
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceY3Al5O12:Ce nanoparticles made by ionic-liquid-assisted particle formation and LiCl-matrix-treated crystallization
Hannah F. Gaiser,
Ana Kuzmanoski and
Claus Feldmann *
*
Institut für Anorganische Chemie, Karlsruhe Institute of Technology (KIT), Engesserstraße 15, D-76131 Karlsruhe, Germany. E-mail: claus.feldmann@kit.edu; Tel: +49-721-60842855
First published on 1st April 2019
Abstract
Y3Al5O12:Ce3+ (YAG:Ce) nanoparticles were prepared by a two-step approach including ionic-liquid-assisted particle formation and LiCl-matrix-treated crystallization. Subsequent to particle formation in [MeBu3N][N(SO2CF3)2] as the ionic liquid (MeBu3N: tributylmethylammonium; N(SO2CF3)2: bis(trifluoromethanesulfonyl)imide), the as-obtained amorphous precursor nanoparticles were crystallized in a LiCl matrix (600 °C, 1 h). The resulting YAG:Ce nanoparticles are well crystallized and exhibit a diameter of about 40 nm. They show bulk-like luminescence and a quantum yield of 51(±3)%. The selected Y![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al ratio and temperature profile turned out to be optimal for the synthesis strategy in terms of particle size and luminescence properties although minor amounts of CeO2 remained. The YAG:Ce nanoparticles can be easily redispersed in the liquid phase and embedded in polymers such as polyester. The course of the reaction and the properties of the nanoparticles are characterized by electron microscopy, dynamic light scattering, infrared spectroscopy, X-ray powder diffraction, and fluorescence spectroscopy.
:Al ratio and temperature profile turned out to be optimal for the synthesis strategy in terms of particle size and luminescence properties although minor amounts of CeO2 remained. The YAG:Ce nanoparticles can be easily redispersed in the liquid phase and embedded in polymers such as polyester. The course of the reaction and the properties of the nanoparticles are characterized by electron microscopy, dynamic light scattering, infrared spectroscopy, X-ray powder diffraction, and fluorescence spectroscopy.
Introduction
Cerium-doped yttrium aluminium garnet (Y3Al5O12:Ce3+, YAG:Ce) is one of the most efficient luminescent materials and is applied, for instance, in phosphor-converted white-light emitting diodes (pc-WLEDs).1 Herein, YAG:Ce converts part of the blue light emitted by the (Ga,In)N-semiconductor chip into yellow emission, which – upon additive colour mixing – results in white light emission. Among a great number of known luminescent materials, YAG:Ce still represents one of the most efficient inorganic materials with a quantum yield of 80–90%.2 Besides efficient luminescence processes, the excellent performance of bulk-YAG:Ce originates from the high lattice energy, the isotropy of cubic lattice, a low defect concentration and low-energy lattice vibrations, which, together, result in minimal non-emissive loss processes.1–3The formation of YAG:Ce nanoparticles – in contrast to the bulk-phase – is a challenge. On the one hand, crystallization and removal of water and hydroxides at low temperatures (<500 °C) are difficult and often lead to highly defective YAG:Ce with quantum yields ≤ 20%. Post-sintering of the as-prepared YAG:Ce nanoparticles, on the other hand, foils the efforts of low-temperature particle formation and causes uncontrolled particle growth and agglomeration. Dispersible, nanosized (≤100 nm) and efficient YAG:Ce nanoparticles, however, are prerequisite to facile liquid-phase deposition, printing of thin films, or an incorporation into polymers.1
Nano- and micron-sized YAG:Ce was typically prepared at high temperature (800–1500 °C) via solid-state synthesis including molten-salts,4 Pechini-type, flame-, plasma- or microwave-driven combustions,5 spray pyrolysis or sintering in silica matrices.6 Liquid-phase approaches comprise sol–gel,7 polyol,8 emulsion synthesis,9 or co-precipitation processes.10 The as-prepared, defective nanoparticles most often require post-synthesis calcination (1000–1200 °C) to adjust efficient luminescence processes. The highest quantum yields of dispersible YAG:Ce nanoparticles (∅ ≤ 100 nm) were yet reported with 50–60%.6b,9a,11 These nanoparticles were either obtained by high-temperature sintering (≥1000 °C),9a calcination in difficult-to-remove SiO2 matrices,6 or by solvothermal synthesis at elevated pressure (70 bar),11 which typically results in significant agglomeration and comparably broad size distributions.
