
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous-flow synthesis of 3,5-disubstituted pyrazoles via sequential alkyne homocoupling and Cope-type hydroamination†
Sándor B. Ötvös *abc,
Ádám Georgiádesa,
Dániel Ozsvára and
Ferenc Fülöp*ab
*abc,
Ádám Georgiádesa,
Dániel Ozsvára and
Ferenc Fülöp*ab
aInstitute of Pharmaceutical Chemistry, University of Szeged, Interdisciplinary Excellence Center, Eötvös u. 6, H-6720 Szeged, Hungary. E-mail: otvossandor@pharm.u-szeged.hu; fulop@pharm.u-szeged.hu; Fax: +36-62-545-705; Tel: +36-62-545-768 Tel: +36-62-545-562
bMTA-SZTE Stereochemistry Research Group, Hungarian Academy of Sciences, Eötvös u. 6, H-6720 Szeged, Hungary
cInstitute of Chemistry, University of Graz, NAWI Graz, Heinrichstrasse 28, A-8010 Graz, Austria
First published on 13th March 2019
Abstract
A flow chemistry-based approach is presented for the synthesis of 3,5-disubstituted pyrazoles via sequential copper-mediated alkyne homocoupling and Cope-type hydroamination of the intermediary 1,3-diynes in the presence of hydrazine as nucleophilic reaction partner. The proposed multistep methodology offers an easy and direct access to valuable pyrazoles from cheap and readily available starting materials and without the need for the isolation of any intermediates.
Introduction
Nitrogen-containing heterocycles have well-known and indisputable importance in synthetic and pharmaceutical chemistry. Among their five-membered counterparts, pyrazoles are of outstanding importance with impressively diversified applications.1 As a core structure, the pyrazole motif constitutes a myriad of pharmaceutically interesting compounds with a wide spectrum of biological activities, such as antihiperglycemic, analgesic, anti-inflammatory, antidepressant, antipyretic, antibacterial, antiviral, antituberculotic and sedative-hypnotic effects (Scheme 1).2 Besides their biological importance, pyrazoles are versatile building blocks in organic chemistry, and they are frequently used as ligands and chelating agents in coordination chemistry and catalysis.3 Furthermore, pyrazoles and their derivatives are widely employed in the agrochemical industry as insecticides, fungicides, and herbicides.4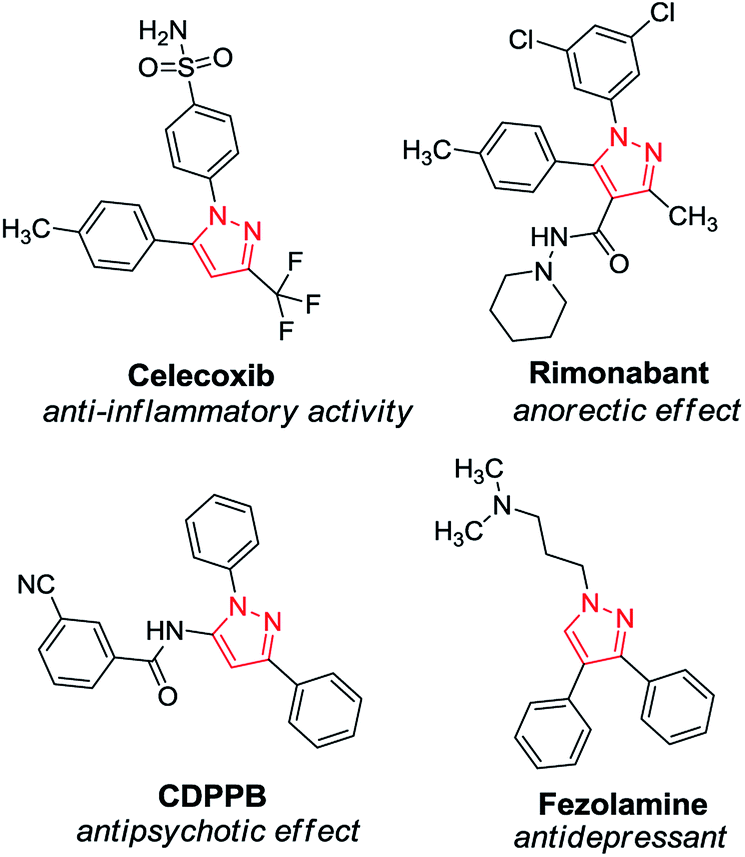 | ||
| Scheme 1 Examples for pharmacologically important pyrazoles. | ||
Because of the outstanding importance of pyrazoles, their syntheses have attracted much attention. The most typical methods for the preparation of substituted pyrazoles involve (i) cyclocondensation of 1,3-dicarbonyls or α,β-unsaturated carbonyls with hydrazine derivatives and (ii) [3 + 2] cycloaddition of alkynes or olefins with 1,3-dipoles such as nitrile imines, diazoalkanes, and sydnones.1b,5 Even multisubstituted pyrazoles can be achieved by using the aforementioned