
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMerging catalyst-free synthesis and iodine catalysis: one-pot synthesis of dihydrofuropyrimidines and spirodihydrofuropyrimidine pyrazolones†
Ya-Yun Zheng,
Kai-Xiang Feng,
Ai-Bao Xia *,
Jie Liu,
Cheng-Ke Tang,
Zhan-Yu Zhou and
Dan-Qian Xu*
*,
Jie Liu,
Cheng-Ke Tang,
Zhan-Yu Zhou and
Dan-Qian Xu*
Catalytic Hydrogenation Research Centre, State Key Laboratory Breeding Base of Green Chemistry-Synthesis Technology, Zhejiang University of Technology, Hangzhou, 310014, China. E-mail: xiaaibao@zjut.edu.cn; chrc@zjut.edu.cn
First published on 27th March 2019
Abstract
A new and efficient one-pot strategy combining catalyst-free synthesis and iodine catalysis has been developed for the synthesis of dihydrofuropyrimidines and spirodihydrofuropyrimidine pyrazolones. This approach affords products in moderate to high yields (up to 96%) with excellent diastereoselectivities (up to >25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr). The reaction is simple to carry out and is metal-free.
:1 dr). The reaction is simple to carry out and is metal-free.
Furo fused pyrimidines1 and dihydrofuro fused pyrimidines,2 derivatives of the pyrimidine species,3 are prevalent heteroaromatic core structures found in a broad variety of natural products, pharmaceuticals, and other biologically active molecules (Scheme 1). In particular, dihydrofuropyrimidine derivatives have shown a diverse range of useful biological and pharmacological activities such as anticancer, anti-inflammatory, antiproliferative, and antirheumatic activities. The substituent group on the dihydrofuran moiety significantly influences the biological activity. However, efficient procedures for the synthesis of dihydrofuropyrimidines are not well developed. In 1987, Taylor et al. reported intramolecular Diels–Alder reactions of 1,2,4-triazines to synthesize dihydrofuro fused pyrimidines under very high temperature condition (Scheme 2A).4 Alternatively, dihydrofuropyrimidine derivatives can be prepared in relatively low yields from pre-synthesized 2,4-dichloro-dihydrofuropyrimidines via further substitution reactions and coupling reactions (Scheme 2B). However, several steps are required to prepare highly functionalized starting materials with different furan groups.2a,5 Both of these methods have some well-known drawbacks: (a) harsh reaction