Aiming at crystalline and dispersible YAG:Ce nanoparticles, we here suggest a novel two-step approach (Fig. 1). In a first step, amorphous precursor nanoparticles were obtained with optimal conditions of particle formation in ionic liquids (ILs). As a second step, the as-prepared, amorphous precursor nanoparticles were crystallized in a LiCl matrix at 600 °C. The resulting crystalline YAG:Ce nanoparticles were examined in detail in regard of particle size, dispersibility, chemical composition and luminescence properties.
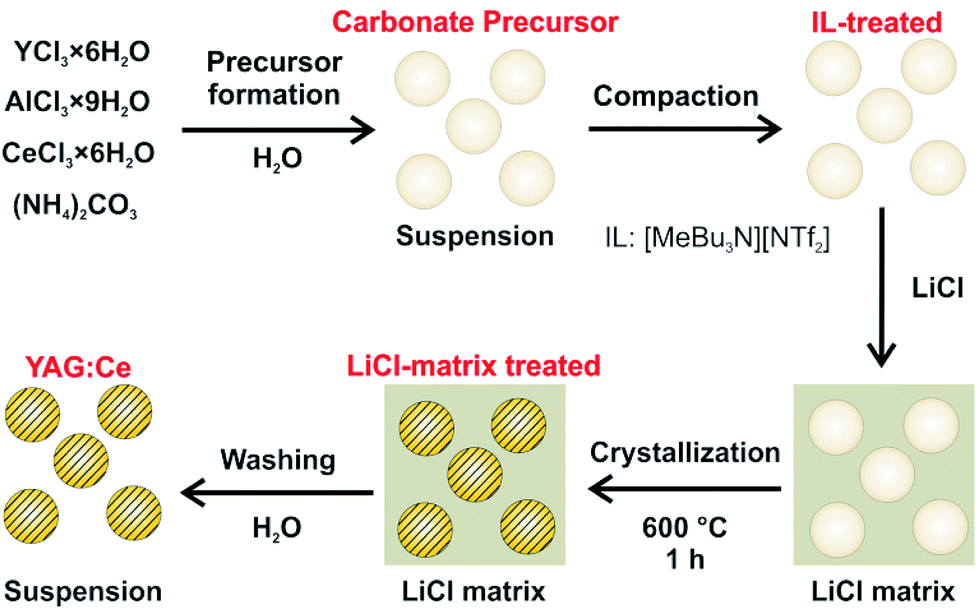 | ||
| Fig. 1 Scheme illustrating the two-step synthesis of dispersible YAG:Ce nanoparticles, including IL-assisted particle formation and LiCl-matrix-treated crystallization. | ||
Experimental
Synthesis
Analytical methods
Results and discussion
Ionic-liquid assisted particle formation
The here proposed two-step approach intends to separate particle formation and crystallization in order to optimize both independently (Fig. 1). Controlling particle size and particle agglomeration is of course essential in both steps of the reaction. In this regard, Y(NO3)3·6H2O, Al(NO3)3·9H2O, Ce(NO3)3·6H2O and (NH4)2CO3 were used as the starting materials and mixed in water to obtain a colourless, fluffy carbonate precursor. Fourier-transform infrared spectroscopy (FT-IR), as expected, indicates the characteristic carbonate-related vibrations (ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O): 1600–1400, ν(C–O): 1100–1000 cm−1, Fig. 2). Due to the aqueous treatment, moreover, a broad vibration related to water is visible (ν(O–H): 3700–3000). Energy-dispersive X-ray (EDX) analysis, finally, already indicates a Ce:Y:Al ratio of 0.04:0.75:1 with excessive Y in relation to the intended ratio in Y3Al5O12:Ce (5 mol%) of 0.03:0.57:1, which was naturally also the ratio introduced with the starting materials.