methods. However, they typically involve numerous drawbacks, such as harsh reaction conditions, selectivity issues, and the necessity of environmentally unfriendly reactants and/or special starting materials.6 In the past few years, a wide array of modern synthetic methods have been reported to overcome the above difficulties and to efficiently obtain pyrazoles with diverse substitution patterns.7 For example, intermolecular Cope-type hydroamination of 1,3-diynes with hydrazine was recently shown as a powerful strategy for the synthesis of 3,5-disubstituted pyrazoles.8 The reaction occurs via nucleophilic attack of hydrazine onto the diyne core and results in an allene intermediate, which undergoes electrophilic cyclization to yield a new C–N bond leading to the desired pyrazole motif (Scheme 2).9 The transformation requires no catalyst and proceeds smoothly without any additives (such as strong bases or acids) under reasonably mild conditions. As concerns the components of the Cope-type hydroamination, hydrazine and its derivatives are readily available, while the 1,3-diyne starting material can be accessed by catalytic oxidative dimerization of terminal acetylenes, which is typically performed by means of copper catalysis. The latter process utilizes either soluble or heterogeneous catalyst sources in the presence of various bases and ligands.10
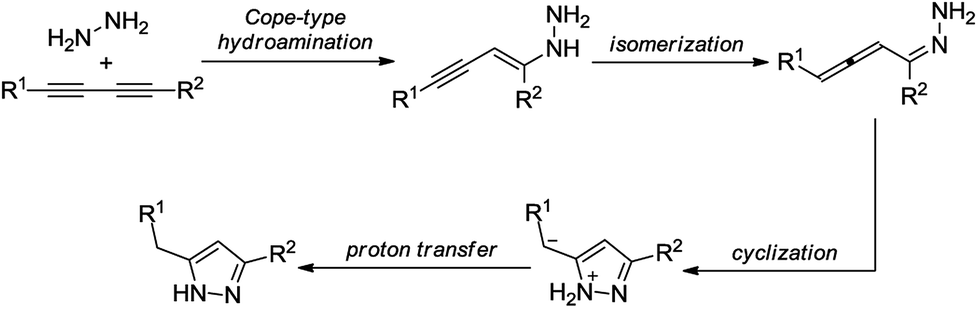 | ||
| Scheme 2 Plausible mechanism of the reaction between 1,3-diyne and hydrazine.9 | ||
Flow chemistry offers numerous benefits for chemical syntheses,11 including multistep processes,12 but there are only a few instances in which these gains have been exploited for the synthesis of substituted pyrazoles and their derivatives.13 On the basis of the above findings, we envisioned that continuous-flow chemistry may be an ideal platform for a telescoped reaction sequence of catalytic alkyne homocoupling and the Cope-type hydroamination of the resulting 1,3-diynes to yield valuable 3,5-disubstituted pyrazoles directly from terminal acetylenes as cheap and readily available starting materials. The proposed integrated synthesis method would permit a facile access to target pyrazoles without the isolation of any intermediates and with significantly less purification issues. Moreover, both alkyne dimerization and the Cope-type hydroamination require long reaction times,8,10 and thus the complete batch synthesis is a rather lengthy process. We, therefore, also aimed to utilize the technological benefits of flow chemistry to achieve chemical intensification as compared with earlier batch processes.14
Thus, we are reporting our results on the continuous-flow synthesis of 3,5-disubstituted pyrazoles via sequential alkyne homocoupling and Cope-type hydroamination.