conditions and the requirement of multistep synthetic sequences; (b) the use of poisonous or expensive reagents such as COCl2, POCl3, and transition metal Pd; and (c) low yields and limited substrate scope. Therefore, it is highly desirable to find an efficient and general synthetic methodology for the construction of various substituted dihydrofuropyrimidines, from easily accessible raw materials, under simple and mild reaction conditions, and without the use of potentially toxic transition metals.
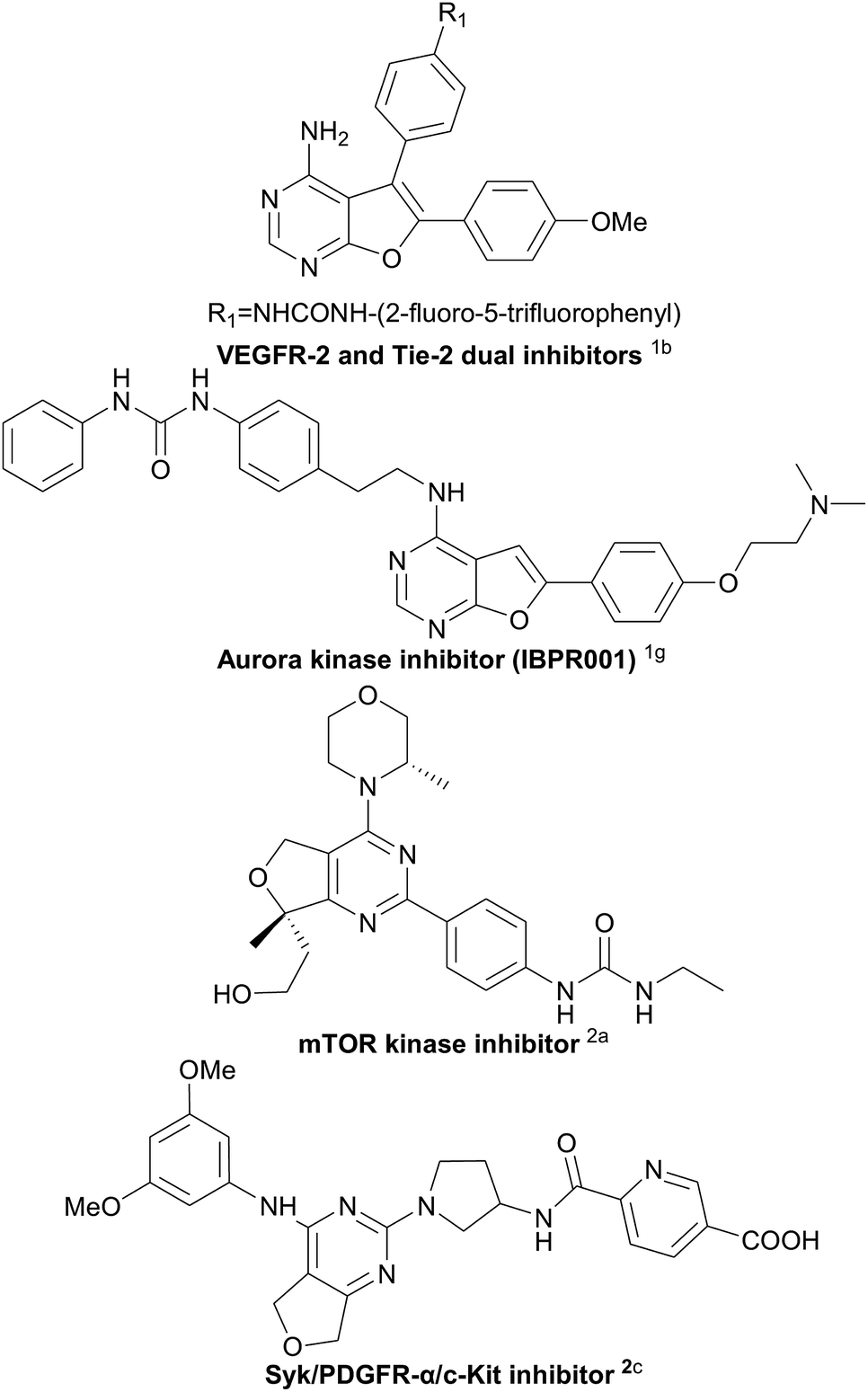 | ||
| Scheme 1 Selected important biologically active heterocycles containing fused pyrimidine moieties. | ||
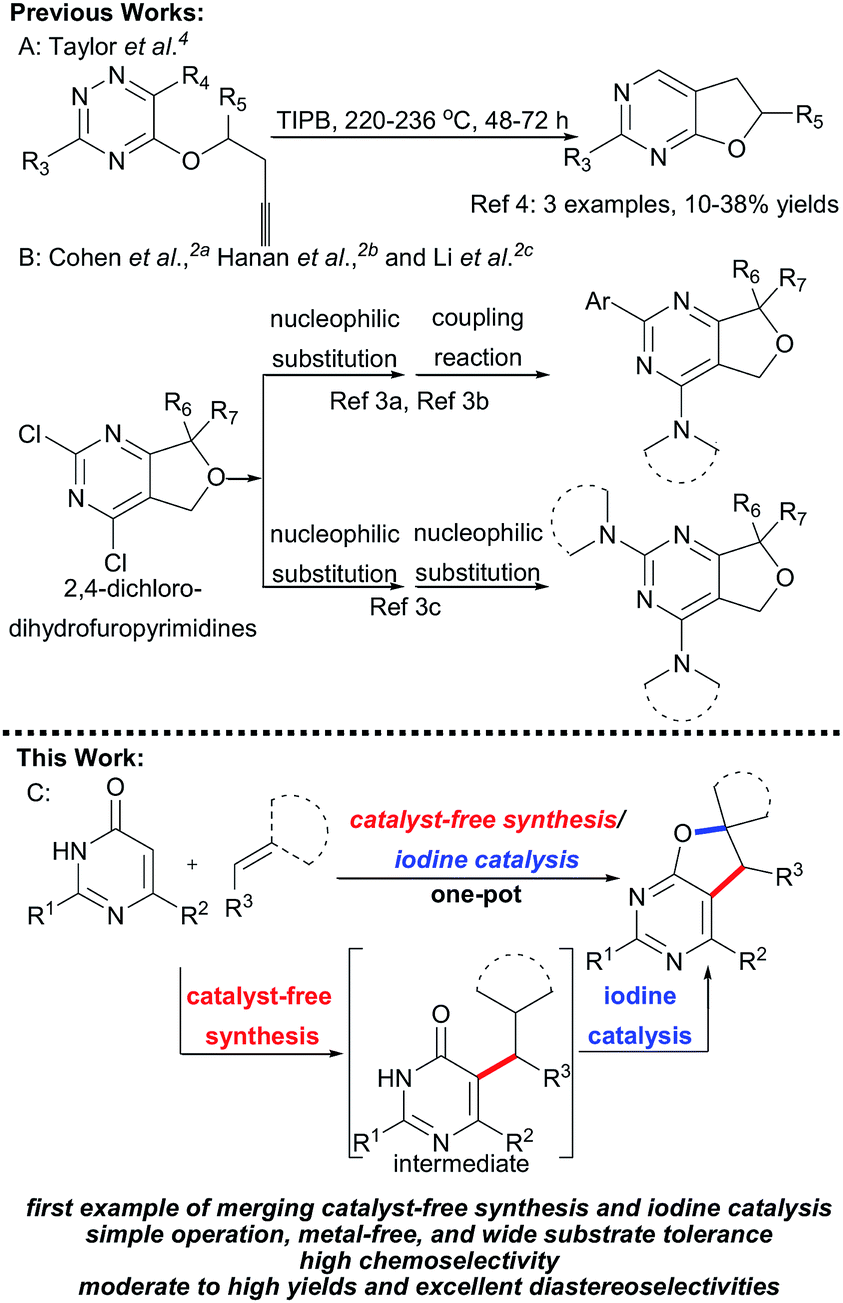 | ||
| Scheme 2 General methods for the synthesis of dihydrofuropyrimidine derivatives. | ||
In past decades, catalyst-free synthesis has been extensively studied and applied to the construction of complex molecules, as it tends to be inexpensive and environmentally friendly.6a,b,c For example, Deb, Baruah, and co-workers described two procedures for the synthesis of 1,3-oxazines6d and pyrimidine ring compounds6f under the catalyst-free conditions. In 2017, the group of Brahmachari presented a catalyst-free protocol for the construction of functionalized pyridodipyrimidines.6e In recent years, iodine catalysis has also emerged as an efficient, metal-free methodology for chemical bond construction which has low toxicity, is environmentally benign, and is readily available.7 Very recently, we presented a new and efficient catalytic strategy that combined organocatalysis and iodine catalysis for the one-pot sequential catalytic synthesis of spirodihydrobenzofurans.8 The success of that approach suggested that it might be possible to prepare the biologically important dihydrofuro fused pyrimidines using a new strategy that combines catalyst-free synthesis and iodine catalysis. Herein, we report a facile and efficient methodology for the synthesis of dihydrofuro fused pyrimidine derivatives using 2,6-diaminopyrimidin-4(3H)-ones,9 versatile building blocks in organic synthesis which can be easily prepared,10 nitroolefins, and unsaturated pyrazolones (Scheme 2C), in which the intermolecular reaction can be easily induced under very mild and catalyst free conditions, and the subsequent step of intramolecular cyclization can then be easily realized via iodine catalysis.