O): 1600–1400, ν(C–O): 1100–1000 cm−1, Fig. 2). Due to the aqueous treatment, moreover, a broad vibration related to water is visible (ν(O–H): 3700–3000). Energy-dispersive X-ray (EDX) analysis, finally, already indicates a Ce:Y:Al ratio of 0.04:0.75:1 with excessive Y in relation to the intended ratio in Y3Al5O12:Ce (5 mol%) of 0.03:0.57:1, which was naturally also the ratio introduced with the starting materials.
 | ||
| Fig. 2 Course of the reaction analysed by FT-IR spectroscopy with spectra of the carbonate precursor nanoparticles, IL-treated precursor nanoparticles, and LiCl-matrix-treated YAG:Ce nanoparticles. | ||
The amorphous and colourless carbonate precursor, thereafter, was dispersed in [MeBu3N][N(SO2CF3)2] as the IL and rapidly heated in a microwave oven to 200 °C (Fig. 1). After ethanol-driven removal of the IL, FT-IR still shows the presence of the carbonate precursor (ν(CO): 1600–1400, ν(C–O): 1200–1000 cm−1) and water (ν(O–H): 3700–3000) in addition to surface-adhered IL (fingerprint: 1400–1100, Fig. 2). The latter finding is validated when comparing to the FT-IR spectra of the pure IL. Moreover, it is to be noted that the carbonate precursor is still amorphous and colourless subsequent to the IL treatment and does not show any specific Bragg peak (Fig. 3a and b). EDX analysis confirms the Y-rich composition and a Ce:Y:Al ratio of 0.04:0.75:1 as already determined for the carbonate precursor (0.04:0.71:1).
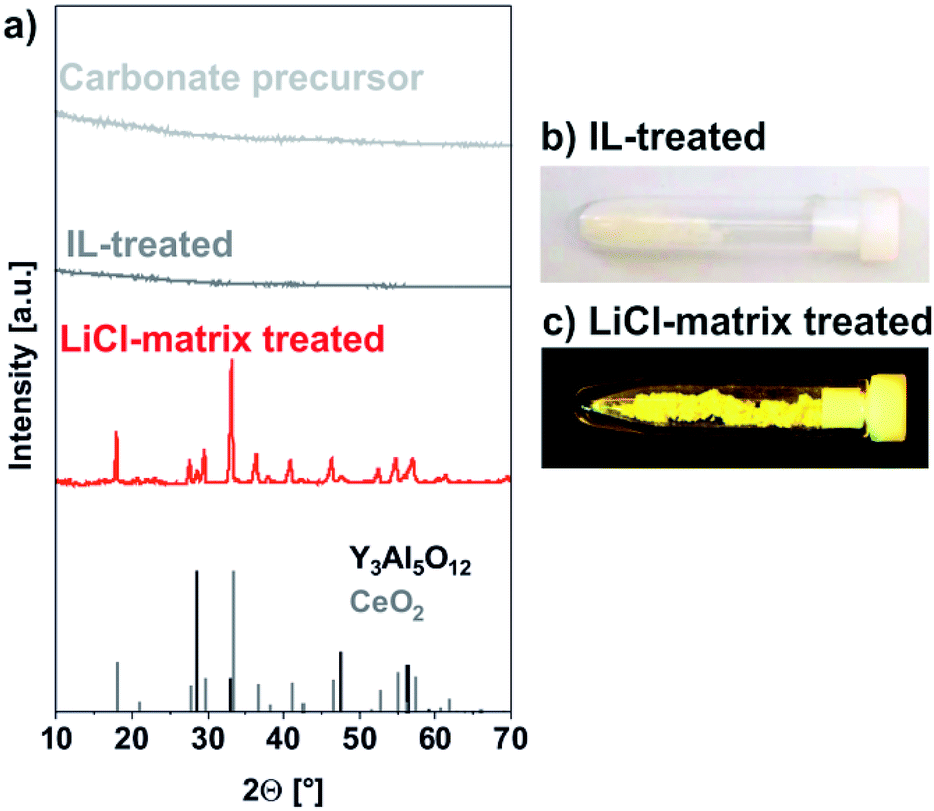 | ||
| Fig. 3 Course of the reaction analysed by X-ray powder diffraction with (a) XRD patterns of carbonate precursor nanoparticles, IL-treated precursor nanoparticles, LiCl-matrix-treated YAG:Ce nanoparticles (references: Y3Al5O12 – ICDD no. 088-2048/black line pattern; CeO2 – ICDD no. 081-0792/grey line pattern); (b and c) photos of IL-treated and LiCl-matrix treated powder samples in daylight. | ||
The IL treatment results in a compaction of the fluffy carbonate precursor and the formation of well-defined nanoparticles. Such IL-assisted treatment was already reported as promising for particle compaction and particle shaping,14 which can be related to the high boiling point and the weakly coordinating properties of ILs.15 In this regard, scanning electron microscopy (SEM) shows uniform, non-agglomerated nanoparticles with a mean diameter of 33 ± 6 nm (Fig. 5a). This diameter matches well with dynamic light scattering (DLS) indicating a mean hydrodynamic diameter of 39 ± 7 nm in diethylene glycol (DEG) (Fig. 5a). DEG was selected as dispersant due to its stabilizing properties in order to avoid agglomeration during the redispersion process.16 Moreover, such dispersions in polyols are relevant as they offer the opportunity of direct polymerization and nanoparticle incorporation in polyesters.17
 | ||
| Fig. 4 Enlarged view of the XRD pattern to indicate minor amount of CeO2 (references: Y3Al5O12 – ICDD no. 088-2048 (black line pattern); CeO2 – ICDD no. 081-0792 (grey line pattern)). | ||
 | ||
| Fig. 5 Size and size distribution of (a) the carbonate precursor nanoparticles after IL-treatment (200 °C, 1 h), (b) YAG:Ce nanoparticles after LiCl-matrix treatment (600 °C, 1 h) with: size distribution according to SEM (statistical evaluation of >100 nanoparticles), size distribution according to DLS (suspensions in DEG), and SEM images. | ||
