
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: PKM2 overexpression protects against 6-hydroxydopamine-induced cell injury in the PC12 cell model of Parkinson's disease via regulation of the brahma-related gene 1/STAT3 pathway
Lei Jiang†
a,
Yuanlin Gao†a,
Gaiying Wangb and
Jie Zhong *c
*c
aThe First Ward of Neurology Department, Kaifeng Central Hospital, Kaifeng 475000, Henan Province, China
bThe Second Ward of Neurology Department, Kaifeng Central Hospital, Kaifeng 475000, Henan Province, China
cDepartment of Nursing, Kaifeng Central Hospital, No. 85 of Hedao Street, Kaifeng 475000, Henan Province, China. E-mail: zhongjiekf@163.com; Fax: +86-371-25672640; Tel: +86-371-25672640
First published on 14th May 2019
Abstract
According to published estimates, pyruvate kinase isoform M2 (PKM2) was expressed in low amounts in patients with Parkinson's disease (PD). However, the function and molecular mechanism of PKM2 in PD remain largely unknown. The main purpose of our study was to reveal the function and mechanism of PKM2 in the in vitro model of PD. Here, we show that PKM2 decreased in PC12 cells after 6-hydroxydopamine (6-OHDA) treatment, which inhibited PC12 cell survival and induced its apoptosis. PKM2 overexpression is required for 6-OHDA-induced PC12 cell survival. Moreover, up-regulated PKM2 expression suppressed PC12 cell apoptosis and caspase-3 activity compared with the 6-OHDA treatment alone group. Increased brahma-related gene 1 (Brg1) and p-STAT3 expression was observed in PKM2-overexpressed PC12 cells compared to those in 6-OHDA treated PC12 cells. Further studies suggested that Brg1 knockdown impeded the high expression of p-STAT3, which was induced by PKM2 overexpression. Finally, the STAT3 inhibitor reversed the effects of PKM2 on cell survival and apoptosis in 6-OHDA-induced PC12 cells. Our results suggest that PKM2 was involved in 6-OHDA-induced PC12 cell injury by mediating the Brg1/STAT3 pathway.
1. Introduction
Parkinson's disease (PD), a progressive disease that affects 2–3% proportion of the population aged more than 65 years, is the most common degenerative disorder occurring in the central nervous system after Alzheimer's disease.1–3 Currently, about 6 million people are suffering from PD all over the world.4 The disease is characterized by cognitive decline, non-motor symptoms and the loss of motor function because of the progressive degeneration of midbrain dopaminergic neurons.3,4 The common features of PD include postural instability, bradykinesia, tremor and rigidity.5 There has been some evidence suggesting that synuclein, including Lewy bodies, are features of PD.6 The monoamine oxidases, including A and B subtypes that act as the drug targets, and anti-parkinsonian drug including rasagiline [N-propargyl-1-(R)-aminoindan; Azilect(R)] and zonisamide play significant roles in the treatment of PD in the literature.7–9 Currently, although many symptomatic drugs have been used to relieve PD symptoms, attenuating neuron degeneration and death is limited.10,11 Thus, studies focusing on neuron damage and apoptosis may have important implications on PD.There are four pyruvate kinase (PK) isoforms, namely, PKR, PKL, PKM1 and PKM2. PKR and PKL are encoded by PKLR gene and are commonly expressed in the erythrocytes and liver, respectively, whereas PKM1 and PKM2 are encoded by PKM gene and are widely expressed in different types of cells and tissues.12 The PKM gene encoded PKM1 or PKM2 depends on the splicing of a single exon, Pkm1 (includes exon 9) and Pkm2 (exon 9 replaces exon 10).13 Acting as a key glycolytic enzyme, pyruvate kinase isozyme type M2 (PKM2) contributes towards pyruvate production and ATP generation and is always implicated in tumor-related cellular processes such as tumor growth and metabolism.14 It is becoming increasingly evident that PKM2 acts as a glycolytic enzyme and plays significant roles in glucose metabolism.15 Moreover, protein kinase PKM2 was involved in cell gene transcription, immunity, signaling and proliferation.15 Recent evidence has suggested that PKM2 plays a critical role in neurogenesis;16 however, the function and mechanism of PKM2 in PD is not fully understood.
Brahma-related gene 1 (Brg1), also named as SMARCA4, is a chromatin remodeling factor and an ATPase catalytic subunit of the SWI/SNF (SWI/sucrose non-fermentable) complex.17,18 Accumulating evidence indicates that Brg-1 was broadly expressed and involved in erythropoiesis,19 zygote genome activation,20 cardiac development21,22 and neuronal development.23,24 Also, Brg-1 plays an important role in cell proliferation and apoptosis and is implicated in the regulation of various gene transcriptions.17,22 Evidence suggests that Brg1 could promote proliferation and inhibit apoptosis in neural crest cells by inhibiting Ask1 and Cdkn1a.25 A recent study has indicated that PKM2 could mediate STAT3 activation and that Brg1 plays an important role in STAT3 recruitment and mediated STAT3 activation.26,27 However, the regulation of PKM2 on Brg1/STAT3 signal pathway is unclear. In the current study, we found that PKM2 was reduced in 6-OHDA-induced PC12 cells and PKM2 overexpression promoted cell survival and suppressed cell proliferation in 6-OHDA-treated PC12 cells, whereas the Brg1 and STAT3 signal was involved in the action of PKM2.