The one-pot reaction was initially performed with 2,6-diaminopyrimidin-4(3H)-one 1a and nitroolefin 2a as model substrates to demonstrate the feasibility of the designed approach. The overall synthesis proceeded smoothly, when the catalyst-free synthesis was conducted at 50 °C and the intramolecular cyclization was performed in the presence of I2 and tert-butyl hydroperoxide in CH3CN at room temperature, the reaction proceeded smoothly, affording the desired product 3a in 82% isolated yield (Table 1, entry 1). A representative range of iodine sources were then tested for the second cyclization step (entries 2–6). The yield could be improved to 86% by using potassium iodide (entry 3). The second step was tested further using KI and a range of oxidants: TBPB, DTBP, NaClO2, K2S2O8, and even H2O2 (entries 7–11). Although the reaction tolerated all of these oxidants, none produced as high a yield with KI as TBHP had. This combination was then examined in other solvents (entries 3 and 12–16) and it was found that CH3CN was best. With these optimal conditions in hand, further investigation was performed to study the influence of the reaction temperature on the catalyst-free synthesis step (entries 17–18). When the reaction was conducted at 40 or 60 °C, the product 3a was obtained at 55% and 73% yields, respectively. Interestingly, at room temperature, the first catalyst-free synthesis step went well in MeOH to give the final product 3a in 65% yield (entry 19). Further screening of the reaction conditions (entries 20–23) showed that the combination of TBAI and H2O2 in the second step, at room temperature, afforded product 3a in 77% yield (entry 23).
|
||||
|---|---|---|---|---|
| Entry | Iodine source | Oxidant | Solvent | Yieldb (%) |
| a Unless otherwise stated, all the reactions were conducted in solvent (1 mL) using 1a (0.1 mmol) and 2a (0.1 mmol) with stirring for 24 h at 50 °C, this was followed by the addition of iodine source (0.01 mmol) and oxidant (0.2 mmol), and the solution was then stirred for 24 h at room temperature.b Isolated yield.c The reaction was conducted in the presence of 20 mol% iodine source.d First step was conducted at 40 °C for 24 h.e First step was conducted at 60 °C for 24 h.f First step was conducted at room temperature for 24 h.g The dr value is >25:1 for all the isolated products. |
||||
| 1 | I2 | TBHP | CH3CN | 82 |
| 2c | NaI | TBHP | CH3CN | 84 |
| 3c | KI | TBHP | CH3CN | 86 |
| 4c | NH4I | TBHP | CH3CN | 72 |
| 5c | nBu4NI | TBHP | CH3CN | 80 |
| 6c | NIS | TBHP | CH3CN | 83 |
| 7c | KI | TBPB | CH3CN | 54 |
| 8c | KI | DTBP | CH3CN | 23 |
| 9c | KI | NaClO2 | CH3CN | 28 |
| 10c | KI | K2S2O8 | CH3CN | 32 |
| 11c | KI | H2O2 | CH3CN | 76 |
| 12c | KI | TBHP | DMF | Trace |
| 13c | KI | TBHP | THF | 82 |
| 14c | KI | TBHP | 1,4-Dioxane | Trace |
| 15c | KI | TBHP | MeOH | 68 |
| 16c | KI | TBHP | H2O | 63 |
| 17c,d | KI | TBHP | CH3CN | 55 |
| 18c,e | KI | TBHP | CH3CN | 73 |
| 19c,f | KI | TBHP | MeOH | 65 |
| 20c,f | NH4I | TBHP | MeOH | 50 |
| 21c,f | nBu4NI | TBHP | MeOH | 70 |
| 22c,f | NIS | TBHP | MeOH | 52 |
| 23c,f | nBu4NI | H2O2 | MeOH | 77 |

With the optimized conditions in hand, the substrate scope was then investigated (Table 2).11 The reaction was found to be compatible with nitroolefins bearing both electron-withdrawing and electron-donating groups. Substrates with electron-withdrawing substituent groups, such as F, Cl, Br, NO2, or CF3 on the phenyl ring, afforded moderate to high yields, ranging from 54% to 88% (3b–m). It was worth noting that, at room temperature, nitroalkenes bearing electron-rich motifs such as MeO, or Me on the phenyl core, heterocyclic nitroolefins, an aliphatic nitroolefin and α-methyl-substituted nitroolefin all tolerated this transformation, producing the desired dihydrofuropyrimidines in 25% to 82% yields (3n–v). For example, by using heterocyclic nitroolefins, products 3s and 3t were obtained in 57% and 75% yields, respectively. Aliphatic nitroolefin bearing a butyl group provided 3u in 25% yield. Furthermore, the α-methyl-substituted nitroolefin was also able to participate in this transformation to afford product 3v with 54% yield.
|
|---|