LiCl-matrix-treated crystallization
Subsequent to the IL-assisted particle formation, the carbonate precursor nanoparticles were mixed and pesteled with LiCl in a ratio of 1:10. As a result, the nanoparticles were separated from each other and embedded in a LiCl matrix (Fig. 1). This solid mixture was then heated to 600 °C in a tube oven for 60 minutes in order to decompose the carbonate precursor and to crystallize the YAG phase. Moreover, the separation of the nanoparticles in the LiCl matrix allows avoiding particle growth and particle agglomeration. Subsequent to this LiCl-matrix treatment, LiCl was removed by simple rinsing with demineralized water which was performed twice. After each washing step, the nanoparticles were collected by centrifugation. The characteristic intense yellow colour of obtained powder samples already indicates successful crystallization of YAG:Ce (Fig. 3c).
This LiCl-matrix treatment indeed results in an effective decarboxylation of the precursor nanoparticles as indicated by significantly reduced intensities of the ν(CO), and ν(C–O) vibrations (Fig. 2). In fact, only lattice vibrations (ν(Al–O): 900–500 cm−1) and vibrations of surface-adhered water stemming from the washing procedure remain (ν(O–H): 3700–3100 cm−1; δ(O–H): 1620, 1450 cm−1).
In contrast to the amorphous carbonate precursor obtained after IL-treatment, the LiCl-matrix-treated nanoparticles turned out as highly crystalline (Fig. 3a). Thus, the observed Bragg peaks are well in accordance with the garnet phase. Just a weak Bragg peak at 28.5° of 2-theta points to the presence of CeO2 (Fig. 4). Here, it must be noticed that all efforts to adjust the ratio of the starting materials either resulted in low amounts of either CeO2 (Y:Al ≤ 0.6:1) or YAlO3:Ce (Y:Al > 0.6:1) as a side phase. Most interestingly, no Bragg peaks of LiCl were observed, indicating that the washing procedure guarantees complete removal of all LiCl (Fig. 3a and 4). Even flame colouring with the as-obtained YAG:Ce nanoparticles did not result in any characteristic red and orange emission lines of lithium.18 Finally, a slight shift to smaller two-theta values in comparison to bulk-YAG points to the incorporation of the larger Ce3+ (103 pm) on lattice sites of Y3+ (89 pm), resulting in increased lattice distance (Fig. 4).
The particle size of the LiCl-matrix-treated YAG:Ce nanoparticles was again verified by SEM (Fig. 5b). Accordingly, a mean diameter of 41 ± 9 nm was observed, which is only slightly larger as compared to the IL-treated carbonate precursor nanoparticles (33 ± 6 nm). Although crystallized at 600 °C in the LiCl-matrix, the YAG:Ce nanoparticles are still spherical and non-agglomerated, which indicates the feasibility of the LiCl-matrix sintering. After redispersion in DEG, the YAG:Ce nanoparticles exhibit a larger hydrodynamic diameter of 93 ± 15 nm (Fig. 5b) as compared to the IL-treated precursor nanoparticles (39 ± 7 nm), which points to at least some agglomeration. Nevertheless, the YAG:Ce nanoparticles turned out as redispersible and still have a mean diameter < 100 nm.