2. Methods
2.1 Cell culture, treatment and transfection
Rat pheochromocytoma cell line PC12 was obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained at 5% CO2/95% humidified air in DMEM, in which 10% fetal bovine serum and 5% horse serum was added at 37 °C. For constructing the PD model in vitro, 6-OHDA (100 μM, Sigma, St. Louis, MO, USA) was added to PC12 cells for 24 h. Then, the PC12 cells were transfected with pcDNA3.1-PKM2, the control pcDNA3.1 vector, co-transfected with pcDNA3.1-PKM2 and Brg1 siRNA or treated with pcDNA3.1-PKM2 and STAT3 inhibitor S3I-201 (100 μM) for 48 h, and then exposed to 100 μM 6-OHDA for 24 h.2.2 Cell survival assay
PC12 cells were transfected with pcDNA3.1-PKM2, the control pcDNA3.1 vector or co-transfected with pcDNA3.1-PKM2 and Brg1 siRNA or treated with pcDNA3.1-PKM2 and STAT3 inhibitor S3I-201 (100 μM) for 48 h. Cells were then plated into 96-well plates at a density of 1 × 104 cells per well. The cells were then exposed to 100 μM 6-OHDA for an additional 24 h. After the incubation period, the MTT solution (0.5 mg ml−1) was added to PC12 cells for another 4 h at 37 °C. The absorbance was recorded using a microplate reader at 595 nm.2.3 Cell apoptosis assay
Cell apoptosis was measured by a Cell Death Detection ELISA kit (Roche, Mannheim, Germany) following the manufacturer's instructions. Histone-associated DNA fragments (mono- and oligo-nucleosomes) from cell lysates were quantified.282.4 Caspase-3 activity assay
Caspase-3 activity in PC12 cells was assayed by the caspase-3 activity assay kit (Beyotime) according to the manufacturer's instructions. The caspase-3 substrate Ac-DEVD-pNA (0.2 mM) was added into PC12 cells, and caspase-3 activity was determined using a microplate reader at 405 nm.2.5 Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and the reverse transcription kit (PrimeScript™ II 1st strand cDNA synthesis kit, Takara Bio, Inc., Otsu, Japan) were used to obtain the total RNA extraction and prepare cDNA, respectively, according to the manufacturers' instructions. RT-qPCR was performed using IQ SYBR Green Super Mix kit (Bio-Rad, Hercules, CA, USA) and an ABI Prism 7500 sequence detection system (Applied Biosystems, Foster City, CA, USA). The mRNA expression levels were calculated using the 2−ΔΔCT method and normalized to β-actin.2.6 Western blot
