
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDown-regulation of lncRNA GAS5 attenuates neuronal cell injury through regulating miR-9/FOXO3 axis in cerebral ischemic stroke
Lijun Wang,
Yanliang Niu,
Gangrui He and
Jianping Wang *
*
Department of Neurology, The Fifth Affiliated Hospital of Zhengzhou University, No. 3, Kangfu Street, Erqi District, Zhengzhou 450000, China. E-mail: wangjianping2019@163.com; Tel: +86-0317-66916091
First published on 23rd May 2019
Abstract
Cerebral ischemic stroke is a leading cause of neurological disability worldwide. Previous study reported that long noncoding RNA (lncRNA) growth arrest-specific transcript 5 (GAS5) was highly expressed in ischemic stroke. However, the mechanism underlying GAS5 in an inflammatory injury during an ischemic stroke remains poorly understood. An in vivo mouse model of middle cerebral artery occlusion (MCAO) and an in vitro cell model of oxygen-glucose deprivation (OGD) were established to induce cerebral ischemic stroke condition. The expressions of GAS5, microRNA-9 (miR-9) and forkhead box O3 (FOXO3) were measured by quantitative real-time polymerase chain reaction (qRT-PCR) or western blot analysis, respectively. The neurological injury in vivo was investigated by neurological score and TTC staining. Cell apoptosis and inflammatory injury were analyzed by western blot, flow cytometry and enzyme-linked immunosorbent assay (ELISA), respectively. The interaction between miR-9 and GAS5 or FOXO3 was explored by luciferase activity, RNA pull-down and RNA immunoprecipitation (RIP) assays. GAS5 expression was enhanced in the cerebral ischemic stroke model. Knockdown of GAS5 attenuated the cerebral infarct, neurological injury, apoptosis and inflammatory injury in the mouse MCAO model. miR-9 was bound to GAS5 and its overexpression inhibited cell apoptosis and inflammatory response in OGD-treated bEnd.3 cells, which was attenuated by GAS5. FOXO3 was a target of miR-9 and its restoration reversed the miR-9-mediated suppression of apoptosis and inflammation. Moreover, GAS5 promoted FOXO3 expression by competitively sponging miR-9. GAS5 knockdown alleviated neuronal cell injury by regulating miR-9/FOXO3, providing a new theoretical foundation for cerebral ischemic stroke.
Introduction
Cerebral ischemia is the most common neurological disease and leading cause of death, caused by the blockage of cerebral vessels.1 Despite the great advances in the development of diagnosis and treatment in ischemic stroke, the effective strategies remain dismal.2 Previous study has suggested neuroinflammatory responses as attractive therapeutic target in human stroke lesions.3 Hence, it is promising to explore novel targets for the treatment of cerebral ischemic stroke through addressing inflammatory responses.Noncoding RNAs (ncRNAs), including long ncRNAs (lncRNAs) and microRNAs (miRNAs), have been associated with stroke-related neurovascular damage, such as cerebral dysregulation, brain edema, inflammatory injury and neural cell death.4 LncRNAs were suggested as potential targets in the pathophysiology of ischemic stroke via lncRNA-miRNA-mRNA regulatory networks.5 For example, Yang et al. reported that lncRNA small nucleolar RNA host gene 1 attenuated oxygen-glucose deprivation (OGD)-induced injury in ischemic stroke by regulating miR-338 and hypoxia-inducible factor-1α.6 Moreover, Zhang et al. revealed that lncRNA metastasis-associated lung adenocarcinoma transcript 1 promoted ischemic stroke by regulating murine double minute homolog 2 through activating p53 pathway.7 LncRNA growth arrest-specific 5 (GAS5) has been reported to inhibit the proliferation, migration, invasion, gemcitabine resistance and epithelial–mesenchymal transition in pancreatic cancer.8 Moreover, GAS5 suppressed the cell proliferation and migration through inactivating the phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin signaling pathway in esophageal cancer.9 Notably, previous study suggested that GAS5 was associated with the risk of ischemic stroke.10 However, the role of GAS5 in inflammatory injury during ischemic stroke and its mechanism remain largely unclear.
miRNAs play essential roles in the development and pathology of ischemic stroke by regulating oxidative stress, inflammation, apoptosis and neurogenesis.11 Moreover, miRNAs have been indicated as the key regulators of inflammatory response in ischemic stroke injuries.12 miR-9 is a versatile regulator of neurogenesis that implicates in human brain pathologies, which plays important roles in the brain injuries.13 In addition, previous study showed that miR-9 was lowly expressed and associated with inflammation as well as infarct in ischemic stroke.14
Forkhead box O (FOXO) transcription factors have important roles in the development and therapy of cancers.15 FOXO3, a member of the FOXO family, has an important impact on neurodegenerative disorders, including stroke injury.16 Moreover, FOXO3 is associated with cerebral infarct and cell death in ischemic injury.17 Intriguingly, bioinformatics analysis provided the putative binding sites between miR-9 and GAS5 or FOXO3. Hence, we hypothesized that GAS5 might function as a competing endogenous RNA (ceRNA) for miR-9 to regulate FOXO3 in cerebral ischemic stroke. In this study, we established a mouse model of middle cerebral artery occlusion (MCAO) in vivo and a cellular model of OGD in vitro to induce cerebral ischemic stroke condition. Moreover, we investigated the roles of GAS5 in the inflammatory response after ischemic stroke and explored the regulatory network of GAS5/miR-9/FOXO3.