In addition to the particle size, the mean crystallite diameter was deduced from the width of the Bragg peaks via the Scherer formalism and resulted in 25–30 nm. Within the significance of the measurement, this value is in agreement with the diameter stemming from SEM and points to the single-crystalline nature of the YAG:Ce nanoparticles. Finally, EDX analysis confirms a Ce:Y:Al ratio of 0.04:0.75:1 with certain excess of Y in relation to the intended ratio in Y3Al5O12:Ce (5 mol%) with 0.03:0.57:1. Whereas this ratio is constant for the whole sequence of synthesis, it is different from the ratio introduced with the starting materials ((Y + Ce):Al = 0.6:1). The lack of Al3+ can be ascribed to its small size and its higher solubility in the liquid phase as compared to Y3+. Any decrease of the Y3+ content (i.e., a ratio Y:Al < 0.6:1), however, resulted in significant amounts of CeO2 as a side phase. Such behaviour is often observed for low-temperature synthesis, where the effect of the different radii of the cations is more significant than for conventional solid-state synthesis at high temperatures (800–1500 °C).4–10 Taken together, the here applied Y:Al ratio and temperature treatment turned out as optimum for the presented synthesis strategy in terms of particle size and luminescence properties.
Luminescence characterization
The luminescence properties of the LiCl-matrix-treated YAG:Ce nanoparticles were finally examined by fluorescence spectroscopy. Excitation spectra show weak absorption due to 4f → 5d transition on Ce3+ (330–360 nm, maximum at 342 nm) and strong absorption at 410–520 nm, peaking at 454 nm, related to O2− → Ce3+ charge-transfer transition (Fig. 6a). Emission spectra indicate intense yellow light emission by 5d → 4f transition on Ce3+ at 500–680 nm with its maximum at 567 nm (Fig. 6b).1,3 With these features, excitation and emission of the LiCl-matrix crystallized YAG:Ce nanoparticles are similar to bulk-YAG:Ce.1,2 Upon excitation with a blue-light LED lamp, the bright yellow YAG:Ce powder (Fig. 6c) shows intense white light generation due to additive colour mixing of yellow emission of YAG:Ce and reflected blue light of the LED lamp (Fig. 6d). Furthermore, the absolute quantum yield of LiCl-matrix-treated YAG:Ce powder samples was determined following the procedure described by Friend13 and results in a value of 51(±3)% upon excitation at 454 nm. This value is among the highest quantum yields reported in the literature for YAG:Ce nanoparticles by now.6b,9a,11 | ||
| Fig. 6 Excitation (a) and emission (b) spectra of the as-prepared YAG:Ce nanoparticles with powder samples after LiCl-matrix treatment in daylight (c) and with white-light emission under 465 nm excitation of a blue-light LED (d). | ||
After the examination of the luminescence properties of YAG:Ce powder samples, embedding the nanoparticles in polymers was tested as a proof-of-the-concept. In this regard, the LiCl-matrix-treated YAG:Ce nanoparticles were again dispersed in a polyol(ethylene glycol). Thereafter, citric acid was added to the suspension, and the polymerization was initiated by heating to 150 °C instantaneously in the liquid phase. The resulting polyester phase contains the YAG:Ce nanoparticles with only 2 mg g−1, resulting in a highly transparent, slightly yellowish polymer (Fig. 7). Upon excitation with a mercury discharge lamp at 366 nm or a UV-LED lamp at 405 nm, the YAG:Ce-containing polyester again shows intense white light generation due to additive colour mixing of yellow emission of YAG:Ce and reflected blue light of the respective light source (Fig. 7). The pure polyester without any nanoparticles as a reference, in contrast, did not show any emission at all (Fig. 7). In sum, the as-prepared YAG:Ce nanoparticles can be used to obtain luminescent and transparent polymers.
