DOI:
10.1039/C9RA01781J
(Paper)
RSC Adv., 2019,
9, 15115-15123
Electrocarboxylation of 1-chloro-(4-isobutylphenyl)ethane with a silver cathode in ionic liquids: an environmentally benign and efficient way to synthesize Ibuprofen†
Received
8th March 2019
, Accepted 9th May 2019
First published on 14th May 2019
Abstract
Electrocarboxylation of organic halides is one of the most widely used approaches for valorising CO2. In this manuscript, we report a new greener synthetic route for synthesising 2-(4-isobutylphenyl)propanoic acid, Ibuprofen, one of the most popular non-steroidal anti-inflammatory drugs (NSAIDs). The joint use of electrochemical techniques and ionic liquids (ILs) allows CO2 to be used as a C1-organic building block for synthesising Ibuprofen in high yields, with conversion ratios close to 100%, and under mild conditions. Furthermore, the determination of the reduction peak potential values of 1-chloro-(4-isobutylphenyl)ethane in several electrolytes (DMF, and ionic liquids) and with different cathodes (carbon and silver) makes it possible to evaluate the most “energetically” favourable conditions for performing the electrocarboxylation reaction. Hence, the use of ILs not only makes the electrolytic media greener, but they also act as catalysts enabling the electrochemical reduction of 1-chloro-(4-isobutylphenyl)ethane to be decreased by up to 1.0 V.
1. Introduction
Carbon dioxide (CO2) is known as a greenhouse gas and is the most important contributor to global warming. The chemical properties of CO2 make it an interesting candidate for developing new green sustainable chemistry, since it is abundant, non-toxic, non-flammable, and a low cost material. Moreover, its C1 carbon skeleton is interesting for making longer molecules. Therefore, one of the main challenges for the scientific community is to eliminate or re-use CO2 in order to obtain other types of compounds with high added value, such as carboxylic derivatives. In this sense, different methodologies are currently being developed to minimise its production, such as the carbon capture and storage (CCS) and carbon capture and utilisation (CCU) technologies.1–6
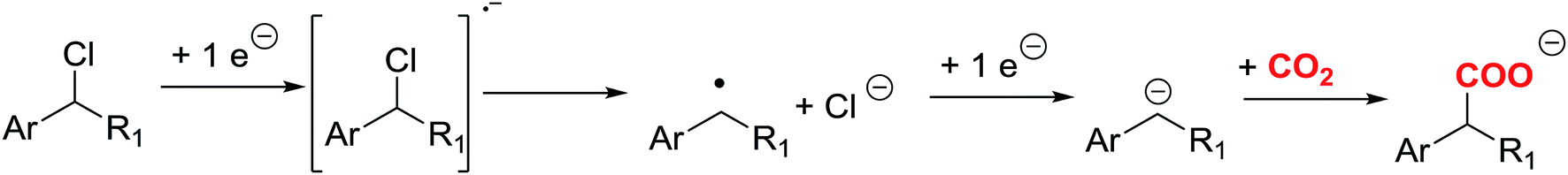
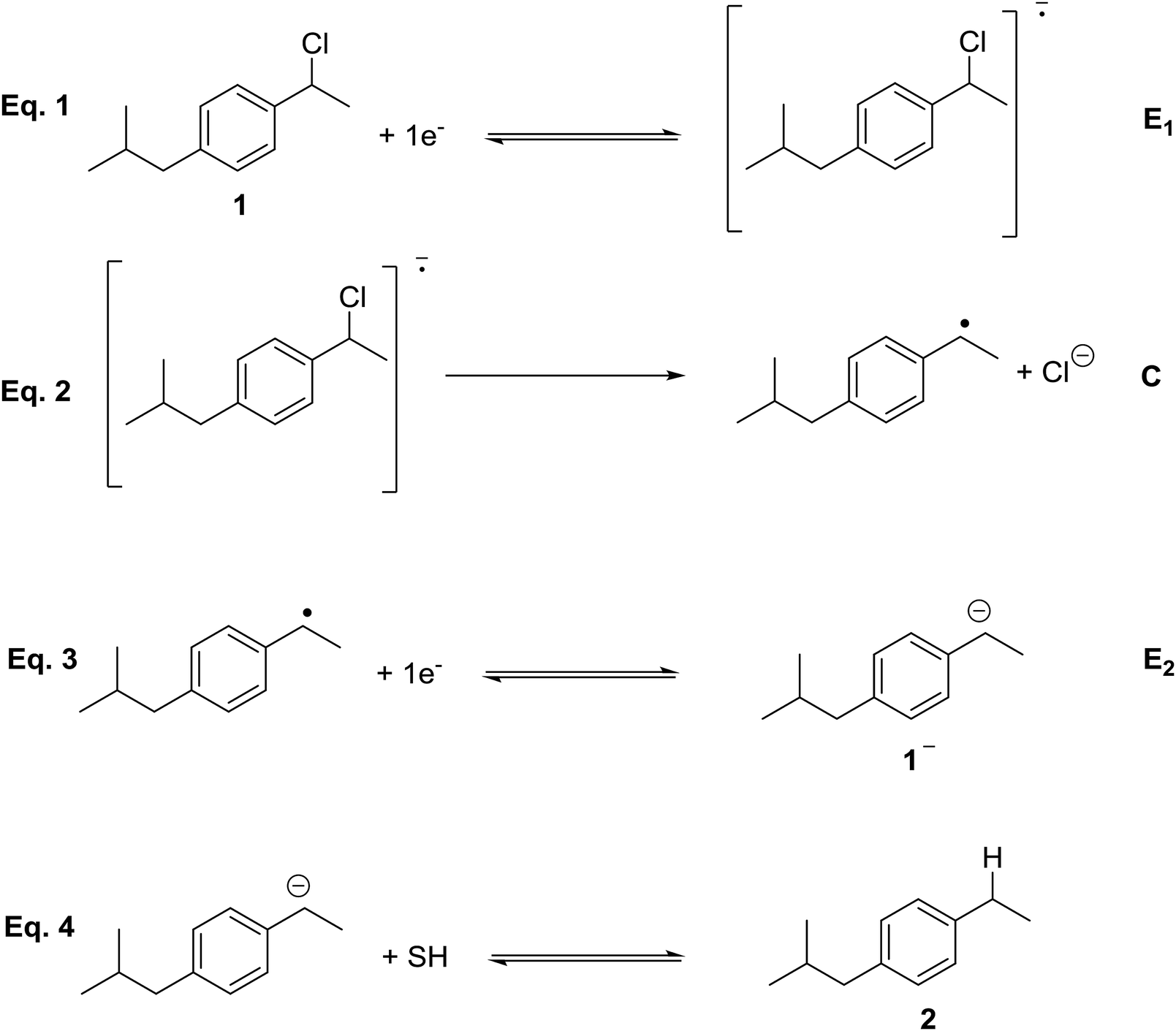
According to the literature,7–9 different organic synthetic routes have been developed using CO2 as a reactant. Direct carboxylation of carbon nucleophile using CO2 as an electrophile is a straightforward route to prepare carboxylic acids. On the other hand, CO2 is highly reactive with carbon nucleophiles, such as Grignard and organolithium reagents. The main disadvantage of those processes is the use of toxic reagents that generate a large amount of waste. An attractive alternative is the use of organic electrochemical techniques for obtaining high valuable carboxylation products, since they can improve environmental conditions. In this sense, electrochemical techniques may offer the possibility to activate the CO2 through a electrocarboxylation process.10–19 Electrocarboxylation of organic halides is one of the most widely used approaches for valorising CO2. In a first step, a one electron transfer process generates the organic radical, which later converts to an anion though a second reduction electron transfer, and a halide anion (Scheme 1).20 The key step of this approach relies on the reduction potential value of the organic halides and on the stability of the organic anion formed after the reduction process. Furthermore, in the recent years, many studies have appeared in which a silver electrode is a good option to reduce the reduction potential in the carbon–halide cleavage reaction.21–29
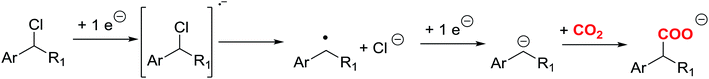 |
| | Scheme 1 Electrochemical carboxylation mechanism of benzyl halides. | |
The electrochemical approach for synthesis of non-steroidal anti-inflammatory drugs (NSAIDs) (e.g. Naproxen30 and Ibuprofen30–32) has been previously performed, obtaining from moderate to good yields. However, the main drawbacks associated with those electrocarboxylation processes were the use of organic solvents, which are well-known to be hazardous and flammable,14 the use of large quantities of supporting electrolyte,16 and either the use of toxic redox mediators, or high reduction potential values. In this sense, in the current manuscript we propose an attractive alternative to these electrocarboxylation processes by: (1) the replacement of conventional electrochemical solvents with Ionic Liquids (ILs). ILs have been widely used as environmentally friendly solvents, electrolytes, as well as catalysts,33–41 and (2) the use of a silver cathode for decreasing the reduction potential values required for the cleavage of the halide–carbon bond, avoiding the use of either toxic mediators. Hence, this manuscript reports a new, more environmentally friendly, approach for synthesising 2-(4-(2-methylpropyl)phenyl)propanoic acid (Ibuprofen) by using green technologies (electrochemistry), green solvents (RTILs), and CO2 feedstock.