Materials and methods
Mouse model of transient focal cerebral ischemia
C57BL/6 mice were used to establish the mouse model of transient focal cerebral ischemia. All mice (male, ten-week-old) were obtained from Vital River Laboratory Animal Technology (Beijing, China). Mice were randomly divided into four groups: vehicle control (sham), MCAO, MCAO + small interfering RNA (siRNA) negative control (si-NC) and MCAO + siRNA against GAS5 (si-GAS5) group. Every group was assigned to minimize animals (n = 6). A lentivirus vector of si-GAS5 or si-NC constructed by GeneCopoeia (Rockville, MD, USA) was introduced into the left lateral ventricle of mice at 24 h before MCAO. The MCAO surgery was conducted as described previously.18 In brief, after anesthesia, a 5-0 surgical nylon monofilament suture was inserted from the external carotid artery up to the internal carotid artery to occlude the middle cerebral artery (MCAO). After 60 min of MCAO surgery, the suture was removed. The sham group was subjected to the same surgery procedures without MCAO. At 1, 3, 7 and 14 d after MCAO, the neurological score was investigated and brain tissues were collected for 2,3,5-triphenyltetrazolium chloride (TTC) staining or molecular analysis. Every animal experiment was performed in strict accordance with the Guidelines for Care and Use of Laboratory Animal and was approved by the Animal Research Committee of the Fifth Affiliated Hospital of Zhengzhou University during this study.Measurement of neurological score and infarct volume
The foot fault test was conducted to evaluate the neurological function of mice after the treatment of MCAO, as previously reported.19,20 The test was performed at 1, 3, 7, and 14 d after the surgery and a low neurological score displayed neurological deficit. At 7 d after stroke, mice were anesthetized, brains were collected, cut into 2 mm-thick slices and stained with 2% TTC solution (Sigma, St. Louis, MO, USA) for 15 min at 37 °C in the dark. The areas of the infarct tissue were analyzed by using Image Lab software (Bio-Rad, Hercules, CA, USA). The infarct tissues were presented in white.Cell culture and treatment
Mouse brain microvascular endothelial cell line (bEnd.3) cells were purchased from American Tissue Culture Collection (Manassas, VA, USA) and cultured in Dulbecco's Modified Eagle Medium (DMEM) (Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (Gibco), 100 U mL−1 penicillin and 100 μg mL−1 streptomycin (Invitrogen, Carlsbad, CA, USA) at 37 °C in a humidified atmosphere with 5% CO2 during the study.To establish the OGD model, bEnd.3 cells were cultured in the glucose-free DMEM and maintained in an anaerobic chamber with 5% CO2 and 95% N2 at 37 °C for 2 h. Then, cells were returned to the normoxic conditions for various recovery times (12, 24 and 48 h). The control group was cultured in the standardized DMEM without depriving of oxygen and glucose.
si-GAS5, si-NC, pcDNA, pcDNA-based GAS5 overexpression vector (GAS5), miR-9 mimic (miR-9), miRNA NC (miR-NC), miR-9 inhibitor (anti-miR-9) and inhibitor NC (anti-miR-NC) were synthesized by Genepharma (Shanghai, China). Cell transfection was performed in bEnd.3 cells for 48 h by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was isolated by using TRIzol reagent (Invitrogen) and reverse transcribed to cDNA by using M-MLV Reverse Transcription Kit (Thermo Fisher, Wilmington, DE, USA) or TaqMan microRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Then, cDNA was used for qRT-PCR using SYBR green (Applied Biosystems) according to the manufacturer's instructions. Every sample was prepared in triplicate and the experiment was repeated three times. The relative expressions of RNAs were calculated using 2−ΔΔCt method with β-actin or U6 small RNA as endogenous control.21 The primers are listed as follows: miR-9 (forward, 5′-GGACGGACAGCGAGAGGAGGCCAAA-3′; reverse, 5′-TTTGGCCTCCTCTCGCTGTCCGTCC-3′), U6 (forward, 5′-CTCGCTTCGGCAGC ACA-3′; reverse, 5′-AACGCTTCACGAATTTGCGT-3′), GAS5 (forward, 5′-GCTTGAGGAGGAGTCTGA-3′; reverse, 5′-GGTCTTCTATTCTAGCACATTG-3′), FOXO3 (forward, 5′-CCTGTCCTACGCTGACCTGAT-3′; reverse, 5′-AGTCCCTTCGTTCTGAACCCG-3′), β-actin (forward, 5′-ATGGTG AAGGTCGGTGTGAA-3′; reverse, 5′-TGGAAGATGGTGATGGG C-3′).Western blot
The protein extraction from brain tissues or bEnd.3 cells was quantified by BCA protein assay kit (Beyotime Biotechnology, Shanghai, China), denatured at 98 °C for 10 min, and then, separated by SDS-PAGE gel electrophoresis and transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5% non-fat milk for 1 h at room temperature, and then incubated overnight at 4 °C with primary antibodies against B-cell lymphoma-2 (Bcl-2) (ab194583, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 dilution, Abcam, Cambridge, UK), Bcl-2-associated x protein (Bax) (ab182733, 1:2000 dilution, Abcam), FOXO3 (ab70315, 1:5000 dilution, Abcam) and β-actin (ab8227, 1:5000 dilution, Abcam). Subsequently, the membranes were incubated with secondary antibody (ab6721, 1:10000 dilution, Abcam), conjugated by horseradish peroxidase for 2 h at room temperature and interacted with enhanced chemiluminescence chromogenic substrate (Beyotime Biotechnology). The protein signals were analyzed with Image Lab software (Bio-Rad) with β-actin as the loading control.
:1000 dilution, Abcam, Cambridge, UK), Bcl-2-associated x protein (Bax) (ab182733, 1:2000 dilution, Abcam), FOXO3 (ab70315, 1:5000 dilution, Abcam) and β-actin (ab8227, 1:5000 dilution, Abcam). Subsequently, the membranes were incubated with secondary antibody (ab6721, 1:10000 dilution, Abcam), conjugated by horseradish peroxidase for 2 h at room temperature and interacted with enhanced chemiluminescence chromogenic substrate (Beyotime Biotechnology). The protein signals were analyzed with Image Lab software (Bio-Rad) with β-actin as the loading control.
Enzyme-linked immunosorbent assay (ELISA)
The releases of inflammatory cytokines [interleukin-1β (IL-1β), IL-6 and tumor necrosis factor-a (TNF-a)] in the brain tissue homogenates or cell culture medium were measured by special commercial ELISA kit (Sigma) following the manufacturer's instructions. Data were expressed as pg mL−1 or pg mg−1 total protein.Cell apoptosis
Flow cytometry was performed to measure cell apoptosis using Annexin V-FITC/PI apoptosis detection kit (Sigma) by flow cytometry. After washing with PBS, treated bEnd.3 cells were resuspended in 200 μL binding buffer, and then double stained with 10 μL Annexin V-FITC for 15 min and 5 μL PI for 15 min in the dark according to the manufacturer's instructions. The apoptotic cells were analyzed by using a flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). Every sample was prepared in triplicate.Luciferase activity assay
The putative binding sites of miR-9 and GAS5 or FOXO3 were predicted by starBase. The sequences of GAS5 or FOXO3 containing wild-type (WT) or mutant-type (MUT) binding sites were amplified, and then inserted into the pmirGLO vectors (Promega, Madison, WI, USA) to conduct luciferase reporter vectors (GAS5-WT, GAS5-MUT, FOXO3-WT or FOXO3-MUT). BEnd.3 cells were co-transfected with 20 ng luciferase reporter vectors, 15 ng control vector and 40 nM miR-9 or miR-NC using Lipofectamine 2000 according to the manufacturer's protocols. The luciferase activity was measured by using luciferase assay kit (Promega) after the transfection for 48 h according to the manufacturer's instructions.RNA pull-down assay
RNA pull-down assay was performed by using Magnetic RNA-protein Pull-Down Kit (Thermo Fisher) according to the manufacturer's protocols. Biotinylated miR-9 or mutated miR-9 and NC were generated by Genepharma and introduced into bEnd.3 cells. After 48 h, cells were collected and lysed, followed by interaction with streptavidin magnetic beads for 4 h at 4 °C. Then, the bound RNAs were eluted and used for the measurement of GAS5 abundance by qRT-PCR.RNA immunoprecipitation (RIP)
RNA-binding protein immunoprecipitation kit (Millipore) was used for RIP assay according to the manufacturer's protocols. In brief, bEnd.3 cells transfected with miR-9 or miR-NC were lysed in RIP buffer containing magnetic beads bound with Ago2 antibody or IgG. The enrichment of FOXO3 was measured by qRT-PCR.Statistical analysis
The data were expressed as the mean ± standard deviation (S.D.). The statistical differences between groups were investigated by the Student's t test or the one-way analysis of variance using SPSS 18.0 software (SPSS, Inc., Chicago, IL, USA). P < 0.05 was regarded as statistically significant.Results
Knockdown of GAS5 inhibits ischemic stroke injury in MCAO model
To explore the role of GAS5 in ischemic stroke, the mouse MCAO model was established. The expression of GAS5 was abnormally enhanced in brain tissues from mice after MCAO for 1, 3 or 7 d (Fig. 1A). However, the abundance of GAS5 was effectively reduced in si-GAS5 group compared with that in si-NC group (Fig. 1B). Moreover, the neurological injury was investigated in every group. Results showed that the MCAO treatment significantly increased the infarct volume and decreased neurological score compared with the sham group, which was reversed by the knockdown of GAS5 (Fig. 1C and D). In addition, the inhibition of Bcl-2 protein and the increase of Bax protein were displayed in MCAO group, which was abated by the depletion of GAS5 (Fig. 1E). Moreover, the protein levels of IL-1β, IL-6 and TNF-α were notably enhanced in brain tissues from MCAO group, which was attenuated by the abrogation of GAS5 (Fig. 1F–H).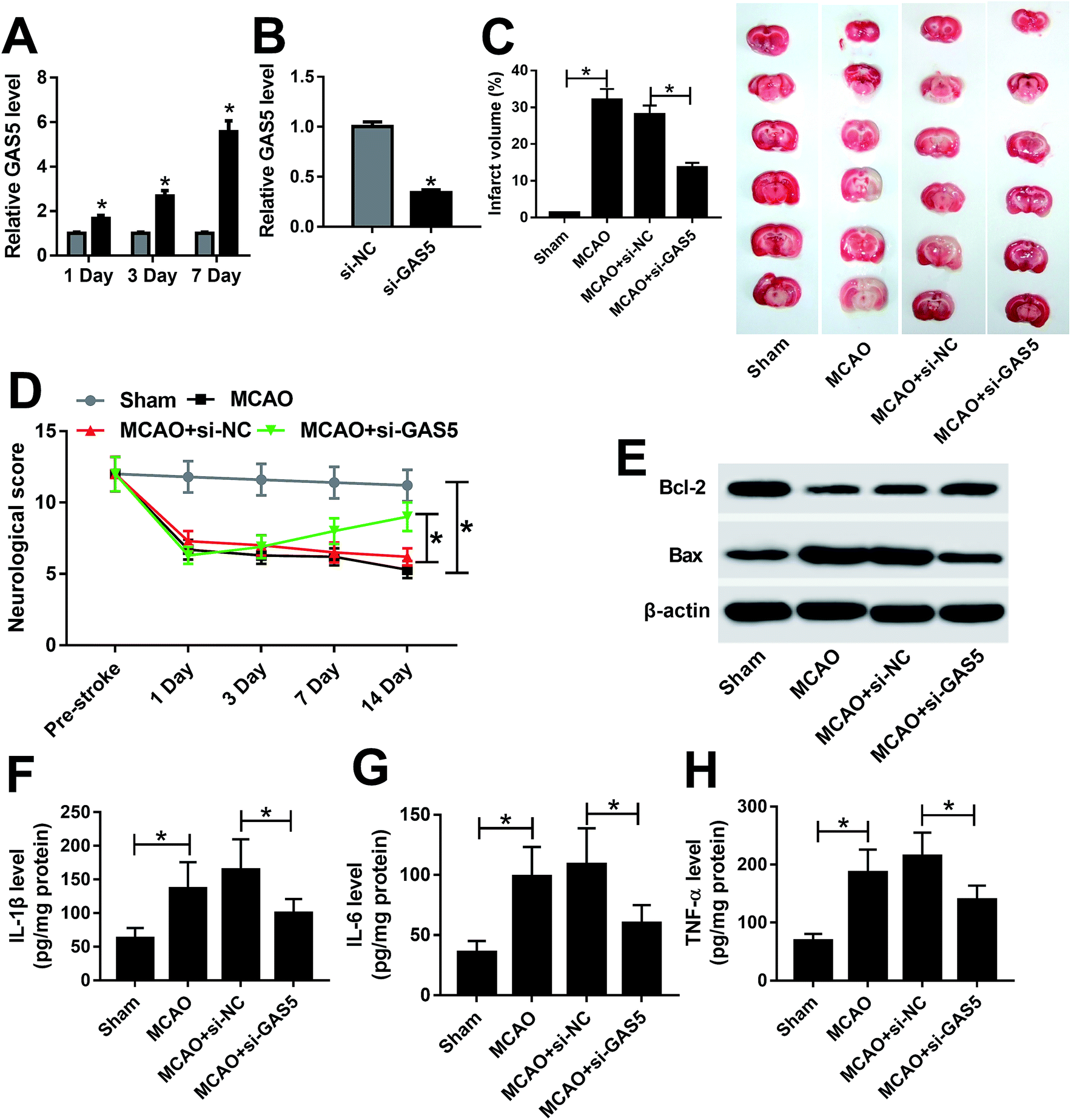 | ||