For determining the protein expression of PKM2, Brg1 and STAT3, Western blot was used. Whole cell extracts, including cytosolic protein extracts and nuclear protein fraction, were obtained from PC12 cells using the lysis buffer as described previously.29 An equal amount of total protein samples (40 μg) was loaded on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrophoretically transferred to PVDF membranes (Millipore, Bedford, MA, USA). Nonfat milk (5%) was used to block the reaction for 1 h at room temperature, and then the primary antibodies were added to the members overnight at 4 °C. Following antibodies were used in this study: PKM2 (ab137852, 1:1000, Abcam, Cambridge, MA, USA), Brg1 (ab4081, 1:1000, Abcam), p-STAT3 (ab76315, 1:5000, Abcam) and STAT3 (ab68153, 1:1000, Abcam). Horseradish peroxidase-conjugated secondary antibody (ab6721, 1:2000, Abcam) was incubated with the members for 1 h at room temperature. The ECL reagent was used to detect the immunoblots.2.7 Statistical analysis
Data represent the means ± SD from three different experiments. The statistical differences were analyzed using one-way analysis of variance (ANOVA), followed by Bonferroni's test. Graphs were plotted using GraphPad Prism 5.0 software. A p-value of <0.05 was considered statistically significant.3. Results
3.1 6-OHDA treatment is associated with the reduction of PKM2 expression
To mimic PD model in vitro, PC12 cells were exposed to 100 μM 6-OHDA for 24 h. PKM2 mRNA and protein expression were examined in control PC12 cells and 6-OHDA-treated PC12 cells, and the results are shown in Fig. 1A and B. We found that PKM2 mRNA level was reduced to 0.49 times in the 6-OHDA group relative to the control group (Fig. 1A, p < 0.05). Moreover, compared with the control group, PKM2 protein expression remarkably decreased in 6-OHDA-treated PC12 cells.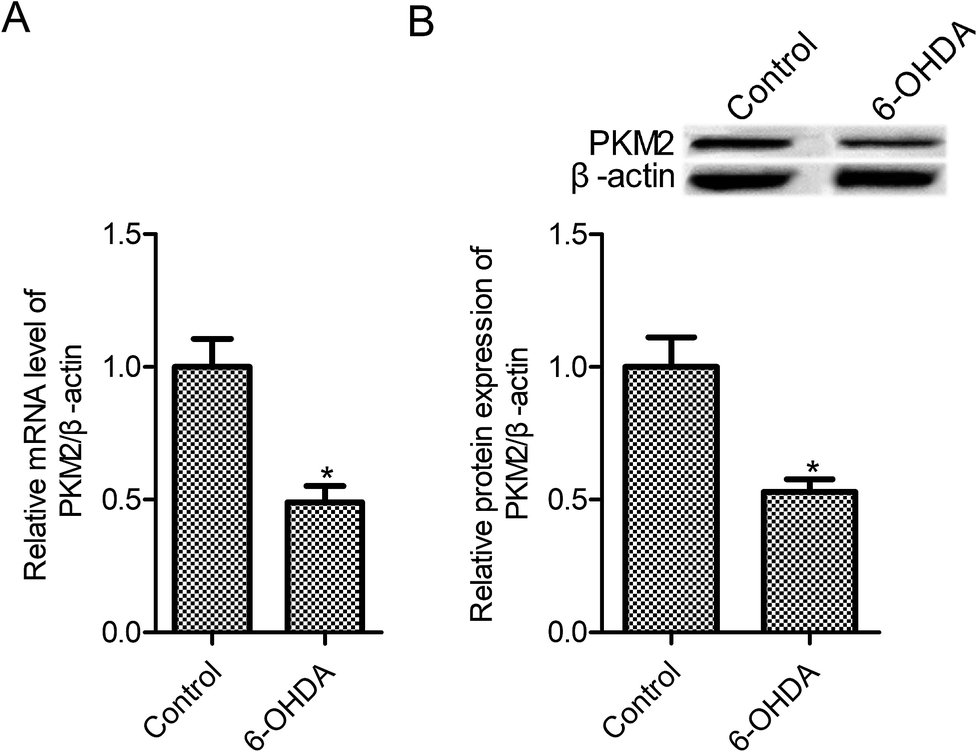 | ||
| Fig. 1 PKM2 level was decreased in 6-OHDA-treated PC12 cells. (A) RT-qPCR was used to measure the PKM2 mRNA level; (B) Western blot was used to measure the PKM2 protein expression. PC12 cells were exposed to 100 μM 6-OHDA for 24 h. Data represent the means ± SD from three different experiments. *P < 0.05 compared with the control group. | ||
3.2 PKM2 overexpression increased 6-OHDA-induced PC12 cell survival
To gain the function of PKM2 in 6-OHDA-induced PC12 cells, PC12 cells were transfected with pcDNA3.1-PKM2 or the control pcDNA3.1 vector for 48 h, and then they were exposed to 100 μM 6-OHDA for 24 h. As shown in Fig. 2A and B, PKM2 mRNA and protein expression increased significantly in pcDNA3.1-PKM2 transfected PC12 cells compared with the control cells (p < 0.05). We next investigated the effect of PKM2 overexpression on the survival of PC-12 cells that were treated by 6-OHDA. We observed that 6-OHDA treatment resulted in PC-12 cell injury and the cell survival was reduced to 39.8% in the 6-OHDA treatment group relative to the control cells (Fig. 2C, p < 0.05). Thus, PKM2 overexpression vector transfection leads to a significant increase in cell survival compared with that of the 6-OHDA-treated cells (Fig. 2C, p < 0.05), whereas there were no significant differences in cell survival between the control vector pcDNA3.1 transfection and the 6-OHDA-treated group (Fig. 2C, p > 0.05).