Results and discussion
The planned synthesis consists of (i) a metal-catalyzed coupling step and (ii) the subsequent non-catalytic hydroamination with hydrazine involving an electrophilic cyclization to yield the pyrazole motif. In the second step, traces of the catalytic metal from the homocoupling may open up unwanted reaction pathways and involve the formation of by-products.15 Therefore, instead of a one-step/one-pot method, a two-step telescoped process seems more feasible with in-line metal removal after the first step. In the first stage of the research, a step-by-step reaction optimization was projected, and after being familiar with both reactions, separate steps were next arranged into a multistep telescoped flow sequence where the intermediary 1,3-diyne is transformed immediately without the need for isolation.Optimization of the 1,3-diyne synthesis
The catalytic dimerization of 4-ethynyltoluene was chosen as a model reaction for the investigation of diyne synthesis (Scheme 3). As concerns the source of the catalytic metal, there are numerous examples for the use of heterogeneous copper catalysts in acetylenic homocouplings. Indeed, these materials exhibit various benefits including reusability and the ease of product isolation.16 However, for the development of a multistep method we preferred soluble copper sources over solid catalysts in order to avoid uncontrollable leaching issues and to create a robust synthesis system.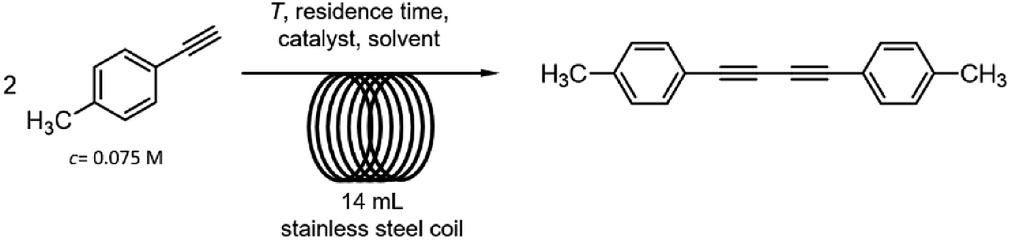 | ||
| Scheme 3 Oxidative homocoupling of 4-ethynyltoluene in a continuous-flow reactor. | ||
In copper-mediated oxidative homocouplings of acetylenes, copper ions of +1 and +2 oxidation states are known to operate cooperatively as active species.17 Therefore, various Cu(II) and Cu(I) salts and complexes were considered as sources for the catalytic metal (Table S1†). Since only homogeneous solutions can be applied in the reaction channels, thorough solubility tests were carried out by using not only the starting alkyne and the copper catalyst but also hydrazine monohydrate, in order to avoid precipitation in both reaction steps. Tests were carried out with DMSO, EtOAc, MeCN, acetone and EtOH as possible solvents. DMSO and EtOH proved to be compatible with the alkyne component, hydrazine and also with [Cu(phen)(PPh3)2]NO3 or CuBr2 as catalyst. Consequently, they were selected as possible solvents for the reactions (see Table S1† for the detailed solubility tests).
The dimerization of 4-ethynyltoluene was next examined with the selected copper sources in a 14 mL stainless steel reaction coil (internal diameter: 0.76 mm, length: 30.5 m) under the following initial conditions: 120 °C and 0.2 mL min−1 flow rate (70 min residence time, Scheme 3). The alkyne was applied in a concentration of 0.075 M (higher concentrations occasionally led to precipitation) in combination with 0.06 equiv. of the copper catalyst in DMSO or EtOH as solvent. With [Cu(phen)(PPh3)2]NO3 as catalyst no conversion was detected either in DMSO or in EtOH even in the presence of 0.5 equiv. of N,N-diisopropylethylamine (DIEA) as a base (Table 1, entries 1–4). In contrast, CuBr2 in combination with N,N,N′,N′-tetramethylethylenediamine (TMEDA) as ligand and DIEA as base (0.5 equiv. each) proved to be superior and offered quantitative conversion and selective diyne formation in DMSO as solvent (Table 1, entry 5). On investigation of the effects of the auxiliaries, it was found that the amount of TMEDA and DIEA can be reduced to 0.25 equiv. without loss in conversion (Table 1, entries 6 and 7). It was also observed that the joint application of the ligand and the base is necessary: conversion was reduced significantly when using only TMEDA or DIEA as the sole additive (Table 1, entries 8 and 9). In the absence of the ligand and base, precipitation and clogging occurred in the reactor tubing (Table 1, entry 10). To our delight, residence time could successfully be reduced from 70 min to 28 and even to 14 min (0.5 and 1 mL min−1 flow rate, respectively) without any decrease in the quantitative conversion (Table 1, entries 11–14).