| Fig. 1 The knockdown of GAS5 inhibits ischemic stroke injury in MCAO model. (A) The expression of GAS5 was measured in brain tissues from MCAO or sham group by qRT-PCR. (B) The level of GAS5 was detected in brain tissues from si-GAS5 or si-NC group after MCAO treatment by qRT-PCR. (C and D) The neurological injury was investigated in each group by TTC staining and neurological score. (E) The expressions of Bcl-2 and Bax were measured in each group by western blot. (F–H) The expression levels of IL-1β, IL-6 and TNF-α protein were examined in each group by ELISA. *p < 0.05. | ||
miR-9 is bound to GAS5
To elucidate the potential mechanism where GAS5 regulated ischemic stroke, the promising bound miRNAs of GAS5 were probed by starBase, which provided the putative binding sites of GAS5 and miR-9 (Fig. 2A). Subsequently, the interaction was explored by luciferase activity and RNA pull-down analyses. Results showed that the up-regulation of miR-9 obviously suppressed luciferase activity in bEnd.3 cells transfected with GAS5-WT, while its efficacy was lost in response to GAS5-MUT group (Fig. 2B). Moreover, the enrichment of GAS5 was significantly elevated in bEnd.3 cells transfected with bio-miR-9-WT compared with that in bio-NC group, while bio-miR-9-MUT group showed little efficacy of GAS5 enrichment (Fig. 2C). Moreover, the expression of miR-9 was measured in bEnd.3 cells transfected with GAS5, pcDNA, si-GAS5 or si-NC. As shown in Fig. 2D, the overexpression of GAS5 effectively decreased miR-9 level, however, its knockdown greatly enhanced miR-9 expression in bEnd.3 cells. | ||
| Fig. 2 miR-9 is bound to GAS5. (A) The putative binding sites of miR-9 and GAS5 were predicted by starBase. (B) Luciferase activity was measured in bEnd.3 cells co-transfected with GAS5-WT or GAS5-MUT and miR-9 or miR-NC. (C) The enrichment of GAS5 was detected in bEnd.3 cells after RNA pull-down assay. (D) The abundance of miR-9 was examined in bEnd.3 cells transfected with pcDNA, GAS5, si-NC or si-GAS5 by qRT-PCR. *p < 0.05. | ||
GAS5 regulates apoptosis and inflammatory injury by sponging miR-9 in OGD-treated cells
After the treatment of OGD, the expression of GAS5 was abnormally enhanced and miR-9 level was decreased in a time dependent manner in bEnd.3 cells (Fig. 3A and B). To explore whether miR-9 was required for GAS5-mediated ischemic stroke injury, bEnd.3 cells were transfected with miR-NC, miR-9, miR-9 and pcDNA or GAS5, and then treated with OGD. The treatment with OGD resulted in a great increase of Bax, loss of Bcl-2 protein, apoptosis and inflammatory cytokines production in bEnd.3 cells, indicating the establishment of ischemic stroke model in vitro (Fig. 3C–G). Moreover, the overexpression of miR-9, especially, reduced Bax protein level but increased Bcl-2 protein expression, which was reversed by the addition of GAS5 in OGD-treated bEnd.3 cells (Fig. 3C). In addition, the up-regulation of miR-9 significantly inhibited the OGD-induced apoptosis of bEnd.3 cells, which was attenuated by the introduction of GAS5 (Fig. 3D). Moreover, the addition of GAS5 weakened the inhibitory effect of miR-9 on the secretion levels of IL-1β, IL-6 and TNF-α in OGD-treated bEnd.3 cells (Fig. 3E–G).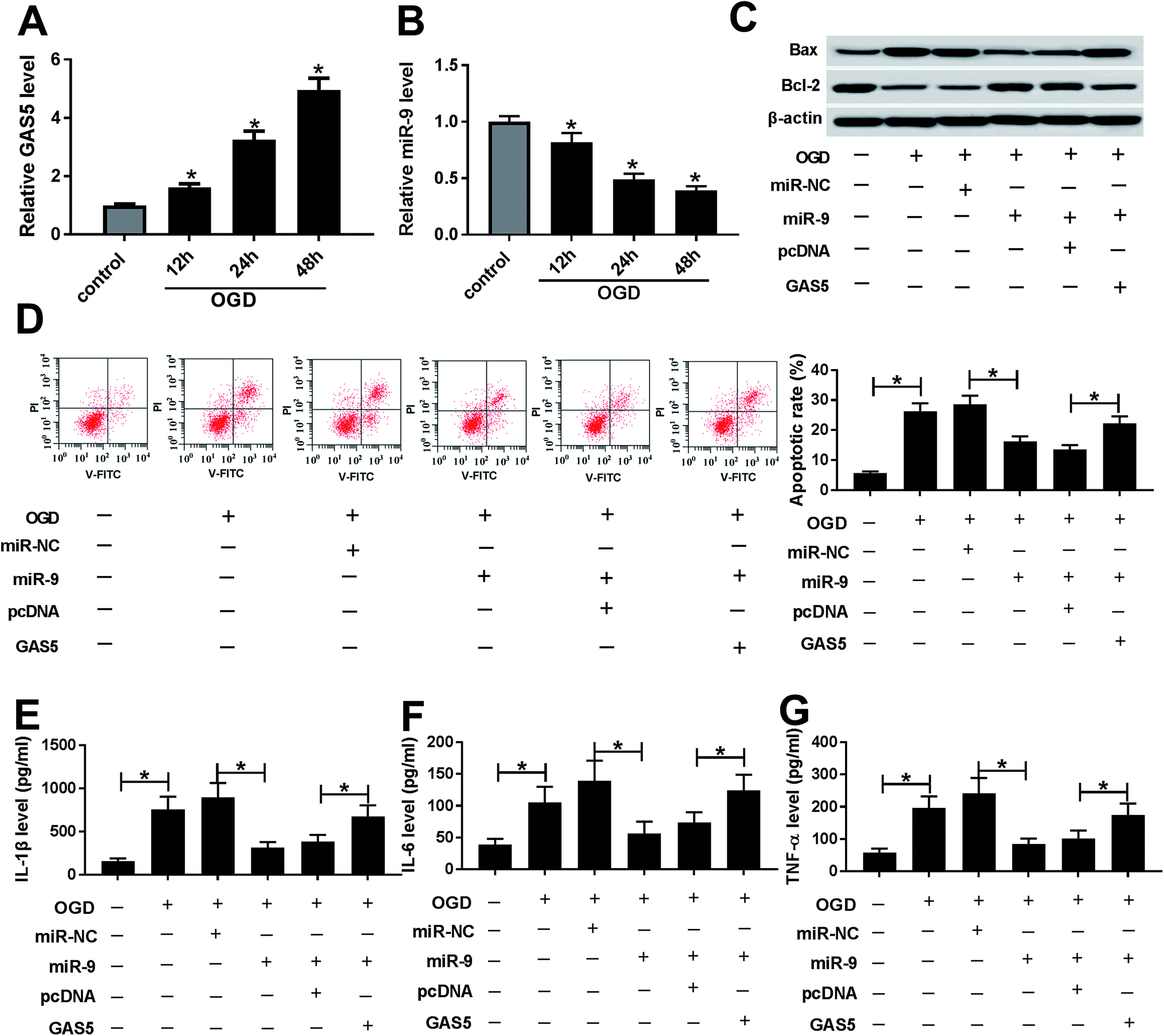 | ||