2. Experimental section
2.1. Materials
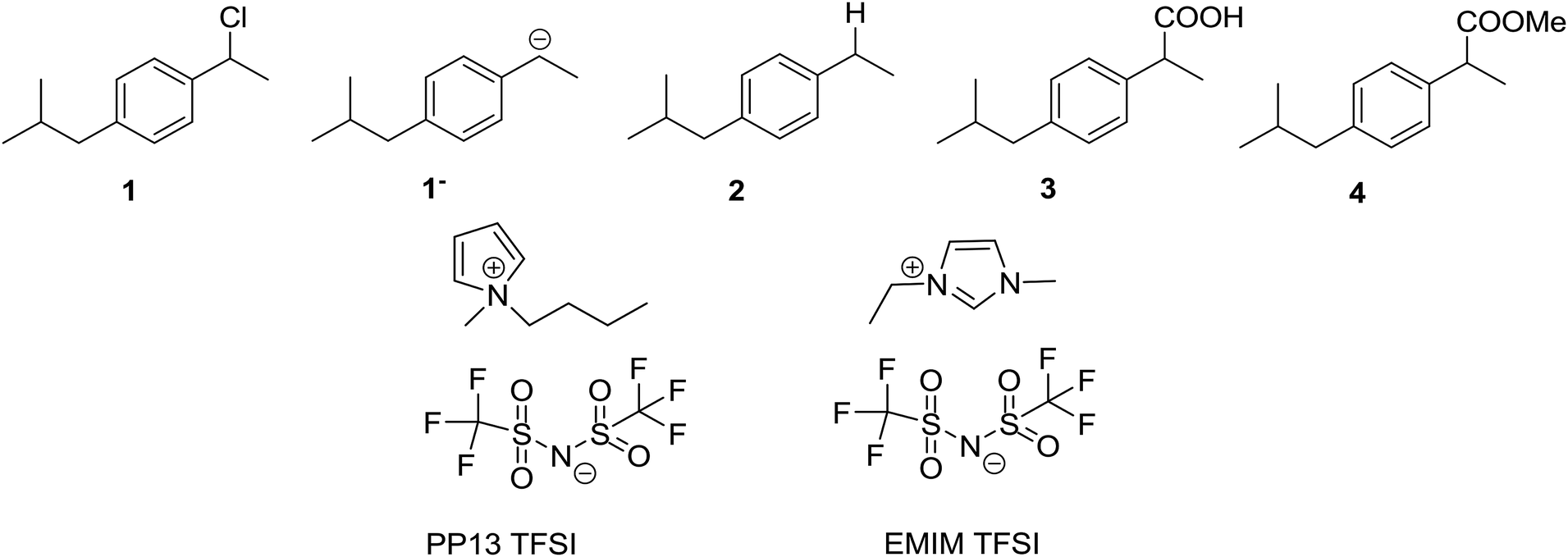
Carbon dioxide, CO2, and nitrogen, N2, were obtained from Carburos Metálicos S.A. (a purity of 99.9999%). 1-Chloro-(4-isobutylphenyl)ethane (1) was synthesised following that in the literature.42 N-Methyl-N-propylpiperidinium bis(trifluoromethanesulfonyl)imide (PP13 TFSI, purity 99.5%, H2O ≤ 0.05%), 1-methyl-1-ethylimidazolium bis(trifluoromethanesulfonyl)imide (EMIM TFSI, purity 99%, H2O ≤ 0.2%) were acquired from Solvionic, and were dried under vacuum using activated molecular sieves for 24 h to make sure that the amount of water was always less than H2O ≤ 0.001% (Scheme 2).
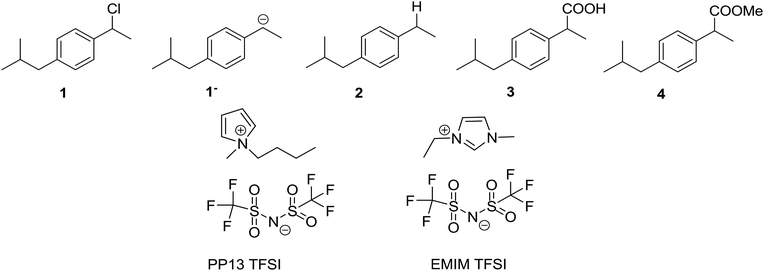 |
| | Scheme 2 Diagram of the structures. | |
2.2. Cyclic voltammetry experiments
An electrochemical conical cell is used for the set-up of the three-electrode system. For CV experiments, the working electrode is a vitreous carbon disk of 1 mm diameter. It is polished using a 1 mm diamond paste. The counter electrode is a Pt disk of <1 mm diameter. All the potentials are reported versus an aqueous saturated calomel electrode (SCE) isolated from the working electrode compartment by a salt bridge. The salt solution of the reference calomel electrode is separated from the electrochemical solution by a salt-bridge ended with a frit, which is made of a ceramic material, allowing ionic conduction between the two solutions and avoiding appreciable contamination. Ideally, the electrolyte solution present in the bridge is the same as the one used for the electrochemical solution, in order to minimise junction potentials. The error associated with the potential values is less than 5 mV. The ohmic drop can be one of the main sources of error when ILs are used as solvents, since they are more resistive media than aprotic polar solvents with 0.1 M concentration of supporting electrolyte.
The number of electrons involved in the first reduction process of 1 was determined by comparison with very well-known one-electron reduction of fluorenone and nitrobenzene (redox probes), in the same medium using the same electrochemical set-up, by terms of cyclic voltammetry. The number of electrons involved in this first electron transfer was also confirmed by controlled-potential electrolysis.43–46
2.3. General procedure for the electrocarboxylation processes
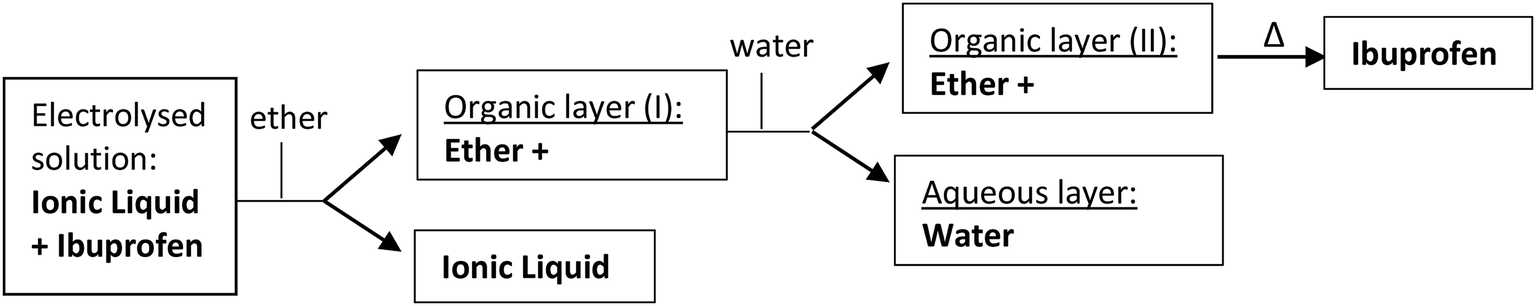
Compound 1 was electrolysed at a negative potential of 0.1 V more negative than the Epc potential value under nitrogen or carbon dioxide saturated solutions. When the reaction is completed, the resulting solution in the electrolysis is extracted into water/ether mixtures. The organic layer is dried with Na2SO4 and evaporated to yield a residue that is analysed by gas chromatography-mass spectrometry, and Proton Nuclear Magnetic Resonance (1H-RMN). When pure ILs are used as electrolyte, the products of the electrolysed solution are extracted with ether, allowing to recover almost an 80% of the IL at the end of the experiment. This organic layer was lately washed with water and dried with Na2SO4 (Scheme 3). Besides, the IL recovered was dried and reused in a new electrochemical process without loss of Ibuprofen yield.
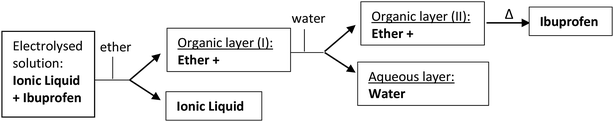 |
| | Scheme 3 Schematic process for Ibuprofen purification and IL recovery after the electrocarboxylation reaction. | |
All products obtained, and the commercial analogues (1-chloro-(4-isobutylphenyl)ethane, 1-ethyl-4-isobutylbenzene, 2-(4-(2-methylpropyl)phenyl)propanoic acid) were characterised by 1H-NMR. Measurements were made using a Bruker DPX360 (360 MHz) (Billerica, MA, USA) spectrometer. Proton chemical shifts were reported in ppm (δ) (CDCl3, δ = 7.26, or CD3CN, δ = 1.94). The J values are reported in Hz.
2.4. Determination of the CO2 concentration in ILs
A thermal mass flowmeter of modular construction with a ‘laboratory style’ pc-board housing (EL-FLOW® Mass Flow Meter/Controller) from Bronkhorst Hi-Tec, was used to monitor the CO2 concentrations in the solution.47 Control valves were integrated to measure and control a gas flow from the lowest range of 0.2 up to 10 ml min−1.
3. Results and discussion
3.1. Electrochemical reduction of 1-chloro-(4-isobutylphenyl)ethane (1) under inert atmosphere
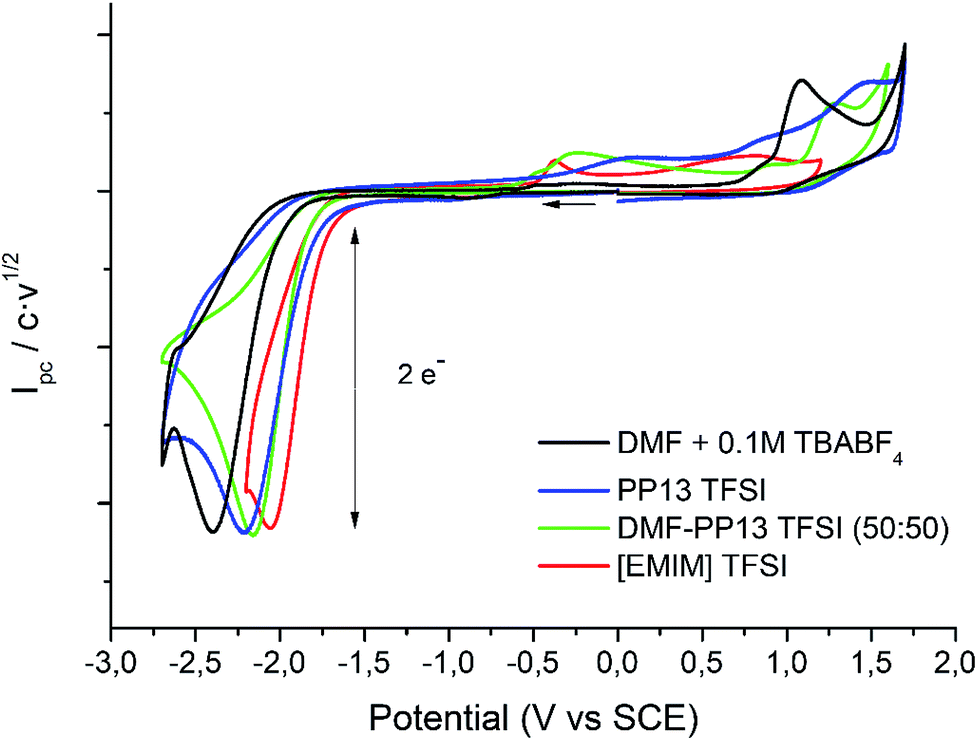
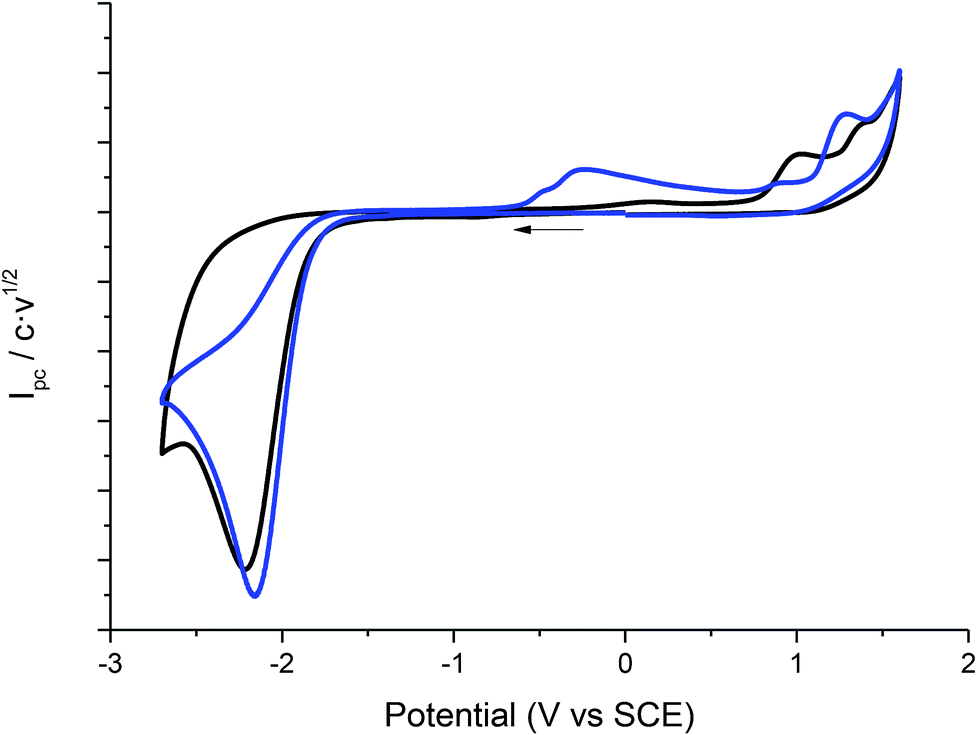
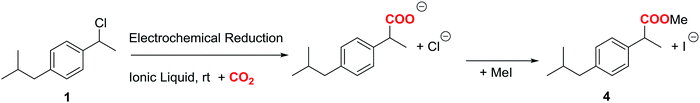
Cyclic voltammetry of compound 1 was recorded in different solvents (DMF + 0.1 M TBABF4, EMIM TFSI, PP13 TFSI, and a DMF–PP13 TFSI (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50)), and scan rates using glassy carbon electrode as a cathode under a N2 atmosphere. In all cases, compound 1 shows a two-electron irreversible reduction peak (Fig. 1), as was expected for these types of compounds. The number of electrons involve in the first reduction process is determined by comparison with the fluorenone, our standard, in the same medium and with the same electrochemical set-up. Hence, in a first cathodic scan, a two electron irreversible reduction wave appears between −1.9 and −2.4 V vs. SCE, depending on the solvent. The peak width value (ΔEp) is always more than 150 mV (at 0.3 V s−1, Table 1), which means that compound 1 shows a slow electron transfer (charge transfer coefficient (α) ca. 0.3).48 The analysis of the peak current values (and their dependence with the concentration and the scan rate), the peak potential values (and their dependence with the concentration) indicates that the chemical reaction coupled to the electron transfer is a first order reaction. In the corresponding anodic counter scan an oxidation peak at ca. 1.1 V is detected, which is attributed to the oxidation of chloride anion.49 A closer look at the CVs also reveals that when EMIM TFSI is used as a solvent, a new peak appears −0.3 V vs. SCE. This new peak is oxidation related to imidazolium moieties formed after the electrochemical reduction of EMIM. This IM radical is immediately reduced at the electrode surface to its anion, and on the CV return its oxidation is observed at ca. 0.1 V vs. SCE.50–54 The cyclic voltammograms performed in PP13 TFSI and DMF–PP13 TFSI also show an irreversible oxidation peak at −0.28 V in the corresponding anodic counter scan. The appearance of this peak indicates that a new electroactive species is present in the solution. Hence, the oxidation peak should be associated with the oxidation of the benzylic anion intermediate 1− (Schemes 4 and 5, Fig. 2). The irreversible wave at Epa = −0.28 V vs. SCE disappears upon the addition of water and acid, which agrees with the presence of the anionic intermediate.
:50)), and scan rates using glassy carbon electrode as a cathode under a N2 atmosphere. In all cases, compound 1 shows a two-electron irreversible reduction peak (Fig. 1), as was expected for these types of compounds. The number of electrons involve in the first reduction process is determined by comparison with the fluorenone, our standard, in the same medium and with the same electrochemical set-up. Hence, in a first cathodic scan, a two electron irreversible reduction wave appears between −1.9 and −2.4 V vs. SCE, depending on the solvent. The peak width value (ΔEp) is always more than 150 mV (at 0.3 V s−1, Table 1), which means that compound 1 shows a slow electron transfer (charge transfer coefficient (α) ca. 0.3).48 The analysis of the peak current values (and their dependence with the concentration and the scan rate), the peak potential values (and their dependence with the concentration) indicates that the chemical reaction coupled to the electron transfer is a first order reaction. In the corresponding anodic counter scan an oxidation peak at ca. 1.1 V is detected, which is attributed to the oxidation of chloride anion.49 A closer look at the CVs also reveals that when EMIM TFSI is used as a solvent, a new peak appears −0.3 V vs. SCE. This new peak is oxidation related to imidazolium moieties formed after the electrochemical reduction of EMIM. This IM radical is immediately reduced at the electrode surface to its anion, and on the CV return its oxidation is observed at ca. 0.1 V vs. SCE.50–54 The cyclic voltammograms performed in PP13 TFSI and DMF–PP13 TFSI also show an irreversible oxidation peak at −0.28 V in the corresponding anodic counter scan. The appearance of this peak indicates that a new electroactive species is present in the solution. Hence, the oxidation peak should be associated with the oxidation of the benzylic anion intermediate 1− (Schemes 4 and 5, Fig. 2). The irreversible wave at Epa = −0.28 V vs. SCE disappears upon the addition of water and acid, which agrees with the presence of the anionic intermediate.
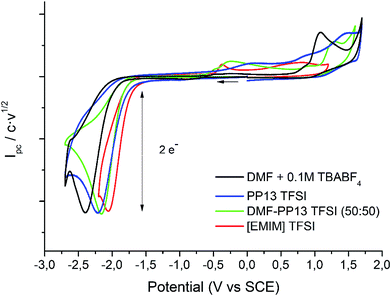 |
| | Fig. 1 Cyclic voltammograms (scan rate 0.5 V s−1) at 25 °C on GC electrode of a solution of 10 mM of 1 in DMF + 0.1 M TBABF4 (black line), PP13TFSI (blue line), DMF–PP13 TFSI (50:50) and EMIM TFSI (red line). | |
Table 1 Cathodic peak potential (Epc in V), ΔEp = |Epc − Epc/2| (in mV), scan rate: 0.3 V s−1, concentration ca. 5 mM, Charge transfer coefficient (α) for 1 in several solvents using glassy carbon as a working electrode at 25 °C under nitrogen atmosphere
| Entry |
Solvent |
Epc (V) |
ΔEpb (mV) |
α |
Number of electrons |
| The DMF solution contains 0.1 M of N-tetrabutylammonium tetrafluoroborate (TBABF4). ΔEp (mV) is the peak width value Epc − Epc/2 at 0.3 V s−1. |
| 1 |
DMFa |
−2.42 |
180 |
0.261 |
1.95 |
| 2 |
EMIM TFSI |
−2.03 |
156 |
0.313 |
2.12 |
| 3 |
DMFa–PP13 TFSI (50–50%) |
−2.18 |
180 |
0.265 |
2.03 |
| 4 |
PP13 TFSI |
−2.15 |
212 |
0.222 |
1.85 |
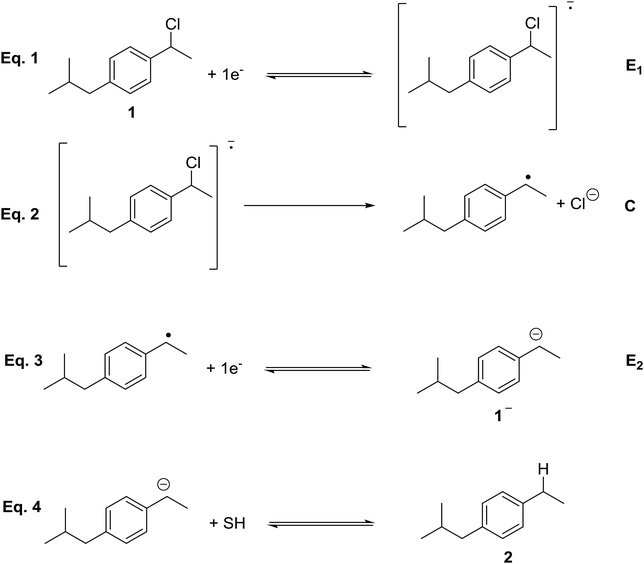 |
| | Scheme 4 Electrochemical reduction mechanism of 1. | |
 |
| | Scheme 5 Electrocarboxylation of 1 under CO2 atmosphere adding MeI as methylated agent at the end of the controlled-potential electrolysis. | |
 |
| | Fig. 2 Cyclic voltammograms (scan rate 0.5 V s−1) at 25 °C on a GC electrode of 10 mM of 1 at CO2 atmosphere (blue line), inert atmosphere (black line) in PP13 TFSI–DMF (1:1). | |
Taking the value Epc value obtained for the electrochemical reduction of 1 in DMF + 0.1 M TBABF4 as a reference value (Table 1), the Epc values obtained in all the ILs are positively shifted, which means that the cation of the IL acts as a catalyst (as was previously pointed out by us and some other authors).35 The reduction potential can be lowered to more than 0.30 V (7 kcal mol−1) with respect to DMF + 0.1 M TBABF4 (Table 1, entries 1–4). This fact can be explained by taking in to account that the IL is a pure ion solvent, whereas the concentration of the counter-cation is considerably lower the DMF solution, where “only” a 0.1 M concentration of tetrabutylammonium cation is present.
In order to determine the nature of the product formed after the first electron transfer of 1, a control potential electrolysis at −2.2 V vs. SCE under inert atmosphere was performed in the above-mentioned electrolytic media (a control potential electrolysis at −2.5 V vs. SCE was performed in DMF). 1-Ethyl-4-isobutylbenzene, 2, was obtained as a unique quantitative product (100%) in DMF, DMF/PP13, and PP13 after the passage of 2F. Overall, the electrochemical reduction mechanism of 1 is a two electron ECE mechanism (Scheme 4). In a first reduction step, a radical anion is formed (electrochemical reaction, E), which undergoes C–Cl cleavage, leading to the corresponding organic radical and halide anion (elementary reaction step, C). The radical is reduced at this potential value, leading to the corresponding anion (electrochemical reaction, E). In a last protonation step the organic anion evolves to 2.
When EMIM TFSI is used as solvent the reactant was recovered at the end of the process, and only decomposition products related to the reduction of EMIM cation were observed. These results can be easily rationalised taking into account that EMIM cation is reduced at −2.3 V, so the solvent is partially reduced at this potential. Thus, EMIM TFSI was discarded for the upcoming electrocarboxylation processes.
3.2. Electrochemical reduction of 1-chloro-(4-isobutylphenyl)ethane (1) under CO2 atmosphere with carbon vitreous working electrode
Fig. 2 shows the cyclic voltammograms of 1 in one of the electrolytic media employed under CO2 atmosphere. In all the cases, a two electron reduction wave was observed at ca. −2.20 V vs. SCE in the initial cathodic scan, whereas an oxidation peak at ca. 1.1 V vs. SCE, which corresponds to the oxidation of chloride ion, is also detected in the ensuing anodic scan.
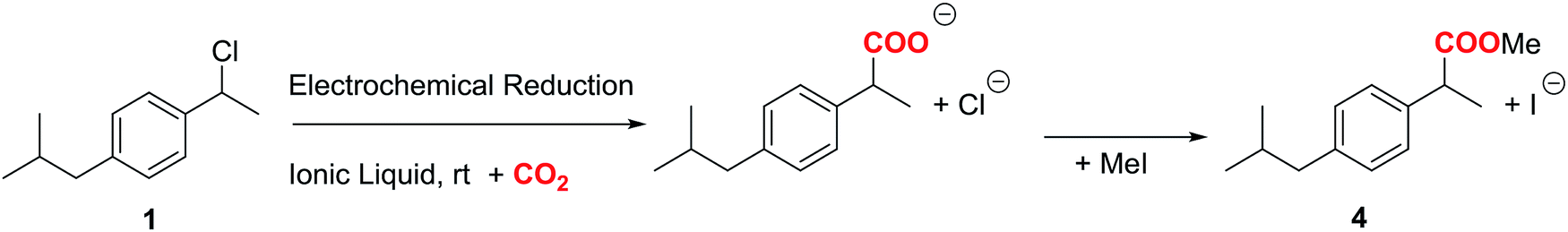
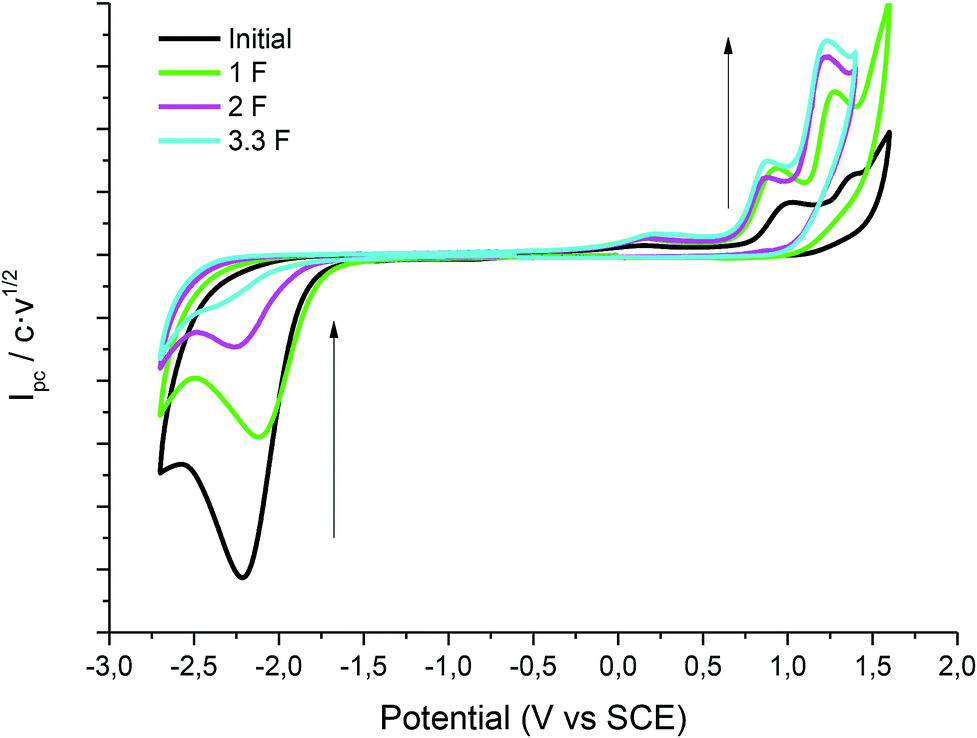
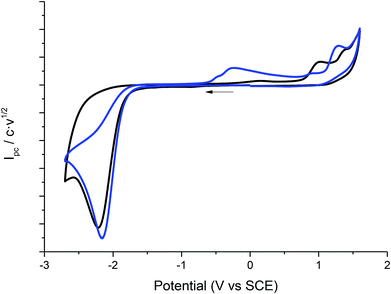
Taking advantage of the long lifetime of 1− in ionic liquid based electrolytes, several electrocarboxylation processes where attempted. The selected electrolyte was previously pre-saturated with CO2 to minimise undesired protonation side reactions. Hence controlled-potential electrolysis of 1 was performed using a graphite bar electrode under a saturated CO2 atmosphere in DMF and ILs. The reduction processes were easily monitored by means of cyclic voltammetry (Fig. 3). In all the cases Ibuprofen was obtained in moderate to good yields (Table 2). The used of PP13 TFSI IL as a co-solvent, not only makes the reaction greener, but also increases the yield up to 75%. This result can be explained due to a higher hydrophobicity of the media, since these electrocarboxylation reactions are very sensitive to the presence of water. Finally, when pure IL electrolyte is used, the product yield increases up to ca. 90% over consumed reactant, with the E-factor being close to 1 (Table 2, entries 5 and 10). Hence, no other products apart from the Ibuprofen and the starting material were recovered. Moreover, a methylated agent, such as iodomethane, was added it is possible to obtain methylated Ibuprofen, 4 (Scheme 5). Finally, note, that for obtaining yields close to 100%, the IL should be previously dried, the use of commercial IL (≤0.05% water content) also leads also to 20% of protonated compound 2.
 |
| | Fig. 3 Cyclic voltammograms (scan rate 0.5 V s−1) at 25 °C on a GC electrode showing controls of the control potential electrolysis of 1 at CO2 atmosphere in DMF–PP13 TFSI (1:1). | |
Table 2 Results of electrocarboxylation of 1 to obtain R–COOH/Me
| Entry |
Solvent |
Eapplied |
C mol−1 |
R–COOH (Ibuprofen isolated yield) |
Reagent recovery (%) |
R–H (%) |
| Yielda (%) |
Conversion rateb (%) |
| Percent yield over isolated product. Percent of Ibuprofen over consumed reactant. The DMF solution contains 0.1 M of N-tetrabutylammonium tetrafluoroborate (TBABF4) dried under vacuum and molecular sieves (less than 0.01% of water). Mixture 1:1 of DMF + 0.1 M TBABF4 and PP13 TFSI dried under vacuum and molecular sieves (less than 0.01% of water). PP13 TFSI commercially available 0.05% of water. PP13 TFSI dried under vacuum and molecular sieves (less than 0.01% of water). |
| 1 |
DMFc |
−2.6 |
3.0 |
61 |
82 |
26 |
13 |
| 2 |
EMIM TFSI |
−2.2 |
3.0 |
— |
— |
— |
— |
| 3 |
DMF-ILd (50–50%) |
−2.4 |
3.3 |
75 |
83 |
10 |
15 |
| 4 |
PP13 TFSIe |
−2.4 |
1.7 |
25 |
57 |
56 |
19 |
| 5 |
PP13 TFSIf |
−2.4 |
1.7 |
47 |
94 |
50 |
3 |
| |
Solvent |
Eapplied |
C mol−1 |
R–COOMe (methylated Ibuprofen isolated yield) |
Reagent recovery (%) |
R–H (%) |
| Yield (%) |
Conversion rate |
| 6 |
DMFc |
−2.6 |
3.0 |
61 |
87 |
30 |
9 |
| 7 |
EMIM TFSI |
−2.2 |
3.0 |
— |
— |
— |
— |
| 8 |
DMF-ILd (50–50%) |
−2.4 |
3.0 |
86 |
86 |
— |
14 |
| 9 |
PP13 TFSIe |
−2.4 |
1.7 |
26 |
67 |
61 |
13 |
| 10 |
PP13 TFSIf |
−2.4 |
1.7 |
48 |
96 |
50 |
2 |
3.3. Electrochemical reduction of 1-chloro-(4-isobutylphenyl)ethane (1) under CO2 atmosphere with silver working electrode
It is well-known that the use of silver cathodes reduces the reduction potential value where the carbon–halide cleavage reaction takes place.21–29 Due to the electrocatalytic effect of silver, it was decided to use a silver electrode as a cathode for the electrocarboxylation process in ILs.
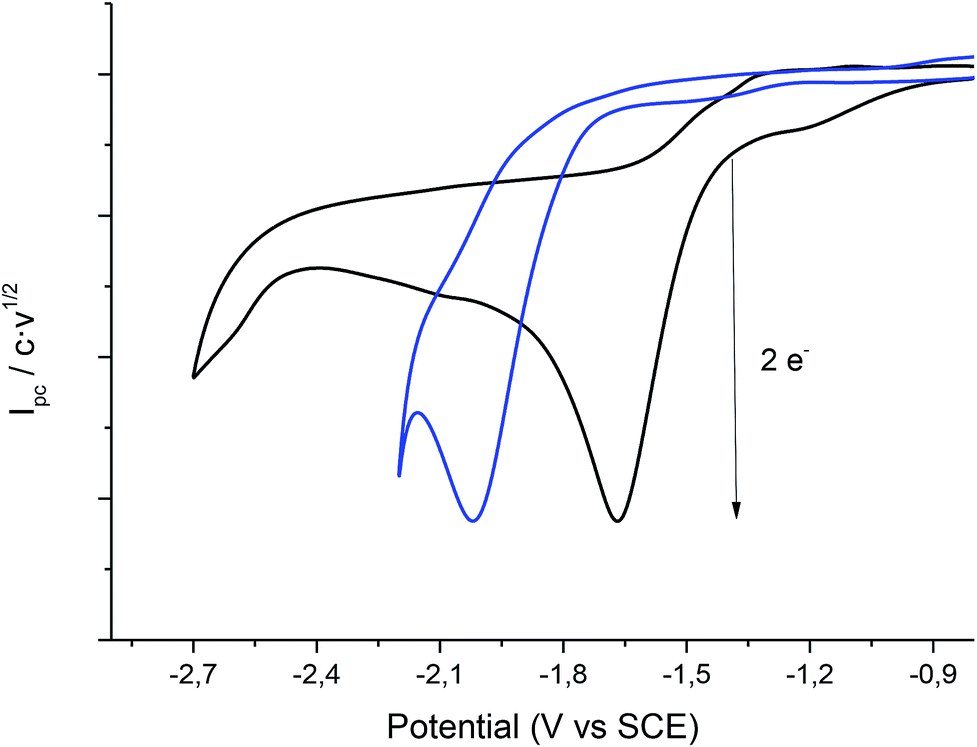
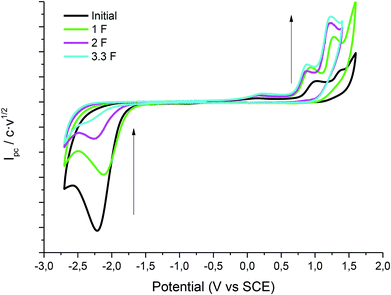
Cyclic voltammetry of compound 1 was recorded, using a silver electrode as a cathode under N2 atmosphere, in EMIM TFSI (to try to improve the results obtained with the carbon electrode) and PP13 TFSI (Fig. 4). In all cases, compound 1 shows the same general behaviour as in the case of using a glassy carbon electrode. Hence, in a first cathodic scan, a two electron irreversible reduction wave appears between −1.7 and −2.0 V vs. SCE depending on the ionic liquid. Taking the Epc value obtained for the electrochemical reduction of 1 in the ionic liquids with glassy carbon electrode as a reference value (Table 1), the Epc values obtained using silver electrode are considerably positively shifted. The reduction potential can be lowered to more than 0.07 V (1.61 kcal mol−1) and 0.49 V (11 kcal mol−1) with respect to the glassy carbon electrode in EMIM TFSI and PP13 TFSI, respectively (Table 3, entries 1 and 2).
 |
| | Fig. 4 Cyclic voltammograms (scan rate 0.5 V s−1) at 25 °C on a silver electrode of a solution of 20 mM of 1 in PP13 TFSI (black line), EMIM TFSI (blue line) under nitrogen atmosphere. | |
Table 3 Cathodic peak potential (Epc in V), ΔEp = |Epc − Epc/2| (in mV), scan rate: 0.5 V s−1, concentration ca. 50 mM, charge transfer coefficient (α) for 1 in several solvents using silver as a working electrode at 25 °C under nitrogen atmosphere
| Entry |
Solvent |
Epc (V) |
ΔEp (mV) |
α |
Number of electrons |
| 1 |
EMIM TFSI |
−1.96 |
95 |
0.46 |
2.3 |
| 2 |
PP13 TFSI |
−1.66 |
130 |
0.37 |
1.9 |
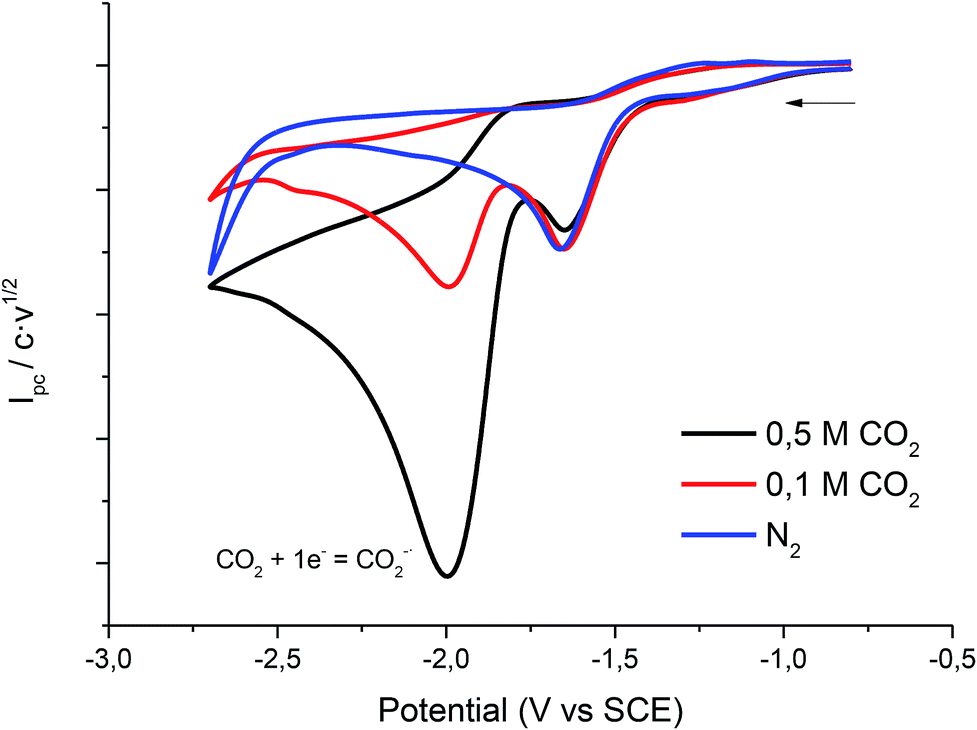
Fig. 5 shows the cyclic voltammogram of 1 in PP13 TFSI under CO2 atmosphere. A two electron reduction wave was observed at ca. −1.7 V vs. SCE, which corresponds to the reduction of 1. Moreover, in the cathodic scan a new reduction peak at −2.00 V vs. SCE is also detected, which is ascribed to the electrochemical reduction of CO2 in the IL. The peak current value of this second reduction peak grows when the concentration of CO2 in the ionic liquid increases.
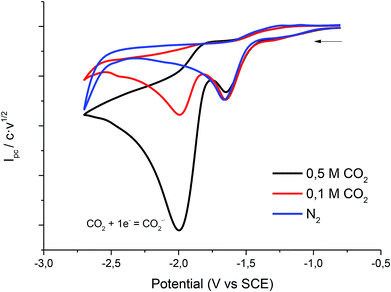 |
| | Fig. 5 Cyclic voltammograms (scan rate 0.5 V s−1) at 25 °C on a silver electrode of 10 mM of 1 at different concentrations of CO2 in PP13 TFSI. | |
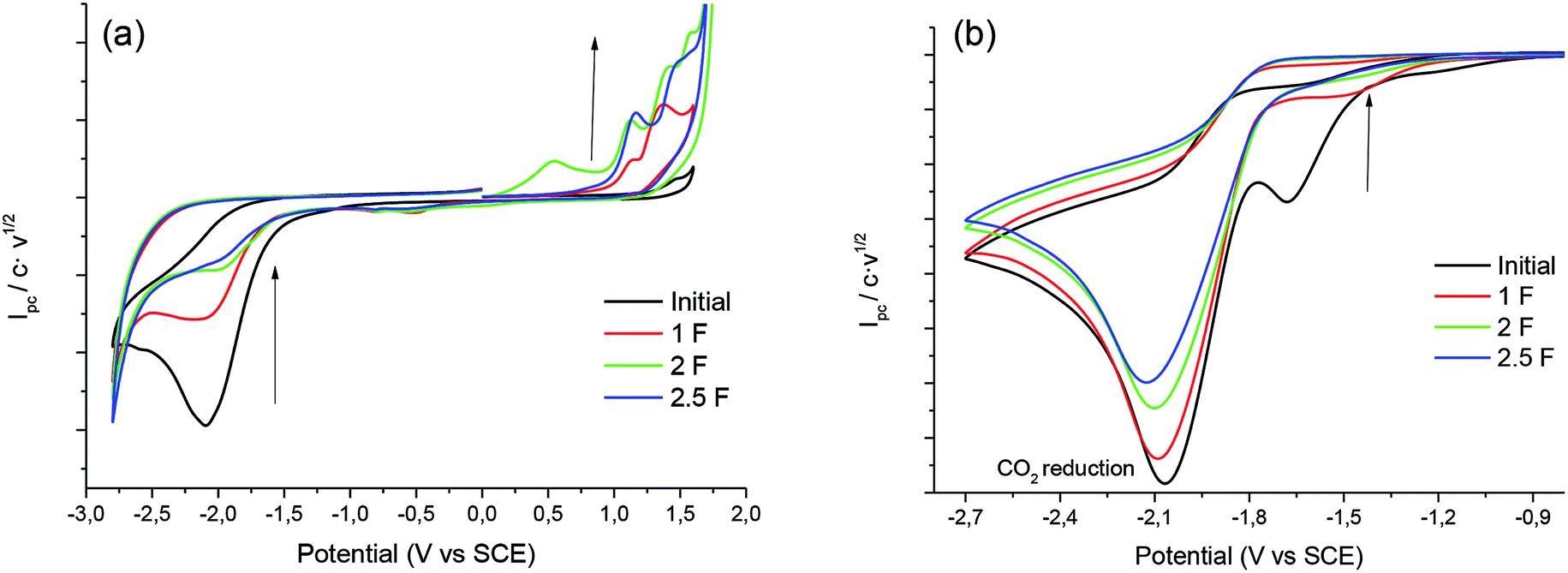
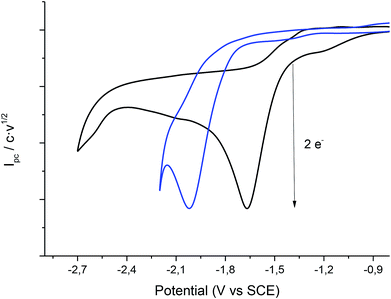
In order to obtain the products 3 and 4, control potential electrolyses at −1.75 V vs. SCE under saturated CO2 atmosphere were performed in the above-mentioned electrolytic media with silver foil (a control potential electrolysis at −2.1 V vs. SCE was performed in EMIM TFSI). The reduction processes were easily monitored by means of cyclic voltammetry with a silver electrode and a glassy carbon electrode (Fig. 6), showing the disappearance of 1. In all the cases Ibuprofen and its methylated analogue (when iodomethane was added as electrophile) were obtained with moderate to good yields (Table 4). As previously mentioned, it should be noted that, for obtaining yields close to 100% the IL should be previously dried. The use of commercial IL (≤0.05% water content) also yields a 20% of protonated compound 2.
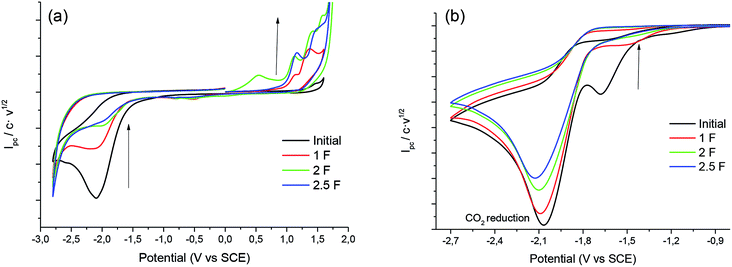 |
| | Fig. 6 Cyclic voltammograms (scan rate 0.5 V s−1) at 25 °C showing controls of the control potential electrolysis of 1 at CO2 atmosphere in PP13 TFSI on Ag electrode (a) GC electrode (b) Ag electrode. | |
Table 4 Results of electrocarboxylation of 1 to obtain R–COOH/Me
| Entry |
Solvent |
Eapplied |
C mol−1 |
R–COOH (Ibuprofen isolated yield) |
Reagent recovery (%) |
R–H (%) |
| Yielda (%) |
Conversion rateb (%) |
| Percent yield over isolated product. Percent of Ibuprofen over consumed reactant. PP13 TFSI commercially available (0.05% of water). PP13 TFSI dried under vacuum and molecular sieves (less than 0.01% of water). |
| 1 |
EMIM TFSI |
−2.10 |
2.3 |
17 |
34 |
50 |
33 |
| 2 |
PP13 TFSIc |
−1.75 |
2.0 |
9 |
38 |
76 |
15 |
| 3 |
PP13 TFSId |
−1.75 |
2.5 |
82 |
85 |
3 |
15 |
| Entry |
Solvent |
Eapplied |
C mol−1 |
R–COOMe (methylated Ibuprofen isolated yield) |
Reagent recovery (%) |
R–H (%) |
| Yield (%) |
Conversion rate |
| 4 |
EMIM TFSI |
−2.10 |
2.3 |
18 |
35 |
49 |
33 |
| 5 |
PP13 TFSIc |
−1.75 |
2.0 |
8 |
35 |
77 |
15 |
| 6 |
PP13 TFSId |
−1.75 |
2.5 |
83 |
83 |
0 |
17 |
Finally, the use of silver as a working electrode also allows the electrocarboxylation synthesis of Ibuprofen to be performed in EMIM TFSI. Furthermore, the electrochemical potential required is lower than in carbon electrodes and within the electrochemical window of the IL. However, when electrochemical synthesis is performed in EMIM TFSI, only 30% of 2 is obtained due to the acidity of the C2–H of the imidazolium moiety.
4. Conclusions
In conclusion, a description is presented of an efficient approach for producing high value compounds using CO2 as building block. The methodology employed is based on electrochemical techniques and ILs, which allow eco-friendly chemistry solutions to be used and maintain the aim of offering a potential long-term strategy for using CO2 feedstocks. The use of dried PP13 TFSI as electrolyte helps to overcome the main drawbacks associated with previous processes reported in the literature, where organic solvents, redox mediators, and large quantities of supporting electrolytes are employed. It is important to highlight the economic feasibility and viability of replacing organic aprotic solvents that contain 0.1 M TBABF4 of supporting electrolyte by ILs. Although the price of a single experiment in IL is double that of DMF + 0.1 M of TBABF4, the fact that the IL can be recovered and reused in almost 80% at the end of the experiment makes this methodology highly viable and attractive. Moreover, changing the nature of the working electrode, the synthesis, in terms of potential applied, is improved and allows the use of ionic liquids with a narrow electrochemical window, such as imidazolium ionic liquids. Controlled potential electrolysis of 1-chloro-(4-isobutylphenyl)ethane under CO2 atmosphere allows Ibuprofen to be obtained in good yields and excellent conversion rates. This methodology offers a new “green” route for the synthesis of different carboxylic acids that can be potential non-steroidal anti-inflammatory drugs (NSAIDs) in a more environmentally friendly way.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by project CTQ2015-65439-R from the MINECO/FEDER. S. M. acknowledges the Autonomous University of Barcelona for her predoctoral research grant (PIF fellowship).
References
- G. Yadav and R. Sen, J. CO2 Util., 2017, 17, 188–206 CrossRef CAS.
- P. Brunel, J. Monot, C. E. Kefalidis, L. Maron, B. Martin-Vaca and D. Bourissou, ACS Catal., 2017, 7, 2652–2660 CrossRef CAS.
- P. Nejat, F. Jomehzadeh, M. M. Taheri, M. Gohari and M. Z. Muhd, Renewable Sustainable Energy Rev., 2015, 43, 843–862 CrossRef CAS.
- R. M. Cuéllar-Franca and A. Azapagic, J. CO2 Util., 2015, 9, 82–102 CrossRef.
- C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- Y. Xu, L. Isom and M. A. Hanna, Bioresour. Technol., 2010, 101, 3311–3319 CrossRef CAS PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 1–15 Search PubMed.
- C. H. Huang and C. S. Tan, Aerosol Air Qual. Res., 2014, 14, 480–499 CrossRef CAS.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- N. Yang, S. R. Waldvogel and X. Jiang, ACS Appl. Mater. Interfaces, 2016, 8, 28357–28371 CrossRef CAS PubMed.
- A. Gennaro, C. M. Sánchez-Sánchez, A. A. Isse and V. Montiel, Electrochem. Commun., 2004, 6, 627–631 CrossRef CAS.
- H. Tateno, K. Nakabayashi, T. Kashiwagi, H. Senboku and M. Atobe, Electrochim. Acta, 2015, 161, 212–218 CrossRef CAS.
- J. Albo, M. Alvarez-Guerra, P. Castaño and A. Irabien, Green Chem., 2015, 17, 2304–2324 RSC.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099 RSC.
- A. Gennaro, A. A. Isse, M.-G. Severin, E. Vianello, I. Bhugun and J.-M. Savéant, J. Chem. Soc., Faraday Trans., 1996, 92, 3963–3968 RSC.
- A. A. Isse, M. G. Ferlin and A. Gennaro, J. Electroanal. Chem., 2005, 581, 38–45 CrossRef CAS.
- A. S. R. Machado, A. V. M. Nunes and M. N. da Ponte, J. Supercrit. Fluids, 2018, 134, 150–156 CrossRef CAS.
- M. A. Scibioh and B. Viswanathan, Proc. Indian Natl. Sci. Acad., 2004, 70, 407–462 CAS.
- I. Reche, I. Gallardo and G. Guirado, RSC Adv., 2014, 4, 65176–65183 RSC.
- R. Matthessen, J. Fransaer, K. Binnemans and D. E. De Vos, Beilstein J. Org. Chem., 2014, 10, 2484–2500 CrossRef PubMed.
- A. A. Isse, S. Gottardello, C. Durante and A. Gennaro, Phys. Chem. Chem. Phys., 2008, 10, 2409–2416 RSC.
- E. E. L. Tanner, C. Batchelor-McAuley and R. G. Compton, J. Phys. Chem. C, 2016, 120, 26442–26447 CrossRef CAS.
- A. A. Isse, A. Galia, C. Belfiore, G. Silvestri and A. Gennaro, J. Electroanal. Chem., 2002, 526, 41–52 CrossRef CAS.
- A. A. Isse, A. De Giusti, A. Gennaro, L. Falciola and P. R. Mussini, Electrochim. Acta, 2006, 51, 4956–4964 CrossRef CAS.
- O. Scialdone, A. Galia, G. Filardo, A. A. Isse and A. Gennaro, Electrochim. Acta, 2008, 54, 634–642 CrossRef CAS.
- O. Lugaresi, A. Minguzzi, C. Locatelli, A. Vertova, S. Rondinini and C. Amatore, Electrocatalysis, 2013, 4, 353–357 CrossRef CAS.
- P. Khalili, K. Y. Tshai and I. Kong, Composites, Part A, 2017, 100, 194–205 CrossRef CAS.
- D. Niu, J. Zhang, K. Zhang, T. Xue and J. Lu, Chin. J. Chem., 2009, 27, 1041–1044 CrossRef CAS.
- A. A. Isse and A. Gennaro, Chem. Commun., 2002, 2798–2799 RSC.
- J. Damodar, S. Krishna Mohan, S. K. Khaja Lateef and S. Jayarama Reddy, Synth. Commun., 2005, 35, 1143–1150 CrossRef CAS.
- J. F. Fauvarque, A. Jutand and M. Francois, J. Appl. Electrochem., 1988, 18, 109–115 CrossRef CAS.
- J. F. Fauvarque, A. Jutand, M. Francois and M. A. Petit, J. Appl. Electrochem., 1988, 18, 116–119 CrossRef CAS.
- R. Liu, P. Zhang, S. Zhang, T. Yan, J. Xin and X. Zhang, Rev. Chem. Eng., 2016, 32, 587–609 CAS.
- I. Reche, I. Gallardo and G. Guirado, Phys. Chem. Chem. Phys., 2015, 17, 2339–2343 RSC.
- H. K. Lim and H. Kim, Molecules, 2017, 22, 536 CrossRef PubMed.
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef CAS.
- J. Li, D. Jia, Z. Guo, Y. Liu, Y. Lyu, Y. Zhou and J. Wang, Green Chem., 2017, 19, 2675–2686 RSC.
- M. Alvarez-Guerra, J. Albo, E. Alvarez-Guerra and A. Irabien, Energy Environ. Sci., 2015, 8, 2574–2599 RSC.
- S. Mena, I. Gallardo and G. Guirado, Catalysts, 2019, 9, 413 CrossRef.
- L. Sun, G. K. Ramesha, P. V. Kamat and J. F. Brennecke, Langmuir, 2014, 30, 6302–6308 CrossRef CAS PubMed.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS PubMed.
- R. A. Kjonaas, P. E. Williams, D. A. Counce and L. R. Crawley, J. Chem. Educ., 2011, 88, 825–828 CrossRef CAS.
- H. Cruz, I. Gallardo and G. Guirado, Electrochim. Acta, 2008, 53, 5968–5976 CrossRef CAS.
- H. Cruz, I. Gallardo and G. Guirado, Green Chem., 2011, 13, 2531–2542 RSC.
- I. Reche, I. Gallardo and G. Guirado, New J. Chem., 2014, 38, 5030–5036 RSC.
- I. Reche, I. Gallardo and G. Guirado, ChemElectroChem, 2014, 1, 2104–2109 CrossRef CAS.
- D. Kodama, K. Sato, M. Watanabe, T. Sugawara, T. Makino and M. Kanakubo, J. Chem. Eng. Data, 2018, 63, 1036–1043 CrossRef CAS.
- L. R. Faulkner and A. J. Bard, Electrochemical Methods: Fundamentals and Applications, University of Texas at Austin, 2nd edn, 2000, vol.
30 Search PubMed.
- I. Gallardo, G. Guirado and J. Marquet, J. Electroanal. Chem., 2000, 488, 64–72 CrossRef CAS.
- E. W. Oliver, D. H. Evans and J. V. Caspar, J. Electroanal. Chem., 1996, 403, 153–158 CrossRef.
- E. W. Oliver and D. H. Evans, J. Electroanal. Chem., 1997, 432, 145–151 CrossRef CAS.
- L. Xiao and K. E. Johnson, J. Electrochem. Soc., 2003, 150, 307–311 CrossRef.
- B. Gorodetsky, T. Ramnial, N. R. Branda and J. A. C. Clyburne, Chem. Commun., 2004, 1972–1973 RSC.
- G. H. Lane, Electrochim. Acta, 2012, 83, 513–528 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01781j |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Jessica Sanchez and
Gonzalo Guirado
,
Jessica Sanchez and
Gonzalo Guirado