| a Condition A: the reactions were conducted in CH3CN (1 mL) using 1a (0.1 mmol) and 2a (0.1 mmol) with stirring for 24 h at 50 °C. This was followed by the addition of KI (0.02 mmol) and TBHP (0.2 mmol), and the solution was then stirred for 24 h at room temperature.b Condition B: the reactions were conducted in MeOH (1 mL) using 1a (0.1 mmol) and 2a (0.1 mmol) with stirring for 24 h at room temperature. This was followed by the addition of nBu4NI (0.02 mmol) and H2O2 (0.2 mmol), and the solution was then stirred for 24 h at room temperature.c Isolated yield.d The dr value is >25:1 for all the products. |
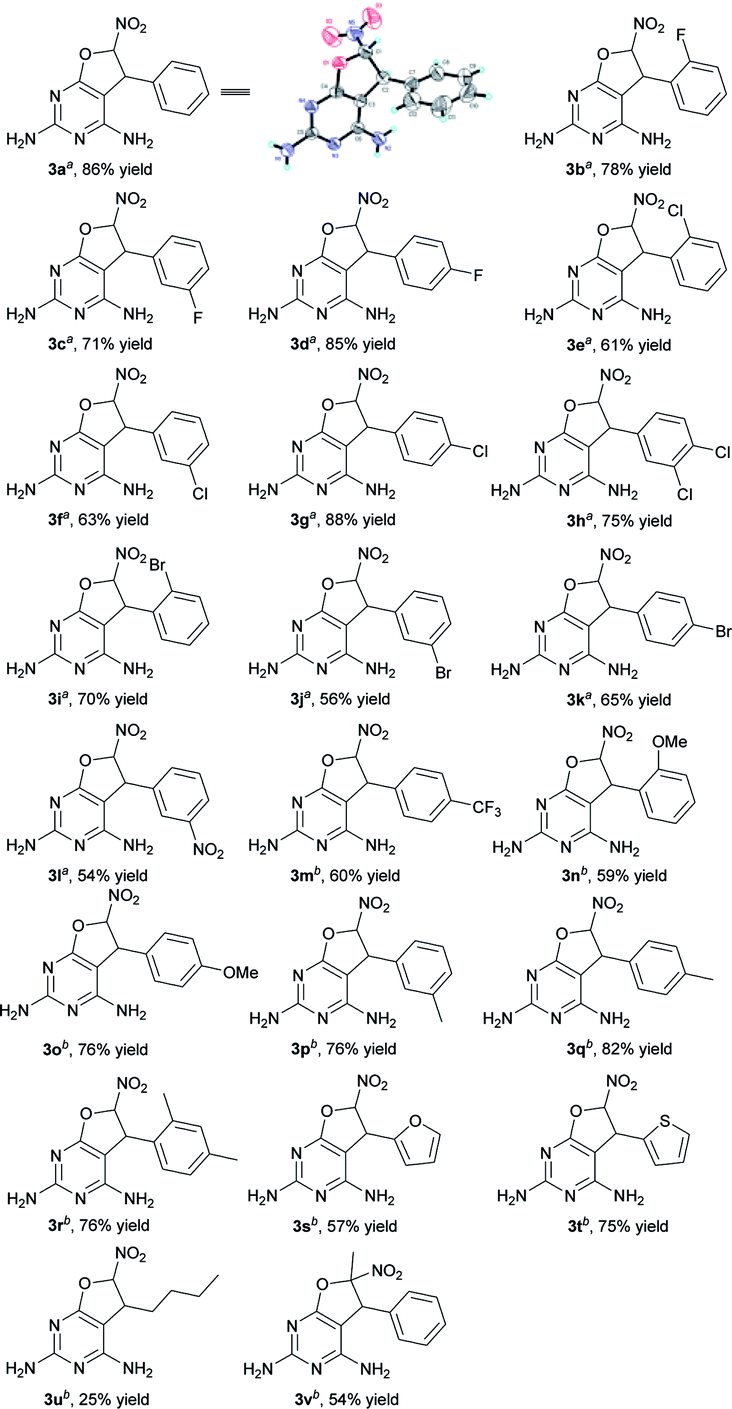 |

Having successfully synthesized an array of dihydrofuropyrimidines, attention was next turned toward the synthesis of biologically interesting spirodihydrofuropyrimidines. In the previous study, it was discovered that 2,6-diaminopyrimidin-4(3H)-one 1a could react with unsaturated pyrazolone 4a to form a spirodihydrofuropyrimidine 5a in 4–91% yields under oxygen atmosphere (Table 3, entries 1 and 2). These results suggested a unique opportunity to develop an one-pot approach for the synthesis of spirodihydrofuropyrimidines in an open air atmosphere. Then iodine sources and solvents were screened with these starting materials (1a and 4a) to optimize the reaction outcome under open air conditions (entries 3–12). The best result was obtained (entry 5, 96% yield) when I2 was utilized as iodine source and 1,4-dioxane was used as the solvent. Next, the influence of temperature on the reaction outcome was studied. The desired product 5a was produced in 80% and 90% yields when the reaction mixture was heated at 40 and 60 °C, respectively (entries 13 and 14).
|
||||
|---|---|---|---|---|
| Entry | Iodine source | Oxidant | Solvent | Yieldb (%) |
| a Unless otherwise stated, all the reactions were conducted in solvent (1 mL) using 1a (0.12 mmol) and 4a (0.1 mmol) with stirring for 24 h at 50 °C. This was followed by the addition of iodine source (0.02 mmol) and oxidant (O2 balloon), and the solution was then stirred for 48 h at room temperature.b Isolated yield.c The reaction was conducted in the presence of 10 mol% iodine source.d First step was conducted at 40 °C for 24 h.e First step was conducted at 60 °C for 24 h.f The dr value is >25:1 for all the isolated products. |
||||
| 1 | KI | O2 | CH3CN | 4 |
| 2 | KI | O2 | 1,4-Dioxane | 91 |
| 3 | KI | Open air | 1,4-Dioxane | 90 |
| 4 | TBAI | Open air | 1,4-Dioxane | 85 |
| 5c | I2 | Open air | 1,4-Dioxane | 96 |
| 6 | NIS | Open air | 1,4-Dioxane | 78 |
| 7c | I2 | Open air | THF | 45 |
| 8c | I2 | Open air | Isopropyl ether | 4 |
| 9c | I2 | Open air | EtOAc | Trace |
| 10c | I2 | Open air | CH3CN | 66 |
| 11c | I2 | Open air | MeOH | Trace |
| 12c | I2 | Open air | H2O | 22 |
| 13c,d | I2 | Open air | 1,4-Dioxane | 80 |
| 14c,e | I2 | Open air | 1,4-Dioxane | 90 |
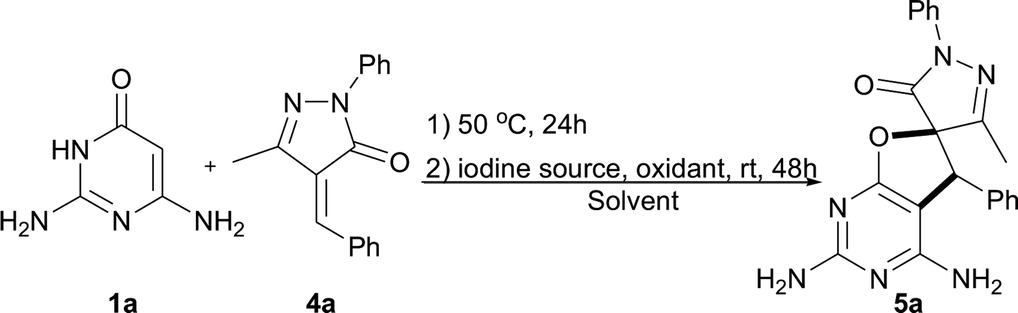
Next, the scope of the spirodihydrofuropyrimidine formation reaction was explored with pyrimidin-4(3H)-ones 1 and a wide variety of unsaturated pyrazolones 4 (Table 4).11 Initially, a number of unsaturated pyrazolones 5 were subjected to the reaction under optimized conditions. In general, a variety of unsaturated pyrazolones bearing both electron-withdrawing (F, Cl, Br, and NO2) and electron-donating (Me and MeO) groups at the phenyl ring were found to be suitable for the reaction, providing the corresponding spirodihydrofuropyrimidines 5a–5r in moderate to excellent yields (50–96%). The reaction of unsaturated pyrazolone containing an ethyl group substitution on the pyrazolone ring proceeded smoothly to afford the desired product 5s in 74% yield. Subsequently, to establish the generality of the one-pot reaction, some other amino substituted pyrimidin-4(3H)-ones were also employed under the optimized reaction conditions. When pyrimidin-4(3H)-one 1b and 1c were introduced as partners with unsaturated pyrazolone 4a, respectively, the desired products 5t and 5u were not formed, because the first step of catalyst-free synthesis did not take place. Substrate 1d reacted well and furnished the spirodihydrofuropyrimidine 5v in 30% yield.
|
|---|
| a All the reactions were conducted in 1,4-dioxane (1 mL) using 1 (0.12 mmol) and 4 (0.1 mmol) with stirring for 24 h at 50 °C. This was followed by the addition of I2 (0.01 mmol) under open air condition, and the solution was then stirred for 48 h at room temperature. Stated yields are isolated yields. The dr value is >25:1 for all the products. |
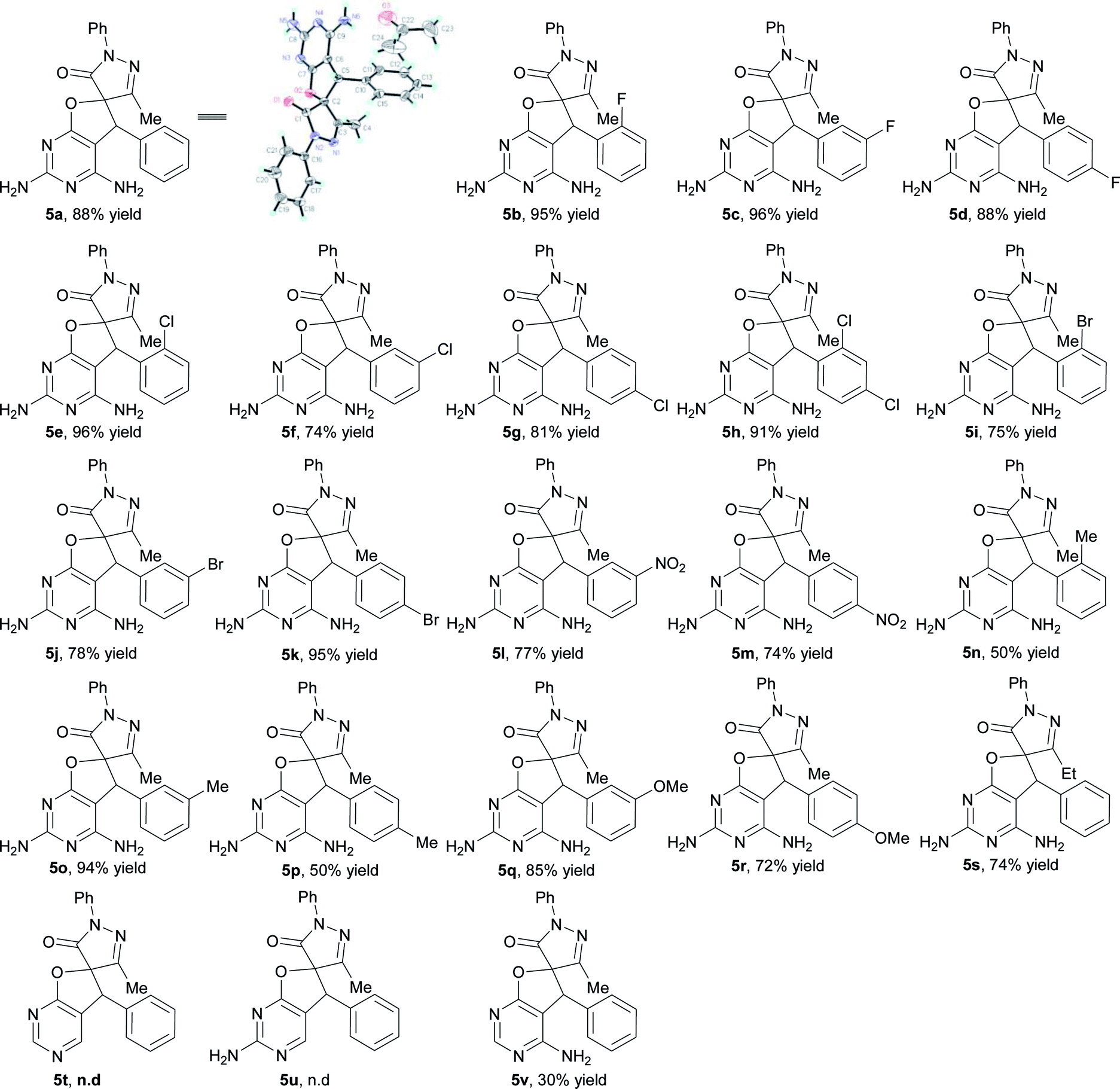 |
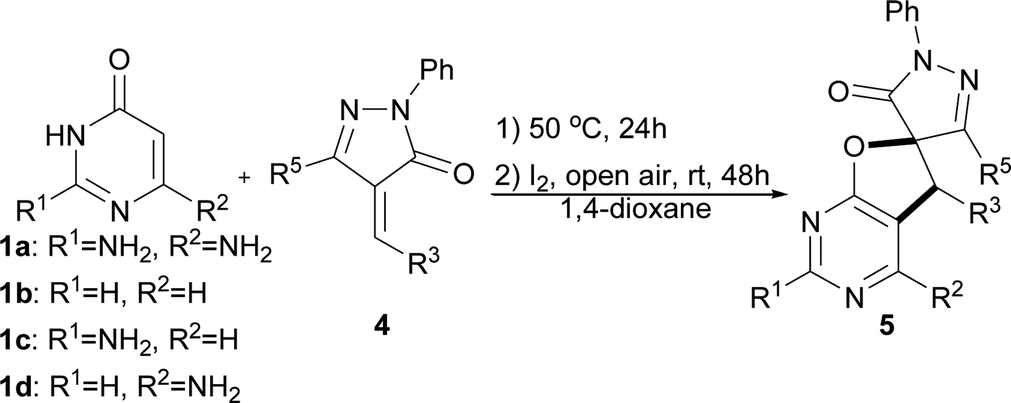
To understand the mechanism of the reaction, some control experiments were performed under modified conditions in 1,4-dioxane (Scheme 3). High yields could still be obtained in the presence of radical scavenger (TEMPO) (Scheme 3a) or under dark conditions for the second I2-catalyzed step (Scheme 3b). Thus, a radical mechanism could be excluded for this kind of transformation. Next, when the product of the first catalyst-free synthesis step, intermediate A, was separated and used for the I2-catalyzed step under the standard conditions, compound 5a was obtained in 89% yield (Scheme 3c).
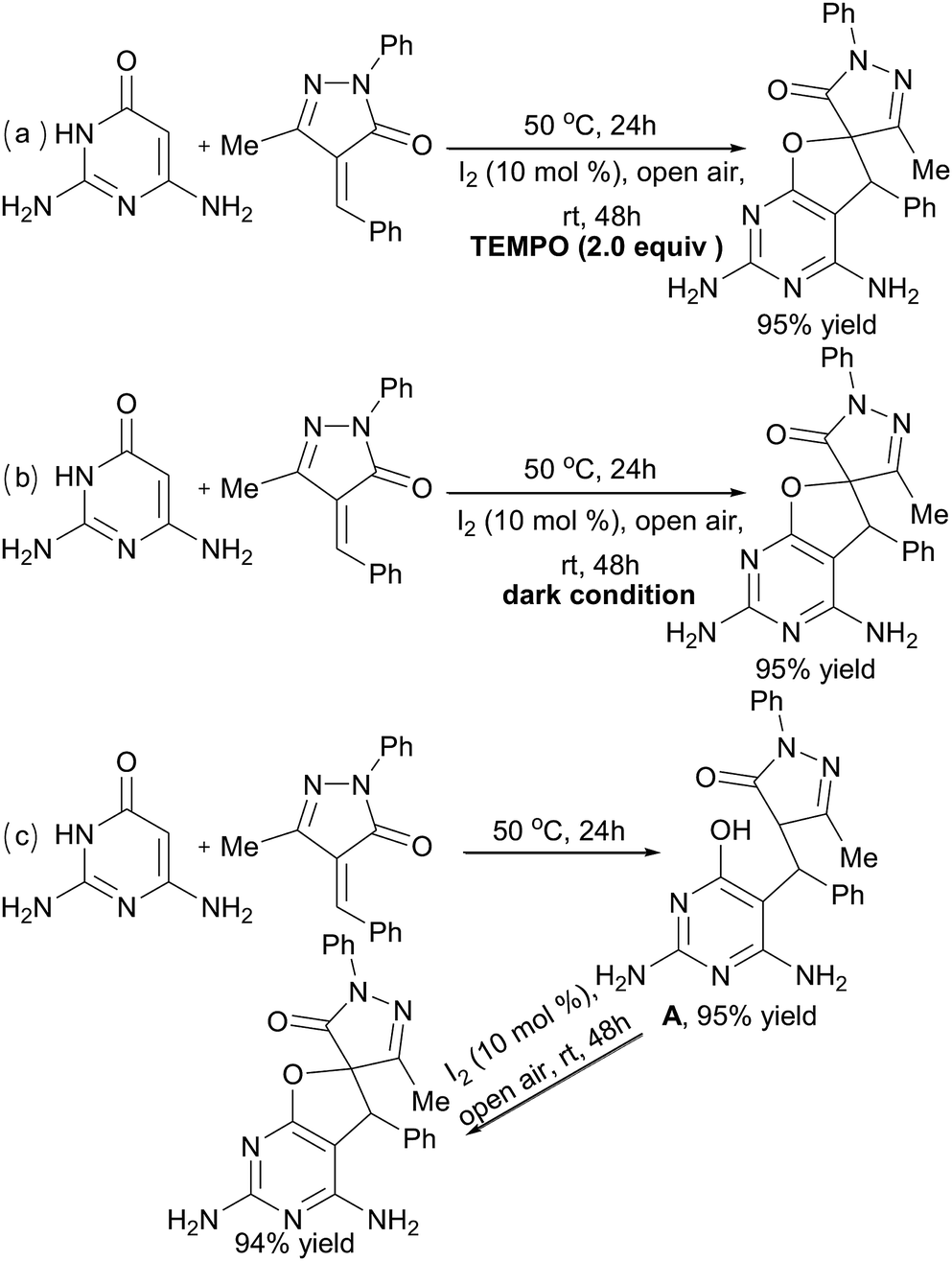 | ||
| Scheme 3 Mechanistic studies. | ||
Then, based on the aforementioned studies,8 a presumptive mechanism for the synthesis of dihydrofuropyrimidines from 2,6-diaminopyrimidin-4(3H)-ones and unsaturated pyrazolones is proposed (Scheme 4). The initial step should be a catalyst-free Michael addition of 1a to 4a, which generates an intermediate A via a transition state (TS). Then, this intermediate A can be transformed into 5a through the I2-catalyzed cyclization (via the iodization reaction and then the nucleophilic substitution). Finally, the HI reacts with O2 to give I2 for the next cycle.