| Fig. 3 GAS5 regulates ischemic stroke injury by sponging miR-9 in OGD-treated cells. (A and B) The expressions of GAS5 and miR-9 were measured in bEnd.3 cells after OGD treatment by qRT-PCR. (C) The protein levels of Bcl-2 and Bax were measured in bEnd.3 cells transfected with miR-NC, miR-9, miR-9 and pcDNA or GAS5 after OGD treatment by western blot. (D) Cell apoptosis was detected in bEnd.3 cells transfected with miR-NC, miR-9, miR-9 and pcDNA or GAS5 after OGD treatment by flow cytometry. (E–G) The secretion levels of IL-1β, IL-6 and TNF-α were examined in bEnd.3 cells transfected with miR-NC, miR-9, miR-9 and pcDNA or GAS5 after OGD treatment by ELISA. *p < 0.05. | ||
FOXO3 is a target of miR-9
To explore the mechanism that allowed miR-9 participation in ischemic stroke, the potential targets of miR-9 were predicted by starBase. The putative binding sites of miR-9 and FOXO3 were shown in Fig. 4A. A luciferase activity assay was performed to confirm this prediction, where the overexpression of miR-9 led to a great loss of luciferase activity in bEnd.3 cells transfected with FOXO3-WT but it failed to affect the activity in FOXO3-MUT group (Fig. 4B). Moreover, the addition of miR-9 resulted in a higher enrichment of FOXO3 in bEnd.3 cells after Ago2 RIP, whereas IgG displayed a little efficacy of enrichment (Fig. 4C). Furthermore, the results of western blot showed that the accumulation of miR-9 significantly reduced the protein level of FOXO3, while the inhibition of miR-9 notably enhanced FOXO3 abundance in bEnd.3 cells (Fig. 4D).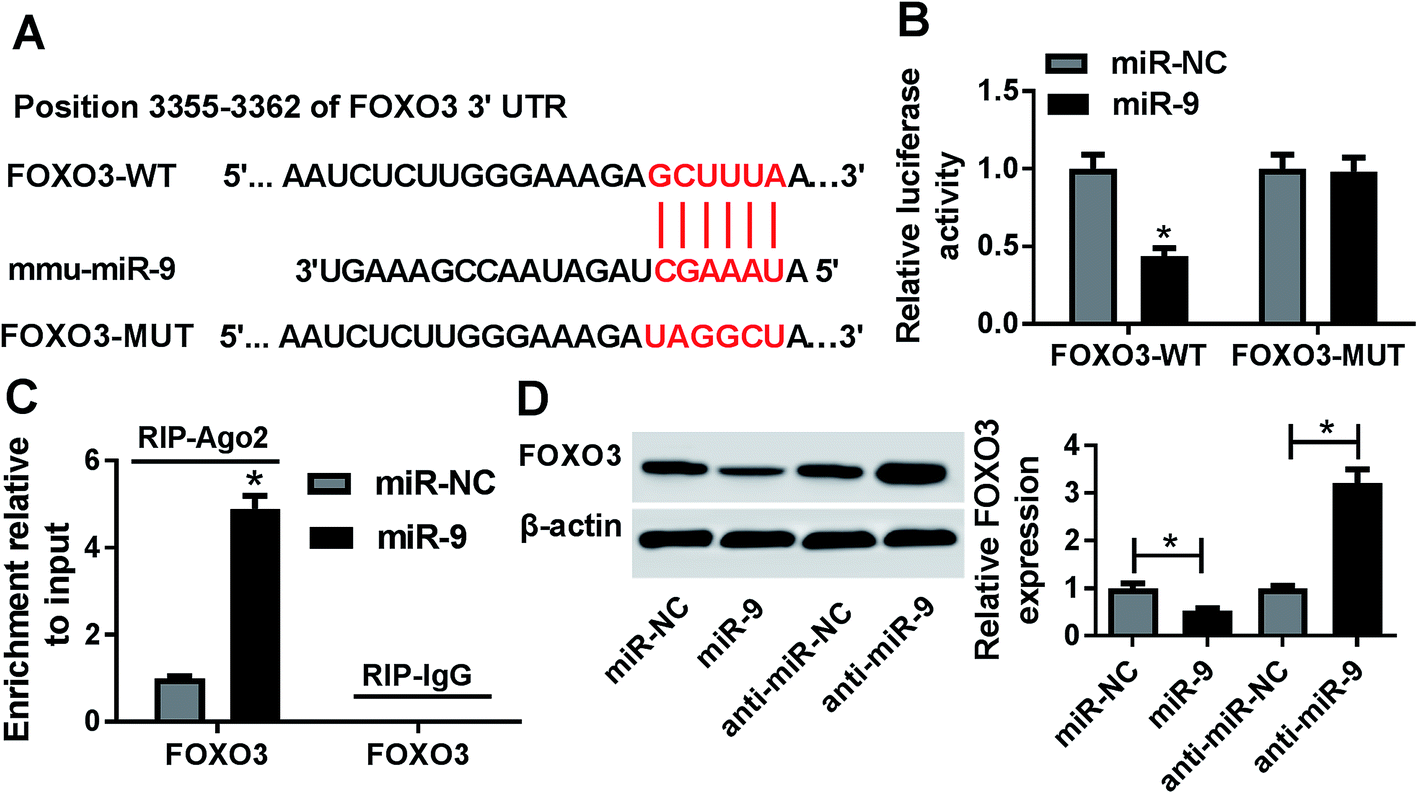 | ||
| Fig. 4 FOXO3 is a target of miR-9. (A) The potential binding sites of miR-9 and FOXO3 were provided by starBase. (B and C) The interaction between miR-9 and FOXO3 was explored in bEnd.3 cells by luciferase activity and RIP assays. (D) The expression of FOXO3 protein was measured in bEnd.3 cells transfected with miR-NC, miR-9, anti-miR-NC or anti-miR-9 by western blot. *p < 0.05. | ||
miR-9 suppresses apoptosis and inflammatory response by targeting FOXO3 in OGD-treated cells
After the treatment with OGD, the expression of FOXO3 mRNA was abnormally increased in a time dependent manner in bEnd.3 cells (Fig. 5A). To explore whether FOXO3 was involved in miR-9-mediated ischemic stroke injury, bEnd.3 cells were transfected with miR-NC, miR-9, miR-9 and pcDNA or FOXO3, and then treated with OGD. As displayed in Fig. 5B, the restoration of FOXO3, especially, reversed the miR-9-induced inhibition of Bax protein level and the up-regulation of Bcl-2 protein expression in OGD-treated bEnd.3 cells. Additionally, the up-regulation of FOXO3 significantly alleviated miR-9-mediated apoptosis suppression in OGD-treated bEnd.3 cells (Fig. 5C). Moreover, the data of ELISA revealed that the addition of FOXO3 obviously counteracted the inhibitory effect of miR-9 on the abundances of IL-1β, IL-6 and TNF-α in OGD-treated bEnd.3 cells (Fig. 5D–F). | ||