| Entry | Catalyst | Additives (equiv.) | Solvent | Flow ratea (mL min−1) | Conv.b (%) | |
|---|---|---|---|---|---|---|
| TMEDA | DIEA | |||||
| a Residence time (in min units) is shown in parentheses.b Determined by 1H NMR spectroscopy of the crude material.c Chemoselectivity of diyne formation was 100%.d No data, clogging occurred. | ||||||
| 1 | [Cu(phen)(PPh3)2]NO3 | 0 | 0 | DMSO | 0.2 (70) | 0 |
| 2 | [Cu(phen)(PPh3)2]NO3 | 0 | 0 | EtOH | 0.2 (70) | 0 |
| 3 | [Cu(phen)(PPh3)2]NO3 | 0 | 0.5 | DMSO | 0.2 (70) | 0 |
| 4 | [Cu(phen)(PPh3)2]NO3 | 0 | 0.5 | EtOH | 0.2 (70) | 0 |
| 5 | CuBr2 | 0.5 | 0.5 | DMSO | 0.2 (70) | 100c |
| 6 | CuBr2 | 0.25 | 0.25 | DMSO | 0.2 (70) | 100c |
| 7 | CuBr2 | 0.1 | 0.1 | DMSO | 0.2 (70) | 30c |
| 8 | CuBr2 | 0.25 | 0 | DMSO | 0.2 (70) | 20c |
| 9 | CuBr2 | 0 | 0.25 | DMSO | 0.2 (70) | 16c |
| 10 | CuBr2 | 0 | 0 | DMSO | 0.2 (70) | N.d.d |
| 11 | CuBr2 | 0.5 | 0.5 | DMSO | 0.5 (28) | 100c |
| 12 | CuBr2 | 0.5 | 0.5 | DMSO | 1 (14) | 100c |
| 13 | CuBr2 | 0.5 | 0.5 | DMSO | 1.5 (10.4) | 65c |
| 14 | CuBr2 | 0.5 | 0.5 | DMSO | 2 (7) | 54c |
Optimization of the Cope-type hydroamination
After having established the best conditions for 1,3-diyne synthesis, the Cope-type hydroamination was studied utilizing preformed 1,4-di-p-tolylbuta-1,3-diyne as model substrate. A 0.0375 M DMSO solution of the diyne (half of the concentration of the starting alkyne employed in the first coupling step) was combined in a T-piece with a hydrazine stream (60 wt% aqueous solution containing 0.1125 M hydrazine in DMSO) and was passed through a 14 mL reaction coil (internal diameter: 0.76 mm, length: 30.5 m) at 120 °C utilizing various flow rates (Scheme 4). The need for the relatively large excess of hydrazine is explained by the fact that it not only plays a role as a reaction partner in the actual hydroamination, but it also participates in the final proton transfer leading to the pyrazole product (Scheme 2).9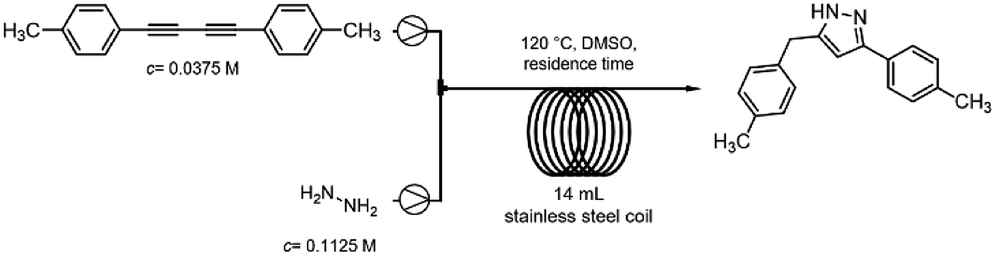 | ||
| Scheme 4 Continuous-flow reactor set-up for the hydroamination of 1,4-di-p-tolylbuta-1,3-diyne. | ||
The thermal transformation was found to be rather sensitive to residence times applied. When both pumps were operated at a flow rate of 0.5 mL min−1 (14 min residence time), a conversion of merely 10% occurred (Fig. 1). Upon increasing the residence time to 70 min (both pumps at 0.1 mL min−1), a satisfying conversion of 89% was measured with exclusive pyrazole formation. Lower flow rates were not investigated so that the residence times of the individual reaction steps remain compatible in the telescoped system.
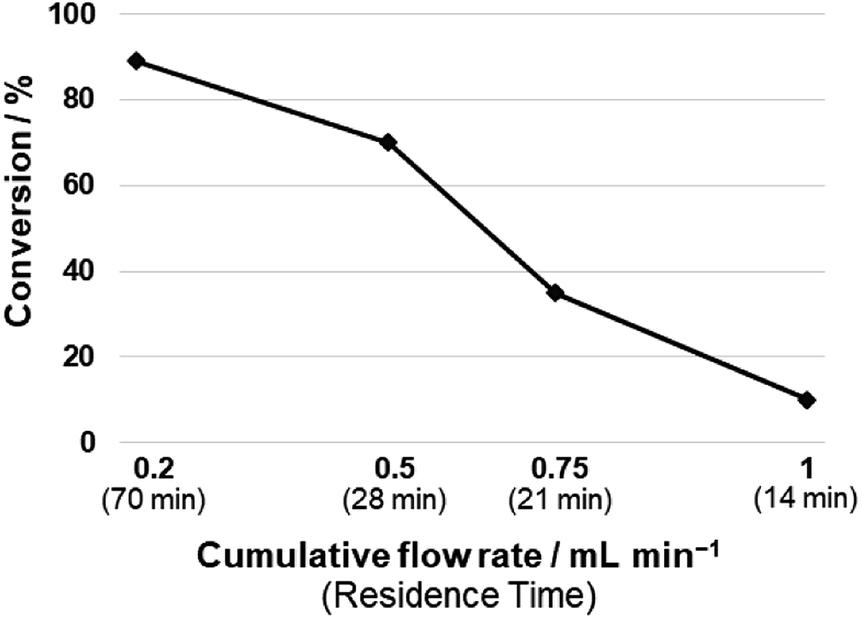 | ||
| Fig. 1 Investigation of the effects of residence time on the hydroamination of 1,4-di-p-tolylbuta-1,3-diyne with hydrazine. Conditions: 1 equiv. diyne (c = 0.0375 M), 3 equiv. hydrazine (c = 0.1125 M), DMSO as solvent, 120 °C, pumps operated at the same flow rate (see Scheme 4). (Chemoselectivity of pyrazole formation was 100% in all reactions). | ||
Telescoped pyrazole synthesis
After getting familiar with the optimum conditions of both the copper-catalyzed alkyne dimerization and the Cope-type hydroamination/cyclization, next individual reaction steps were combined (Scheme 5). To be able to synchronize the residence times in the reactor components, a 3.5 mL heated coil was used for the alkyne homocoupling reaction. The solution of 4-ethynyltoluene (0.075 M) was pumped continuously at 0.1 mL min−1 flow rate which gave a residence time of 35 min. A subsequent column packed with a thiourea-based scavenger resin was installed to remove copper species after the homocoupling took place. The resulting diyne solution was next combined with a hydrazine stream (60 wt% aqueous solution containing 0.1125 M hydrazine in DMSO) at 0.1 mL min−1. Pyrazole formation took place during passage through a 14 mL heated coil at a cumulative flow of 0.2 mL min−1, which corresponded to a residence time of 70 min.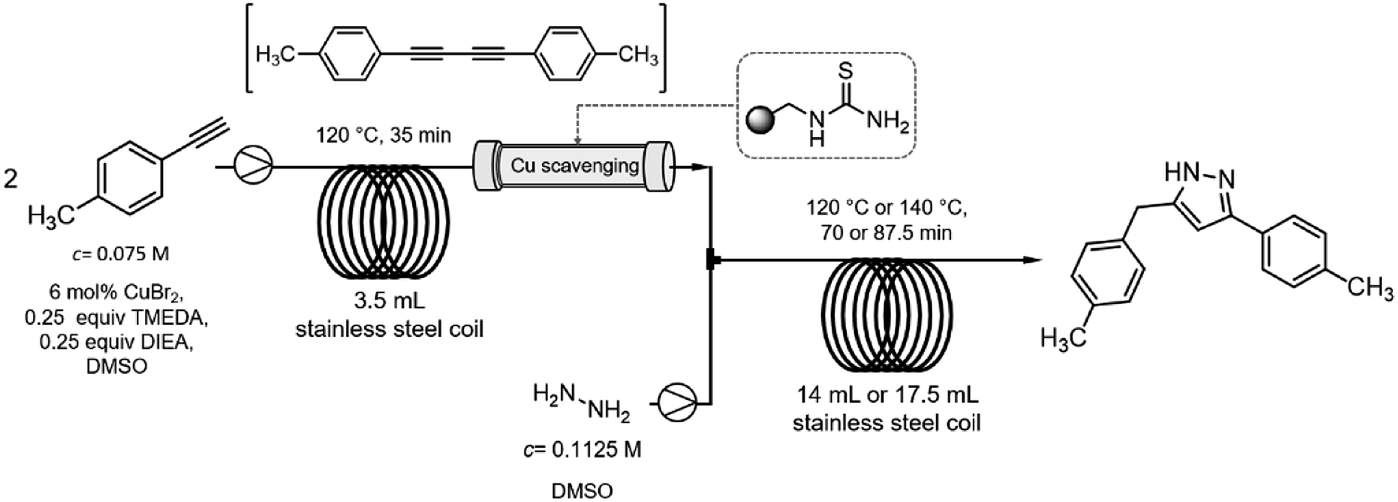 | ||
| Scheme 5 Schematic representation of the two-step continuous-flow pyrazole synthesis. | ||
When both coils were heated at 120 °C similarly to step-by-step optimizations, all akyne starting material got consumed and the corresponding pyrazole was formed to an extent of 76% (together with 24% of 1,4-di-p-tolylbuta-1,3-diyne; Table 2, entry 1). To improve pyrazole formation, the hydroamination coil was replaced with a 17.5 mL one affording a residence time of 87.5 min at a flow rate of 0.2 mL min−1. The increased residence time improved pyrazole formation to 86% at 120 °C; and, finally, increasing the temperature to 140 °C resulted in 98% pyrazole formation without traces of unreacted alkyne in the crude material (Table 2, entries 2 and 3).
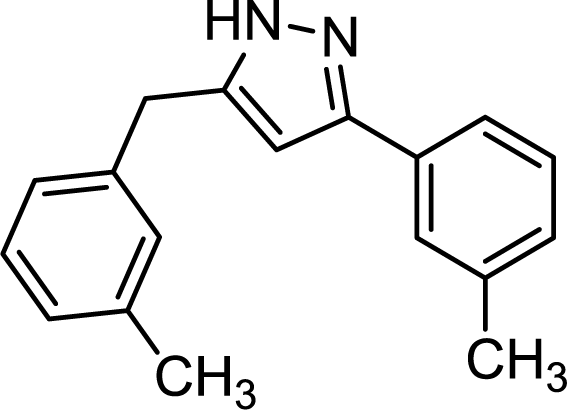
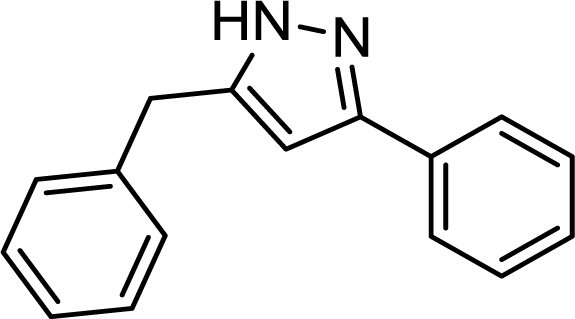
With optimized conditions for the telescoped pyrazole synthesis in hand, the reactivity of different alkynes was next investigated. Excellent results were achieved with phenylacetylene, its methyl-substituted derivatives, and with 3-ethynylthiophene (Table 3). The starting alkynes were quantitatively converted to the corresponding diynes (no traces of unreacted alkyne were found in the crude material), and after the subsequent hydroamination/cyclization the corresponding pyrazoles were obtained with yields of 90–98%. The reactions were chemoselective, no sideproduct formation was detected, isolated yields were in the range of 84–90% after chromatographic purification. It should be noted that aliphatic alkynes such as 3-cyclohexyl-1-propyne and oct-1-yne constitute a limitation yet. In these cases, diyne formation was not complete possibly because of the lack of conjugation, and, in addition, conversion in hydroamination/cyclization was also low resulting in non-acceptable yields.
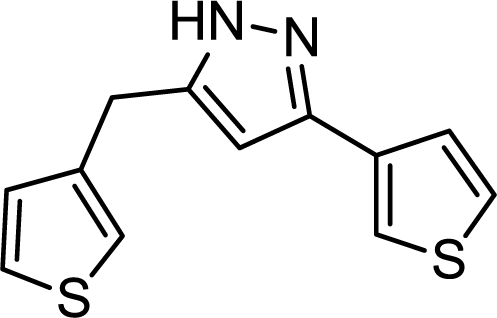
In the case of 3-ethynylthiophene, a scale-up experiment was carried out by simply prolonging the operation time without any change in the optimized conditions. We were delighted to find that the flow system was stable for an extended period of 16 h giving 0.52 g of pure 3-(thiophen-3-yl)-5-(thiophen-3-ylmethyl)-1H-pyrazole corresponding to an isolated yield of 81%. As a result of the in-line copper scavenging, the crude product obtained in the scale-up experiment contained only a negligible copper amount of 0.3 ppm as determined by ICP-MS.
Conclusion
A two-step continuous-flow methodology has been developed for the synthesis of 3,5-disubsituted pyrazole derivatives from terminal alkynes and hydrazine monohydrate as readily available starting materials. The process relied on (i) the copper-mediated homocoupling of the alkyne component in the presence of CuBr2 as a simple soluble catalyst source, and (ii) the subsequent Cope-type hydroamination of the in situ formed 1,3-diyne with hydrazine as nucleophilic reaction partner. After an initial step-by-step optimization procedure, individual reaction components were organized into an uninterrupted flow sequence where, after in-line copper removal, the intermediary diyne was transformed further after being combined with a hydrazine stream. The developed process proved to be applicable for a range of aromatic alkynes and permitted a simple and easy access to a diverse set of pyrazoles without the isolation of any intermediates. With a cumulative residence time of around 2 h, the flow methodology involved a significant chemical intensification as compared with the corresponding batch reaction.Experimental section
General information
Reagents and materials were commercially available and were used as received. Analytical thin-layer chromatography was performed on Merck silica gel 60 F254 plates and flash column chromatography on Merck silica gel 60. Compounds were visualized by means of UV or KMnO4. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance DRX 400 spectrometer, in CDCl3 as solvent, with TMS as internal standard, at 400.1 and 100.6 MHz, respectively. Copper concentration was determined by inductively coupled plasma mass spectrometry (ICP-MS), using an Agilent 7700x-type instrument equipped with a collision cell after digestion with concentrated HNO3. The determination was carried out on the isotope 63Cu, with He as the collision gas.General procedure for the flow reactions
Reactions were carried out in a home-made flow reactor consisting of HPLC pumps (JASCO PU-880 and Knauer AZURA P 2.1 S), stainless steel coils (internal diameter: 0.76 mm, length: 7.5, 30.5 or 38.5 m), and a commercially available 50 bar backpressure regulator (IDEX P-455 BPR Assembly 750, PEEK). The parts of the system were connected with stainless steel and PEEK capillary tubing (254 μm internal diameter). The reaction coils were immersed in a heated oil bath.After each reaction, 20 mL water or, in the case of homocouplings, 20 mL 0.1 M aqueous ethylenediaminetetraacetic acid (EDTA) solution was added to the collected mixtures and the aqueous phase was extracted with 3 × 30 mL of EtOAc. The combined organic phases were washed with brine (2 × 10 mL), dried over Na2SO4, and concentrated in vacuo. Column chromatography purification was carried out with mixtures of n-hexane/EtOAc as eluent. Between two flow experiments, the reactor was washed with DMSO–MeOH–DMSO, respectively, 10 min each at flow rates of 1 mL min−1.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Hungarian Research Foundation (OTKA No. K 115731). The financial support of the GINOP-2.3.2-15-2016-00014 project are acknowledged. Ministry of Human Capacities, Hungary grant 20391-3/2018/FEKUSTRAT is acknowledged. SBÖ acknowledges the Premium Post Doctorate Research Program of the Hungarian Academy of Sciences. ÁG was supported by the UNKP-17-3 New National Excellence Program of the Ministry of Human Capacities. Financial supports are highly appreciated. We are grateful to Prof. Árpád Molnár for proofreading our manuscript.References
-
(a) L. Yet, in Comprehensive Heterocyclic Chemistry III, ed. C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, pp. 1–141 Search PubMed
; (b) K. Karrouchi, S. Radi, Y. Ramli, J. Taoufik, Y. Mabkhot, F. Al-aizari and M. h. Ansar, Molecules, 2018, 23, 134 CrossRef PubMed
-
(a) A. Ansari, A. Ali, M. Asif and Shamsuzzaman, New J. Chem., 2017, 41, 16–41 RSC
-
(a) C. Pettinari, A. Tăbăcaru and S. Galli, Coord. Chem. Rev., 2016, 307, 1–31 CrossRef CAS
-
(a) G. P. Lahm, D. Cordova and J. D. Barry, Bioorg. Med. Chem., 2009, 17, 4127–4133 CrossRef CAS PubMed
- S. Fustero, M. Sánchez-Roselló, P. Barrio and A. Simón-Fuentes, Chem. Rev., 2011, 111, 6984–7034 CrossRef CAS PubMed
-
(a) V. K. Rao, R. Tiwari, B. S. Chhikara, A. N. Shirazi, K. Parang and A. Kumar, RSC Adv., 2013, 3, 15396–15403 RSC
-
(a) Y. Yang, Z.-L. Hu, R.-H. Li, Y.-H. Chen and Z.-P. Zhan, Org. Biomol. Chem., 2018, 16, 197–201 RSC
-
(a) M. Patel, R. K. Saunthwal and A. K. Verma, Acc. Chem. Res., 2017, 50, 240–254 CrossRef CAS PubMed
- Y. Wang, D. Wei, W. Zhang, Y. Wang, Y. Zhu, Y. Jia and M. Tang, Org. Biomol. Chem., 2014, 12, 7503–7514 RSC
-
(a) K. S. Sindhu and G. Anilkumar, RSC Adv., 2014, 4, 27867–27887 RSC
-
(a) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed
-
(a) R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25 CrossRef CAS
-
(a) J. Britton and T. F. Jamison, Angew. Chem., Int. Ed., 2017, 56, 8823–8827 CrossRef CAS PubMed
-
(a) V. Hessel, D. Kralisch, N. Kockmann, T. Noël and Q. Wang, ChemSusChem, 2013, 6, 746–789 CrossRef CAS PubMed
-
(a) S. R. Chemler and P. H. Fuller, Chem. Soc. Rev., 2007, 36, 1153–1160 RSC
-
(a) S. B. Ötvös, Á. Georgiádes, R. Mészáros, K. Kis, I. Pálinkó and F. Fülöp, J. Catal., 2017, 348, 90–99 CrossRef
- J. Jover, P. Spuhler, L. Zhao, C. McArdle and F. Maseras, Catal. Sci. Technol., 2014, 4, 4200–4209 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01590f |
| This journal is © The Royal Society of Chemistry 2019 |