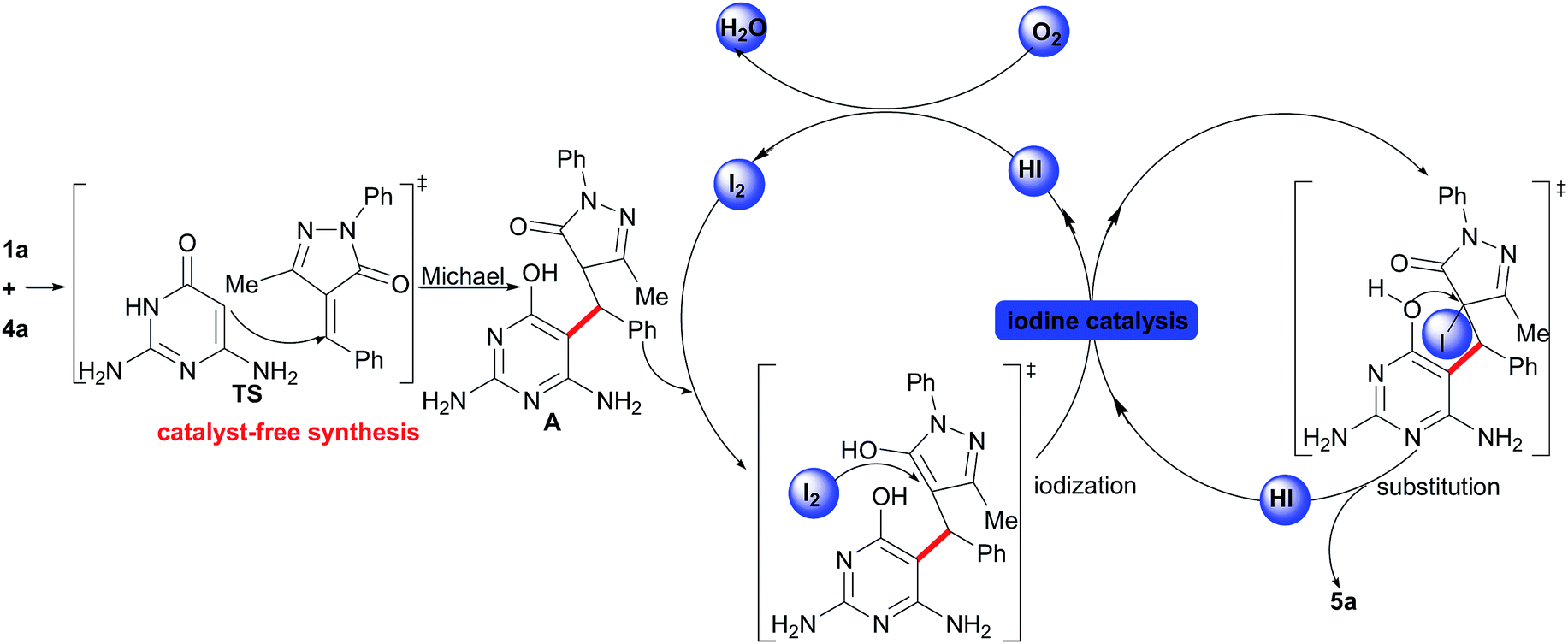 | ||
| Scheme 4 A proposed mechanism for the one-pot reaction. | ||
Conclusions
In summary, a novel strategy that combines catalyst-free synthesis and iodine catalysis in the presence of oxidant was developed for the one-pot synthesis of dihydrofuropyrimidines and spirodihydrofuropyrimidine pyrazolones from 2,6-diaminopyrimidin-4(3H)-ones, nitroolefins, and unsaturated pyrazolones. Dihydrofuro fused pyrimidine derivatives were synthesized in moderate to good yields with excellent diastereoselectivities using TBHP, or H2O2 as the oxidants. Spirodihydrofuropyrimidine pyrazolones were produced in high to excellent yields with good functional group tolerance and excellent diastereoselectivities under open air conditions (molecular oxygen as the oxidant). This reaction features simple operation and is metal-free. Further studies on the applications of this methodology to organic synthesis are ongoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Zhejiang Natural Science Foundation (LY18B020017), Zhejiang Key Laboratory of Green Pesticides and Cleaner Production Technology.Notes and references
- For selected examples, see: (a) A. Gangjee, X. Guo, S. F. Queener, V. Cody, N. Galitsky, J. R. Luft and W. Pangborn, J. Med. Chem., 1998, 41, 1263 CrossRef CAS PubMed; (b) Y. Miyazaki, S. Matsunaga, J. Tang, Y. Maeda, M. Nakano, R. J. Philippe, M. Shibahara, W. Liu, H. Sato, L. Wang and R. T. Nolte, Bioorg. Med. Chem. Lett., 2005, 15, 2203 CrossRef CAS PubMed; (c) A. Gangjee, Y. Zeng, M. Ihnat, L. A. Warnke, D. W. Green, R. L. Kisliuk and F. T. Lin, Bioorg. Med. Chem., 2005, 13, 5475 CrossRef CAS PubMed; (d) Y. Miyazaki, J. Tang, Y. Maeda, M. Nakano, L. Wang, R. T. Nolte, H. Sato, M. Sugai, Y. Okamoto, A. T. Truesdale, D. F. Hassler, E. N. Nartey, D. R. Patrick, M. L. Ho and K. Ozawa, Bioorg. Med. Chem. Lett., 2007, 17, 1773 CrossRef CAS PubMed; (e) E. F. DiMauro, J. Newcomb, J. J. Nunes, J. E. Bemis, C. Boucher, J. L. Buchanan, W. H. Buckner, A. Cheng, T. Faust, F. Hsieh, X. Huang, J. H. Lee, T. L. Marshall, M. W. Martin, D. C. McGowan, S. Schneider, S. M. Turci, R. D. White and X. Zhu, Bioorg. Med. Chem. Lett., 2007, 17, 2305 CrossRef CAS PubMed; (f) Y. Miyazaki, Y. Maeda, H. Sato, M. Nakano and G. W. Mellor, Bioorg. Med. Chem. Lett., 2008, 18, 1967 CrossRef CAS PubMed; (g) H.-Y. Shiao, M. S. Coumar, C.-W. Chang, Y.-Y. Ke, Y.-H. Chi, C.-Y. Chu, H.-Y. Sun, C.-H. Chen, W.-H. Lin, K.-S. Fung, P.-C. Kuo, C.-T. Huang, K.-Y. Chang, C.-T. Lu, J. T. A. Hsu, C.-T. Chen, W.-T. Jiaang, Y.-S. Chao and H.-P. Hsieh, J. Med. Chem., 2013, 56, 5247 CrossRef CAS PubMed.
- (a) F. Cohen, P. Bergeron, E. Blackwood, K. K. Bowman, H. Chen, A. G. DiPasquale, J. A. Epler, M. F. T. Koehler, K. Lau, C. Lewis, L. Liu, C. Q. Ly, S. Malek, J. Nonomiya, D. F. Ortwine, Z. Pei, K. D. Robarge, S. Sideris, L. Trinh, T. Truong, J. Wu, X. Zhao and J. P. Lyssikatos, J. Med. Chem., 2011, 54, 3426 CrossRef CAS PubMed; (b) E. J. Hanan, M. Baumgardner, M. C. Bryan, Y. Chen, C. Eigenbrot, P. Fan, X.-H. Gu, H. La, S. Malek, H. E. Purkey, G. Schaefer, S. Schmidt, S. Sideris, I. Yen, C. Yu and T. P. Heffron, Bioorg. Med. Chem. Lett., 2016, 26, 534 CrossRef CAS PubMed; (c) X. Li, Y. Huang, J. Cheng, L. Zhang, F. Mao, J. Zhu, C. Sheng and J. Li, Bioorg. Med. Chem., 2018, 26, 4375 CrossRef CAS PubMed.
- (a) J. Teixidó, J. I. Borrell, C. Colominas, X. Deupí, J. L. Matallana, J. L. Falcó and B. Martinez-Teipel, J. Org. Chem., 2001, 66, 192 CrossRef; (b) A. Rosowsky, R. A. Forsch and S. F. Queener, J. Med. Chem., 2002, 45, 233 CrossRef CAS PubMed; (c) Y. Deng, Y. Wang, C. Cherian, Z. Hou, S. A. Buck, L. H. Matherly and A. Gangjee, J. Med. Chem., 2008, 51, 5052 CrossRef CAS PubMed; (d) M. Mannaghani and R. H. Nia, J. Heterocycl. Chem., 2017, 54, 1700 CrossRef.
- E. C. Taylor and J. L. Pont, J. Org. Chem., 1987, 52, 4287 CrossRef CAS.
- (a) P. Bergeron, F. Cohen, A. Estrada, M. Koehler, W. Lee, C. Ly, J. Lyssikatos, Z. Pei and X. Zhao, PCT Int. Appl., 2010151601, 2010; (b) K. M. Boy, J. M. Guernon, J. E. Macor, R. E. Olson, J. Shi, L. A. Thompson, Y.-J. Wu, L. Xu, Y. Zhang and D. S. Zuev, PCT Int. Appl., 2011014535, 2011; (c) B.-K. Kim, J.-H. Lui, B.-K. Lee, O. H. Kwon, Y.-K. Kim, C.-W. Kim, H.-S. Kim, J.-H. Seo, C.-J. Shin, E.-S. Yu, S.-J. Lee, B. K. Choi and K. Y. Hwang, PCT Int. Appl., 2015105316, 2015; (d) H.-J. Zhou and W. David, US Pat., 9828363B2, 2017; (e) S.-S. Lee and D.-M. Kang, Eur. Pat., 3056498B1, 2018.
- For selected reviews, see: (a) M. B. Gawande, V. D. Bonifácio, R. Luque, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 5522 RSC; (b) M. B. Gawande, V. D. Bonifácio, R. Luque, P. S. Branco and R. S. Varma, ChemSusChem, 2014, 7, 24 CrossRef CAS PubMed; (c) A. Sarkar, S. Santra, S. K. Kundu, A. Hajra, G. V. Zyryanov, O. N. Chupakhin, V. N. Charushin and A. Majee, Green Chem., 2016, 18, 4475 RSCFor selected examples, see: (d) M. L. Deb, C. D. Pegu, P. J. Borpatra, P. J. Saikia and P. K. Baruah, Green Chem., 2017, 19, 4036 RSC; (e) G. Brahmachari, K. Nurjamal, I. Karmakar, S. Begam, N. Nayek and B. Mandal, ACS Sustainable Chem. Eng., 2017, 5, 9494 CrossRef CAS; (f) M. L. Deb, P. J. Borpatra and P. K. Baruah, Green Chem., 2019, 21, 69 RSC.
- For selected reviews, see: (a) A. N. French, S. Bissmire and T. Wirth, Chem. Soc. Rev., 2004, 33, 354 RSC; (b) P. T. Parvatkar, P. S. Parameswaran and S. G. Tilve, Chem.–Eur. J., 2012, 18, 5460 CrossRef CAS PubMed; (c) D. Liu and A. Lei, Chem.–Asian J., 2015, 10, 806 CrossRef CAS PubMed; (d) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328 CrossRef CAS PubMedfor selected examples, see: (e) R. Yan, X. Li, X. Yang, X. Kang, L. Xiang and G. Huang, Chem. Commun., 2015, 51, 2573 RSC; (f) G.-W. Wang and J. Gao, Org. Lett., 2009, 11, 2385 CrossRef CAS PubMed.
- C.-K. Tang, Z.-Y. Zhou, A.-B. Xia, L. Bai, J. Liu, D.-Q. Xu and Z.-Y. Xu, Org. Lett., 2018, 20, 5840 CrossRef CAS PubMed.
- For selected recent representative advances based on 2,6-diaminopyrimidin-4(3H)-one, see: (a) F. Zhang, C. Li and X. Liang, Green Chem., 2018, 20, 2057 RSC; (b) C. Li and F. Zhang, Synlett, 2017, 28, 1315 CrossRef CAS; (c) M. Camarasa, C. Barnils, R. P. Bellacasa, J. Teixidó and J. I. Borrell, Mol. Diversity, 2013, 17, 525 CrossRef CAS PubMed.
- (a) F. Ye, L. Chen, L. Hu, T. Xiao, S. Yu, D. Chen, Y. Wang, G. Liang, Z. Liu and S. Wang, Bioorg. Med. Chem. Lett., 2015, 25, 1556 CrossRef CAS PubMed; (b) V. E. Saraev, I. M. Zviagin, R. G. Melik-Oganjanyan, Y. V. Sen'ko, S. M. Desenko and V. A. Chebanov, J. Heterocycl. Chem., 2017, 54, 318 CrossRef CAS.
- CCDC 1893720 contains the supplementary crystallographic data for compound 4a and CCDC 1893721 contains the supplementary crystallographic data for compound 5a.†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, data, and NMR spectra of all compounds. CCDC 1893720 and 1893721. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra01665a |
| This journal is © The Royal Society of Chemistry 2019 |