| Fig. 5 miR-9 suppresses ischemic stroke injury by targeting FOXO3 in OGD-treated cells. (A) The expression of FOXO3 mRNA was measured in OGD-treated bEnd.3 cells by qRT-PCR. (B) The protein levels of Bax and Bcl-2 were detected in bEnd.3 cells transfected with miR-NC, miR-9, miR-9 and pcDNA or FOXO3 after OGD treatment by western blot. (C) Cell apoptosis was detected in bEnd.3 cells transfected with miR-NC, miR-9, miR-9 and pcDNA or FOXO3 after OGD treatment by flow cytometry. (D–F) The secretion levels of IL-1β, IL-6 and TNF-α were examined in bEnd.3 cells transfected with miR-NC, miR-9, miR-9 and pcDNA or FOXO3 after OGD treatment by ELISA. *p < 0.05. | ||
GAS5 promotes FOXO3 expression by competitively sponging miR-9
To further elucidate the potential ceRNA network addressed by GAS5, luciferase activity was analyzed in bEnd.3 cells by transfecting FOXO3-WT. Results showed that the introduction of GAS5 reversed the miR-9-mediated inhibition of luciferase activity in the cells (Fig. 6A). Furthermore, western blot assay showed that the overexpression of GAS5 resulted in an obvious increase of FOXO3 protein level, which was attenuated by the addition of miR-9 (Fig. 6B). Meanwhile, the knockdown of GAS5 significantly reduced the expression of FOXO3 protein and it was restored by the depletion of miR-9 in bEnd.3 cells (Fig. 6C).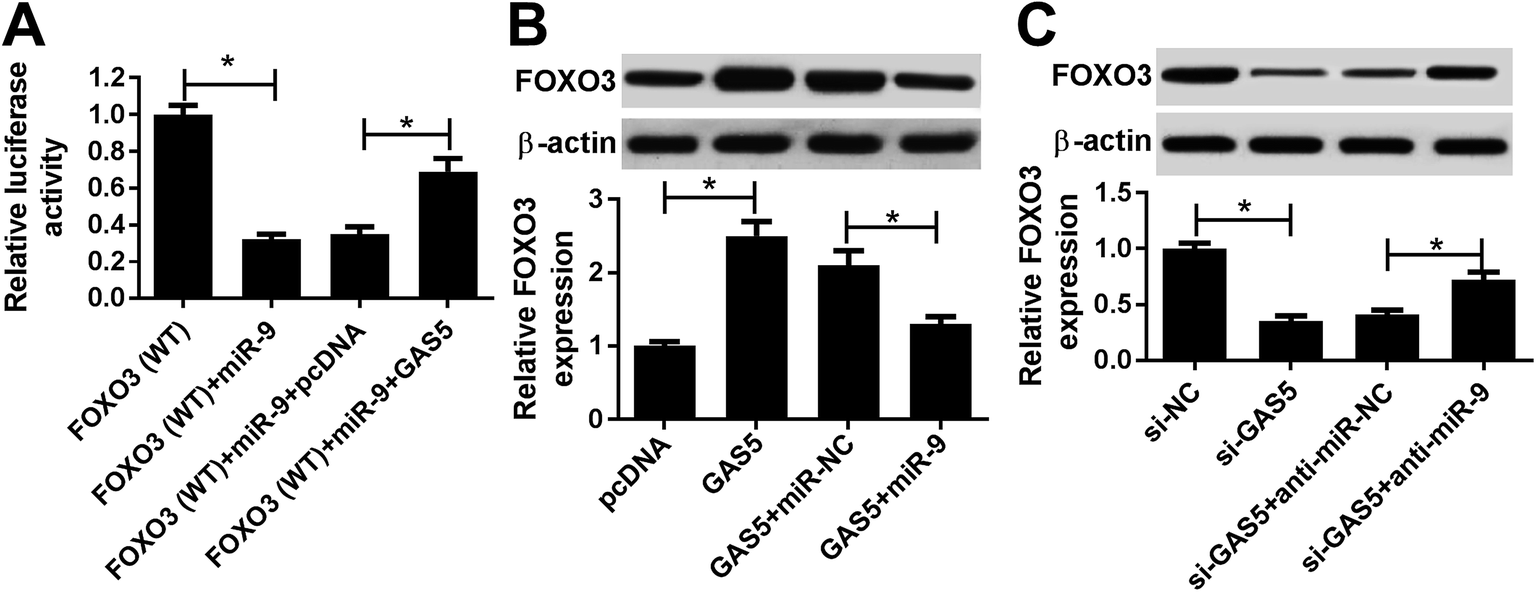 | ||
| Fig. 6 FOXO3 is regulated by GAS5 and miR-9. (A) Luciferase activity was measured in bEnd.3 cells transfected with FOXO3-WT, FOXO3-WT + miR-9, FOXO3 + miR-9 + pcDNA or FOXO3-WT + miR-9 + GAS5. (B) The expression of FOXO3 protein was detected in bEnd.3 cells transfected with pcDNA, GAS5, GAS5 and miR-NC or miR-9. (C) The protein level of FOXO3 was analyzed in bEnd.3 cells transfected with si-NC, si-GAS5, si-GAS5 and anti-miR-NC or anti-miR-9. *p < 0.05. | ||
Discussion
In recent years, great advances have been gained in the development of neuroprotective agents in cerebral ischemic stroke treatment, while the effective strategies are still limited.22 ncRNAs have been suggested to play important roles in stroke and neuroprotection.23 Emerging evidence suggested that lncRNAs played essential roles in cerebral ischemia through addressing inflammatory response, cell cycle and apoptosis, proved in the mice model.24 However, the role of GAS5 in cerebral ischemic stroke and underlying mechanism are largely unclear. In this study, we first demonstrated that GAS5 functioned as a ceRNA for miR-9 to regulate FOXO3, contributing to ischemic stroke injury in vivo and in vitro (Fig. 7). | ||
| Fig. 7 The schematic diagram of mechanism mediated by GAS5 in this study. This research established MCAO model in vivo and OGD-induced bEnd.3 cells in vitro as ischemic stroke injury model. GAS5 promoted ischemic stroke injury by acting as a ceRNA for miR-9 to derepress FOXO3. | ||