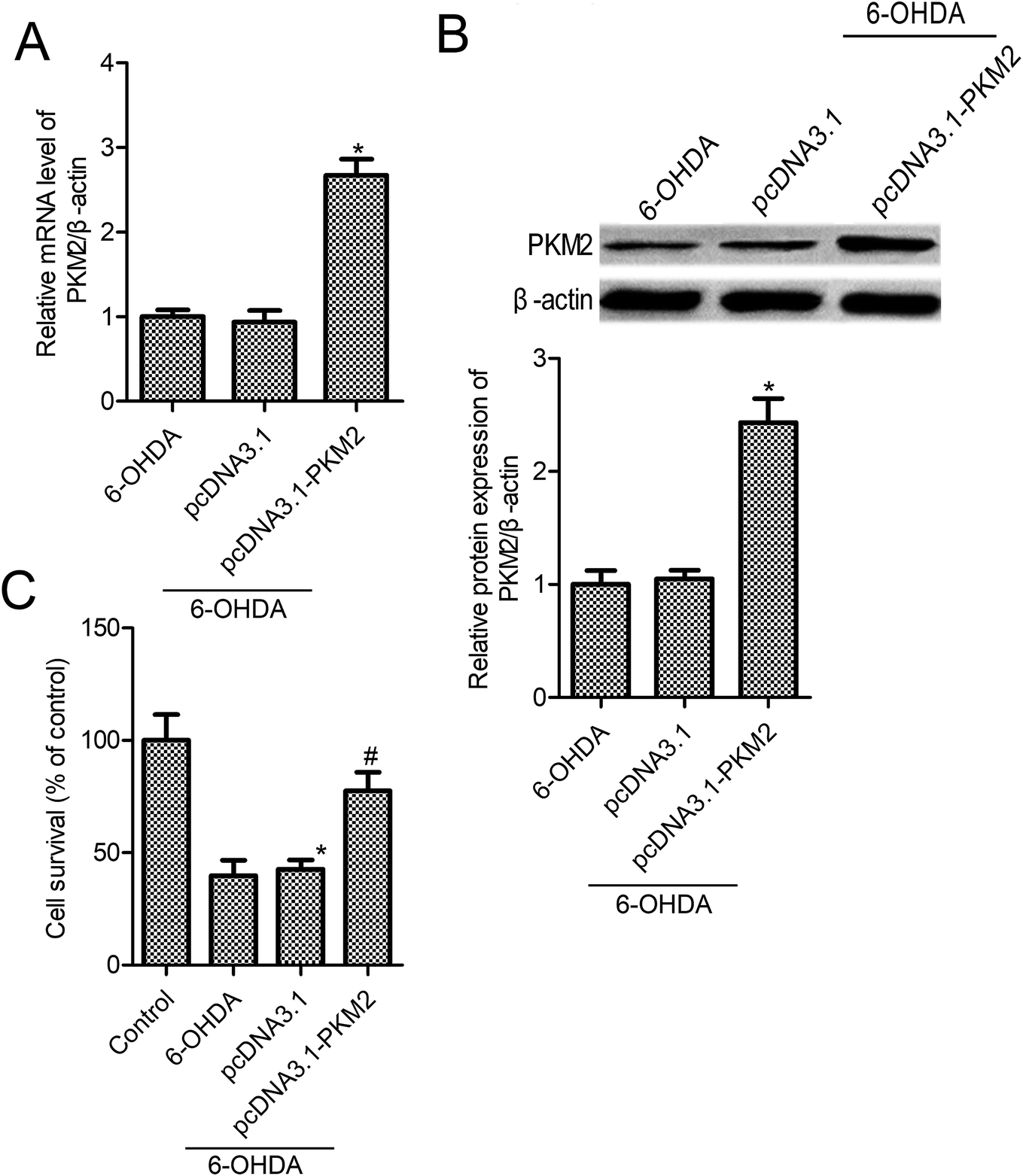 | ||
| Fig. 2 Up-regulation PKM2 expression suppressed 6-OHDA-induced PC12 cell injury. (A) RT-qPCR was used to measure PKM2 mRNA level; (B) Western blot was used to measure PKM2 protein expression; (C) PC12 cell survival was measured by the MTT assay. PC12 cells were exposed to 100 μM 6-OHDA for 24 h or transfected with pcDNA3.1-PKM2 or the control pcDNA3.1 vector for 48 h, and then exposed to 100 μM 6-OHDA for 24 h. Data represent the means ± SD from three different experiments. *P < 0.05 compared with the control group; #P < 0.05 compared with the 6-OHDA group. | ||
3.3 PC12 cell apoptosis was impeded by pcDNA3.1-PKM2 transfection in 6-OHDA-induced PC12 cells
The above results confirmed that PKM2 overexpression promoted cell survival in 6-OHDA-induced PC12 cells. To further find out whether PKM2 overexpression could protect against PC-12 cells from 6-OHDA-induced injury, we next detected the effects of PKM2 on cell apoptosis and caspase-3 activity in 6-OHDA-treated PC-12 cells. The results from Cell Death Detection ELISA assay showed that 6-OHDA treatment promoted PC12 cell apoptosis, whereas cell apoptosis was decreased in pcDNA3.1-PKM2 transfection PC-12 cells compared with that of the group treated with 6-OHDA alone (Fig. 3A, p < 0.05). Furthermore, studies confirmed the same trend between cell apoptosis and caspase-3 activity. PcDNA3.1-PKM2 transfection reduced caspase-3 activity compared with that of the 6-OHDA group (Fig. 3B, p < 0.05).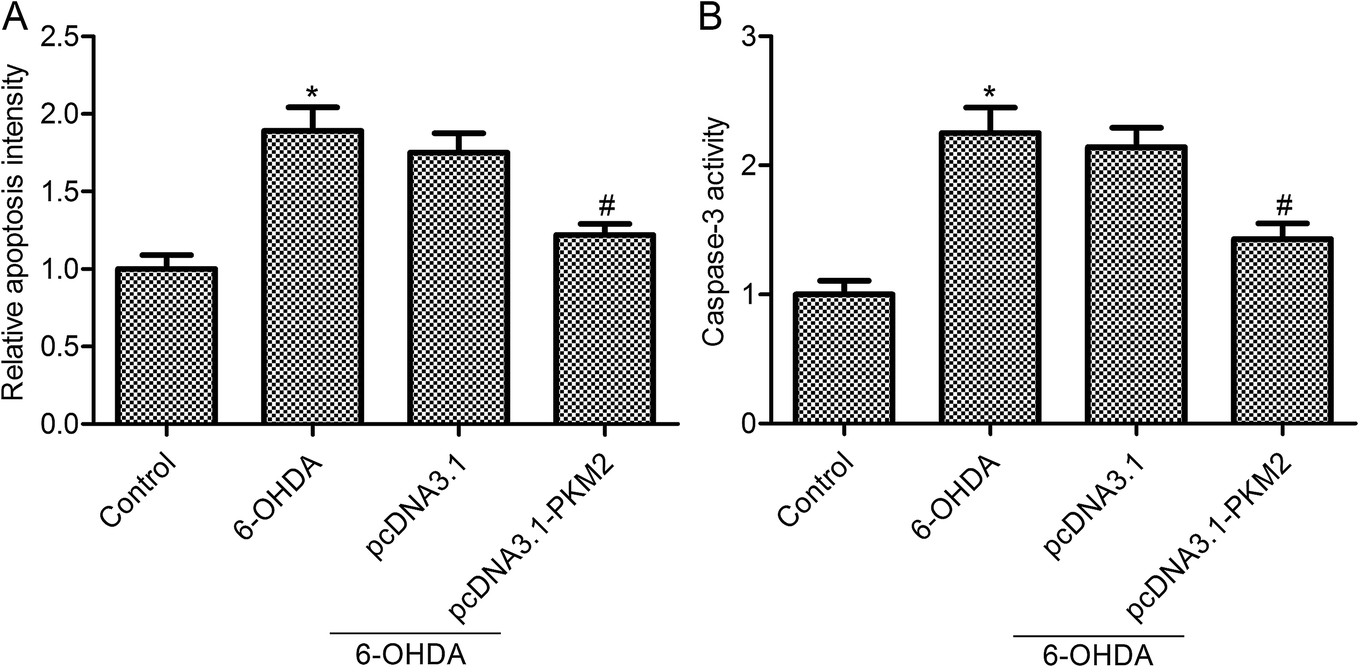 | ||
| Fig. 3 PKM2 overexpression inhibited 6-OHDA-induced PC12 cell apoptosis. (A) Cell apoptosis was determined by Cell Death Detection ELISA kit; (B) caspase-3 activity assay kit was used to evaluate the caspase-3 activity. PC12 cells were exposed to 100 μM 6-OHDA for 24 h or transfected with pcDNA3.1-PKM2 or the control pcDNA3.1 vector for 48 h and then exposed to 100 μM 6-OHDA for 24 h. Data represent the means ± SD from three different experiments. *P < 0.05 compared with the control group; #P < 0.05 compared with the 6-OHDA group. | ||
3.4 Increased Brg1 and pSTAT3 expression in PKM2-overexpressed PC12 cells
The immunoblotting data showed that the expression of Brg1 and pSTAT3 was markedly decreased by 6-OHDA treatment, whereas the total STAT3 expression was not obviously changed after 6-OHDA treatment (Fig. 4). Moreover, we observed that Brg1 and pSTAT3 expression were increased in PKM2-overexpressed PC12 cells (Fig. 4). To further investigate the regulating effect of PKM2 on STAT3 activation, the Brg1 gene expression was knocked down by transfecting Brg1 siRNA into PC12 cells. First, we observed that Brg1 siRNA transfection significantly suppressed Brg1 protein expression, and we also found that STAT3 activation, which was induced by PKM2 overexpression, was impeded in Brg1-silenced PC12 cells. These results suggested that PKM2-increased p-STAT3 expression depended on the high expression of Brg1 in 6-OHDA-treated PC12 cells.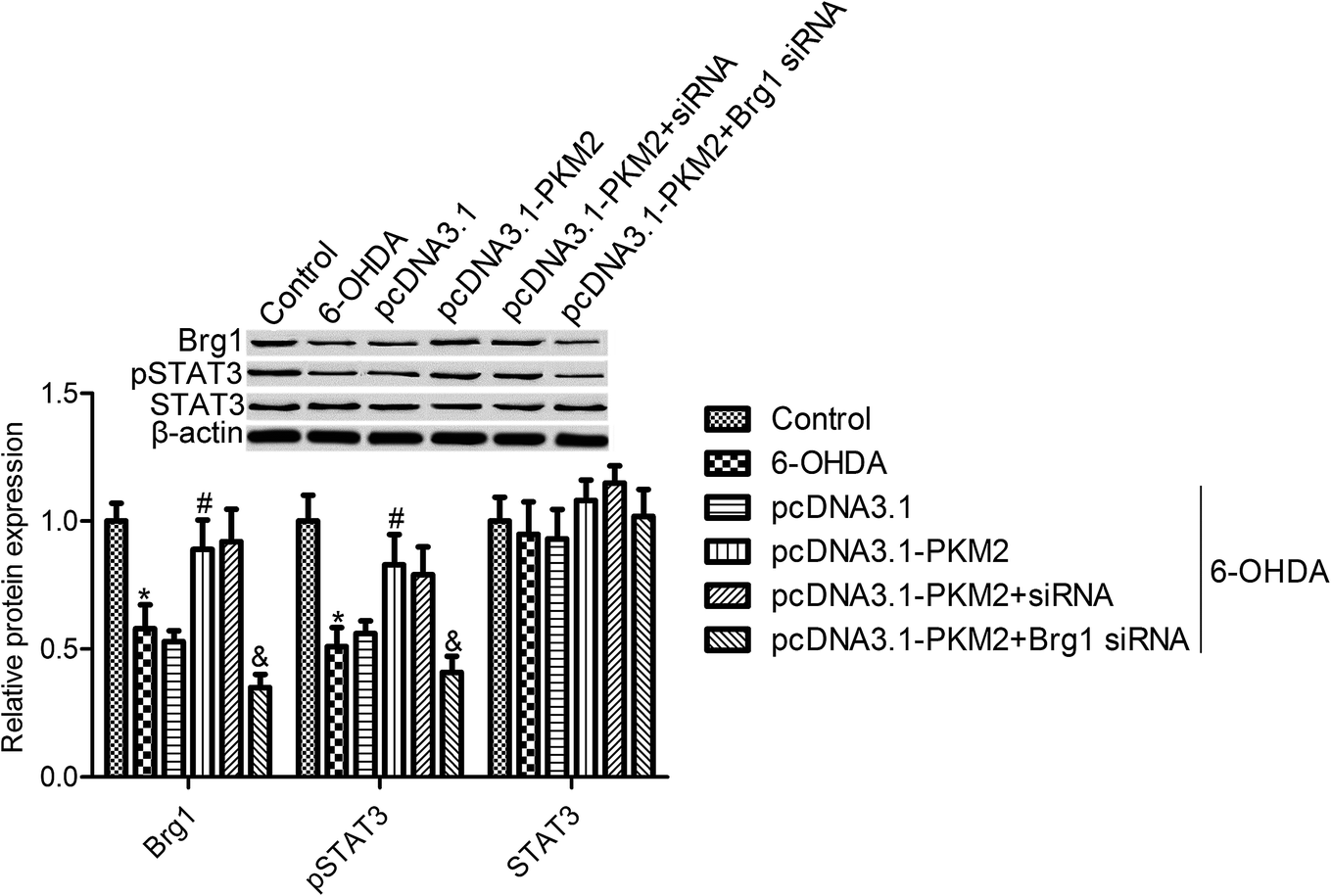 | ||
| Fig. 4 PKM2 overexpression increased Brg1 and pSTAT3 expression in 6-OHDA treated PC12 cells. Brg1, p-STAT3 and STAT3 expression were determined by Western blots. PC12 cells were exposed to 100 μM 6-OHDA for 24 h or transfected with pcDNA3.1-PKM2 or the control pcDNA3.1 vector for 48 h or co-transfected with pcDNA3.1-PKM2 and Brg1 siRNA, and then exposed to 100 μM 6-OHDA for 24 h. Data represent the means ± SD from three different experiments. *P < 0.05 compared with the control group; #P < 0.05 compared with the 6-OHDA group. | ||
3.5 STAT3 inhibition reversed the effects of PKM2 on 6-OHDA-induced PC12 cell injury
To further investigate the effect of STAT3 activity on PKM2 function in 6-OHDA-induced PC12 cells, STAT3 was inhibited by S3I-201 (Calbiochem, La Jolla, CA, USA), a novel inhibitor of STAT3. Both cell survival and apoptosis were measured in 6-OHDA induced PC12 cells, which were treated with pcDNA3.1-PKM2 and STAT3 inhibitors. MTT assay confirmed that STAT3 inhibition suppressed PC12 cell survival compared with the PKM2 overexpression group (Fig. 5A). Also, STAT3 inhibition reversed the effects of PKM2 overexpression on cell apoptosis and caspase-3 activity in 6-OHDA-treated PC12 cells (Fig. 5B and C). These results suggested that the STAT3 signal pathway was involved in the protection effects of PKM2 on 6-OHDA-induced PC12 cell injury.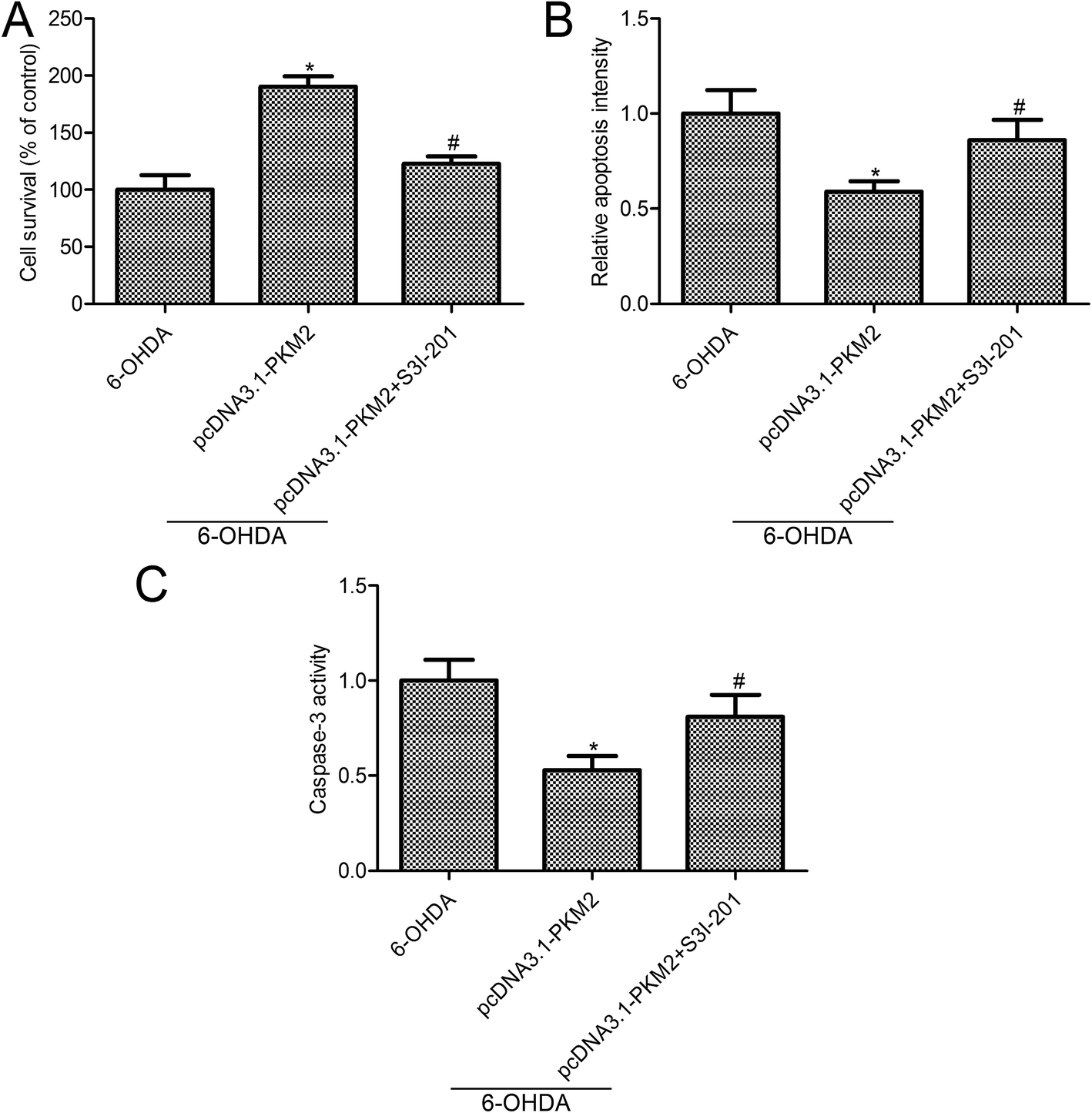 | ||
| Fig. 5 STAT3 inhibition reversed the effects of PKM2 on 6-OHDA-induced PC12 cells. (A) PC12 cell survival was measured by the MTT assay; (B) cell apoptosis was determined by Cell Death Detection ELISA kit; (C) caspase-3 activity assay kit was used to evaluate the caspase-3 activity. PC12 cells were exposed to 100 μM 6-OHDA for 24 h or transfected with pcDNA3.1-PKM2 for 48 h or treated with pcDNA3.1-PKM2 and STAT3 inhibitor S3I-201 (100 μM) for 48 h, and then exposed to 100 μM 6-OHDA for 24 h. Data represent the means ± SD from three different experiments. *P < 0.05 compared with the 6-OHDA group; #P < 0.05 compared with the pcDNA3.1-PKM2 group. | ||