 | ||
| Fig. 7 YAG:Ce nanoparticles after embedding in polyester (2 mg YAG:Ce per 1 g of polyester) in daylight and with 366 nm excitation (Hg discharge lamp) or 405 nm excitation (UV-LED). Pure polyester (without nanoparticles) shown as reference. | ||
Conclusions
In summary, Y3Al5O12:Ce3+ (YAG:Ce) nanoparticles with a diameter of 30–50 nm were prepared by a two-step approach. This includes microwave-assisted compaction of a pre-formed carbonate precursor in ionic liquids (at 200 °C), followed by LiCl-matrix-treated crystallization (at 600 °C). Mixing of the IL-treated carbonate precursor with LiCl (1:10) and heating in the resulting LiCl matrix, on the one hand, allows decomposing the carbonate precursor and crystallizing the YAG phase, and on the other hand, guarantees the separation of the nanoparticles in the LiCl matrix to avoid particle growth and particle agglomeration. Subsequent to the crystallization, the LiCl matrix can be easily removed by rinsing with water, resulting in colloidally stable, dispersible YAG:Ce suspensions.
The YAG:Ce nanoparticles show intense luminescence with a quantum yield of 51(±3)%, which is among the highest values obtained for YAG:Ce nanoparticles. Due to the good dispersibility, the obtained YAG:Ce nanoparticles can be embedded in polymers by polymerization instantaneously in the liquid phase. As a proof-of-the-concept, this is shown for polyester, which shows bright white light emission with only 2 mg YAG:Ce nanoparticles per 1 g of polyester upon excitation with 366 to 465 nm light sources (mercury discharge lamp, UV-LED, blue-light LED) due to additive colour mixing of yellow emission of YAG:Ce and reflected blue light of the respective light source. The presented two-step approach can be optionally transferred to other, high-lattice-energy luminescent materials, such as Y2O3:Eu, YVO4:Eu, BaMgAl10O17:Eu, BaMgAl10O17:Eu,Mn or Zn2SiO4:Mn.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Deutsche Forschungs-gemeinschaft (DFG) for funding within the priority program SPP1708 “Synthesis near Room Temperature”.Notes and references
- S. Ye, F. Xiao, Y. X. Pan, Y. Y. Ma and Q. Y. Zhang, Mater. Sci. Eng., R, 2010, 71, 1 CrossRef.
- V. Bachmann, C. Ronda and A. Meijerink, Chem. Mater., 2009, 21, 2077 CrossRef CAS.
- S. Shionoya, W. M. Yen and H. Yamamoto, Phosphor Handbook, CRC Press, Boca Raton, 2006 Search PubMed.
- (a) H. K. Yang, H. M. Noh and J. H. Jeong, Solid State Sci., 2014, 27, 43 CrossRef CAS; (b) K. Zhang, W. B. Hu, Y. T. Wu and H. Z. Liu, Inorg. Mater., 2008, 44, 1218 CrossRef CAS; (c) M. Li, D. Zhou, C. P. Li and Z. Zhao, Mater. Sci. Semicond. Process., 2016, 44, 101 CrossRef CAS; (d) L. Gan, Z. Y. Mao, F. F. Xu, Y. C. Zhu and X. J. Liu, J. Am. Ceram. Soc., 2012, 95, 49 CrossRef.
- (a) M. Borlaf, R. Kubrin, V. Aseev, A. Y. Petrov, N. Nikonorov and T. Graule, J. Am. Chem. Soc., 2017, 100, 3784 CAS; (b) A. Boukerika, L. Guerbous and N. Brihi, J. Alloys Compd., 2014, 614, 383 CrossRef CAS; (c) C. Shen, C. Zhong and J. Ming, J. Exp. Nanosci., 2013, 8, 54 CrossRef CAS; (d) S. Mukherjee, V. Sudarsan, R. K. Vatsa and A. K. Tyagi, J. Lumin., 2008, 129, 69 CrossRef; (e) H. M. H. Fadlalla, C. C. Tang, S. Y. Wei and X. X. Ding, J. Lumin., 2008, 128, 1655 CrossRef CAS; (f) B. Masenelli, O. Mollet, O. Boisron, B. Canut, G. Ledoux, J. M. Bluet, P. Melinon, C. Dujardin and S. Huant, Nanotechnology, 2013, 24, 165703 CrossRef CAS PubMed; (g) A. Bhaskar, H. Y. Chang, T. H. Chang and S. Y. Cheng, Mater. Lett., 2012, 78, 124 CrossRef CAS.
- (a) L. Mancic, K. Marinkovic, B. A. Marinkovic, M. Dramicanin and O. Milosevic, J. Eur. Ceram. Soc., 2010, 30, 577 CrossRef CAS; (b) A. Revaux, G. Dantelle, N. George, R. S. Seshadri, T. Gacoin and J. P. Boilot, Nanoscale, 2011, 3, 2015 RSC.
- (a) G. Dantelle, D. Testemale, E. Homeyer, A. Cantarano, S. Kodjikian, C. Dujardin, J.-L. Hazemann and A. Ibanez, RSC Adv., 2018, 8, 26857 RSC; (b) X. He, X. Liu, R. Li, B. Yang, K. Yu, M. Zeng and R. Yu, Sci. Rep., 2016, 6, 22238 CrossRef CAS PubMed; (c) Y. H. Song, G. S. Han, E. K. Ji, M. J. Lee, Y. L. Song, D. S. Kong, M. K. Jung, B. W. Jeong, H. S. Jung and S. H. Yoon, J. Mater. Chem. C, 2015, 3, 6148 RSC; (d) A. Boukerika, L. Guerbous and N. Brihi, J. Alloys Compd., 2014, 614, 383 CrossRef CAS; (e) A. Aboulaich, J. Deschamps, R. Deloncle, A. Potdevin, B. Devouard, G. Chadeyron and R. Mahiou, New J. Chem., 2012, 36, 2493 RSC; (f) X. Zhou, K. Zhou, Y. Li, Z. Wang and Q. Feng, J. Lumin., 2012, 132, 3004 CrossRef CAS; (g) H. J. Byun, W. S. Song, Y. S. Kim and H. Yang, J. Phys. D, 2010, 43, 195401 CrossRef.
- (a) M. Odziomek, F. Chaput, F. Lerouge, M. Sitarz and S. Parola, J. Mater. Chem. C, 2017, 5, 12561 RSC; (b) H. Yang and Y. S. Kim, J. Lumin., 2008, 128, 1570 CrossRef CAS.
- (a) L. Song, Y. Dong, Q. Shao and J. Jiang, J. Mater. Sci., 2018, 53, 15196 CrossRef CAS; (b) J. D. Furman, G. Gundiah, K. Page, N. Pizarro and A. K. Cheetham, Chem. Phys. Lett., 2008, 465, 67 CrossRef CAS.
- (a) W. Shi, A. Feng, H. Tang, Z. Ding, Y. Ma, M. Wu and G. Li, Opt. Mater., 2013, 35, 609 CrossRef CAS; (b) K. Zhang, W. Hu, Y. Wu and H. Liu, Ceram. Int., 2009, 35, 719 CrossRef CAS; (c) V. Pankratov, L. Grigorjeva, D. Millers and T. Chudoba, Radiat. Meas., 2007, 42, 679 CrossRef CAS.
- A. Revaux, G. Dantelle, D. Decanini, A. M. Haghri-Gosnet, T. Gacoin and J. P. Boilot, Opt. Mater., 2011, 33, 1124 CrossRef CAS.
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS PubMed.
- J. C. de Mello, H. F. Wittmann and R. H. Friend, Adv. Mater., 1997, 9, 230 CrossRef CAS.
- (a) M. Baghbanzadeh, L. Carbone, P. D. Cozzoli and C. O. Kappe, Angew. Chem., Int. Ed., 2011, 50, 11312 CrossRef CAS PubMed; (b) M. Antonietti, D. Kuang, B. Smarsly and Y. Zhou, Angew. Chem., Int. Ed., 2004, 43, 4989 CrossRef PubMed.
- (a) G. Bühler and C. Feldmann, Angew. Chem., Int. Ed., 2006, 45, 4864 CrossRef PubMed; (b) P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Weinheim, 2008 Search PubMed.
- H. Dong, Y. C. Chen and C. Feldmann, Green Chem., 2015, 17, 4107 RSC.
- M. S. Ata, Y. Liu and I. Zhitomirsky, RSC Adv., 2014, 4, 22716 RSC.
- D. C. Harris and C. A. Lucy, Quantitative Chemical Analysis, Freeman Publishers, New York, 9th edn, 2016, p. 529 Search PubMed.
| This journal is © The Royal Society of Chemistry 2019 |