To explore the potential role of GAS5 in ischemic stroke, the MCAO model in vivo and OGD model in vitro were established. Former work suggested that GAS5 was highly expressed in MCAO and OGD model and its knockdown attenuated OGD-induced apoptosis.25 Similarly, we also revealed that GAS5 was elevated in both MCAO-treated mice and OGD-treated cells, contributing to the apoptosis in ischemic stroke. Moreover, this work revealed that the inhibition of GAS5 reduced the infarct and enhanced the neurological score, which suggested that the knockdown of GAS5 played a neuroprotective effect in ischemic stroke. However, there was no evidence in support of the interaction between GAS5 and inflammatory injury in ischemic stroke. Former work uncovered that GAS5 was associated with pro-inflammatory mediators in varying cells.26 Therefore, we hypothesized that GAS5 also facilitated the production of inflammatory response in ischemic stroke. Unsurprisingly, this study showed that GAS5 positively regulated pro-inflammatory cytokine expression in vivo and in vitro. These data indicated that GAS5 contributed to the inflammatory injury in ischemic stroke. However, the potential mechanism needs more investigation. This work displayed the importance of lncRNA-miRNA regulatory network in brain after ischemic stroke.27 For example, GAS5 has been reported to promote ischemic stroke injury through regulating Notch1 pathway by sponging miR-137.25 To figure out whether GAS5 acts as a miRNA sponge in ischemic stroke, the potential miRNA was explored and the results showed miR-9 containing the putative binding sites. Subsequently, the interaction between GAS5 and miR-9 was validated by the luciferase activity and RNA pull-down analyses.
Previous efforts indicated miR-9 as a neuroprotective target by regulating inflammation and survival in ischemic brain.28 Moreover, miR-9 was reported to be lowly expressed in MCAO and OGD models and its overexpression reduced OGD-induced apoptosis by regulating B cell lymphoma2-like protein 11 in ischemic stroke.29 Additionally, miR-9 could suppress the inflammatory response and infarct in acute ischemic stroke.14 Similarly, this study also revealed that miR-9 was reduced in OGD-treated bEnd.3 cells and its up-regulation alleviated OGD-induced apoptosis and inflammatory injury. Furthermore, the introduction of GAS5 reversed the miR-9-mediated inhibition of ischemic stroke injury, which suggested that GAS5 regulated the ischemic stroke by sponging miR-9. Functional miRNA is known to regulate related target expressions. Previous works displayed that miR-9 negatively regulated FOXO3 expression in different conditions.30,31 Hence, we assumed that the regulatory network of miR-9/FOXO3 might play a pivotal role in ischemic stroke process. The interaction between miR-9 and FOXO3 was identified by the luciferase activity and RIP assays.
Former work exhibited that FOXO3 reversed the miR-122-mediated inhibition of neuronal cell death in ischemic stroke.32 Herein, we showed that FOXO3 expression was elevated in OGD-treated model in vitro, which is also in agreement with previous study.17 Moreover, the restoration of FOXO3 abated the miR-9-mediated suppression of ischemic stroke injury, which indicated that miR-9 mediated ischemic stroke progression by targeting FOXO3. Additionally, FOXO3 expression was promoted by GAS5 and weakened by miR-9. This reflected that GAS5 might act as a ceRNA for miR-9 to derepress FOXO3 expression in bEnd.3 cells. However, there were some limitations in this study. First, the clinical effect of GAS5 on patients with ischemic stroke needs to be further investigated in future. Furthermore, the promising signaling pathway addressed by GAS5 should be explored for better understanding the mechanism in a further study.
In conclusion, the expressions of GAS5 were increased in cerebral ischemic stroke model in vivo and in vitro. The knockdown of GAS5 attenuated MCAO- or OGD-induced ischemic stroke injury, possibly by the ceRNA regulatory network of GAS5/miR-9/FOXO3, providing a new potential strategy for the treatment of cerebral ischemic stroke.
Conflicts of interest
The authors have no conflict of interest to declare.References
- Y. Li, W. Zhong, Z. Jiang and X. Tang, Brain Res. Bull., 2018, 144, 46–57 CrossRef PubMed.
- C. Zerna, G. Thomalla, B. C. V. Campbell, J. H. Rha and M. D. Hill, Lancet, 2018, 392, 1247–1256 CrossRef.
- I. Wimmer, T. Zrzavy and H. Lassmann, J. Neuroimmunol., 2018, 323, 10–18 CrossRef CAS PubMed.
- X. J. Zhang, M. Hamblin and K. J. Yin, Neuroscientist, 2018, 4, 1–5 Search PubMed.
- X. Guo, J. Yang, B. Liang, T. Shen, Y. Yan, S. Huang, J. Zhou, J. Huang, L. Gu and L. Su, Cell. Physiol. Biochem., 2018, 50, 2157–2175 CrossRef CAS PubMed.
- X. Yang and X. H. Zi, Brain Res., 2019, 1714, 174–181 CrossRef CAS PubMed.
- T. Zhang, H. Wang, Q. Li, J. Fu, J. Huang and Y. Zhao, Cell. Physiol. Biochem., 2018, 50, 2216–2228 CrossRef CAS PubMed.
- B. Liu, S. Q. Wu, J. Ma, S. Yan, Z. G. Xiao, L. H. Wan, F. Zhang, M. Y. Shang and A. W. Mao, Mol. Ther.--Nucleic Acids, 2018, 13, 472–482 CrossRef CAS PubMed.
- G. Wang, J. Sun, H. Zhao and H. Li, Med. Sci. Monit., 2018, 24, 7689–7696 CrossRef PubMed.
- Z. S. Zheng, S. Y. Liu, C. H. Wang and X. M. Han, J. Stroke Cerebrovasc. Dis., 2018, 27, 3535–3541 CrossRef PubMed.
- G. W. Li, K. C. Morris-Blanco, M. S. Lopez, T. Yang, H. P. Zhao, R. Vemuganti and Y. M. Luo, Prog. Neurobiol., 2018, 163–164, 59–78 CrossRef CAS PubMed.
- S. Khoshnam, W. Winlow and M. Farzaneh, J. Neuropathol. Exp. Neurol., 2017, 76, 548–561 CrossRef CAS PubMed.
- M. Coolen, S. Katz and L. Bally-Cuif, Front. Cell. Neurosci., 2013, 7, 220 Search PubMed.
- Y. P. Liu, J. J. Zhang, R. F. Han, H. X. Liu, D. Sun and X. Liu, J. Clin. Neurosci., 2015, 22, 291–295 CrossRef CAS PubMed.
- A. C. De Brachène and J. B. Demoulin, Cell. Mol. Life Sci., 2016, 73, 1159–1172 CrossRef PubMed.
- K. Maiese, Z. Z. Chong and Y. C. Shang, Curr. Neurovasc. Res., 2007, 4, 295–302 CrossRef CAS PubMed.
- J. Song, J. Park, Y. Oh and J. E. Lee, Oxid. Med. Cell. Longevity, 2015, 2015, 426069 Search PubMed.
- Y. S. Tian, D. Zhong, Q. Q. Liu, X. L. Zhao, H. X. Sun, J. Jin, H. N. Wang and G. Z. Li, J. Neurosurg., 2018, 1–12 Search PubMed.
- G. Metz and I. Whishaw, J. Neurosci. Methods, 2002, 115, 169–179 CrossRef PubMed.
- D. Guo, J. Ma, L. Yan, T. Li, Z. Li, X. Han and S. Shui, Cell. Physiol. Biochem., 2017, 43, 182–194 CrossRef CAS PubMed.
- K. J. Livak and T. D. Schmittgen, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- Y. Luo, H. Tang, H. Li, R. Zhao, Q. Huang and J. Liu, Eur. J. Med. Chem., 2018, 162, 132–146 CrossRef PubMed.
- J. Saugstad, Front. Neurol., 2015, 6, 50 Search PubMed.
- C. Y. Liu, J. Yang, C. C. Zhang, M. L. Liu, X. K. Geng, X. M. Ji, H. S. Du and H. Zhao, Neurosci. Lett., 2018, 665, 123–129 CrossRef CAS PubMed.
- F. H. Chen, L. X. Zhang, E. Wang, C. F. Zhang and X. T. Li, Biochem. Biophys. Res. Commun., 2018, 496, 184–190 CrossRef CAS PubMed.
- C. R. Keenan, M. J. Schuliga and A. G. Stewart, Can. J. Physiol. Pharmacol., 2015, 93, 203–206 CrossRef CAS PubMed.
- J. Liu, K. S. Zhang, B. Hu, S. G. Li, Q. Li, Y. P. Luo, Y. Wang and Z. F. Deng, BioMed Res. Int., 2018, 2018, 8354350 Search PubMed.
- W. A. Li, A. Efendizade and Y. Ding, Neurol. Res., 2017, 1–9 Search PubMed.
- N. Wei, L. Xiao, R. Xue, D. D. Zhang, J. Zhou, H. Y. Ren, S. Guo and J. J. Xu, Mol. Neurobiol., 2016, 53, 6809–6817 CrossRef CAS PubMed.
- Y. Zhang, L. Li, C. Yu, V. Senyuk, F. Li, J. G. Quigley, T. Y. Zhu and Z. J. Qian, Sci. Rep., 2018, 8, 6519 CrossRef PubMed.
- H. Y. Zhang, Z. Zhang, S. R. Wang, S. Q. Zhang and J. Y. Bi, Biomed. Pharmacother., 2018, 102, 626–632 CrossRef CAS PubMed.
- D. Guo, J. Ma, T. F. Li and L. Yan, Exp. Cell Res., 2018, 369, 34–42 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |