett., 2009, 11, 3974–3977 CrossRef CAS PubMed.
- C. Chen, Y. Pan, H. Zhao, X. Xu, J. Xu, Z. Zhang, S. Xi, L. Xu and H. Li, Org. Chem. Front., 2018, 5, 415–422 RSC.
- K. Padala and M. Jeganmohan, Chem. Commun., 2013, 49, 9651–9653 RSC.
- K. Padala and M. Jeganmohan, Chem.–Eur. J., 2014, 20, 4092–4097 CrossRef CAS.
- N. Y. More, K. Padala and M. Jeganmohan, J. Org. Chem., 2017, 82, 12691–12700 CrossRef CAS.
- K. Raghuvanshi, K. Rauch and L. Ackermann, Chem.–Eur. J., 2015, 21, 1790–1794 CrossRef CAS.
- K. Raghuvanshi, D. Zell and L. Ackermann, Org. Lett., 2017, 19, 1278–1281 CrossRef CAS PubMed.
- T. Okada, K. Nobushige, T. Satoh and M. Miura, Org. Lett., 2016, 18, 1150–1153 CrossRef CAS PubMed.
- T. Sarkar, S. Pradhan and T. Punniyamurthy, J. Org. Chem., 2018, 83, 6444–6453 CrossRef CAS PubMed.
- P. B. De, S. Banerjee, S. Pradhan and T. Punniyamurthy, Org. Biomol. Chem., 2018, 16, 5889–5898 RSC.
- E. Kianmehr and S. B. Nasab, Eur. J. Org. Chem., 2019, 1038–1044 CrossRef CAS.
- C.-L. Sun, J. Liu, Y. Wang, X. Zhou, B.-J. Li and Z.-J. Shi, Synthesis, 2011, 883–886 CAS.
- C.-J. Hu, X.-H. Zhang, Q.-P. Ding, T. Lv, S.-P. Ge and P. Zhong, Tetrahedron Lett., 2012, 53, 2465–2468 CrossRef CAS.
- Y. Li, Y.-J. Ding, J.-Y. Wang, Y.-M. Su and X.-S. Wang, Org. Lett., 2013, 15, 2574–2577 CrossRef CAS PubMed.
- Z. Wang and C. Kuang, Adv. Synth. Catal., 2014, 356, 1549–1554 CrossRef CAS.
- S. K. Santra, A. Banerjee, N. Khatun and B. K. Patel, Eur. J. Org. Chem., 2015, 2015, 350–356 CrossRef CAS.
- J. Wu, K. L. M. Hoang, M. L. Leow and X.-W. Liu, Org. Chem. Front., 2015, 2, 502–505 RSC.
- K. K. Yu, Y. Guo, Y. H. Hu, Z. Xu, H. W. Liu, D. H. Liao and Y. F. Ji, Asian J. Org. Chem., 2016, 5, 1219–1224 CrossRef.
- J. Guo, H. He, Z. Ye, K. Zhu, Y. Wu and F. Zhang, Org. Lett., 2018, 20, 5692–5695 CrossRef CAS PubMed.
- J. Song, J. Cui, H. Liang, Q. Liu, Y. Dong and H. Liu, Asian J. Org. Chem., 2018, 7, 341–345 CrossRef CAS.
- J. Lan, H. Xie, X. Lu, Y. Deng, H. Jiang and W. Zeng, Org. Lett., 2017, 19, 4279–4282 CrossRef CAS PubMed.
- C. Lin, Z. Chen, Z. Liu and Y. Zhang, Adv. Synth. Catal., 2018, 360, 519–532 CrossRef CAS.
- R. Ueno, S. Natsui and N. Chatani, Org. Lett., 2018, 20, 1062–1065 CrossRef CAS PubMed.
-
(a) M. B. Gawande, A. Goswami, F.-X. Felpin, T. Asefa, X. Huang, R. Silva, X. Zou, R. Zboril and R. S. Varma, Chem. Rev., 2016, 116, 3722–3811 CrossRef CAS PubMed;
(b) H. Saeidian, M. Abdoli and R. Salimi, C. R. Chim., 2013, 16, 1063–1070 CrossRef CAS.
- Y. Wang, A. V. Gulevich and V. Gevorgyan, Chem.–Eur. J., 2013, 19, 15836–15840 CrossRef CAS.
- F. Wang, Q. Hu, C. Shu, Z. Lin, D. Min, T. Shi and W. Zhang, Org. Lett., 2017, 19, 3636–3639 CrossRef CAS.
- F. Wang, Z. Lin, W. Yu, Q. Hu, C. Shu and W. Zhang, RSC Adv., 2018, 8, 16378–16382 RSC.
- A. Ahmad, H. S. Dutta, B. Khan, R. Kant and D. Koley, Adv. Synth. Catal., 2018, 360, 1644–1649 CrossRef CAS.
- A. Pradal, P. Y. Toullec and V. Michelet, Org. Lett., 2011, 13, 6086–6089 CrossRef CAS PubMed.
- J.-J. Dai, W.-T. Xu, Y.-D. Wu, W.-M. Zhang, Y. Gong, X.-P. He, X.-Q. Zhang and H.-J. Xu, J. Org. Chem., 2015, 80, 911–919 CrossRef CAS PubMed.
- Y. Liu, W. Zhang, K. Xie and Y. Jiang, Synlett, 2017, 28, 1496–1500 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *d
*d