4. Discussion
PD, a common, debilitating, neurological disorder, is growing fast in the developed world.30 PD causes a significant loss of dopaminergic neurons in substantia nigra.31 PD is always associated with irreversible neural cell impairment and damage. To reveal the related mechanism in neuron damage, an experimental PC12 in vitro cell model of PD was established using 6-OHDA, which is commonly used to mimic PD neurodegeneration.10 In the current study, we observed that 6-OHDA treatment markedly suppressed cell survival and promoted cell apoptosis. The current study evaluated the neuroprotection role of PKM2 and the related mechanism that involved the Brg 1/STAT3 pathway. For this purpose, the effects of PKM2 on cell viability, apoptosis and Brg1, STAT3 expression were instigated in 6-OHDA-treated PC12 cells.It is now increasingly clear that the global gene expression was altered in postmortem brains of PD patients, which were analyzed by a meta-genome-wide expression study (GWES) analysis.32 Evidence also demonstrated that the pyruvate kinase PKM is expressed in the brain.33 Moreover, pyruvate kinase isozyme M2 (PKM2) expression was decreased in PD participants.34 We speculate that PKM2 may be involved in the neuron loss of PD. RT-PCR and Western blot assay showed that PKM2 expression was reduced in 6-OHDA-treated PC12 cells at mRNA and protein levels. PKM2 is a member of pyruvate kinase family and is widely expressed in cancer cells and normal proliferating cells.8 Currently, abundant evidence has focused on the function and mechanism of PKM2 on cancer cells.35,36 There is evidence suggesting that the crystal structures and kinetic coupling related mutants (H391Y and R399E) play roles in cell growth and migration.37 Matsui et al. showed that PKM2 is always expressed in tumor cells in a low active form, and PKM2 activation by an activator inhibited the tumor cell growth.38 The studies revealed PKM2 function and mechanism in neurons is limited. Venkatesan et al. suggested that the impairment of hippocampal neurogenesis is associated with PKM2 inhibition in the setting of methamphetamine abuse.16 In the current study, we found that PKM2 overexpression contributed to 6-OHDA-treated PC12 cell survival. However, the function of PKM2 structure, its mutants and active sites in neurons is unclear; thus, additional experiments are required for confirmation.
PKM2 has been shown to be implicated in ERK1/2, STAT-3 and PI3K/AKT phosphorylation.39,40 Wang et al. observed that Brg1 plays a significant role in trans-differentiation between distinct neural cell types and is involved in the process of neural stem cell (NSC) differentiation to distinct neural cell types.41 Moreover, there is evidence indicating that the key subunit of the SWI/SNF chromatin-remodeling complex Brg1 (SMARCA4) could bind to STAT3 and control the STAT3 activation26 and involvement in neuron development and survival.41 Also, Xu et al. indicated that Brg1 and p-STAT3 levels were reduced in diabetic rats and increased Brg1 and p-STAT3 protein expression, leading to improved cardiac function.26 Moreover, evidence suggests that the increase in the Brg1 and p-STAT3 expression plays the role of cardioprotection function in cultured H9C2 cells.41 In the present study, we demonstrated that PKM2 overexpression significantly promoted Brg1 and p-STAT3 expression in 6-OHDA-treated PC12 cells. We also confirmed that PKM2-activated STAT3 signal is dependent on the high level of Brg1. However, there is additional evidence indicating that Brg1 knockdown increased p-STAT3 expression in cancer cells.23 Thus, the relationship between Brg1 and p-STAT3 is different in different types of cells.
5. Conclusion
In conclusion, our study demonstrated that PKM2 overexpression markedly promoted the survival of 6-OHDA-treated PC12 cells and inhibited cell apoptosis through the Brg1/STAT3 signal pathway. However, in vivo tissue studies of animals and patients were needed to reveal the neuroprotection effects of PKM2 on PD.Conflicts of interest
The authors declare that they have no conflict of interest.References
- W. Dauer and S. Przedborski, Neuron, 2003, 39, 889–909 CrossRef CAS PubMed.
- W. Poewe, K. Seppi, C. M. Tanner, G. M. Halliday, P. Brundin, J. Volkmann, A. E. Schrag and A. E. Lang, Nat. Rev. Dis. Primers, 2017, 3, 13 Search PubMed.
- Z. S. Agim and J. R. Cannon, BioMed Res. Int., 2015, 672838, 20 Search PubMed.
- O. B. Tysnes and A. Storstein, J. Neural Transm., 2017, 124, 901–905 CrossRef PubMed.
- J. D. Gazewood, D. R. Richards and K. Clebak, Am. Fam. Physician, 2013, 87, 267–273 Search PubMed.
- H. Javed, M. F. Nagoor Meeran, S. Azimullah, A. Adem, B. Sadek and S. K. Ojha, Front. Pharmacol., 2018, 9, 1555 CrossRef PubMed.
- J. Tong, G. Rathitharan, J. H. Meyer, Y. Furukawa, L. C. Ang, I. Boileau, M. Guttman, O. Hornykiewicz and S. J. Kish, Brain, 2017, 140, 2460–2474 CrossRef PubMed.
- O. Bar-Am, O. Weinreb, T. Amit and M. B. Youdim, J. Neurochem., 2010, 112, 1131–1137 CrossRef CAS PubMed.
- M. M. Hossain, B. Weig, K. Reuhl, M. Gearing, L. J. Wu and J. R. Richardson, Exp. Neurol., 2018, 308, 111–119 CrossRef CAS PubMed.
- A. Chierchia, N. Chirico, L. Boeri, I. Raimondi, G. A. Riva, M. T. Raimondi, M. Tunesi, C. Giordano, G. Forloni and D. Albani, Eur. J. Pharm. Biopharm., 2017, 121, 113–120 CrossRef CAS PubMed.
- O. A. Levy, C. Malagelada and L. A. Greene, Apoptosis, 2009, 14, 478–500 CrossRef PubMed.
- W. Yang and Z. Lu, J. Cell Sci., 2015, 128, 1655–1660 CrossRef CAS PubMed.
- T. Noguchi, H. Inoue and T. Tanaka, J. Biol. Chem., 1986, 261, 13807–13812 CAS.
- X. Yang, H. Chen, M. Zhu, R. Zhu, B. Qin, H. Fang, M. Dai, A. Sang and X. Liu, Cell. Mol. Neurobiol., 2015, 35, 1175–1186 CrossRef CAS PubMed.
- B. Wang, S. Liu, B. Fan, X. Xu, Y. Chen, R. Lu, Z. Xu and X. Liu, J. Headache Pain, 2018, 19, 018–0836 CrossRef PubMed.
- A. Venkatesan, L. Uzasci, Z. Chen, L. Rajbhandari, C. Anderson, M. H. Lee, M. A. Bianchet, R. Cotter, H. Song and A. Nath, Mol. Brain, 2011, 4, 1756–6606 Search PubMed.
- C. Simone, J. Cell. Physiol., 2006, 207, 309–314 CrossRef CAS PubMed.
- C. Xiao, L. Gao, Y. Hou, C. Xu, N. Chang, F. Wang, K. Hu, A. He, Y. Luo, J. Wang, J. Peng, F. Tang, X. Zhu and J. W. Xiong, Nat. Commun., 2016, 7, 13787 CrossRef CAS PubMed.
- S. J. Bultman, T. C. Gebuhr and T. Magnuson, Genes Dev., 2005, 19, 2849–2861 CrossRef CAS PubMed.
- S. J. Bultman, T. C. Gebuhr, H. Pan, P. Svoboda, R. M. Schultz and T. Magnuson, Genes Dev., 2006, 20, 1744–1754 CrossRef CAS PubMed.
- K. Stankunas, C. T. Hang, Z. Y. Tsun, H. Chen, N. V. Lee, J. I. Wu, C. Shang, J. H. Bayle, W. Shou, M. L. Iruela-Arispe and C. P. Chang, Dev. Cell, 2008, 14, 298–311 CrossRef CAS PubMed.
- C. T. Hang, J. Yang, P. Han, H. L. Cheng, C. Shang, E. Ashley, B. Zhou and C. P. Chang, Nature, 2010, 466, 62–67 CrossRef CAS PubMed.
- B. Eroglu, G. Wang, N. Tu, X. Sun and N. F. Mivechi, Dev. Dyn., 2006, 235, 2722–2735 CrossRef CAS PubMed.
- S. Seo, G. A. Richardson and K. L. Kroll, Development, 2005, 132, 105–115 CrossRef CAS PubMed.
- W. Li, Y. Xiong, C. Shang, K. Y. Twu, C. T. Hang, J. Yang, P. Han, C. Y. Lin, C. J. Lin, F. C. Tsai, K. Stankunas, T. Meyer, D. Bernstein, M. Pan and C. P. Chang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1738–1743 CrossRef CAS PubMed.
- J. Xu, S. Lei, Y. Liu, X. Gao, M. G. Irwin, Z. Y. Xia, Z. Hei, X. Gan, T. Wang and Z. Xia, J. Diabetes Res., 2013, 2013, 716219 Search PubMed.
- X. Zhu, L. Sun, J. Lan, L. Xu, M. Zhang, X. Luo, J. Gong, G. Wang, X. Yuan, J. Hu and J. Wang, Oncotarget, 2016, 7, 36501–36509 Search PubMed.
- G. Wang, X. Li, F. Huang, J. Zhao, H. Ding, C. Cunningham, J. E. Coad, D. C. Flynn, E. Reed and Q. Q. Li, Cell. Mol. Life Sci., 2005, 62, 881–893 CrossRef CAS PubMed.
- J. Lee, U. Kang, E. K. Seo and Y. S. Kim, Int. Immunopharmacol., 2016, 34, 155–164 CrossRef CAS PubMed.
- M. J. Haney, N. L. Klyachko, Y. Zhao, R. Gupta, E. G. Plotnikova, Z. He, T. Patel, A. Piroyan, M. Sokolsky, A. V. Kabanov and E. V. Batrakova, J. Controlled Release, 2015, 207, 18–30 CrossRef CAS PubMed.
- L. M. de Lau and M. M. Breteler, Lancet Neurol., 2006, 5, 525–535 CrossRef PubMed.
- B. Zheng, Z. Liao, J. J. Locascio, K. A. Lesniak, S. S. Roderick, M. L. Watt, A. C. Eklund, Y. Zhang-James, P. D. Kim, M. A. Hauser, E. Grunblatt, L. B. Moran, S. A. Mandel, P. Riederer, R. M. Miller, H. J. Federoff, U. Wullner, S. Papapetropoulos, M. B. Youdim, I. Cantuti-Castelvetri, A. B. Young, J. M. Vance, R. L. Davis, J. C. Hedreen, C. H. Adler, T. G. Beach, M. B. Graeber, F. A. Middleton, J. C. Rochet and C. R. Scherzer, Sci. Transl. Med., 2010, 2, 3001059 Search PubMed.
- K. Imamura and T. Tanaka, J. Biochem., 1972, 71, 1043–1051 CrossRef CAS PubMed.
- J. A. Potashkin, J. A. Santiago, B. M. Ravina, A. Watts and A. A. Leontovich, PLoS One, 2012, 7, 27 CrossRef PubMed.
- T. L. Dayton, T. Jacks and M. G. Vander Heiden, EMBO Rep., 2016, 17, 1721–1730 CrossRef CAS PubMed.
- N. Azoitei, A. Becher, K. Steinestel, A. Rouhi, K. Diepold, F. Genze, T. Simmet and T. Seufferlein, Mol. Cancer, 2016, 15, 015–0490 CrossRef PubMed.
- T. J. Chen, H. J. Wang, J. S. Liu, H. H. Cheng, S. C. Hsu, M. C. Wu, C. H. Lu, Y. F. Wu, J. W. Wu, Y. Y. Liu, H. J. Kung and W. C. Wang, Commun. Biol., 2019, 2, 105 CrossRef PubMed.
- Y. Matsui, I. Yasumatsu, T. Asahi, T. Kitamura, K. Kanai, O. Ubukata, H. Hayasaka, S. Takaishi, H. Hanzawa and S. Katakura, Bioorg. Med. Chem., 2017, 25, 3540–3546 CrossRef CAS PubMed.
- X. Gao, H. Wang, J. J. Yang, J. Chen, J. Jie, L. Li, Y. Zhang and Z. R. Liu, J. Biol. Chem., 2013, 288, 15971–15979 CrossRef CAS PubMed.
- C. Wang, J. Jiang, J. Ji, Q. Cai, X. Chen, Y. Yu, Z. Zhu and J. Zhang, Sci. Rep., 2017, 7, 017–03031 CrossRef PubMed.
- Y. Wang, H. Li, H. Huang, S. Liu, X. Mao, S. Wang, S. S. Wong, Z. Xia and M. G. Irwin, Clin. Sci., 2016, 130, 801–812 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |