
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA photochemical and theoretical study of the triplet reactivity of furano- and pyrano-1,4-naphthoquionones towards tyrosine and tryptophan derivatives†
Rodolfo I. Teixeira a,
Juliana S. Goularta,
Rodrigo J. Corrêaa,
Simon J. Gardena,
Sabrina B. Ferreiraa,
José Carlos Netto-Ferreirab,
Vitor F. Ferreirac,
Paula Mirod,
M. Luisa Marind,
Miguel A. Mirandad and
Nanci C. de Lucas*a
a,
Juliana S. Goularta,
Rodrigo J. Corrêaa,
Simon J. Gardena,
Sabrina B. Ferreiraa,
José Carlos Netto-Ferreirab,
Vitor F. Ferreirac,
Paula Mirod,
M. Luisa Marind,
Miguel A. Mirandad and
Nanci C. de Lucas*a
aInstituto de Química – Universidade Federal do Rio de Janeiro, Cidade Universitária, RJ, Brazil. E-mail: nancicl@iq.ufrj.br
bDepartamento de Química – Universidade Federal Rural do Rio de Janeiro, Antiga Rio São Paulo, RJ, Brazil
cUniversidade Federal Fluminense, Faculdade de Farmácia, Departamento de Tecnologia Farmaceûtica, Niterói, Santa Rosa, Brazil
dInstituto de Tecnología Química, Universitat Politècnica de València-Consejo Superior de Investigaciones Científicas, Valencia, Spain
First published on 30th April 2019
Abstract
The photochemical reactivity of the triplet state of pyrano- and furano-1,4-naphthoquinone derivatives (1 and 2) has been examined employing nanosecond laser flash photolysis. The quinone triplets were efficiently quenched by L-tryptophan methyl ester hydrochloride, L-tyrosine methyl ester hydrochloride, N-acetyl-L-tryptophan methyl ester and N-acetyl-L-tyrosine methyl ester, substituted phenols and indole (kq ∼109 L mol−1 s−1). For all these quenchers new transients were formed in the quenching process. These were assigned to the corresponding radical pairs that resulted from a coupled electron/proton transfer from the phenols, indole, amino acids, or their esters, to the excited state of the quinone. The proton coupled electron transfer (PCET) mechanism is supported by experimental rate constants, isotopic effects and theoretical calculations. The calculations revealed differences between the hydrogen abstraction reactions of phenol and indole substrates. For the latter, the calculations indicate that electron transfer and proton transfer occur as discrete steps.
Introduction
Many natural and synthetic naphthoquinones are known to possess varied and potent biological properties including trypanocidal,1–3 antifungal,4–8 leishmanicidal,9–12 anti-inflammatory,13–16 and antitumor activities.17–22 In this sense, quinones are important cytotoxic compounds and some examples have been clinically used for cancer chemotherapy, for example, anthracycline antibiotics and mitomycin-C.23–25 In addition α-lapachone, a natural 1,4-naphthoquinone, and its derivatives present promising biological activity for example topoisomerase II-mediated DNA cleavage,26,27 trypanocidal,28,29 and antineoplastic activity.30–32The mechanism of action of these quinones is related to redox cycling, which can lead to the formation of reactive oxygen species that can damage cellular macromolecules and lead to cell death.33–36 In particular, cell death can be induced by apoptotic or necrotic signaling pathways induced by the combined use of a photosensitizer and light. Photosensitizing mechanisms can involve photooxidation of nucleic acid or protein components by the sensitizer yielding the corresponding radical pair. These radicals can lead to sensitizer-protein photo-binding and to the formation of other reactive oxygen species (ROS), such as superoxide anion, hydrogen peroxide and hydroxyl radical (type I mechanism). In addition, the excited photosensitizer can undergo triplet–triplet energy transfer to molecular oxygen, resulting in formation of singlet oxygen (1O21Δg, type II mechanism).36,37
Several mechanisms account for the photosensitization processes involving biomolecules, especially proteins and peptides, that result in cellular damage.36,38–40 Specifically targeting amino acids in metabolic enzymes is a promising strategy for cancer therapy and compounds that target tryptophan have been introduced in clinical trials.41 In this sense, the reactions of tryptophan (Trp) and tyrosine (Tyr) with photosensitizers have received considerable attention in the field of proteic photooxidation as these aromatic amino acid residues are easily oxidized.36,38,40,42,43 As a consequence, photosensitizers that direct photooxidation of these amino acid residues are of interest as they can play a role in photo-induced degradation of enzymes involved in cancer.40
The photophysics and photochemistry of 1,4-naphthoquinones have been extensively investigated both in organic solvents and in aqueous solution.44–47 Photoexcitation of 1,4-naphthoquinones leads to efficient intersystem crossing to the triplet state48 and from there, to reactions involving energy and hydrogen/electron transfer.49–58 Therefore, naphthoquinones can be used as potential photosensitizers, for example it has been shown that α-lapachone can photosensitize the one-electron oxidation of amino acid derivatives and it is also an efficient photosensitizer for formation of singlet oxygen.59
The synthetic tetrahydropyrano- and dihydrofurano-naphthoquinone compounds shown in Scheme 1 are similar to the natural product α-lapachone. They have shown potential as antifungal,60 antitubercular,61 trypanocidal,62 and antitumor63 agents as well as inhibitors of dengue virus replication.64 In a previous study, we reported that naphthoquinones 1 and 2 undergo intersystem crossing to the triplet state and that they are very efficient singlet oxygen generators, with experimental quantum yields in the range 0.7–1.65 Thus, these compounds can be considered as powerful type II photosensitizers. Additionally, it is important to study their photochemical reactivity with respect to type I photosensitization as the direct interaction of the triplet excited state with biological substrates can lead to the formation of radical species that result in the degradation of the biological material. Therefore, the present study has investigated the type I photosensitizer properties of the furano- and pyrano-1,4-naphthoquinones (Scheme 1) with respect to model biological substrates i.e. derivatives of tryptophan and tyrosine, by laser flash photolysis and theoretical calculations.
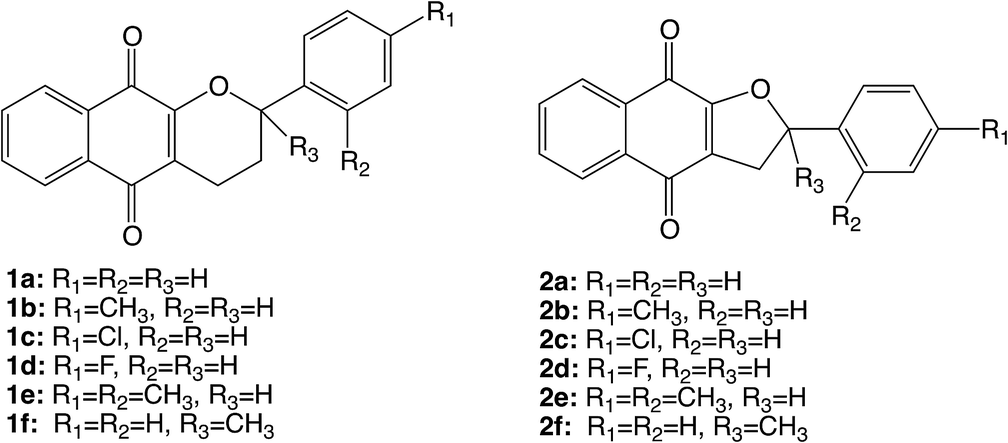 | ||
| Scheme 1 Structures of the furano- and pyrano-1,4-naphthoquinones. | ||
Experimental
A. General
L-Tyrosine methyl ester hydrochloride (TyrMe), L-tryptophan methyl ester hydrochloride (TrpMe), acetonitrile, 2-propanol (all spectroscopic grade), phenol, 4-cyanophenol, 4-methoxyphenol, indole, methylviologen and deuterated water were purchased from Sigma Aldrich or Tedia. Water was Milli-Q grade. N-Acetyl L-tryptophan methyl ester (NATrpME)66 and N-acetyl L-tyrosine methyl ester (NATyrME)67 were prepared by literature procedures. The naphthoquinone derivatives were prepared as described in the literature.60,61,68B. Quenching of naphthoquinones
Laser flash photolysis studies were carried out by using the 3rd harmonic of a Nd:YAG SL404G-10 Spectron Laser Systems or on a LuzChem Instrument model mLFP122 with an excitation wavelength of 355 nm. The energy was adjusted to ∼17 μJ per pulse. The detecting light source was obtained from a pulsed Lo255 Oriel Xenon lamp. The laser flash photolysis system consisted of the pulsed laser, the Xe lamp, a 77200 Oriel monochromator, a photomultiplier equipment (Oriel, model 70705) and a TDS-640A Tektronix oscilloscope.Samples were contained in 10 × 10 mm cells made of Suprasil quartz and were deaerated for at least 20 min with dry, oxygen-free, nitrogen or argon prior to the experiments. All laser flash photolysis experiments were performed in acetonitrile solution, unless otherwise indicated in the text. The concentration of naphthoquinones was adjusted to yield an absorbance of ∼0.3 at the excitation wavelength (355 nm). Stock solutions of quenchers were prepared so that it was only necessary to add microliter volumes to the sample cell in order to obtain appropriate concentrations of the quencher. The rate constants for the reaction of triplet naphthoquinones with the different quenchers employed in this study were obtained from Stern–Volmer plots, following eqn (1).
| kobs = ko + kq[Q] | (1) |
C. Computational methods
All calculations were performed using the Gaussian 09.C package of programs.69 Geometries and properties were calculated with (U)B3LYP/6-311++G(d,p)//(U)B3LYP/6-31+G(d). Solvent effects were implicitly included by use of the IEFPCM method and acetonitrile as solvent.69 The DFT method was used as it reasonably eliminates spin contamination70 and values for 〈S2〉 were consistent with two unpaired electrons. The optimized structures were confirmed as energy minima or as transition states by vibrational analysis (absence or presence of a single imaginary frequency that corresponded to vibration along the reaction coordinate).Results and discussion
A. Quenching of naphthoquinones
The characterization of the triplet excited state of the naphthoquinone series shown in Scheme 1 has been previously reported by us.65 Upon laser excitation, in acetonitrile, the quinones 1 and 2 resulted in the formation of the corresponding triplet excited state with ΦISC close to 1. Triplets of 1 show absorption maxima at 390 and 450 nm, and for 2 at 370 nm and an apparent shoulder at about 450 nm. For examples of triplet–triplet absorption spectra see Fig. S1 in the ESI.†The transient absorption spectra for the 1 and 2 series recorded in 2-propanol are quite different from that observed in acetonitrile. As an example, Fig. 1 shows the spectra for 1a in 2-propanol, whereas the Fig. S2 in the ESI† shows the spectra for 2a. This solvent can act as a hydrogen donor and the spectra at long delay times show a band with maximum at ∼370 nm (for both 1 and 2 series). The decay associated with this band shows a fast component related to the triplet state and a residual absorption which is due to the semiquinone radical resulting from a hydrogen abstraction reaction from 2-propanol by the triplet 1a (inset Fig. 1).71 The formation of the semiquinone radical was confirmed by experiments in the presence of methylviologen. Electron donation from a ketyl radical to methylviologen can generate the methylviologen cation radical which shows absorption bands at 398 nm and 603 nm (Fig. 2, eqn (2)).49,72–74 In a similar manner, photoinduced hydrogen abstraction from the furanose sugar ring of deoxyribose results in the formation of sugar radicals that can result in DNA cleavage.75
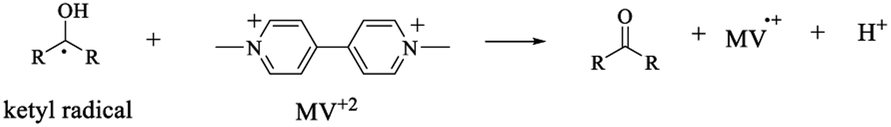 | (2) |
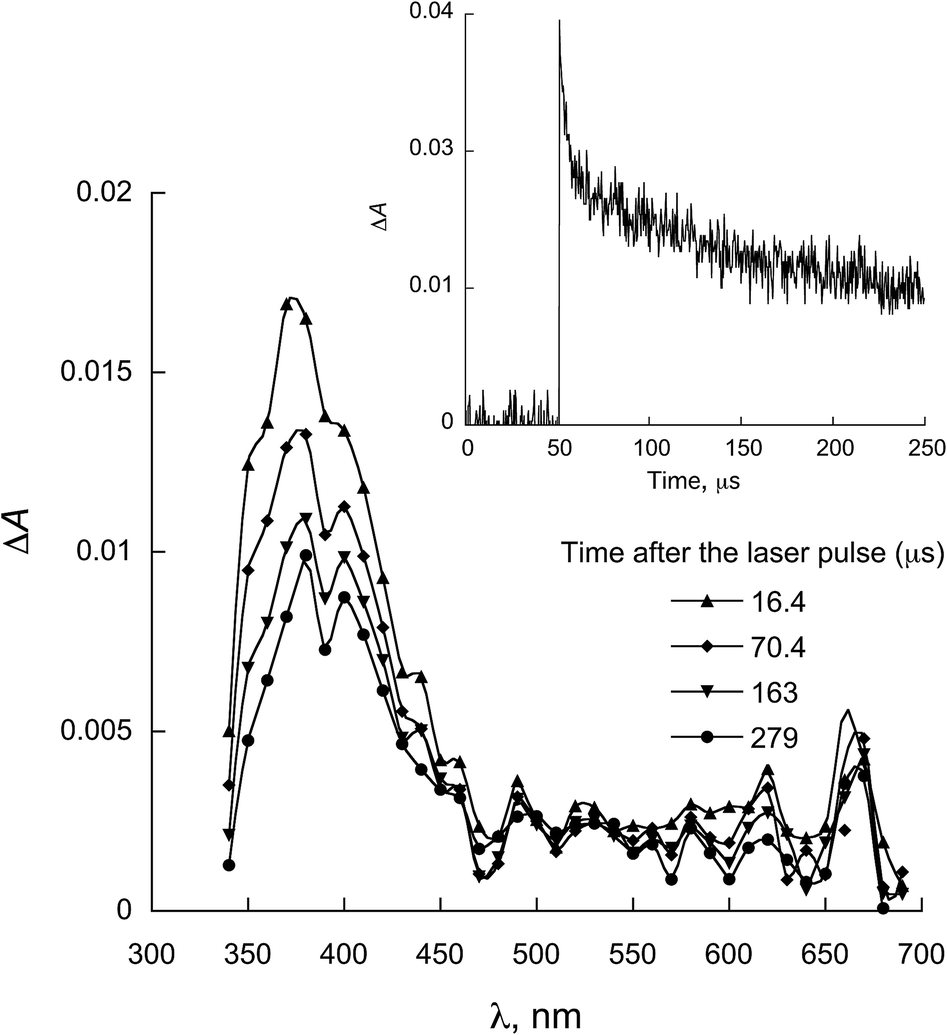 | ||
| Fig. 1 Transient absorption spectrum recorded upon excitation (355 nm) of 1a in 2-propanol. Inset: decay at 380 nm. | ||
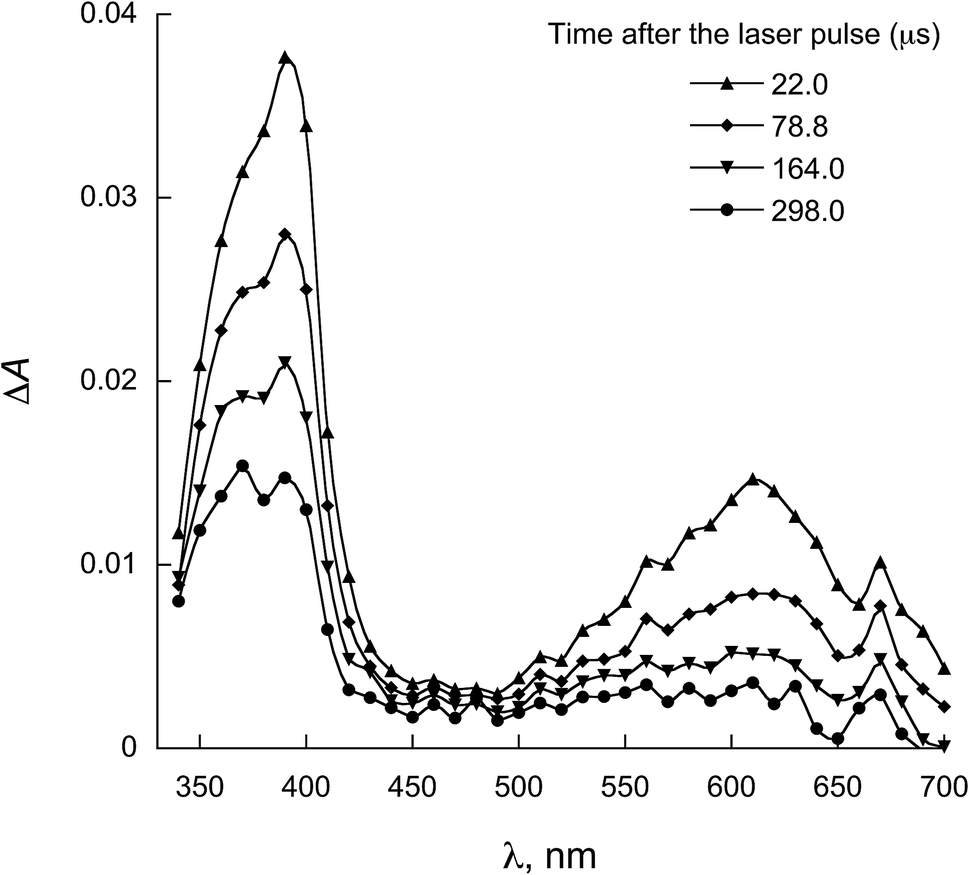 | ||
| Fig. 2 Transient absorption spectra obtained upon 355 nm excitation of 1a in 2-propanol containing methylviologen. | ||
Oxidative damage is a principal cause of cellular death and maximizing photooxidation reactions is one of the main objectives of photodynamic therapy (PDT). We have previously shown that compounds 1 and 2 can act as type II photosensitizers and generate singlet oxygen. However, the concentration of oxygen can vary within the cellular medium and therefore to consider only the use of singlet oxygen as a means for promoting protein damage is simplistic.76 In addition to amino-acids being oxidized by singlet oxygen at physiological pH,77 other mechanisms involving electron and hydrogen transfer processes can contribute to photoinduced oxidation of these molecules.38,39 L-Tryptophan methyl ester hydrochloride (TrpMe), L-tyrosine methyl ester hydrochloride (TyrMe), N-acetyl L-tryptophan methyl ester (NATrpME) and N-acetyl L-tyrosine methyl ester (NATyrME) have been employed, as models for amino acid constituents of proteins, to act as quenchers for the study of the type I photosensitizer reactivity of the naphthoquinone (1 and 2) triplet excited states in acetonitrile.36 Indole and phenols were also employed as quenchers in order to compare their reactivity with the biomolecules.
In all cases, linear quenching plots following eqn (1) were obtained and the resulting quenching rate constants (kq) are of the order of 108 to 109 L mol−1 s−1, as shown in Table 1. Fig. 3 and 4 show representative Stern–Volmer plots and kinetic traces with and without quencher, NATrpME and NATyrME, for 1c (for more examples of quenching plots see Fig. S3–S7 in the ESI†). The large values for the quenching rate constants for the amino acid derivatives, as well as their similarity to those obtained for phenol or indole, indicate that analogous quenching mechanisms may be involved.
| akq × 109/L mol−1 s−1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| TyrMe | TrpMe | NATyrME | NATrpME | Phenol | 4-CN phenol | 4-MeO phenol | Indole | |
a Error ± 10%.b CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (9:1, v/v).c CH3CN:D2O (9:1, v/v). :H2O (9:1, v/v).c CH3CN:D2O (9:1, v/v). |
||||||||
| 1a | 1.22 | 3.45 | 0.14 | 2.20 | 0.49 | 0.22 | 1.92 | 0.90 |
| 1b | 1.28 | 2.39 | 0.69 | 2.10 | 0.77 | 0.33 | 1.10 | 1.26 |
| 1c | 1.53 | 3.25 | 0.82 | 1.75 | 0.74 | 0.21 | 1.95 | 3.37 |
| 1d | 1.42 | 1.84 | 0.85 | 1.30 | 0.98 | 0.20 | 1.08 | 1.39 |
| 1e | 1.04 | 3.65 | 0.72 | 1.70 | 0.73 | 0.25 | 1.89 | 1.99 |
| 1f | 1.02 | 1.92 | 0.80 | 2.29 | 0.84 | 0.45 | 0.96 | 1.62 |
| 0.89b | 1.90b | 0.71b | 1.85b | |||||
| 0.78c | 1.80c | 0.59c | 1.27c | |||||
| 2a | 1.01 | 3.01 | 0.53 | 1.55 | 0.45 | 0.14 | 2.57 | 2.91 |
| 2b | 0.86 | 2.22 | 0.36 | 1.43 | 0.25 | 0.06 | 1.66 | 2.38 |
| 0.76b | 1.77b | 0.99b | 1.38b | |||||
| 0.59c | 1.40c | 0.61c | 1.22c | |||||
| 2c | 1.17 | 3.63 | 0.61 | 1.25 | 0.57 | 0.04 | 1.92 | 1.60 |
| 2d | 1.15 | 3.05 | 0.60 | 1.19 | 0.25 | 0.10 | 1.65 | 3.00 |
| 2e | 1.42 | 2.53 | 0.55 | 1.20 | 0.49 | 0.09 | 1.37 | 2.18 |
| 2f | 1.37 | 2.16 | 0.58 | 3.95 | 0.41 | 0.12 | 1.00 | 3.56 |
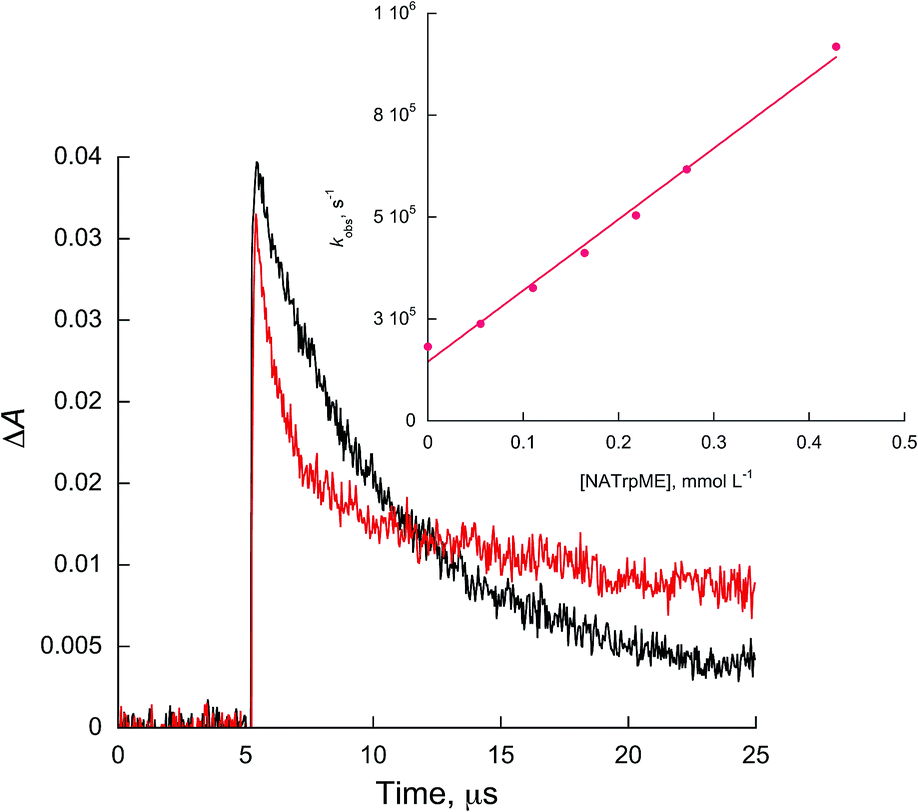 | ||
| Fig. 3 Kinetic traces recorded at 450 nm for 1c without (black) and with 0.27 mmol L−1 of NATrpME (red) in ACN. Inset: quenching plots of 1c by NATrpME. | ||
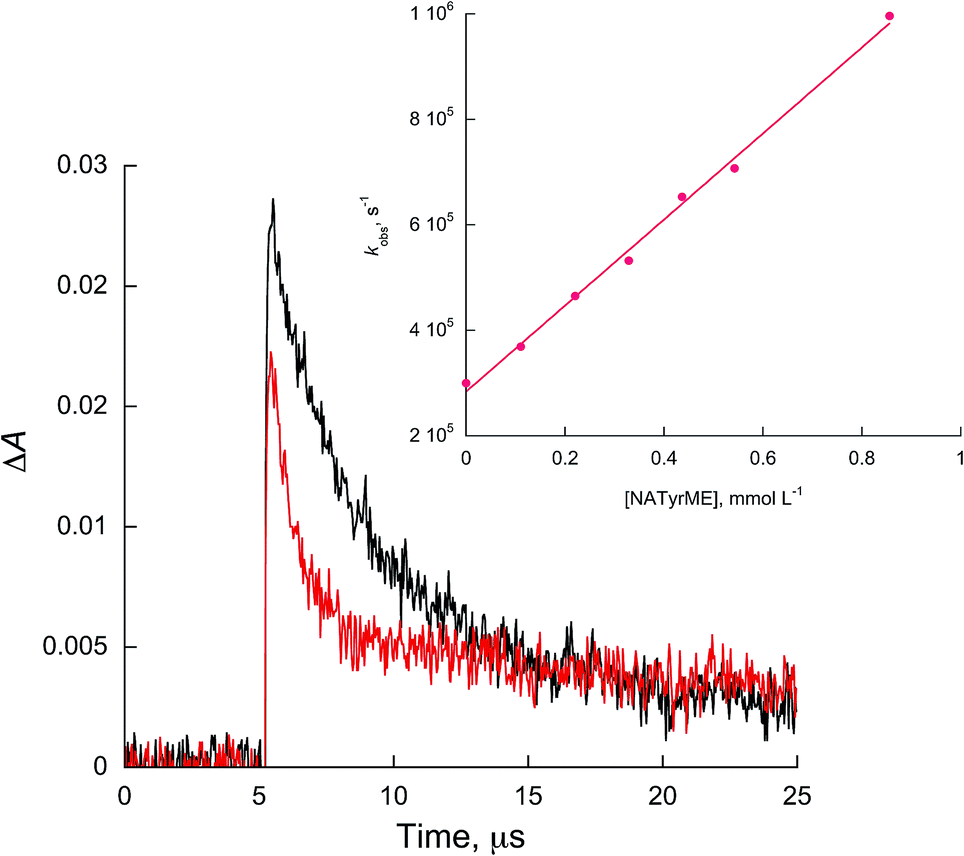 | ||
| Fig. 4 Kinetic traces recorded at 450 nm for 1c in acetonitrile without (black) and with 0.54 mmol L−1 of NATyrME (red). Inset: quenching plots of 1c by NATyrME. | ||
Fig. 5 and 6 show the transient absorption spectra recorded upon laser excitation of an acetonitrile solution of 1a and 2a in the presence of 0.28 mmol L−1 of NATrpME. A strong absorption band at aprox. 370 nm can be attributed to the corresponding semiquinone radical derived from the quinone and a broad absorption in the 450–550 nm region to an indolyl-like radical (eqn (3)) as indicated in the literature.78–80 Thus, the 450 nm absorption on the 500 ns time scale is due to the triplet excited state of 1a (see Fig. S1 in the ESI†). On the other hand, at longer time scales we can observe an absorption between 450–550 nm with maximum at 510 nm due to the tryptophanyl-like radical. In fact, experiments using indole as quencher revealed the formation of a similar transient, i.e. the indolyl radical, which results from the N–H hydrogen abstraction (for further examples see Fig. S8 and S9 in the ESI†). The inset in Fig. 6 shows the kinetic trace at 370 nm, that clearly indicates the fast component in this region related to the triplet and the longer component related to the semiquinone radical formed from the hydrogen abstraction. When the kinetic trace is record at 450 nm the short and the long-lived components related to the triplet and to the indolyl radical, respectively, are both observed.
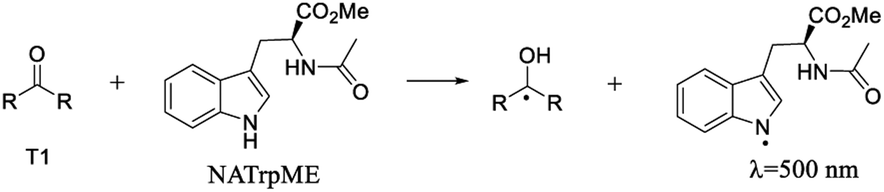 | (3) |
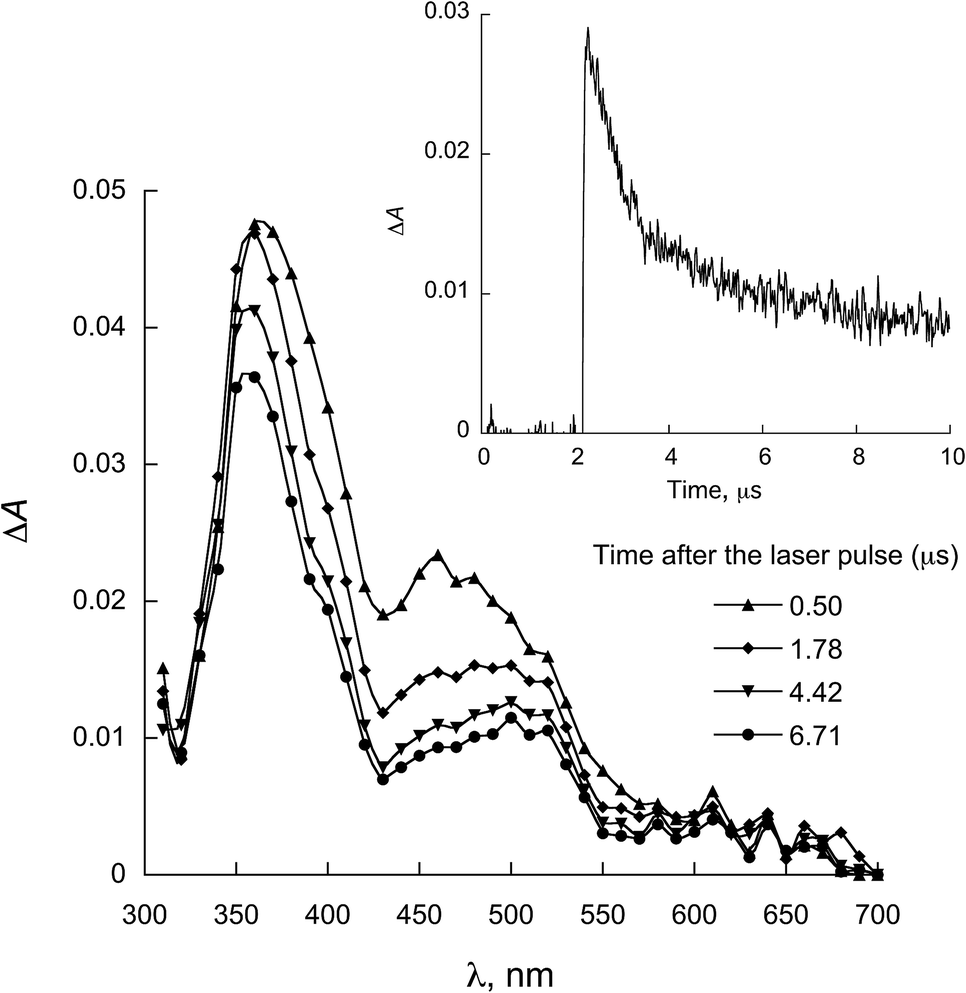 | ||
| Fig. 5 LFP spectra observed for 1a in the presence of 0.28 mmol L−1 NATrpMe in ACN (λexc = 355 nm). Inset: decay at 450 nm. | ||
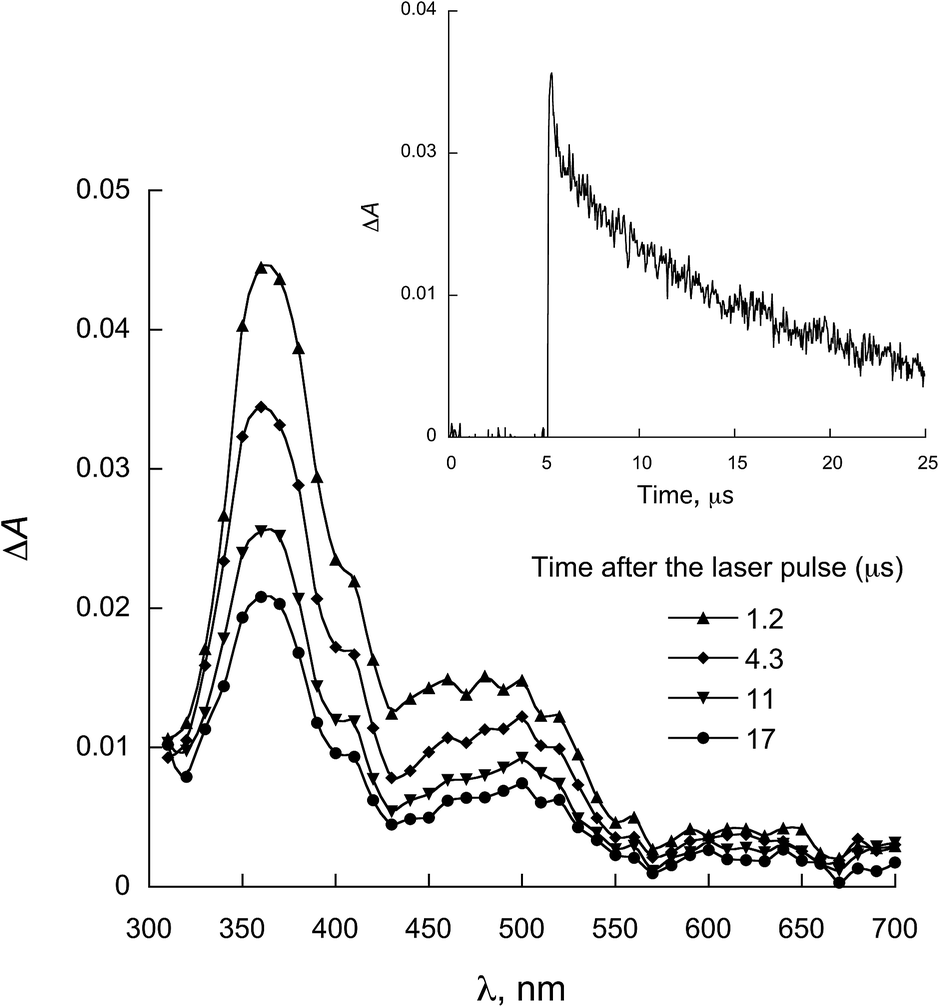 | ||
| Fig. 6 LFP spectra observed for 2a in the presence of 0.28 mmol L−1 NATrpME in ACN (λexc = 355 nm). Inset: decay at 370 nm. | ||
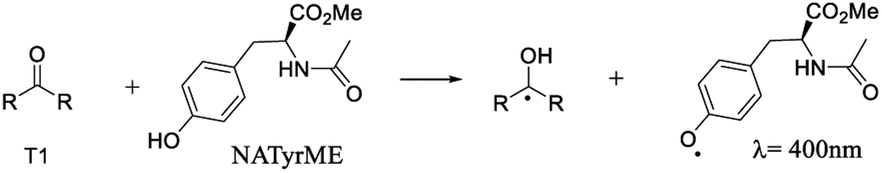
When NATyrME was employed as a quencher, a strong absorption band in the 340–420 nm region was readily observed. Fig. 7 and 8 show representative examples of the transient absorption spectra obtained for 1b and 2a in the presence of NATyrME. It is well known that carbonyl triplets readily abstract hydrogen from phenols and that this reaction leads to the formation of the corresponding phenoxyl radical that have strong absorption bands in the 370–480 nm region (405 nm for phenol) depending upon the nature of the phenol substituent.81–83 As the ketyl radical and the phenoxyl radical absorption are in the same region, the strong absorption band observed can be attributed to the overlap of the absorptions of the corresponding semiquinone radical and the tyrosinyl radical as a result of phenolic hydrogen abstraction from NATyrME (eqn (4)). This conclusion is supported by the fact that the same transients are observed when phenol is used as a quencher (see Fig. S10 and S11 in ESI†). It is also known that the reactivity of phenols toward the carbonyl triplets is also dependent upon the nature of the phenolic substituent.82 Thus, phenols with an electron donating substituent, such as 4-MeO, react faster than phenols with an electron withdrawing substituent, such as 4-CN, as seen from the quenching rate constants in Table 1.
 | (4) |
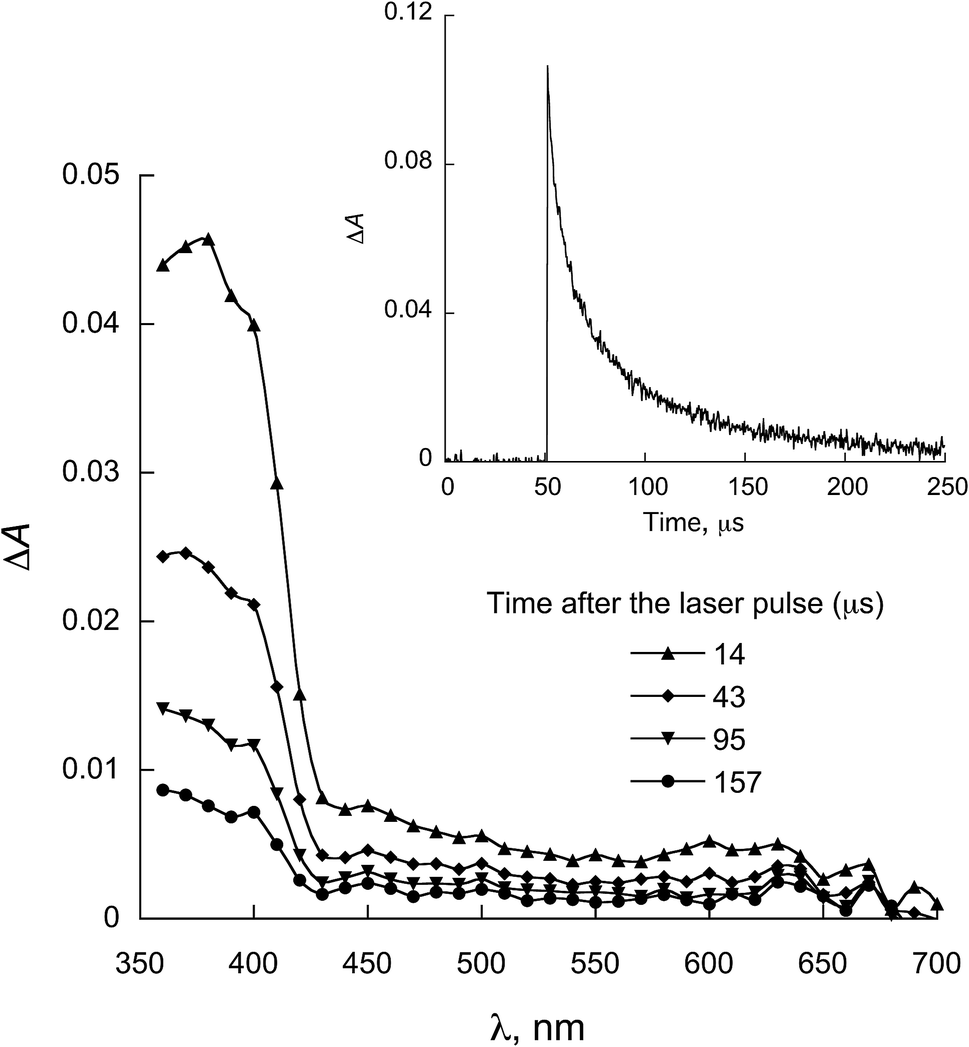 | ||
| Fig. 7 LFP spectra observed for 1b in the presence of 0.58 mmol L−1 NATyrME in ACN (λexc = 355 nm). Inset: decay at 380 nm. | ||
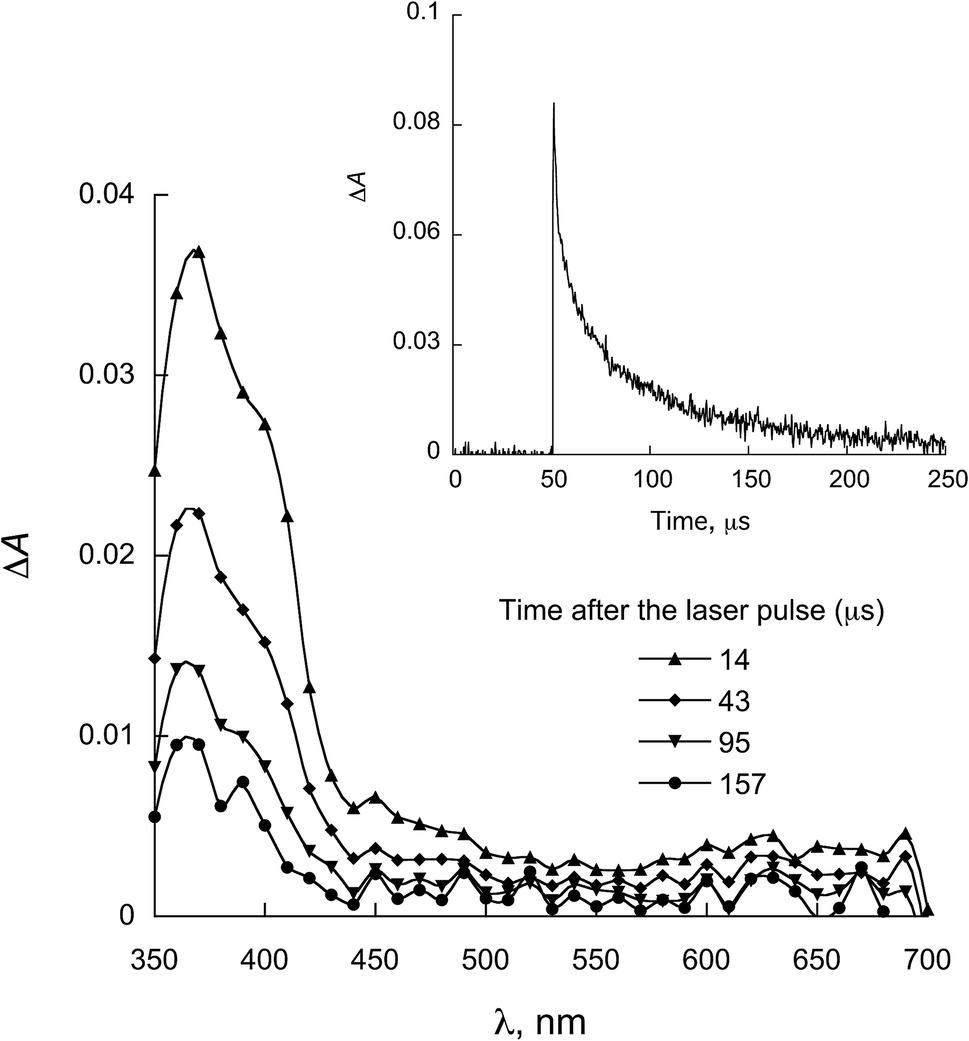 | ||
| Fig. 8 LFP spectra observed for 2a in the presence of 0.58 mmol L−1 NATyrME in ACN (λexc = 355 nm). Inset: decay at 380 nm. | ||
The similarity of the values for the quenching rate constants of triplet naphthoquinones by NATrpME and indole and by NATyrME and phenol as well as the formation of the radical pair naphthoquinone semiquinone/indolyl (or phenoxyl) radicals suggests that similar reaction mechanisms may be operating in both quenching processes. A series of previous experimental and theoretical studies from our groups,58,71,84–87 have clearly indicated that a proton coupled electron transfer (PCET) mechanism88–96 operates in the hydrogen atom transfer reaction for both phenols and indole.
To further investigate the PCET process, two representative quinones, 1f and 2b (six and five-membered ring 1,4-naphthoquinones, respectively) were chosen in order to investigate the isotope effect upon the quenching rate constants for the reactions of the naphthoquinones with L-tyrosine methyl ester hydrochloride (TyrMe) and L-tryptophan methyl ester hydrochloride (TrpMe) as well as the respective N-acetyl derivatives NATyrME and NATrpME. For these studies, mixtures of CH3CN/H2O and CH3CN/D2O (9:1 v/v) were employed as the solvent. Isotope effects for 1f/TyrMe: 1.15; 1f/TrpMe: 1.06; 1f/NATyrME: 1.20; 1f/NATrpME: 1.46; 2b/TyrMe: 1.28; 2b/TrpMe: 1.26; 2b/NATyrME: 1.62; 2b/NATrpME: 1.13 were observed (see Table 1 for the rate constant values). These values indicate that stretching of the O–H bond in TyrMe or NATyrME or of the N–H bond in TrpMe or NATrpME is not important in the transition state, strongly indicating that an asynchronous, electron first, PCET mechanism is operating.97–102
With respect to the compounds used in the present study, the substituted phenyl groups are not conjugated with the quinone system and the electronic effect of the substituted phenyl group is therefore limited to a possible inductive effect upon the furano- or pyrano- ring oxygen. In a general manner, the quenching rate constants by indolic quenchers and 4-methoxyphenol are larger than the quenching rate constants for the tyrosine derivatives, phenol and 4-cyanophenol reflecting the more electron rich nature of the indolic derivatives. Further, the quenching rate constants for a given quencher were similar for the respective quinone 1 or 2, indicating that ring size had a minimal effect. The reactivity of 1 and 2, as detailed in Table 1 is similar to that reported for α-lapachone.59 However, rate constants for tryptophan derivatives are 2 to 3 times larger than the previously reported values for α-lapachone (1.3 × 109 L mol−1 s−1 for TrpMe). The quenching rate constants observed in the present study are similar to those observed for 1,4-naphthoquinone,103 although larger than those observed for alkyl derivatives such as 2,3-dimethyl-1,4-naphthoquinone.103
In the present study, laser flash photolysis experiments in the presence of biological model quenchers have established that the naphthoquinones 1 and 2 can readily act as type I photosensitizers. In a biological context, the radicals formed in this process could significantly increase the formation of reactive oxygen species (ROS). This combined with the type II photosensitizer properties of the quinones 1 and 2, to generate singlet oxygen, could result in oxidative stress and cell death. Additionally, type I photosensitization via PCET from tyrosine or tryptophan could contribute to protein damage and enzyme inactivation,104–107 as a consequence of further transformation of the amino acid radicals via oxidation and/or dimerization reactions.108
B. Theoretical calculations
Theoretical calculations were used to further investigate the hydrogen abstraction processes from amino acid derivatives by quinones 1 and 2. As the reactive site of the tyrosine and tryptophan derivatives are respectively the phenolic OH bond and the indolic NH bond, these latter compounds (which were also experimentally investigated) were used as models for the calculations.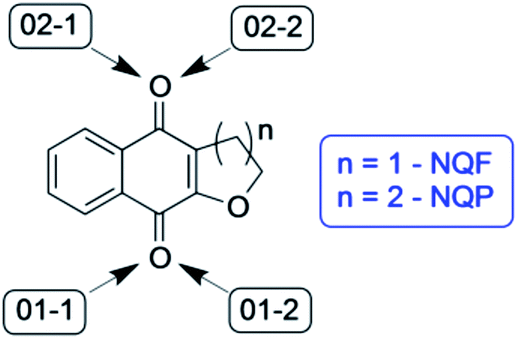 | ||
| Fig. 9 Identification of the four possible sites for hydrogen abstraction by NQF and NQP. | ||
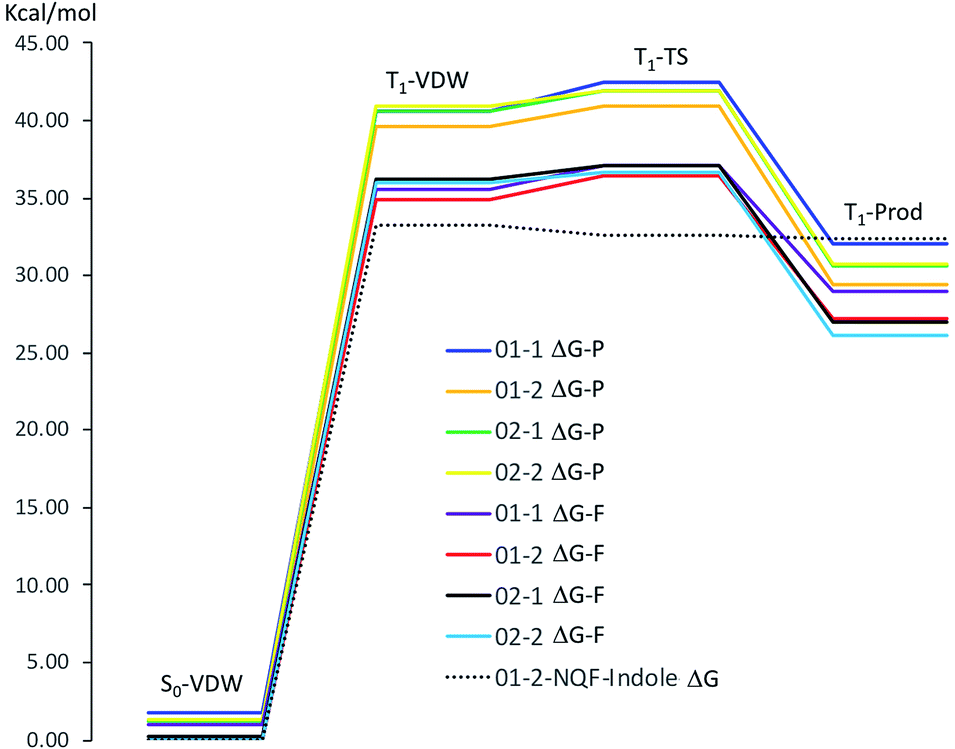 | ||
| Fig. 10 Gibbs free energy of the respective stationary points along the reaction coordinates for hydrogen abstraction from phenol by NQP and NQF (ΔG-P, NQP coordinate; ΔG-F, NQF coordinate; 01−1(2) and 02−1(2) as defined in Fig. 9) and for hydrogen abstraction from indole by NQF (01−2). | ||
| ΔG (ΔH) (kcal mol−1) | S0-VDW | T1-VDW | T1-TS | T1-prod | ΔΔG‡ (ΔΔH‡) (kcal mol−1) |
|---|---|---|---|---|---|
| NQP | |||||
| 01−1 | 1.80 (1.04) | 40.66 (40.26) | 42.48 (41.09) | 32.05 (31.55) | 1.82 (0.83) |
| 01−2 | 0.00 (0.00) | 39.64 (39.43) | 40.99 (39.29) | 29.46 (29.17) | 1.35 (−0.15) |
| 02−1 | 1.23 (0.43) | 40.66 (40.84) | 41.91 (40.83) | 30.59 (30.25) | 1.25 (−0.01) |
| 02−2 | 1.30 (0.24) | 40.96 (40.96) | 41.89 (40.21) | 30.70 (30.05) | 0.93 (−0.75) |
|
|||||
| NQF | |||||
| 01−1 | 1.02 (1.24) | 35.58 (36.07) | 37.13 (37.38) | 28.97 (29.70) | 1.55 (1.31) |
| 01−2 | 0.00 (0.00) | 34.95 (34.83) | 36.43 (35.16) | 27.23 (27.61) | 1.48 (0.33) |
| 02−1 | 0.21 (0.52) | 36.19 (36.65) | 37.09 (37.07) | 27.02 (27.84) | 0.90 (0.43) |
| 02−2 | 0.02 (0.11) | 35.95 (36.35) | 36.62 (35.70) | 26.09 (26.67) | 0.67 (−0.65) |
The phenol-NQP reaction coordinate is approximately 4 kcal mol−1 vertically shifted in comparison to the phenol-NQF reaction coordinate due to the difference in energy of the respective T1 states of the quinones (T1NQP ΔG(ΔH) = 40.42(41.24) kcal mol−1, T1NQF ΔG(ΔH) = 36.49(36.87) kcal mol−1 relative to the respective S0 states). The respective T1-VDW complexes are almost isoenergetic with the relaxed T1 state of the respective quinone. The small energy differences between the four potential sites for hydrogen abstraction (≤1.3 kcal mol−1) in the T1-VDW complexes indicates that all sites can participate in the hydrogen abstraction reaction. Very small free energy activation barriers (ΔΔG‡) are observed for both NQP and NQF hydrogen abstraction from phenol. These barriers are associated with a decrease in entropy of the system as a result of greater organization of the transition state relative to the T1-VDW complex.
Table 3 details the partial electronic charges associated with the molecular fragments for the stationary points along the hydrogen abstraction reaction coordinate for each of the four possible molecular arrangements of the phenol–quinone reaction complex. The table also details the variation in the phenoxyl–hydrogen (PhO⋯H) and hydrogen–quinone (H⋯Oq) bond lengths along the respective reaction coordinates.
| Chelpg (e)a O⋯H (Å) | NQF-phenol | NQP-phenol | ||||||
|---|---|---|---|---|---|---|---|---|
| S0-VDW | T1-VDW | T1-TS | T1-prod | S0-VDW | T1-VDW | T1-TS | T1-prod | |
| a Any small variation from a total charge of zero electrons is a consequence of limiting the number of significant figures associated with the fragmented partial charges. | ||||||||
| NQ-01-1 | 0.033 | 0.042 | −0.639 | −0.575 | 0.049 | 0.048 | −0.569 | −0.560 |
| H | 0.445 | 0.477 | 0.528 | 0.519 | 0.423 | 0.467 | 0.517 | 0.501 |
| Phenol (–H) | −0.478 | −0.519 | 0.111 | 0.055 | −0.472 | −0.515 | 0.052 | 0.059 |
| PhO⋯H | 0.982 | 0.989 | 1.075 | 1.728 | 0.982 | 0.990 | 1.054 | 1.741 |
| H⋯Oq | 1.836 | 1.744 | 1.391 | 0.995 | 1.827 | 1.738 | 1.441 | 0.993 |
| NQ-01-2 | 0.100 | 0.112 | −0.536 | −0.518 | 0.105 | 0.118 | −0.451 | −0.481 |
| H | 0.389 | 0.407 | 0.486 | 0.494 | 0.372 | 0.377 | 0.456 | 0.451 |
| Phenol (–H) | −0.489 | −0.519 | 0.049 | 0.023 | −0.477 | −0.495 | −0.005 | 0.030 |
| PhO⋯H | 0.982 | 0.990 | 1.067 | 1.705 | 0.983 | 0.991 | 1.043 | 1.767 |
| H⋯Oq | 1.863 | 1.755 | 1.427 | 1.001 | 1.876 | 1.760 | 1.500 | 0.997 |
| NQ-02-1 | 0.067 | 0.060 | −0.535 | −0.506 | 0.086 | 0.084 | −0.575 | −0.515 |
| H | 0.398 | 0.430 | 0.462 | 0.443 | 0.372 | 0.403 | 0.453 | 0.445 |
| Phenol (–H) | −0.466 | −0.490 | 0.073 | 0.063 | −0.458 | −0.487 | 0.122 | 0.070 |
| PhO⋯H | 0.984 | 0.987 | 1.059 | 1.751 | 0.984 | 0.987 | 1.049 | 1.758 |
| H⋯Oq | 1.795 | 1.768 | 1.426 | 0.992 | 1.796 | 1.766 | 1.450 | 0.991 |
| NQ-02-2 | 0.059 | 0.070 | −0.504 | −0.564 | 0.070 | 0.071 | −0.496 | −0.529 |
| H | 0.451 | 0.406 | 0.485 | 0.516 | 0.436 | 0.412 | 0.485 | 0.497 |
| Phenol (–H) | −0.510 | −0.475 | 0.019 | 0.048 | −0.506 | −0.484 | 0.011 | 0.031 |
| PhO⋯H | 0.986 | 0.989 | 1.065 | 1.719 | 0.984 | 0.988 | 1.049 | 1.752 |
| H⋯Oq | 1.791 | 1.757 | 1.422 | 0.996 | 1.802 | 1.763 | 1.465 | 0.993 |
Comparison of the fragmented charges for the T1-VDW complex and the S0-VDW complex is instructive in that the differences are very minor, revealing that neither electron or proton transfer spontaneously occurs. An orientation effect is observed when comparing NQ-01-1 and NQ-01-2 (for both NQF and NQP) in that the latter reveals a larger partial positive charge associated with the quinone fragment as a consequence of increased hydrogen bonding to the phenoxyl proton despite a slightly longer H⋯Oq bond length. This is consistent with the energetics detailed in Table 2 where the structures NQ-01-2 (NQF and NQP) are the thermodynamically more stable molecular arrangements. The largest change in partial charge distribution is observed in the transition state. For both NQF and NQP, in all molecular geometries, partial electron transfer has occurred increasing the electron density upon the quinone and reducing the electron density of the phenoxyl fragment. Proton transfer is incomplete in the TS as seen from the slightly stretched PhO⋯H bond length and the considerably reduced H⋯Oq bond length as compared with the respective T1-VDW structures. On passing through the TS, proton and electron transfer are completed to give the triplet product with short H⋯Oq and long PhO⋯H bond lengths. A residual partial positive charge on the phenoxyl radical reveals a hydrogen bonding interaction with the semi-quinone radical. Thus hydrogen abstraction from phenol by the triplet quinone is best characterized as an asynchronous PCET mechanism.88–96
| S0-VDW | T1-VDW | T1-TS | TRP | T1-π01c | T1-π02c | |
|---|---|---|---|---|---|---|
| a Calculations: (U)B3LYP/6-311++G(d,p)//6-31+G(d), IEFPCM, solvent = acetonitrile.b Indole fragment less the “hydrogen atom”.c π–π T1-complex between NQF and indole, 01 and 02 differ in the respective orientations of the indole and NQF units (see ESI). Energies are given relative to S0-VDW. | ||||||
| Thermochema (kcal mol−1) | ||||||
| ΔH | 0.00 | 32.08 | 30.19 | 31.06 | 36.45 | 37.53 |
| ΔG | 0.00 | 33.28 | 32.63 | 32.36 | 38.23 | 39.39 |
|
||||||
| Charges (e)a | ||||||
| NapQfuran | 0.045 | −0.838 | −0.729 | −0.569 | −0.363 | −0.376 |
| H | 0.385 | 0.394 | 0.497 | 0.462 | 0.358 | 0.385 |
| Indole(–H)b | −0.430 | 0.444 | 0.232 | 0.107 | 0.005 | −0.009 |
|
||||||
| Bond lengths (Å) | ||||||
| Indole N⋯H | 1.019 | 1.071 | 1.286 | 1.678 | 1.012 | 1.012 |
| NapQ⋯H | 1.985 | 1.588 | 1.206 | 1.023 | ||
With respect to changes in electron density (Fig. 11 and Table 4), in the ground state a VDW complex between indole and the quinone reveals that the indole moiety has greater electron density than the quinone moiety as a consequence of electron pair donation from the quinone to form a hydrogen bond with the indole. This serves as a reference point for describing the relative changes in electron density of the stationary points along the reaction coordinate. Experimentally, what is observed is the diffusion-controlled encounter between the triplet excited state quinone and the hydrogen donor to give an exciplex. This is simulated as the T1-VDW encounter complex. The electrostatic charge differential is at a maximum in the T1-VDW complex where electron transfer from indole to the T1 quinone has already been observed to occur, but proton transfer has not proceeded to any extent. The transfer of the proton to the radical anion of the quinone in the transition state reduces the deficit of electron density on the indole radical and results in the formation of the triplet radical pair of products (TRP).
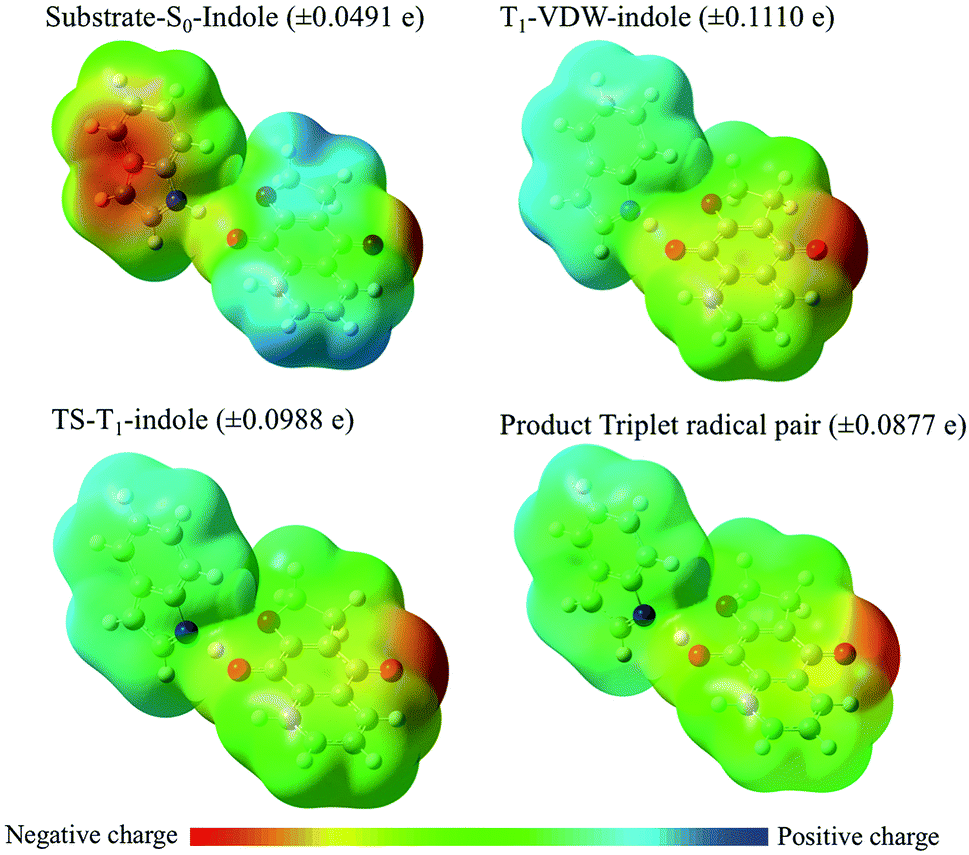 | ||
| Fig. 11 Electrostatic potential maps for the hydrogen abstraction reaction coordinate from indole by NQF calculations: (U)B3LYP/6-311++G(d,p)//6-31+G(d). Maximum and minimum surface charges are given in parenthesis for each structure. | ||
Notably, a significantly smaller degree of electron transfer is observed in the π–π stacking exciplexes (T1-π01 and T1-π02) in comparison to the hydrogen bonded T1-indole–quinone (01−2) exciplex (T1-VDW). This major difference reveals that the degree of electron transfer depends upon the respective orientations of electron donor and acceptor (molecular geometry of the VDW complex) and specific molecular orbital interactions for electron/proton transfer.
The larger rate constants (Table 1) for hydrogen transfer from indole or tryptophan derivatives to the quinone triplets is consistent with the advanced electron transfer from the indole moiety prior to proton transfer in a thermodynamically barrier-less transition state.
Conclusion
The laser flash photolysis technique was used to show that compounds 1 and 2 can react as type 1 photosensitizers, in that they readily participate in hydrogen abstraction reactions to generate radical species. Experiments using amino acid derivatives as quenchers have shown that the respective quinone triplet excited state can oxidize tyrosine and tryptophan derivatives to produce the respective ketyl/tyrosyl (or tryptophanyl) radical pairs. The quenching rate constants, isotopic effect experiments, and theoretical calculations indicate that a PCET mechanism is involved in the reaction with the amino acid derivatives. Calculations further revealed that the degree of electron transfer preceding proton transfer was dependent upon the nature of the quencher with distinct differences for phenol and indole reactivity, consistent with the larger rate constants for indolic quenching of the quinone triplet. For the former, transition states involving coupled electron/proton transfer with small activation energy barriers were found whilst for the latter, electron transfer readily occurred from indole in the T1-VDW encounter complex and proton transfer occurred through a barrier-less transition state as a consequence of the coulombic interaction between the quinone radical anion and the indole radical cation. The potential for these quinones to act as both type I and type II photosensitizers is of interest for biological applications involving the production of ROS and other reactive intermediates that may provoke cellular responses.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
The authors thank the following Brazilian agencies Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) for financial assistance. The authors also thank the Generalitat Valenciana (Prometeo Program).Notes and references
- I. Sieveking, P. Thomas, J. C. Estévez, N. Quiñones, M. A. Cuéllar, J. Villena, C. Espinosa-Bustos, A. Fierro, R. A. Tapia, J. D. Maya, R. López-Muñoz, B. K. Cassels, R. J. Estévez and C. O. Salas, Bioorg. Med. Chem., 2014, 22, 4609–4620 CrossRef CAS PubMed.
- A. D. R. Louvis, N. A. A. Silva, F. S. Semaan, F. D. C. Da Silva, G. Saramago, L. C. S. V. De Souza, B. L. A. Ferreira, H. C. Castro, J. P. Salles, A. L. A. Souza, R. X. Faria, V. F. Ferreira and D. D. L. Martins, New J. Chem., 2016, 40, 7643–7656 RSC.
- L. S. Lara, C. S. Moreira, C. M. Calvet, G. C. Lechuga, R. S. Souza, S. C. Bourguignon, V. F. Ferreira, D. Rocha and M. C. S. Pereira, Eur. J. Med. Chem., 2018, 144, 572–581 CrossRef CAS PubMed.
- C. S. Medeiros, N. T. Pontes-Filho, C. A. Camara, J. V. Lima-Filho, P. C. Oliveira, S. A. Lemos, A. F. G. Leal, J. O. C. Brandão and R. P. Neves, Braz. J. Med. Biol. Res., 2010, 43, 345–349 CrossRef CAS PubMed.
- A. Riffel, L. F. Medina, V. Stefani, R. C. Santos, D. Bizani and A. Brandelli, Braz. J. Med. Biol. Res., 2002, 35, 811–818 CrossRef CAS PubMed.
- M. M. M. Santos, N. Faria, J. Iley, S. J. Coles, M. B. Hursthouse, M. L. Martins and R. Moreira, Bioorg. Med. Chem. Lett., 2010, 20, 193–195 CrossRef CAS PubMed.
- V. K. Tandon, H. K. Maurya, M. K. Verma, R. Kumar and P. K. Shukla, Eur. J. Med. Chem., 2010, 45, 2418–2426 CrossRef CAS PubMed.
- J. M. Sánchez-Calvo, G. R. Barbero, G. Guerrero-Vásquez, A. G. Durán, M. Macías, M. A. Rodríguez-Iglesias, J. M. G. Molinillo and F. A. Macías, Med. Chem. Res., 2016, 25, 1274–1285 CrossRef.
- M. J. Teixeira, Y. M. de Almeida, J. R. Viana, J. G. Holanda Filha, T. P. Rodrigues, J. R. C. Prata, I. C. B. Coelho, V. S. Rao and M. M. L. Pompeu, Phyther. Res., 2001, 15, 44–48 CrossRef CAS.
- G. de S. V. Tavares, D. V. C. Mendonça, D. P. Lage, J. da T. Granato, F. M. Ottoni, F. Ludolf, M. A. Chávez-Fumagalli, M. C. Duarte, C. A. P. Tavares, R. J. Alves, E. S. Coimbra and E. A. F. Coelho, Basic Clin. Pharmacol. Toxicol., 2018, 123, 236–246 CrossRef CAS PubMed.
- A. A. D. S. Naujorks, A. O. Da Silva, R. D. S. Lopes, S. De Albuquerque, A. Beatriz, M. R. Marques and D. P. De Lima, Org. Biomol. Chem., 2015, 13, 428–437 RSC.
- M. V. de Araújo, C. C. David, J. C. Neto, L. A. P. L. de Oliveira, K. C. J. da Silva, J. M. dos Santos, J. K. S. da Silva, V. B. C. de A. Brandão, T. M. S. Silva, C. A. Camara and M. S. Alexandre-Moreira, Exp. Parasitol., 2017, 176, 46–51 CrossRef PubMed.
- M. Dong, D. Liu, Y. H. Li, X. Q. Chen, K. Luo, Y. M. Zhang and R. T. Li, Planta Med., 2017, 83, 631–635 CAS.
- H. Ju Woo, D. Y. Jun, J. Y. Lee, H. S. Park, M. H. Woo, S. J. Park, S. C. Kim, C. H. Yang and Y. H. Kim, J. Ethnopharmacol., 2017, 205, 103–115 CrossRef PubMed.
- I. Milackova, M. S. Prnova, M. Majekova, R. Sotnikova, M. Stasko, L. Kovacikova, S. Banerjee, M. Veverka and M. Stefek, J. Enzyme Inhib. Med. Chem., 2015, 30, 107–113 CrossRef CAS PubMed.
- A. S. Soares, F. L. Barbosa, A. L. Rüdiger, D. L. Hughes, M. J. Salvador, A. R. Zampronio and M. É. A. Stefanello, J. Nat. Prod., 2017, 80, 1837–1843 CrossRef CAS PubMed.
- N. Hatae, J. Nakamura, T. Okujima, M. Ishikura, T. Abe, S. Hibino, T. Choshi, C. Okada, H. Yamada, H. Uno and E. Toyota, Bioorg. Med. Chem. Lett., 2013, 23, 4637–4640 CrossRef CAS PubMed.
- M. Marastoni, C. Trapella, A. Scotti, A. Fantinati, V. Ferretti, E. Marzola, G. Eleonora, R. Gavioli and D. Preti, J. Enzyme Inhib. Med. Chem., 2017, 32, 865–877 CrossRef CAS PubMed.
- H. Y. Qiu, P. F. Wang, H. Y. Lin, C. Y. Tang, H. L. Zhu and Y. H. Yang, Chem. Biol. Drug Des., 2018, 91, 681–690 CrossRef CAS PubMed.
- L. Romão, V. P. Do Canto, P. A. Netz, V. Moura-Neto, Â. C. Pinto and C. Follmer, Anticancer Drugs, 2018, 29, 520–529 CrossRef PubMed.
- P. Poma, M. Labbozzetta, M. Notarbartolo, M. Bruno, A. Maggio, S. Rosselli, M. Sajeva and P. Zito, PLoS One, 2018, 13, 1–11 CrossRef PubMed.
- F. Prati, C. Bergamini, M. T. Molina, F. Falchi, A. Cavalli, M. Kaiser, R. Brun, R. Fato and M. L. Bolognesi, J. Med. Chem., 2015, 58, 6422–6434 CrossRef CAS PubMed.
- U. Galm, M. H. Hager, S. G. Van Lanen, J. Ju, J. S. Thorson and B. Shen, Chem. Rev., 2005, 105, 739–758 CrossRef CAS PubMed.
- D. A. Gewirtz, Biochem. Pharmacol., 1999, 57, 727–741 CrossRef CAS PubMed.
- S. E. Wolkenberg and D. L. Boger, Chem. Rev., 2002, 102, 2477–2495 CrossRef CAS PubMed.
- P. Krishnan and K. F. Bastow, Cancer Chemother. Pharmacol., 2001, 47, 187–198 CrossRef CAS PubMed.
- B. Frydman, L. J. Marton, J. S. Sun, K. Neder, D. T. Witiak, A. A. Liu, H. M. Wang, Y. Mao, H. Y. Wu, M. M. Sanders and L. F. Liu, Cancer Res., 1997, 57, 620–627 CAS.
- A. V. Pinto, V. F. Ferreira, R. S. Capella, B. Gilbert, M. C. R. Pinto and J. S. Da Silva, Trans. R. Soc. Trop. Med. Hyg., 1987, 81, 609–610 CrossRef CAS PubMed.
- K. C. G. G. De Moura, F. S. Emery, C. Neves-Pinto, M. do C. F. R. R. Pinto, A. P. Dantas, K. Salomão, S. L. De Castro, A. V. Pinto, S. L. De Castro and A. V. Pinto, J. Braz. Chem. Soc., 2001, 12, 325–338 CrossRef CAS.
- E. J. S. Salustiano, C. D. Netto, R. F. Fernandes, A. J. M. da Silva, T. S. Bacelar, C. P. Castro, C. D. Buarque, R. C. Maia, V. M. Rumjanek and P. R. R. Costa, Invest. New Drugs, 2010, 28, 139–144 CrossRef CAS PubMed.
- S. G. Renou, S. E. Asís, M. I. Abasolo, D. G. Bekerman and A. M. Bruno, Pharmazie, 2003, 58, 690–695 CAS.
- H. W. Moore, Science, 1977, 197, 527–532 CrossRef CAS PubMed.
- R. Docampo, F. S. Cruz, A. Boveris, R. P. A. Muniz and D. M. S. Esquivel, Biochem. Pharmacol., 1979, 28, 723–728 CrossRef CAS.
- G. Powis, Pharmacol. Ther., 1987, 35, 57–162 CrossRef CAS PubMed.
- D. M. Santos, M. M. M. M. Santos, R. Moreira, S. Sola, C. M. P. P. Rodrigues, S. Solá and C. M. P. P. Rodrigues, Mol. Neurobiol., 2013, 47, 313–324 CrossRef CAS PubMed.
- M. S. Baptista, J. Cadet, P. Di Mascio, A. A. Ghogare, A. Greer, M. R. Hamblin, C. Lorente, S. C. Nunez, M. S. Ribeiro, A. H. Thomas, M. Vignoni and T. M. Yoshimura, Photochem. Photobiol., 2017, 93, 912–919 CrossRef CAS PubMed.
- H. Abrahamse and M. R. Hamblin, Biochem. J., 2016, 473, 347–364 CrossRef CAS PubMed.
- M. J. Davies and R. J. W. W. Truscott, J. Photochem. Photobiol., B, 2001, 63, 114–125 CrossRef CAS.
- H. Østdal, M. J. Davies and H. J. Andersen, Free Radical Biol. Med., 2002, 33, 201–209 CrossRef.
- D. I. Pattison, A. S. Rahmanto and M. J. Davies, Photochem. Photobiol. Sci., 2012, 11, 38–53 RSC.
- E. Ananieva, World J. Biol. Chem., 2015, 6, 281 CrossRef PubMed.
- E. Silva, P. Barrias, E. Fuentes-Lemus, C. Tirapegui, A. Aspee, L. Carroll, M. J. Davies and C. López-Alarcón, Free Radical Biol. Med., 2019, 131, 133–143 CrossRef CAS PubMed.
- C. Castaño, M. Vignoni, P. Vicendo, E. Oliveros and A. H. Thomas, J. Photochem. Photobiol., B, 2016, 164, 226–235 CrossRef PubMed.
- O. Brahmia and C. Richard, Photochem. Photobiol. Sci., 2003, 2, 1038–1043 RSC.
- H. Görner, J. Phys. Chem. A, 2007, 111, 2814–2819 CrossRef PubMed.
- H. Gorner, Photochem. Photobiol., 2005, 81, 376–383 CrossRef PubMed.
- T. Itoh, Chem. Rev., 1995, 95, 2351–2368 CrossRef CAS.
- J. M. Bruce, A. Chaudhry and K. Dawes, J. Chem. Soc., Perkin Trans. 1, 1974, 288–294 RSC.
- R. I. Teixeira, I. C. dos Santos, S. J. Garden, P. F. Carneiro, V. F. Ferreira and N. C. de Lucas, ChemistrySelect, 2017, 2, 11732–11738 CrossRef CAS.
- L. J. Mitchell, W. Lewis and C. J. Moody, Green Chem., 2013, 15, 2830 RSC.
- Y. Ando and K. Suzuki, Chem.–Eur. J., 2018, 24, 15955–15964 CrossRef CAS PubMed.
- Y. Ando, T. Matsumoto and K. Suzuki, Synlett, 2017, 28, 1040–1045 CrossRef CAS.
- Y. Ando, A. Hanaki, R. Sasaki, K. Ohmori and K. Suzuki, Angew. Chem., Int. Ed., 2017, 56, 11460–11465 CrossRef CAS PubMed.
- Q. Zhou, Y. Wei, X. Liu, L. Chen, X. Zhou and S. Liu, Photochem. Photobiol., 2018, 94, 61–68 CrossRef CAS PubMed.
- A. M. Szymczak, R. Podsiadły, K. Podemska and J. Sokołowska, Color. Technol., 2013, 129, 284–288 CAS.
- H. Görner, J. Photochem. Photobiol., A, 2011, 224, 135–140 CrossRef.
- A. Bose, D. Dey and S. Basu, J. Photochem. Photobiol., A, 2007, 186, 130–134 CrossRef CAS.
- D. Jornet, F. Bosca, J. M. Andreu, L. R. Domingo, R. Tormos and M. A. Miranda, J. Photochem. Photobiol., B, 2016, 155, 1–6 CrossRef CAS PubMed.
- J. C. Netto-Ferreira, V. Lhiaubet-Vallet, B. O. Bernardes, A. B. B. Ferreira and M. Á. Miranda, Phys. Chem. Chem. Phys., 2008, 10, 6645–6652 RSC.
- C. P. V. Freire, S. B. Ferreira, N. S. M. De Oliveira, A. B. J. Matsuura, I. L. Gama, F. D. C. Da Silva, M. C. B. V. De Souza, E. S. Lima and V. F. Ferreira, MedChemComm, 2010, 1, 229–232 RSC.
- S. B. Ferreira, F. D. C. Da Silva, F. A. F. M. Bezerra, M. C. S. Lourenço, C. R. Kaiser, A. C. Pinto and V. F. Ferreira, Arch. Pharm., 2010, 343, 81–90 CAS.
- E. N. da Silva Júnior, M. C. B. V. de Souza, M. C. Fernandes, R. F. S. Menna-Barreto, M. do C. F. R. Pinto, F. de Assis Lopes, C. A. de Simone, C. K. Z. Andrade, A. V. Pinto, V. F. Ferreira and S. L. de Castro, Bioorg. Med. Chem., 2008, 16, 5030–5038 CrossRef PubMed.
- F. D. R. Ferreira, S. B. Ferreira, A. J. Araújo, J. D. B. Marinho Filho, C. Pessoa, M. O. Moraes, L. V. Costa-Lotufo, R. C. Montenegro, F. D. C. Da Silva, V. F. Ferreira, J. G. Da Costa, F. C. De Abreu and M. O. F. Goulart, Electrochim. Acta, 2013, 110, 634–640 CrossRef CAS.
- E. C. B. Da Costa, R. Amorim, F. C. Da Silva, D. R. Rocha, M. P. Papa, L. B. De Arruda, R. Mohana-Borges, V. F. Ferreira, A. Tanuri, L. J. Da Costa and S. B. Ferreira, PLoS One, 2013, 8, 1–11 Search PubMed.
- N. C. De Lucas, R. J. Corrêa, S. J. Garden, G. Santos, R. Rodrigues, C. E. M. Carvalho, S. B. Ferreira, J. C. Netto-Ferreira, V. F. Ferreira, P. Miro, M. L. Marin, M. A. Miranda, R. Rodrigues, C. E. M. Carvalho, S. B. Ferreira, J. C. Netto-Ferreira, V. F. Ferreira, P. Miro, M. L. Marin and M. A. Miranda, Photochem. Photobiol. Sci., 2012, 11, 1201–1209 RSC.
- E. L. Jackson, J. Am. Chem. Soc., 1952, 74, 837–838 CrossRef CAS.
- H. T. Huang and C. Niemann, J. Am. Chem. Soc., 1951, 73, 1541–1548 CrossRef CAS.
- F. de C. da Silva, S. B. Ferreira, C. R. Kaiser, A. C. Pinto and V. F. Ferreira, J. Braz. Chem. Soc., 2009, 20, 1478–1482 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- B. Ahrens, M. G. Davidson, V. T. Forsyth, M. F. Mahon, A. L. Johnson, S. A. Mason, R. D. Price and P. R. Raithby, J. Am. Chem. Soc., 2001, 123, 9164–9165 CrossRef CAS.
- J. C. Netto-Ferreira, B. Bernardes, A. B. B. Ferreira and M. Á. Miranda, Photochem. Photobiol. Sci., 2008, 7, 467–473 RSC.
- J. C. Scaiano, Acc. Chem. Res., 1982, 15, 252–258 CrossRef CAS.
- N. C. de Lucas, M. T. Silva, C. Gege and J. C. Netto-Ferreira, J. Chem. Soc., Perkin Trans. 2, 1999, 2795–2801 RSC.
- N. C. de Lucas, C. P. Ruis, R. I. Teixeira, L. L. Marcal, S. J. Garden, R. J. Correa, S. Ferreira, J. C. Netto-Ferreira and V. F. Ferreira, J. Photochem. Photobiol., A, 2013, 276, 16–30 CrossRef CAS.
- S. Monti and I. Manet, Chem. Soc. Rev., 2014, 43, 4051–4067 RSC.
- I. O. L. Bacellar, T. M. Tsubone, C. Pavani and M. S. Baptista, Int. J. Mol. Sci., 2015, 16, 20523–20559 CrossRef CAS PubMed.
- M. J. Davies, Biochem. Biophys. Res. Commun., 2003, 305, 761–770 CrossRef CAS PubMed.
- Y. P. Tsentalovich, O. A. Snytnikova and R. Z. Sagdeev, J. Photochem. Photobiol., A, 2004, 162, 371–379 CrossRef CAS.
- J. C. Netto-Ferreira, V. Lhiaubet-Vallet, A. R. da Silva, A. M. da Silva, A. B. B. Ferreira and M. A. Miranda, J. Braz. Chem. Soc., 2010, 21, 966–972 CrossRef CAS.
- G. Merényi, J. Lind and X. Shen, J. Phys. Chem., 1988, 92, 134–137 CrossRef.
- P. K. Das, M. V. Encinas and J. C. Scaiano, J. Am. Chem. Soc., 1981, 103, 4154–4162 CrossRef CAS.
- P. K. Das and S. N. Bhattacharyya, J. Phys. Chem., 1981, 85, 1391–1395 CrossRef CAS.
- P. K. Das, M. V. Encinas, S. Steenken and J. C. Scaiano, J. Am. Chem. Soc., 1981, 103, 4162–4166 CrossRef CAS.
- N. C. De Lucas, R. J. Correa, A. C. C. Albuquerque, C. L. Firme, S. J. Garden, A. R. Bertoti and J. C. Netto-Ferreira, J. Phys. Chem. A, 2007, 111, 1117–1122 CrossRef CAS PubMed.
- N. C. de Lucas, M. M. Elias, C. L. Firme, R. J. Corrêa, S. J. Garden, J. C. Netto-Ferreira and D. E. Nicodem, J. Photochem. Photobiol., A, 2009, 201, 1–7 CrossRef CAS.
- J. Pérez-Prieto, S.-E. Stiriba, F. Boscá, A. Lahoz, L. R. Domingo, F. Mourabit, S. Monti and M. A. Miranda, J. Org. Chem., 2004, 69, 8618–8625 CrossRef PubMed.
- O. B. Morozova, M. S. Panov, N. N. Fishman and A. V Yurkovskaya, Phys. Chem. Chem. Phys., 2018, 20, 21127–21135 RSC.
- C. T. Saouma and J. M. Mayer, Chem. Sci., 2014, 5, 21–31 RSC.
- C.-C. Hsieh, C.-M. Jiang and P.-T. Chou, Acc. Chem. Res., 2010, 43, 1364–1374 CrossRef CAS PubMed.
- D. C. Miller, K. T. Tarantino and R. R. Knowles, Top. Curr. Chem., 2016, 374, 1–59 CrossRef CAS PubMed.
- C. J. Gagliardi, B. C. Westlake, C. A. Kent, J. J. Paul, J. M. Papanikolas and T. J. Meyer, Coord. Chem. Rev., 2010, 254, 2459–2471 CrossRef CAS.
- J. W. Darcy, B. Koronkiewicz, G. A. Parada and J. M. Mayer, Acc. Chem. Res., 2018, 51, 2391–2399 CrossRef CAS PubMed.
- A. A. Pizano, D. A. Lutterman, P. G. Holder, T. S. Teets, J. Stubbe and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 39–43 CrossRef CAS PubMed.
- E. C. Gentry and R. R. Knowles, Acc. Chem. Res., 2016, 49, 1546–1556 CrossRef CAS PubMed.
- S. Hammes-Schiffer, J. Am. Chem. Soc., 2015, 137, 8860–8871 CrossRef CAS PubMed.
- J.-M. Seveant, Annu. Rev. Anal. Chem., 2014, 7, 537–560 CrossRef CAS PubMed.
- J. M. Mayer, I. J. Rhile, F. B. Larsen, E. A. Mader, T. F. Markle and A. G. DiPasquale, Photosynth. Res., 2006, 87, 3–20 CrossRef CAS PubMed.
- J. J. Concepcion, M. K. Brennaman, J. R. Deyton, N. V. Lebedeva, M. D. E. Forbes, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2007, 129, 6968–6969 CrossRef CAS PubMed.
- T. Irebo, O. Johansson and L. Hammarstrom, J. Am. Chem. Soc., 2008, 130, 9194–9195 CrossRef CAS PubMed.
- J. Ravensbergen, C. L. Brown, G. F. Moore, R. N. Frese, R. van Grondelle, D. Gust, T. A. Moore, A. L. Moore and J. T. M. Kennis, Photochem. Photobiol. Sci., 2015, 14, 2147–2150 RSC.
- T. F. Markle, J. W. Darcy and J. M. Mayer, Sci. Adv., 2018, 4, eaat5776 CrossRef PubMed.
- G. F. Manbeck, E. Fujita and J. J. Concepcion, J. Am. Chem. Soc., 2016, 138, 11536–11549 CrossRef CAS PubMed.
- I. Amada, M. Yamaji, M. Sase and H. Shizuka, J. Chem. Soc., Faraday Trans., 1995, 91, 2751 RSC.
- J. Craggs, S. H. Kirk and S. I. Ahmad, J. Photochem. Photobiol., B, 1994, 24, 123–128 CrossRef CAS.
- D. Ouyang and K. Hirakawa, J. Photochem. Photobiol., B, 2017, 175, 125–131 CrossRef CAS PubMed.
- T. Hageman, H. Wei, P. Kuehne, J. Fu, R. Ludwig, L. Tao, A. Leone, M. Zocher and T. K. Das, Pharm. Res., 2019, 1–10 CAS.
- A. H. Thomas, B. N. Zurbano, C. Lorente, J. Santos, E. A. Roman and M. Laura Dántola, J. Photochem. Photobiol., B, 2014, 141, 262–268 CrossRef CAS PubMed.
- B. A. Kerwin and R. L. Remmele, J. Pharm. Sci., 2007, 96, 1468–1479 CrossRef CAS PubMed.
- J. Pérez-Prieto, F. Boscá, R. E. Galian, A. Lahoz, L. R. Domingo and M. A. Miranda, J. Org. Chem., 2003, 68, 5104–5113 CrossRef PubMed.
- N. C. de Lucas, H. S. Fraga, C. P. Cardoso, R. J. Corrêa, S. J. Garden and J. C. Netto-Ferreira, Phys. Chem. Chem. Phys., 2010, 12, 10746–10753 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional transient absorption spectra and quenching plots. Atomic coordinates and thermodynamics for theoretical calculations. See DOI: 10.1039/c9ra01939a |
| This journal is © The Royal Society of Chemistry 2019 |